
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-volatile iron carbonyls as versatile precursors for the synthesis of iron-containing nanoparticles†
John
Watt
a,
Grant C.
Bleier
a,
Mariah J.
Austin
a,
Sergei A.
Ivanov
b and
Dale L.
Huber
*a
aSandia National Laboratories, Albuquerque, NM, USA 87185. E-mail: Dale.Huber@sandia.gov
bLos Alamos National Laboratory, Los Alamos, NM, USA 87545
First published on 17th March 2017
Abstract
The most commonly used method for the formation of well-defined iron and iron-containing heterometallic nanoparticles is the thermal decomposition of iron pentacarbonyl (Fe(CO)5). However, iron pentacarbonyl is highly toxic and volatile, which introduces safety concerns and drastically diminishes control over the reaction stoichiometry. Here we alleviate these issues by beginning with an easy-to-handle solid, triiron dodecacarbonyl (Fe3(CO)12). The issue of poor solubility of this cluster is addressed by its reaction with amine, which renders the cluster fully soluble in common high boiling point solvents. This reaction generates non-volatile anionic iron carbonyl species in solution which are subsequently used as the nanoparticle precursor. We demonstrate that the thermolysis of this novel precursor solution yields well-defined Fe, Fe1−xCox, and Fe1−xPtx nanoparticles. In addition, the same approach overcomes the solubility issue of another poorly soluble iron carbonyl compound, diiron nonacarbonyl (Fe2(CO)9). By using these precursors in an array of nanoparticle-forming reactions, we demonstrate a convenient replacement for the commonly used Fe(CO)5, producing particles of similar quality, but without the drawbacks of the precursor volatility and high toxicity.
Magnetic nanoparticles have enjoyed a large amount of research interest due to their great potential in a range of applications including waste remediation, data storage, and magnetic resonance imaging.1 Zero-valent iron nanoparticles are particularly attractive due to the high magnetic moment (Ms = 222 Am2 kg−1 @ 273 K) and high susceptibility of bulk iron. Due to low magnetocrystalline anisotropy, iron nanoparticles display superparamagnetic behavior at much larger sizes than other magnetic materials, which makes them excellent candidates for such applications as MRI contrast agents, bioseparation and as recoverable catalysts.2 Likewise, magnetically soft heterometallic iron-cobalt alloy nanoparticles (Fe1−xCox) are of significant interest as they possess the highest room temperature saturation magnetization of all known metallic alloys (Ms ≈ 240 Am2 kg−1 @ 273 K).3 Iron–platinum (Fe1−xPtx) nanoparticles, on the other hand, possess a high magnetocrystalline anisotropy (K ≈ 6.6 × 106 J m−3) and large coercivity (Hc) when annealed to the chemically ordered tetragonal L10 phase. As such, these nanoparticles have been extensively studied for their potential application in ultra-high density magnetic storage applications and as components in exchange-coupled composite magnets.4
Common solution-phase approaches to the synthesis of iron and heterometallic iron-containing nanoparticles include the reduction of iron salts and/or the thermolysis of iron complexes.4c,5 Perhaps, the most widely used approach is the thermal decomposition of iron pentacarbonyl, Fe(CO)5. This compound is inexpensive and readily available commercially, which makes it a popular choice for nanoparticle synthesis. While Fe(CO)5 has proven valuable for the small-scale research synthesis, its continued use, especially for potential scale up efforts, is severely limited. It is pyrophoric with an auto-ignition temperature of 49 °C and occupational exposure limits are set to an incredibly low 0.1 ppm.6
Fe(CO)5 has a low boiling point (103 °C) and under high temperature nanoparticle synthesis conditions unknown quantities can be boiled off, significantly decreasing yield in general and changing iron content in the case of heterometallic particle synthesis. Furthermore, the formation of decomposition products under storage introduces the need for a purification step.7 It is easy then to see that at larger scales, Fe(CO)5 would present significant technical and safety burdens. The development of a simple, less toxic, and non-volatile precursor that could substitute directly for Fe(CO)5 would be of significant benefit for further synthetic investigations of iron and iron-containing heterometallic nanoparticles.
Here we report the synthesis of well-defined Fe, Fe1−xCox, and Fe1−xPtx nanoparticles from the low-volatility solid and commercially available iron-carbonyl cluster, triiron dodecacarbonyl Fe3(CO)12. Fe3(CO)12 is stable under ambient conditions, easy to handle, and much less toxic than Fe(CO)5.8 It decomposes at high temperatures, increasing the range of available reaction temperatures, which would allow for improvements in crystallinity.9 Furthermore, it can be used as received directly from a supplier, removing the need for purification, and thereby simplifying the overall synthetic procedure. The cluster is commonly employed as a catalyst in organic transformations,10 but it is poorly soluble in high boiling point organic solvents and therefore there are only a few examples of its use as a precursor for nanoparticle synthesis. Shpaisman et al. formed nanocrystalline Fe particles by thermally decomposing Fe3(CO)12 under argon flow at high temperatures, and Amara et al. formed Fe and Fe3O4 nanoparticles by a solvothermal decomposition synthesis.8,11 Choplin et al. formed iron-containing heterometallic particles from carbonyl-containing heteropolynuclear precursor clusters.12 However, the lack of precursor solubility made size and shape control difficult.
