
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reductive dissolution of supergrowth carbon nanotubes for tougher nanocomposites by reactive coagulation spinning†
A. J.
Clancy
 ,
D. B.
Anthony
,
S. J.
Fisher
,
H. S.
Leese
,
C. S.
Roberts
and
M. S. P.
Shaffer
*
,
D. B.
Anthony
,
S. J.
Fisher
,
H. S.
Leese
,
C. S.
Roberts
and
M. S. P.
Shaffer
*
Imperial College London, Department of Chemistry, Frankland Road, London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
First published on 31st May 2017
Abstract
Long single-walled carbon nanotubes, with lengths >10 μm, can be spontaneously dissolved by stirring in a sodium naphthalide N,N-dimethylacetamide solution, yielding solutions of individualised nanotubide ions at concentrations up to 0.74 mg mL−1. This process was directly compared to ultrasonication and found to be less damaging while maintaining greater intrinsic length, with increased individualisation, yield, and concentration. Nanotubide solutions were spun into fibres using a new reactive coagulation process, which covalently grafts a poly(vinyl chloride) matrix to the nanotubes directly at the point of fibre formation. The grafting process insulated the nanotubes electrically, significantly enhancing the dielectric constant to 340% of the bulk polymer. For comparison, samples were prepared using both Supergrowth nanotubes and conventional shorter commercial single-walled carbon nanotubes. The resulting nanocomposites showed similar, high loadings (ca. 20 wt%), but the fibres formed with Supergrowth nanotubes showed significantly greater failure strain (up to ∼25%), and hence more than double the toughness (30.8 MJ m−3), compared to composites containing typical ∼1 μm SWCNTs.
1. Introduction
Single walled carbon nanotubes (SWCNTs) consist of a seamless cylinder of sp2 hybridised carbon, between 0.5 nm–5 nm in diameter, with superlative intrinsic mechanical,1 optical, and electronic properties.2 The length of SWCNTs can vary dramatically from <100 nm to tens of centimetres.3 The so-called ultra-long (>10 μm) SWCNTs4 have several predicted benefits derived from their high aspect ratio, particularly in nanocomposites, including increased thermal and electrical conductivity,5 improved matrix stress transfer,6 and increased pull-out toughening.1 Several routes have been developed to synthesise ultra-long SWCNTs such as Si-substrate-directed chemical vapour deposition (CVD),3 flaked substrate “carpet” growth,7 and the “Supergrowth” water vapour enhanced CVD8 route. Supergrowth nanotubes (SG-CNTs, Fig. 1) can be grown up to millimetre lengths, contain negligible catalytic impurities, and can now be produced commercially at up to the tonne-scale annually.9 The diameter of SG-CNTs varies from 2–5 nm, controlled by the amount of catalyst deposited; below around 3 nm, as used in this study, the majority are single wall nanotubes.10 | ||
| Fig. 1 SEM micrographs of as-received SG-CNTs. Additional SEM for length distribution can be found in the ESI (Fig. S1†). | ||
As-synthesised, SWCNTs generally form bundles, due to their high surface energy, which are mechanically and electronically inferior to the ideal individualised species. Debundling is usually performed through solution processing, which overcomes the inter-SWCNT van der Waals forces to form an alternative, (meta)stable state. High shear forces from ultrasonication can supply energy to separate nanotubes, and the suspension can be stabilised by the presence of a surfactant, polymer,11 or tuned solvent12 to trap the nanotubes in a dispersed state, usually kinetically. The SWCNT's sp2 framework is damaged by these high-shear methods, introducing defects and shortening of SWCNTs.13 In addition, only a fraction of the SWCNTs are individualised, requiring subsequent ultracentrifugation to obtain a predominantly individualised dispersion. In particular long SWCNTs cannot easily be processed using solution routes designed for typical, shorter (ca. 1 μm) SWCNTs. Although shear-based processing routes are known to cut SG-CNTs to the micron-scale (0.1 μm–3 μm),14 the shortened nanotubes still do not disperse well and instead a “mesh structure” forms, even with the most conventionally efficient dispersion routes,15 possibly due to the presence of residual long-SWCNTs adhering to the shortened nanotubes. Current composites using SWCNTs with lengths 10 μm, therefore, either involve direct manipulation of dry constructs,16 or use partially-dispersed dendritic agglomerates,6,15 limiting versatility and performance.