Here, we demonstrate a significant increase in the solubility of Fe3(CO)12 by reacting it with an excess of a long-chain primary alkylamine, 1-dodecylamine (DDA), under an inert atmosphere and mild heating. Multi-metal center iron carbonyl clusters have been reported to form anionic cluster species on surfaces or in amine solvents, usually via disproportionation.13 Here, the formation of such an anionic iron complex leads to the complete dissolution of original Fe3(CO)12 in common solvents employed in nanoparticle synthesis (e.g., 1-octadecene) (ODE), thereby eliminating the multiple synthetic steps that are often required for other complex iron nanoparticle precursors.3b,14 This easily prepared, non-volatile iron cluster solution is then used here as the actual precursor in thermal decomposition reactions to form well-defined Fe, Fe1−xCox, and Fe1−xPtx nanoparticles, with the DDA as stabilizing agent. The presented approach to the synthesis of iron and iron-containing heterometallic nanoparticles can also be extended to another poorly soluble iron carbonyl cluster, Fe2(CO)9.
When Fe3(CO)12 is heated in an excess of DDA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) under nitrogen (Fig. 1a), a color change from green to deep red is observed indicating the formation of the anionic [HFe3(CO)11]− (Fig. 1a).15 The UV-Vis absorption profile of Fe3(CO)12 in chloroform shows two absorption peaks at 456 and 607 nm, as well as multiple absorption bands stretching into the UV (Fig. 1b). After heating in the presence of excess DDA (Fig. 1c), the strong band at 607 nm blue shifts to 545 nm with a strong second absorption peak now clearly visible at 319 nm, which is consistent with the formation of [HFe3(CO)11]−. Previously, this deep-red cluster anion has been identified as a disproportionation product of Fe3(CO)12 on the surface of alumina or magnesia.15 In these studies, Fe3(CO)12 dissociates at a surface and could be liberated by the use of a counteraction, e.g., NEt4+. In the present case, the anionic complex forms in situ due to the presence of DDA, simplifying the solubilization process. The presence of [HFe3(CO)11]− in solution has been confirmed by electrospray ionization mass spectrometry (ESI-MS), revealing the [HFe3(CO)11]− (m/z = 476.7) ion (and its decarbonylation products) to be the majority of species present in the precursor solution (Fig. S1†). UV-vis spectroscopy measurements (Fig. S2†) are also in agreement with ESI-MS: the absorption profile of the as-prepared precursor solution matched closely with that of the [HFe3(CO)11]− cluster anion isolated using high performance liquid chromatography (HPLC).
:30) under nitrogen (Fig. 1a), a color change from green to deep red is observed indicating the formation of the anionic [HFe3(CO)11]− (Fig. 1a).15 The UV-Vis absorption profile of Fe3(CO)12 in chloroform shows two absorption peaks at 456 and 607 nm, as well as multiple absorption bands stretching into the UV (Fig. 1b). After heating in the presence of excess DDA (Fig. 1c), the strong band at 607 nm blue shifts to 545 nm with a strong second absorption peak now clearly visible at 319 nm, which is consistent with the formation of [HFe3(CO)11]−. Previously, this deep-red cluster anion has been identified as a disproportionation product of Fe3(CO)12 on the surface of alumina or magnesia.15 In these studies, Fe3(CO)12 dissociates at a surface and could be liberated by the use of a counteraction, e.g., NEt4+. In the present case, the anionic complex forms in situ due to the presence of DDA, simplifying the solubilization process. The presence of [HFe3(CO)11]− in solution has been confirmed by electrospray ionization mass spectrometry (ESI-MS), revealing the [HFe3(CO)11]− (m/z = 476.7) ion (and its decarbonylation products) to be the majority of species present in the precursor solution (Fig. S1†). UV-vis spectroscopy measurements (Fig. S2†) are also in agreement with ESI-MS: the absorption profile of the as-prepared precursor solution matched closely with that of the [HFe3(CO)11]− cluster anion isolated using high performance liquid chromatography (HPLC).
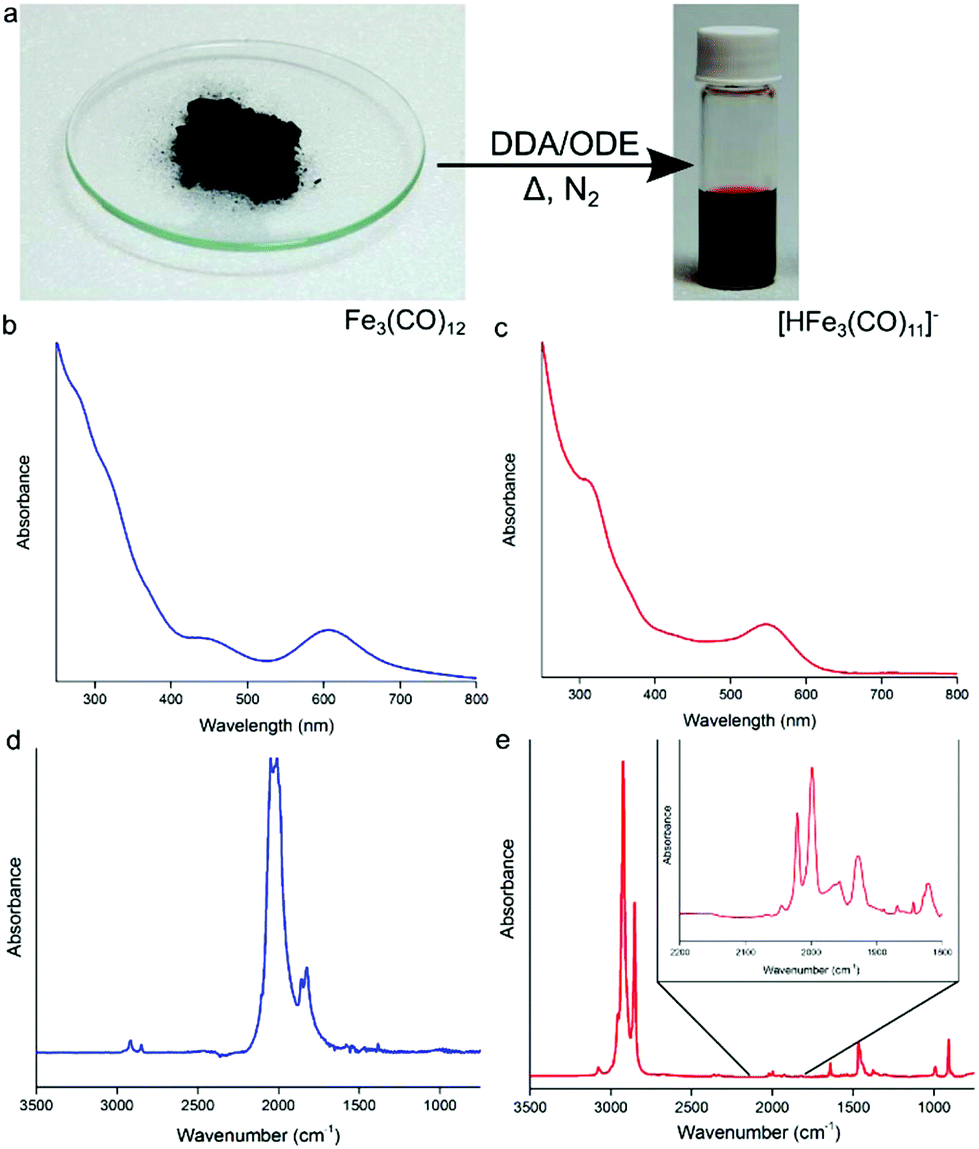 | ||
| Fig. 1 (a) Schematic representation of the conversion of dark-green Fe3(CO)12, upon heating in an excess of 1-dodecylamine (DDA) in 1-octadecene (ODE) under an inert atmosphere, into dark-red [HFe3(CO)11]− anion that is fully soluble in the DDA/ODE mixture. (b) UV-Vis spectra of Fe3(CO)12 dissolved in CHCl3. (c) UV-Vis spectrum of [HFe3(CO)11]− in ODE/DDA mixture (d) FTIR spectrum of Fe3(CO)12 in a KBr pellet. (e) FTIR spectrum of the as-formed [HFe3(CO)11]− cluster in a DDA/ODE mixture. | ||
Results of FTIR spectroscopy are also consistent with the formation of the protonated iron carbonyl cluster anion. Fig. 1d shows the FTIR absorption spectrum of pure solid Fe3(CO)12 in the CO ligand stretching vibrations region that consists of two strong bands at 2050 cm−1 and 2010 cm−1 and weaker ones at 1860 cm−1 and 1825 cm−1.16Fig. 1e shows the FTIR absorption spectrum of the [HFe3(CO)11]− anion in ODE/DDA solution. Present is a strong absorption band at ∼2800 cm−1 corresponding to C–H stretches of excess DDA. The continued presence of terminal and bridging CO stretches (see Fig. 1e inset) points to the multi-metal iron center of the anionic cluster being maintained and is consistent with the formation of [HFe3(CO)11]−.13c,15b
Once prepared, the precursor solution with [HFe3(CO)11]− is diluted in ODE and used as the iron source for nanoparticle synthesis, with the results shown in Fig. 2. The synthesis occurred at 200 °C under an inert atmosphere ensuring the formation of zero-valent iron. However, partial oxidation of particles during sample preparation for size analysis was observed leading to Fe/Fe3O4 core–shell nanoparticles (vide infra). Transmission electron microscopy (TEM) imaging (Fig. 2a) reveals well-dispersed particles of 9.3 ± 0.9 nm in diameter with regions of hexagonal and bilayer packing. The raw scattering data of a Small Angle X-ray scattering (SAXS) measurement together with a model curve and residuals are displayed in Fig. 2b. A spherical particle model with Gaussian size distribution (as observed from TEM) was used in the analysis of the scattering curves, giving an average nanoparticle size of 9.5 nm ± 1.0 nm, which is in good agreement with TEM results. We then show that good nanoparticle size control could be obtained. By varying the reaction temperature, oxidized iron nanoparticles of 11.1 ± 2.0 nm and 7.2 ± 0.7 nm in diameter could be formed at 180 °C and 280 °C, respectively (Fig. S3†).