An alternative solution dispersion route for SWCNTs relies on charging the nanotubes, either with protons from superacids,17 or electrons to form negatively charged ‘nanotubide’ anions.18 Both routes have been shown to spontaneously dissolve SWCNTs; however, only superacid dissolution has been previously reported to dissolve individual, long SWCNTs (here 10 μm–100 μm), and then only at very low concentrations (50 ppm).4 Reductive dissolution is a thermodynamically-driven process, in contrast to the kinetically stable dispersions obtained from ultrasonication; dissolution occurs without the application of shear forces, although the mixtures may be stirred to accelerate the process. The solutions consist of truly individualised19 SWCNTs and in principle damage to the carbon framework or length can be avoided. SWCNT reduction to nanotubide can be performed through several routes including Birch reduction by group I metals in liquid ammonia,19 addition of molten metal,20 or electrochemically.21 The resulting nanotubide salt dissolves spontaneously in certain aprotic, polar solvents to form SWCNT solutions,18 in some cases, with sufficiently high concentrations to form lyotropic liquid crystals.22N,N-Dimethylacetamide (DMAc) is capable of dissolving SWCNTs and does not degrade in the presence of reducing agents such as sodium naphthalide (NaNp), facilitating a simple one-step reduction/dissolution.23 Nanotubide anions can be functionalised by reaction with a host of electrophiles, leading to covalent addition to the nanotube sidewall, most commonly organohalides.24 The charge added to the SWCNT during functionalisation is depleted in this reaction, although a fraction of residual charge may remain.20
The incorporation of SWCNTs into structural and multifunctional polymer composites has received particular attention;25 however, transferring the exceptional intrinsic properties of SWCNTs into real world macroscopic materials has proved challenging. Shear mixing and other simple matrix addition methods suffer from cutting and damage, weak interfaces, and poor final SWCNT individualisation. There are studies indicating that the use of reduced nanotubes may alleviate these issues, in epoxy26 and chlorinated polypropylene.27 The anisotropy of nanotubes creates a further challenge, since alignment is needed to maximise performance. A variety of routes to forming aligned nanotube fibres have been developed, including various dry, melt, and wet spinning methods.28 The most relevant, here, is the coagulation of SWCNT dispersions by injection into a polymer-containing anti-solvent bath; usually aqueous surfactant dispersions have been injected into poly(vinyl alcohol) solutions to produce nanotube reinforced composite fibres.29 One report describes spinning nanotubide dispersion into a water-based coagulant30 to produce pure nanotube fibres, though with relatively low strengths (<100 MPa) likely due to poor packing and alignment following rapid quenching. A more controlled, non-aqueous polymeric coagulant should offer more opportunity to optimise the microstructure.
Poly(vinyl chloride) (PVC) is one of the most ubiquitous polymers in the world and exists in flexible and rigid forms depending on processing route. When compared to flexible PVC, rigid PVC31 has higher tensile strength (40–70 vs. 6–25 MPa) and Young's modulus (1–3.5 vs. 0.05–0.15 GPa), however, the improved strength is accompanied by a substantially reduced strain to failure (30–80 vs. 150–400%) leading to poor toughness32 (∼0.3 MJ m−3). Carbon nanotubes (CNTs) have been introduced to form CNT/PVC composites to increase electrical/thermal conductivity,33 manipulate PVC phase behaviour,34 and increase thermal stability,35 however, addition of neat nanotubes has not led to notable mechanical improvement,36 thought to be due to poor interface between matrix and nanotube.37 While PVC has been grafted onto multi-walled carbon nanotubes (MWCNTs),37,38 these materials have not been studied in a bulk composite. The related polymer, chlorinated-poly(propylene), has been covalently grafted to MWCNTs27 through reductive functionalisation, generating composites with impressive reinforcing efficiency, attributed to a strong interface; however, these composites were limited to low loading fractions (0.6 wt%) and random planar orientations.
Here, we extend reductive chemistries to SG-CNTs, dissolved as individualised species. The results are benchmarked against standard sonication methods. The utility of SG-CNTs versus standard micron-scale SWCNTs is demonstrated by investigating the effect of SWCNT length upon mechanical behaviour through the synthesis of a series of SWCNT/PVC composite fibres. Increasing the aspect ratio should increase the extent of SWCNT reinforcement by improving stress transfer efficiency. Further, the reactive fibres are spun via a new reactive coagulation spinning process in organic solvent, intended to create a strong composite interface in situ.
2. Experimental
2.1 Materials
Supergrowth SWCNTs (Batch HTO0209-DC14a-4) were provided by Prof. Kenji Hata of The National Institute of Advanced Industrial Science and Technology (AIST, Japan), and were used as received. SG-CNTs were supplied as vertical arrays of nanotubes ca. 250 μm–500 μm tall (Fig. 1 and ESI Fig. S1,† measured with ImageJ, NIH, v1.45s); the initial length of the SG-CNTs is assumed to be equivalent to the height of the grown forest. Elicarb SWCNTs (P929 Batch K108511/g) were provided by Thomas Swan Ltd, UK. Tuball SWCNTs (Batch 4-18032014) were purchased from OCSiAl, Russia. Elicarb SWCNTs and Tuball SWCNTs were purified reductively according to a previously reported NaNp/DMAc purification procedure39 using 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10:1 charging stoichiometry respectively. Methyl-iso-butyl-ketone (MiBK) and tetrahydrofuran (THF) were purchased from VWR UK Ltd (UK) and all other chemicals were purchased from Sigma Aldrich Ltd (UK). Commercial anhydrous N,N-dimethylacetamide was dried further with ∼10 vol% of activated 4 Å molecular sieves for a minimum of two days. PVC was purchased with average Mw ∼ 43 kDa, average Mn ∼ 22 kDa. Dry air (oxygen 20 vol%/nitrogen 80 vol% mix) was purchased from BOC (UK).
:1 and 10:1 charging stoichiometry respectively. Methyl-iso-butyl-ketone (MiBK) and tetrahydrofuran (THF) were purchased from VWR UK Ltd (UK) and all other chemicals were purchased from Sigma Aldrich Ltd (UK). Commercial anhydrous N,N-dimethylacetamide was dried further with ∼10 vol% of activated 4 Å molecular sieves for a minimum of two days. PVC was purchased with average Mw ∼ 43 kDa, average Mn ∼ 22 kDa. Dry air (oxygen 20 vol%/nitrogen 80 vol% mix) was purchased from BOC (UK).
2.2 Characterisation
Scanning electron microscopy (SEM) micrographs were taken with a LEO Gemini 1525 FEGSEM (Zeiss, Germany) controlled by SmartSEM software, with a working distance of ca. 7 mm, accelerating voltage of 10 keV, and a 30 μm aperture using an InLens detector. As-received materials were adhered using silver DAG (Agar Scientific Ltd, UK). Dispersions were prepared for SEM by covering a stub with aluminium foil and drop-casting a dilute solution which was dried in ambient conditions. The dried samples were then submerged in DI water and tetrahydrofuran briefly to remove NaOH and naphthalene, before drying in ambient conditions.Atomic force microscopy (AFM) samples were prepared on silicon substrates (Si wafer chips, Agar Scientific Ltd, UK) which were submerged in freshly prepared 3:1 mixture of H2SO4 (98%) and H2O2 (32%), before washing with copious DI water and drying at 120 °C. Dilute (<1 μg mL−1) dispersions of nanotubide were drop-cast onto the wafer and dried in vacuo at RT for 16 h before exposure to air and soaking for 1 h in DI water and absolute ethanol (HPLC grade). Sonicated SWCNT dispersions were diluted (<1 μg mL−1), dropcast onto the wafer and dried at 150 °C in vacuo for 16 h. The aqueous surfactant AFM samples were briefly submerged in HPLC water to remove excess surfactant before redrying. AFM measurements of nanotubide and NMP samples were performed with tapping mode on a Veeco Multimode VIII AFM with Nanoscope IV Digital Instruments AFM controller (Bruker, USA) using Nanosensor tapping mode probes (Windsor Scientific Ltd, UK). AFM micrographs were processed in NanoScope Analysis (v1.40, Bruker), using 3rd order flattening (5% z-threshold) followed by spike removal (value 3.00). AFM measurements of SDS-dispersed samples were performed with dynamic mode on a hpAFM with AFM Controller (NanoMagnetics Instruments, UK) using Nanosensor tapping mode probes. AFM micrographs were processed in NMI Image Analyzer (v1.4, NanoMagnetics Instruments), with plane correction achieved via the in-built automatic height and line correction functions, followed by scar removal.