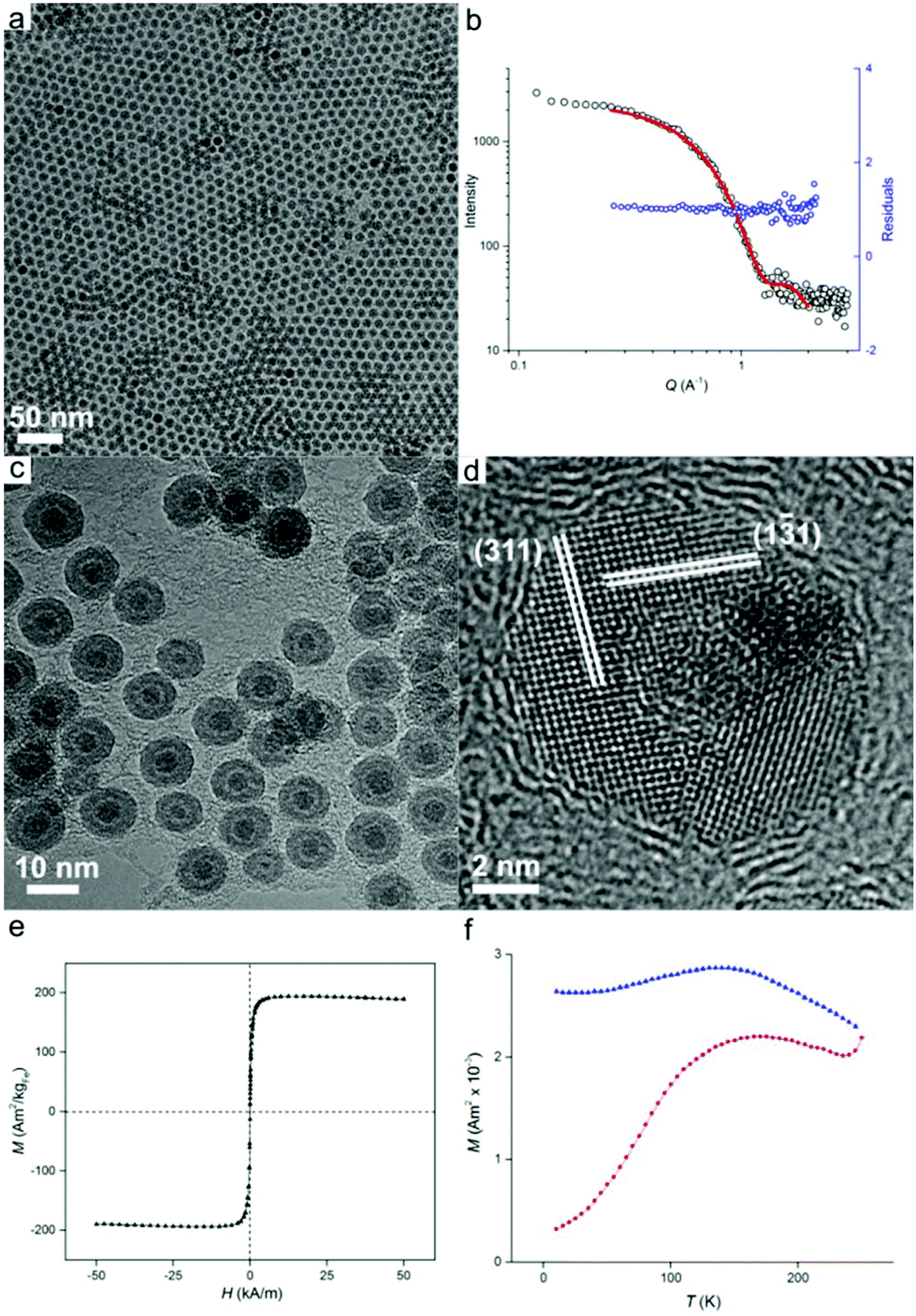 | ||
| Fig. 2 (a) Transmission electron microscopy (TEM) image of Fe nanoparticles synthesized from the [HFe3(CO)11]− complex-containing precursor solution. (b) Small Angle X-ray Scattering (SAXS) data of Fe nanoparticles showing the raw data (black circles) overlaid with the model fit (red line) and residuals (blue circles). (c, d) HRTEM images of synthesized iron nanoparticles in lower (c) and higher (d) magnifications. Magnetite formation on the surface of Fe nanoparticles occurs during sample preparation. (e, f) Results of SQUID measurements on Fe nanoparticles. (e) Plot of magnetization (M) vs. applied field (H) showing the saturation magnetization. (f) Field cooled–zero field cooled (FC–ZFC) measurements establish the blocking temperature of 169 K, which indicates superparamagnetic behavior at room temperature. | ||
Morphology of the oxidized iron nanoparticles is more clearly identifiable using high resolution TEM (Fig. 2c). The contrast difference between the oxide shell and the metal nanoparticle core is evident, together with a thin hollow interlayer produced by the Kirkendall effect during oxidation.17
No further growth of the hollow layer was observed under the electron beam. To calculate the size of the original nanoparticles that are present as zero-valent iron in solution (i.e., before oxidation), the thickness of the oxide layer must be taken into account. Analysis of lattice planes (Fig. 2d) indicates the oxide shell could be indexed to polycrystalline magnetite Fe3O4,18 which was further confirmed by X-ray diffraction analysis (Fig. S4†). Neither HRTEM or XRD showed evidence of bcc Fe, revealing the amorphous state of the iron particle core, which is not unexpected for iron nanoparticles of this size.1b The average thickness of the oxide shell was measured to be 2.9 ± 0.3 nm. Therefore the size of original Fe(0) nanoparticles in solution can be calculated as 7.9 ± 0.7 nm when the expansion of iron upon oxidation is taken into account.19
To investigate the magnetic properties of the unoxidized nanoparticles, an aliquot of the nanoparticle solution was sealed under vacuum (with extra care being paid to avoid oxidation), and its magnetic response was elucidated by means of superconducting quantum interference device (SQUID) magnetometry. The nanoparticles possess a saturation magnetization of 195 Am2 kg−1 at 250 K, which is close to the value for bulk iron (Fig. 2e).20 The particles are superparamagnetic at room temperature with a blocking temperature, TB, of 169 K obtained from temperature-dependent field cooled (FC), zero-field cooled (ZFC) measurements (Fig. 2f). Superparamagnetism was also confirmed by the absence of noticeable remanence in Fig. 2e. The measured blocking temperature of these particles is much higher than what was expected for 7.9 nm bcc Fe nanoparticles.5b This has been previously observed for amorphous iron nanoparticles in this size range which indicates a significantly higher value of magnetocrystalline anisotropy, K.1b,5b