Thermogravimetric analysis (TGA) was run on a Pyris 1 (PerkinElmer Inc., USA) unless stated, under 60 mL min−1 nitrogen. The samples were held for 30 min at 100 °C as a drying step before heating at 10 °C min−1 to 700 °C.
Tensile tests of SWCNT composite fibres were based on standard ISO BSI11566 for single carbon fibres. Tensile specimens were prepared by mounting on a card template (15 mm gauge) using epoxy (Araldite Rapid, Huntsman Corporation, USA) to aid handling. Tensile tests of PVC film was based on standard ISO 527-2 5B. Samples were tested using a TST350 Tensile Stress Tester (Linkam Scientific Ltd, UK) fitted with a 20 N loading cell. Samples were measured with a cross head displacement rate of 17 μm s−1 until failure. Fibre cross-sectional areas were taken from optical microscopy of the fracture surface (true stress); fibre diameter is assumed to be circular.
Statistical Raman spectroscopy was performed on an InVia confocal Raman microscope (Renishaw PLC, UK) with 532 nm (2.37 eV) DPSS diode excitation (30 s, 10% power, over a ca. 150 μm2 region, with >1000 points). Spectra were processed in WiRE v4.1 by 11th order ‘intelligent fitting’ baseline subtraction, and cosmic ray removal and curve fitting single peaks for D-mode (ca. 1340 cm−1) and G-mode (ca. 1560 cm−1).40 Histograms are binned at D/G mode intensity ratio intervals of 0.1. Polarised Raman spectroscopy was performed using a 633 nm laser DPSS diode excitation (30 s, 5% power); the polarisation direction of the laser was changed by applying half wave plates prior to interaction with the sample and analyser. Three polarisation configurations were applied; VV (incident and scattered light polarised parallel to fibre axis), VH (incident light parallel and scattered light perpendicular to fibre axis) and HH (incident and scattering light polarised perpendicular to fibre axis). Measurements at all three configurations were taken at the same location. Alignment (S) was calculated from the scattering intensities of the G mode, using eqn (1) from Puech et al.,41 and the values given are an average of the S values from five spectra with standard deviations presented as errors.
 | (1) |
For Karl Fischer titrations, dry solvent (ca. 10 mL) was placed in a flask and sealed with a Suba seal. A 2 mL aliquot of each solvent was drawn into a syringe and weighed. Samples were injected into a Mettler Toledo DL32 coulometer filled with HYDRANAL-Coulomat AD, and the emptied syringe was weighed to give the weight of solvent added. Results are presented as the mean of the three measurements with an error of one standard deviation.
Capacitance was measured using a Rohde & Schwarz Hamag HM8118, using a 1 cm diameter circular cross section. Electrical conductivity was measured using an Iso-Tech IDM67 in a two-probe configuration with silver paint adhering contacts 1 cm apart to the composite fibre. Measurements were attempted at multiple points along all fibres. Breakdown voltage measurements were performed using a Kiethley Model 2290-5 Power Supply.