Besides the synthesis of homometallic Fe nanoparticles, the solution with [HFe3(CO)11]− anion can also be used in the synthesis of heterometallic particles: Fe1−xCox and Fe1−xPtx. Fe1−xCox nanoparticles were synthesized from the thermal decomposition of a mixture of [HFe3(CO)11]− and Co2(CO)8 in ODE, with typical TEM images of the particles shown in Fig. 3. The particles were measured to be 9.4 ± 1.3 nm and 10.0 nm ± 1.4 nm in size, by TEM and SAXS, respectively (Fig. 3a and b). Results of the energy dispersive X-ray (EDX) spectroscopy are shown in Fig. 3c. After deconvolution of the overlapping CoKα and FeKβ peaks (see inset), a quantitative analysis could be carried out using the Cliff–Lorimer equation.21 The Cliff–Lorimer k factor, which is dimensionless and dependent on the TEM/EDX system used, was experimentally derived to be 0.96. We could then calculate an elemental composition of Fe67Co33 which is within the range known to display maximum magnetic saturation (0.3 < x < 0.4).22 As prepared, Fe67Co33 nanoparticles were measured by SQUID to possess a magnetization of Ms = 142 Am2 kg−1 at 250 K, which is comparable to previous examples of Fe1−xCox nanoparticles (Fig. 3d).3d,23
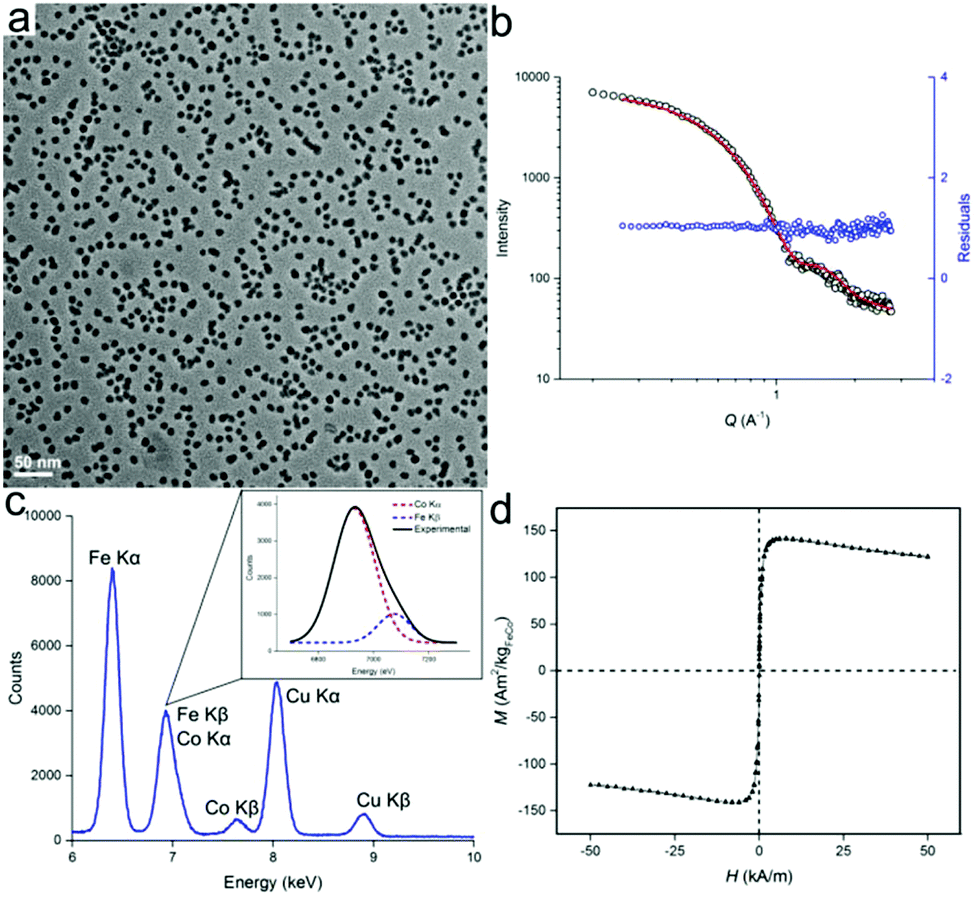 | ||
| Fig. 3 Fe1−xCox nanoparticles from [HFe3(CO)11]− complex-containing precursor solution. (a) TEM image and (b) corresponding SAXS curves. (c) Energy dispersive X-ray (EDX) analysis along with deconvolution of overlapping CoKα and FeKβ peaks (inset) allowing for quantitative analysis of both Fe and Co. (d) SQUID magnetic measurements of FeCo nanoparticles showing magnetization (M) vs. applied field (H). | ||
Fig. 4a shows a typical TEM image of Fe1−xPtx nanoparticles synthesized from a mixture of [HFe3(CO)11]− and Pt(acac)2 prepared in dibenzyl ether (DBE). The nanoparticles have a branched morphology and are 7.7 ± 1.6 nm in size measured along the longest axis. EDX elemental analysis (Fig. S5, Table S2†) established the nanoparticle composition as Fe50Pt50. Selected area electron diffraction (SAED) pattern is given in Fig. 4b, which can be indexed to the face-centered cubic (fcc) crystal structure.24 It is well-established that FePt particles must be thermally annealed to obtain the magnetically hard tetragonal L10 phase.25 Therefore, the as-synthesized nanoparticles were annealed in situ at 750 °C for 30 min using a TEM heating stage, with the results shown in Fig. 4c. The shape of the particles becomes more rounded due to the minimization of their surface area, with the average size being slightly reduced to 7.2 ± 1.1 nm. A SAED pattern (Fig. 4d) shows the emergence of the (001), (110) and (201) reflections of the tetragonal L10 phase. HRTEM experiments were also performed, confirming the transformation to the tetragonal phase upon annealing (Fig. S6†). Particle coalescence is avoided by annealing directly on the TEM substrate.
 | ||
| Fig. 4 Fe1−xPtx nanoparticles synthesized from the [HFe3(CO)11]− complex-containing precursor solution. TEM images of Fe1−xPtx particles (a) before and (c) thermal annealing at 750 °C for 30 min together with corresponding SAEDs (b and d) which can be indexed to the fcc crystal structure. SAED before annealing can be indexed to the fcc crystal structure, whereas after annealing SAED corresponds to the tetragonal L10 phase of Fe1−xPtx. | ||
Similarly to Fe3(CO)12, the non-volatile diiron carbonyl cluster Fe2(CO)9 is also known to react with strong bases, including amines.15 The possibility of using Fe2(CO)9 to form the [HFe3(CO)11]− anionic cluster in solution was also investigated. The Fe2(CO)9 cluster readily dissolves in a DDA/ODE mixture upon heating forming an orange-red solution (see FTIR analysis, Fig. S7†). The thermal decomposition of this precursor solution also yields metallic iron, Fe1−xCox, and Fe1−xPtx nanoparticles, similar to those obtained from the Fe3(CO)12 case. To the best of our knowledge, prior to this work, only Kramer et al. used Fe2(CO)9 dissolved in ionic liquids to obtain Fe and Fe2O3 nanoparticles. However, only small particles were formed (∼4 nm) and they subsequently readily agglomerated.26 Here, the oxidized iron nanoparticles were measured to be 10.8 ± 0.9 nm and 10.2 nm ± 1.3 nm, by TEM and SAXS, respectively (Fig. S8 and 9†). The size of Fe1−xCox alloy nanoparticles was found to be 9.6 ± 1.6 nm and 10.3 nm ± 1.6 nm, by TEM and SAXS, respectively (Fig. S10a and b†). From EDX analysis, they were shown to possess Fe55Co45 composition (Fig. S10c and d†). The Fe1−xPtx nanoparticles were 7.4 ± 0.5 nm in size (from SAXS analysis) and had a composition of Fe35Pt65, which is within the requirements for tetragonal ordering (Fig. S11 and 12, Table S3†).27 The size and composition of pure iron and iron-containing heterometallic nanoparticles obtained from Fe2(CO)9 were similar to that formed using Fe3(CO)12 indicating the versatility of our approach for the solubilization of multi-nuclear iron carbonyl clusters. The lighter color of the final reaction solution formed with Fe2(CO)9 indicates a lower concentration of [HFe3(CO)11]− (Fig. S7†). This explains the subtle variations in reaction temperatures required for the formation of well-defined nanoparticles (see Experimental section). This change in reaction kinetics with [HFe3(CO)11]− concentration also indicates that it is the major species responsible for nanoparticle formation. While the solutions formed from both iron carbonyl compounds used here are not simple stoichiometric compounds, this study supports our previous observation that non-stoichiometric precursors can produce extremely reproducible results, if consistently prepared.15c
Conclusions
In summary, we have demonstrated that by the straightforward mixing of insoluble iron carbonyls Fe3(CO)12 or Fe2(CO)9 with an amine surfactant, a precursor solution containing the [HFe3(CO)11]− anionic cluster can be formed. This solution, in turn, can be used as a direct replacement for iron pentacarbonyl in a variety of nanoparticle forming reactions, including the thermolytic synthesis of Fe, Fe1−xCox, and Fe1−xPtx nanoparticles. Compared to more commonly used techniques, the use of solutions of these non-volatile cluster solutions as iron precursors allow for safer reactions and improved synthetic control for the formation of iron and iron-containing heterometallic nanoparticles.Acknowledgements
Supported by the Laboratory Directed Research and Development program at Sandia National Laboratories. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science. Sandia National Laboratories is a multi-program laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000. Los Alamos National Laboratory, an affirmative action equal opportunity employer, is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy under contract DE-AC52-06NA25396.Notes and references
- (a) D. L. Huber, Small, 2005, 1, 482–501 CrossRef CAS PubMed; (b) T. C. Monson, Q. Ma, T. E. Stevens, J. M. Lavin, J. L. Leger, P. V. Klimov and D. L. Huber, Part. Part. Syst. Charact., 2013, 30, 258–265 CrossRef CAS; (c) L. M. Lacroix, N. F. Huls, D. Ho, X. L. Sun, K. Cheng and S. H. Sun, Nano Lett., 2011, 11, 1641–1645 CrossRef CAS PubMed; (d) A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed; (e) C. J. Meledandri and D. F. Brougham, Anal. Methods, 2012, 4, 331–341 RSC.