2.3 Procedures
:Na 10:1, varied for different charging ratios) was taken and diluted with DMAc to give the desired concentration. The solution was poured over the SWCNTs and stirred with a glass stirrer bar vigorously (sufficient to vortex the liquid) for 24 h (Elicarb/Tuball SWCNTs) or 2 weeks (SG-CNTs) to give the nanotubide solution. Solutions were centrifuged (1000g, 30 min) and decanted by hand to remove undissolved materials. A ‘quenched control’ SG-CNT sample was reduced (C:Na 6:1) and left unstirred over 48 h.
3. Results and discussion
3.1 SG-CNT dissolution
To dissolve the SG-CNTs, NaNp/DMAc solution was poured over the dried powder at a SWCNT loading (the mass of raw SWCNTs added per unit volume of solution) of 1 mg mL−1. The charging ratio, defined by the C:Na stoichiometry ratio, where higher numbers refer to a lower degree of charging, was varied from 20:1 to 2:1. In all cases, the SG-CNT powder swelled immediately upon addition of the NaNp solution indicating successful charge transfer; dissolution of the SWCNTs (visually confirmed by the formation of a stable black solution) required approximately 2 weeks of rapid stirring (sufficient to observe a vortex). The result contrasts with the dissolution of typical micron-length SWCNTs (Tuball, Elicarb and HiPco) upon simple stirring overnight. Previous experiments23 with shorter nanotubes showed that dissolution of some, generally very short and/or defective species, begins from at low degrees of charging, around C:Na 100:1. In the case of SG-CNTs, dissolution was only observed once the degree of charging was greater than C:Na 20:1 (ESI, Fig. S3†). Thereafter, generally, a higher charge led to a higher concentration, reaching up to 0.74 mg mL−1 at C:Na 4:1, in line with increasing inter-SWCNT Coulombic repulsion. However, when charge was increased from C:Na 4:1 to 2:1, the SG-CNT concentration and yield decreased (0.16 mg mL−1); similar effects in short SWCNT and graphene systems have been previously attributed23,43 to the condensation of sodium cations screening the repulsion required to drive dissolution.
SG-CNTs were dispersed through probe sonication as a baseline for comparison with reductive SWCNT processing. Previous literature on SG-CNTs has often involved sonication in MiBK;14,15 however, here, only low levels of dispersion were obtained, regardless of sonication intensity or duration. Instead, sonication was performed in N-methyl-2-pyrrolidone (NMP), the most common pure solvent for SWCNT shear dispersion, and in aqueous SDS solution, a typical surfactant for nanotube dispersion. Sonication was limited to 30 min to prevent unnecessary damage and SWCNT loading set to 0.1 mg mL−1. Large flocs of nanotubes were clearly visible for both systems even immediately after sonication. Sonication was attempted for an extended 3 hours, however, the solution visually appeared similar to the 30 min sonication sample; accordingly, the 30 min sonication samples were used for further analysis.
SEM micrographs of dropcast nanotubide solution (Fig. 2) illustrate dissolution of the initial aligned forests, leading to the formation of random mats. The NMP sonicated sample also adopts a random orientation, however, the dropcast nanotubide bundle diameters are substantially smaller, indicating a higher degree of exfoliation. AFM (Fig. 3) confirmed that the majority of the nanotubes in the NaNp/DMAc dispersion were individualised (2 nm–3 nm in height, ESI, Fig. S4†), although the length of the individualised nanotubes (1 μm–15 μm) is notably shorter than the measured initial forest heights. While forest height may not directly correlate to true SWCNT length, previous literature has provided indirect evidence to support using forest height as an approximation of nanotube length for pre-deposited catalyst CVD-grown nanotubes,44,45 and this assumption is widely used within the field (see discussion in ESI section 2†). As such, it is likely that the SWCNTs are originally >100 μm long and the aspect ratio is reduced during the processing. Evidence for cutting can be seen in the Raman spectra (Fig. 4) with reductively dissolved SG-CNTs showing a noticeable increase in the D/G mode intensity ratio from 0.123 to 0.283. Simple charging and discharging of the SWCNTs (after 2 days soaking, no stirring) only increased the D/G mode intensity slightly (0.147), attributed to minor damage from uncontrolled discharging.20 Simple stirring of typical, short SWCNTs does not introduce damage, but longer SWCNTs can be broken more easily;46 in this context, D/G ratio is inversely related to nanotube length.47 It is worth noting that damage as measured (semi-qualitatively) with Raman spectroscopy is higher following SWCNT sonication (NMP D/G = 0.407, SDS(aq) D/G = 0.312). In addition, AFM of the NMP sonicated SG-CNTs (Fig. 3) indicated a more significant degree of shortening, without individualisation of the nanotubes. Accurate quantification of sonicated SG-CNT length was complicated by the heavy bundling (bundles 5–35 nm height ∼2–12 CNTs across, ESI, Fig. S5†). The SG-CNTs sonicated in aqueous SDS showed a significantly lower degree of exfoliation from the constituent forests than either NMP dispersion or NaNp dissolution. A number of individualised nanotubes can be observed independent of the agglomerates, but are of sub-micron length and represent only a small fraction of the total sample mass (Fig. 3, ESI, Fig. S6†). Both sonicated samples appear to be consistent with the 0.1–3 μm reported by Kobashi et al.14 for the sonication of SG-CNTs in MiBK.