- (a) D. A. J. Herman, P. Ferguson, S. Cheong, I. F. Hermans, B. J. Ruck, K. M. Allan, S. Prabakar, J. L. Spencer, C. D. Lendrum and R. D. Tilley, Chem. Commun., 2011, 47, 9221–9223 RSC; (b) S. Cheong, P. Ferguson, I. F. Hermans, G. N. L. Jameson, S. Prabakar, D. A. J. Herman and R. D. Tilley, ChemPlusChem, 2012, 77, 135–140 CrossRef CAS; (c) A. B. Salunkhe, V. M. Khot and S. H. Pawar, Curr. Top. Med. Chem., 2014, 14, 572–594 CrossRef CAS PubMed; (d) T. A. P. Rocha-Santos, Trends Anal. Chem., 2014, 62, 28–36 CrossRef CAS; (e) Y.-W. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS PubMed; (f) R. Hudson, A. Riviere, C. M. Cirtiu, K. L. Luska and A. Moores, Chem. Commun., 2012, 48, 3360–3362 RSC.
- (a) C. Desvaux, F. Dumestre, C. Amiens, M. Respaud, P. Lecante, E. Snoeck, P. Fejes, P. Renaud and B. Chaudret, J. Mater. Chem., 2009, 19, 3268–3275 RSC; (b) C. Desvaux, C. Amiens, P. Fejes, P. Renaud, M. Respaud, P. Lecante, E. Snoeck and B. Chaudret, Nat. Mater., 2005, 4, 750–753 CrossRef CAS PubMed; (c) X. B. Su, H. G. Zheng, Z. P. Yang, Y. C. Zhu and A. L. Pan, J. Mater. Sci., 2003, 38, 4581–4585 CrossRef CAS; (d) G. S. Chaubey, C. Barcena, N. Poudyal, C. Rong, J. Gao, S. Sun and J. P. Liu, J. Am. Chem. Soc., 2007, 129, 7214–7215 CrossRef CAS PubMed.
- (a) S. H. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed; (b) K. Elkins, D. Li, N. Poudyal, V. Nandwana, Z. Q. Jin, K. H. Chen and J. P. Liu, J. Phys. D: Appl. Phys., 2005, 38, 2306–2309 CrossRef CAS; (c) S. H. Sun, Adv. Mater., 2006, 18, 393–403 CrossRef CAS.
- (a) S. Cheong, P. Ferguson, K. W. Feindel, I. F. Hermans, P. T. Callaghan, C. Meyer, A. Slocombe, C.-H. Su, F.-Y. Cheng, C.-S. Yeh, B. Ingham, M. F. Toney and R. D. Tilley, Angew. Chem., Int Ed., 2011, 50, 4206–4209 CrossRef CAS PubMed; (b) T. C. Monson, E. L. Venturini, V. Petkov, Y. Ren, J. M. Lavin and D. L. Huber, J. Magn. Magn. Mater., 2013, 331, 156–161 CrossRef CAS; (c) Y. P. Sun, X. Q. Li, J. S. Cao, W. X. Zhang and H. P. Wang, Adv. Colloid Interface Sci., 2006, 120, 47–56 Search PubMed; (d) D. A. J. Herman, S. Cheong-Tilley, A. J. McGrath, B. F. P. McVey, M. Lein and R. D. Tilley, Nanoscale, 2015, 7, 5951–5954 RSC; (e) B. Bian, W. Xia, J. Du, J. Zhang, J. P. Liu, Z. Guo and A. Yan, Nanoscale, 2013, 5, 2454–2459 RSC.
- Occupational Safety and Health Administration [OSHA], Retrieved from; https://www.osha.gov/dts/chemicalsampling/data/CH_247500.html.
- H. G. Cutforth and P. W. Selwood, J. Am. Chem. Soc., 1943, 65, 2414–2415 CrossRef CAS.
- N. Shpaisman, E. R. Bauminger and S. Margel, J. Alloys Compd., 2008, 454, 89–96 CrossRef CAS.
- M. Kin, H. Kura, M. Tanaka, Y. Hayashi, J. Hasaegawa and T. Ogawa, J. Appl. Phys., 2015, 117, 17E714 CrossRef.