 | ||
| Fig. 2 Typical SEM micrograph of SG-CNTs; as-received and dispersions from NaNp/DMAc and NMP sonication. Dispersions were drop-cast on aluminium and washed with acetone and water. Higher magnification micrographs can be found in the ESI (Fig. S2†). | ||
 | ||
| Fig. 3 AFM of SG-CNTs dispersed reductively and through sonication (30 min) in NMP and SDS/water. Scale bars are 1 μm and all micrographs share the same height scale. Height profiles can be found in the ESI (Fig. S4–S6†). | ||
 | ||
| Fig. 4 Histograms of D/G mode intensities from statistical Raman spectroscopy for as received, charge/quenched control, NaNp/DMAc dissolved, and NMP and SDS/H2O sonicated SG-CNTs (30 min). Errors represent standard deviation. | ||
The nanotubide dissolution process does not intrinsically reduce aspect ratio, however, the kinetics depend strongly on SWCNT length and hence entanglement. Rigid rod-like species of length L in high concentration have rotational and translational relaxation times48 proportional to ∼L9 and ∼L respectively. While short SWCNTs are well modelled as rigid rods, very long SWCNTs may be more flexible and better treated as semi-flexible polymers,49 with reptation time also showing a strong length dependence (L3). In either model, SG-CNTs will tend to disentangle much more slowly than shorter SWCNTs. Mechanical stirring accelerates the kinetics of homogenizing for short SWCNTs by dispersing concentration gradients. In contrast, the high aspect ratio SG-CNTs are likely to remain highly entangled with simple dissolution unfeasibly slow. There is therefore a need to break the SG-CNTs before dissolution can occur on a reasonable timescale, hence the requirement for prolonged, strong stirring. Furthermore, the long SG-CNTs are much more easily broken by simple stirring, as they couple more strongly to shear in the solution, since stress transfer is enhanced with increasing SWCNT length.46
3.2 SWCNT/PVC composite fibres
Long SWCNTs offer advantages in many applications, in particular in composites. The mechanical behaviour of composites is expected to be heavily dependent on SWCNT aspect ratio and crystallinity. Three SWCNT sources were used for comparison: SG-CNTs and two commercial grade SWCNTs, Elicarb™ SWCNTs and Tuball™ SWCNTs, typically 1 μm in length (ESI, Fig. S6†).39 As-received SWCNTs were characterised via Raman (ESI, Fig. S7†) and TGA (ESI, Fig. S8†), indicating that the commercial SWCNTs contained higher catalyst content but lower defect concentrations than the SG-CNTs.For composite synthesis, a reactive coagulation process was developed, injecting the DMAc nanotubide solution into a co-flowing solution of reactive polymer solution in a miscible antisolvent. Akin to traditional coagulation fibre spinning, the diffusion of dope solvent into the coagulant causes the collapse of the nanotubes into a fibre, while the polymer from the coagulant bath diffuses into the forming fibre.29,50 However, here the use of a reactive polymer in the coagulant allows the simultaneous covalent grafting of the matrix polymer onto the SWCNTs, forming a strong SWCNT-matrix interface to increase interfacial shear strength, maximising the efficiency of matrix-CNT stress transfer, while wrapping the SWCNTs to limit weak inter-SWCNT interactions. The networking of the grafted PVC may also help to stablise the spinning process, whilst removing the stabilising Coulombic charge from the SWCNTs.
Polyvinyl chloride (PVC) was chosen as the composite fibre matrix, due to the ability of its C–Cl bonds to react with nanotubide;51 although bulk PVC has modest mechanical performance,31,32 it provides a model matrix for exploring the effects of both grafting and SWCNT aspect ratio. The PVC was dissolved in MiBK to form the nanotubide-reactive antisolvent; bundling of the SWCNTs from reaction with residual water is limited due to the low concentration of water in MiBK (281 ppm ± 18 ppm), far below the concentration of the reactive C–Cl bonds (0.64 M in the 5 wt% PVC solution used). Bulk PVC properties were measured using dog-bones cut from a solution cast PVC film as pure PVC fibres could not be synthesised from the wet-spinning route performed here.
To demonstrate the reactivity of PVC towards nanotubide in this reaction system, dilute (0.1 mg mL−1) nanotubide and NMP sonicated SWCNT dispersions were stirred with the PVC/MiBK solution (10× PVC weight versus SWCNT). After washing, the uncharged sonicated nanotubes showed smaller weight losses in TGA (10.6–23.6%, ESI, Fig. S11†) than the nanotubide samples (38.9–43.4 wt%), indicating that the reductive functionalisation contributes significantly.
Viscous, concentrated nanotubide solutions were prepared for all SWCNT types using NaNp/DMAc. Elicarb and Tuball SWCNTs dopes could only be spun at a concentration of 3.5 mg mL−1, while SG-CNTs solutions were successfully spun at low concentration (1.0 mg mL−1), as the high aspect ratio increased the viscosity significantly. In comparison to the high concentration nanotubide dopes used by Jiang et al.30 the lower concentrations used here precluded the possibility of coagulation fibre spinning of pure SWCNT fibres; injection of the dope into pure MiBK produced fibres too weak to remove from the coagulation bath.
In spite of the concentration differences between nanotubide dopes, the final composites from coagulation in the PVC solution contained similar SWCNT loadings of 16.1 to 27.1 wt% determined via TGA (Fig. 5f and ESI eqn (S1)†). The similarity in grafting ratios, independent of dope concentration, implies that only tightly-bound PVC remains in the produced composite fibre, likely