- (a) R. B. King and F. G. A. Stone, J. Am. Chem. Soc., 1960, 82, 4557–4562 CrossRef CAS; (b) H. Yamada, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 1996, 118, 1054–1059 CrossRef CAS; (c) S. Enthaler, M. Haberberger and E. Irran, Chem. – Asian J., 2011, 6, 1613–1623 CrossRef CAS PubMed; (d) G. K. Jarugumilli and S. P. Cook, Org. Lett., 2011, 13, 1904–1907 CrossRef CAS PubMed.
- D. Amara, I. Felner, I. Nowik and S. Margel, Colloids Surf., A, 2009, 339, 106–110 CrossRef CAS.
- A. Choplin, L. Huang, A. Theolier, P. Gallezot, J. M. Basset, U. Siriwardane, S. G. Shore and R. Mathieu, J. Am. Chem. Soc., 1986, 108, 4224–4225 CrossRef CAS.
- (a) W. Hieber and R. Werner, Chem. Ber., 1957, 90, 1116–1120 CrossRef CAS; (b) W. F. Edgell, M. T. Yang, B. J. Bulkin, R. Bayer and N. Koizumi, J. Am. Chem. Soc., 1965, 87, 3080–3088 CrossRef CAS; (c) F. Hugues, J. M. Bassett, Y. B. Taarit, A. Choplin, M. Primet, D. Rojas and A. K. Smith, J. Am. Chem. Soc., 1982, 104, 7020–7024 CrossRef CAS.
- D. Zitoun, C. Amiens, B. Chaudret, M. C. Fromen, P. Lecante, M. J. Casanove and M. Respaud, J. Phys. Chem. B, 2003, 107, 6997–7005 CrossRef CAS.
- (a) W. Hieber and H. Beutner, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1962, 17, 211 CAS–&; (b) F. Hugues, A. K. Smith, Y. B. Taarit, J. M. Basset, D. Commereuc and Y. Chauvin, J. Chem. Soc., Chem. Commun., 1980, 68–70 Search PubMed; (c) H. Kuroda, Pure Appl. Chem., 1992, 64, 1449–1460 CrossRef CAS.
- (a) R. K. Sheline, J. Am. Chem. Soc., 1951, 73, 1615–1618 CrossRef CAS; (b) F. A. Cotton and G. Wilkinson, J. Am. Chem. Soc., 1957, 79, 752–753 CrossRef CAS.
- (a) D. A. J. Herman, S. Cheong, M. J. Banholzer and R. D. Tilley, Chem. Commun., 2013, 49, 6203–6205 RSC; (b) Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- Magnetite is indistinguishable from another iron oxide, maghemite (Fe2O3), with the characterization tools employed here. However for our purposes, the assumption of a magnetite phase is sufficient.
- Y. Zhao, H. Ren, H. Dai and W. Jin, Corros. Sci., 2011, 53, 1646–1658 CrossRef CAS.
- B. D. Cullity, Introduction to Magnetic Materials, Addison-Wesley Publishing Company, Reading, 1972 Search PubMed.
- D. B. Williams and C.B. Carter, Transmission Electron Microscopy: IV Spectrometry, Springer Science, 1996 Search PubMed.
- (a) Y. Fu, X. F. Cheng and Z. Yang, Phys. Status Solidi A, 2006, 203, 963–969 CrossRef CAS; (b) P. Sirvent, E. Berganza, A. M. Aragón, A. Bollero, A. Moure, M. García-Hernández, P. Marín, J. F. Fernández and A. Quesada, J. Appl. Phys., 2014, 115, 17B505 CrossRef; (c) S. Farabi Khaneghahi and S. Sharafi, Adv. Powder Technol., 2014, 25, 211–218 CrossRef CAS.
- (a) V. Tzitzios, G. Basina, D. Niarchos, W. Li and G. Hadjipanayis, J. Appl. Phys., 2011, 109, 07A313 CrossRef; (b) C. Desvaux, P. Lecante, M. Respaud and B. Chaudret, J. Mater. Chem., 2010, 20, 103–109 RSC.
- W. Lv, W. He, X. Wang, Y. Niu, H. Cao, J. H. Dickerson and Z. Wang, Nanoscale, 2014, 6, 2531–2547 RSC.
- (a) D. R. Li, N. Poudyal, V. Nandwana, Z. Q. Jin, K. Elkins and J. P. Liu, J. Appl. Phys., 2006, 99 Search PubMed; (b) A. Delattre, S. Pouget, J. F. Jacquot, Y. Samson and P. Reiss, Small, 2010, 6, 932–936 CrossRef CAS PubMed.
- J. Krämer, E. Redel, R. Thomann and C. Janiak, Organometallics, 2008, 27, 1976–1978 CrossRef.
- M. Delalande, M. J. F. Guinel, L. F. Allard, A. Delattre, R. Le Bris, Y. Samson, P. Bayle-Guillemaud and P. Reiss, J. Phys. Chem. C, 2012, 116, 6866–6872 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental detail, DFT calculations, additional TEM, SAXS, EDX, UV-vis, XRD and FTIR characterization. See DOI: 10.1039/c7nr01028a |
| This journal is © The Royal Society of Chemistry 2017 |