through both wrapping and grafting. Under SEM, unwashed fibres clearly show excess, ungrafted PVC both on the fibre surface and dewetted along bundles in the fracture surface (ESI, Fig. S12†); no such features are present in the washed samples (Fig. 6), suggesting that the majority of the remaining PVC is tightly bound to the surface.
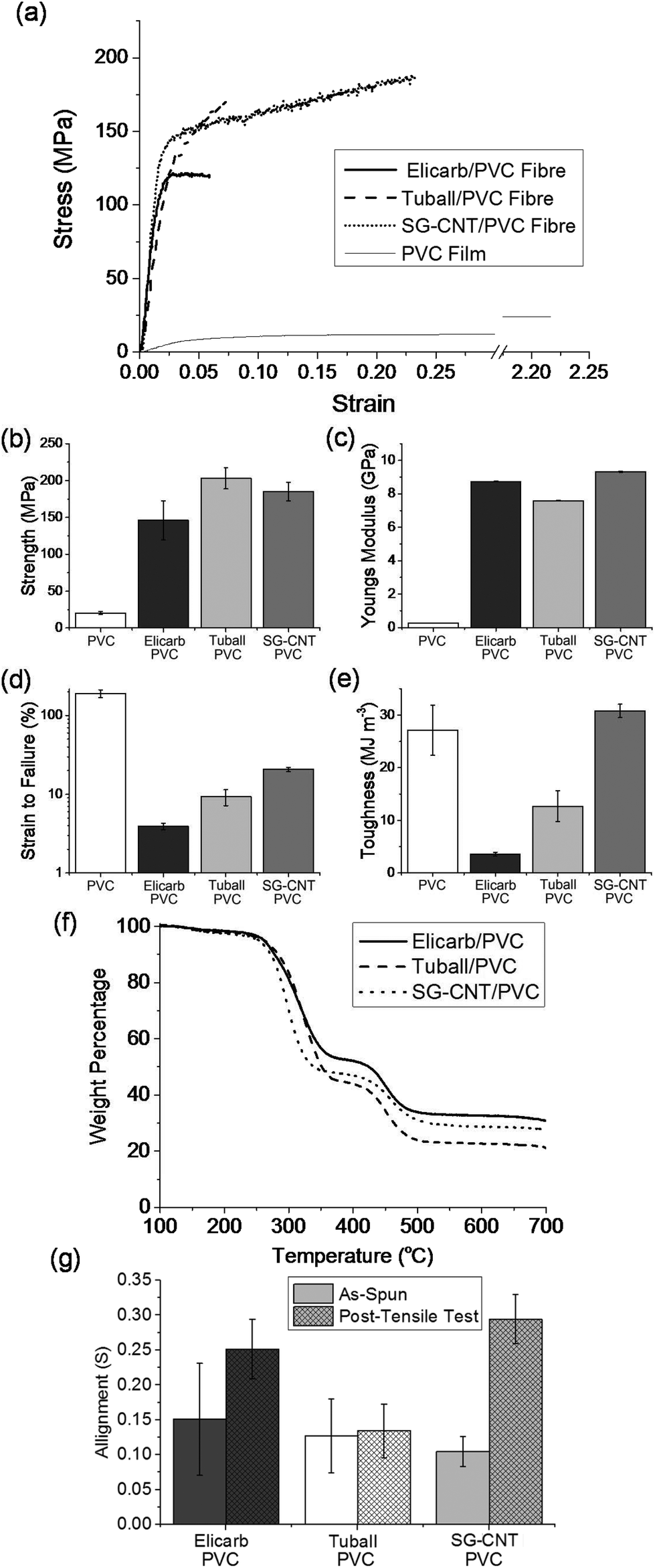 | ||
| Fig. 5 (a) Typical stress–strain curves illustrating the differing mechanical responses, (b) strength, (c) Young's modulus, (d) strain-to-failure, (e) toughness. Error bars represent standard errors, (f) Thermogravimetric profiles of SWCNT/PVC composite fibres under N2. (g) Alignment values (S) from polarised Raman spectroscopy before and after tensile failure. | ||
 | ||
| Fig. 6 SEM micrographs of tensile fracture surfaces for CNT/PVC composites. | ||
The very low electrical conductivity of the composites (<10–7 S cm−1) despite SWCNT volume fractions (φv) significantly higher than the threshold for electrical percolation52 (φv ≈ 1.5d2L−2) also indicates that a dense insulating PVC coating forms on the individual SWCNTs.
Intriguingly, the lack of conductivity combined with an inherent high dielectric constant of the individualised SWCNTs53 can be exploited to form high κ dielectric composites. Films produced from the wet-spun fibres demonstrated a dielectric constant of ca. 9, a 240% increase from a film PVC baseline (ESI, Fig. S13 and Table S3†) with a breakdown voltage >15 MV m−1.
All of the wet-spun fibres had a kidney-shaped cross-section, attributed to differing the relative rates of SWCNT coagulation and DMAc diffusion. Fibre diameters were all around 50 μm, with specific values of 54.4 μm ± 14.7 μm, 53.0 μm ± 8.2 μm, and 42.3 μm ± 8.4 μm for Elicarb and Tuball, and SG-CNT composite fibres, respectively. The lower diameter of the SG-CNT/PVC composite fibres is attributed to the lower SWCNT concentration in the dope. No large scale voids (>5 μm) were observed (Fig. 6); smaller scale cavities on the fracture surface may be intrinsically present or form during failure. The mechanical responses of the fibres, in tension, were initially similar, with Young's modulus around 7.5–9 GPa (Fig. 5c) and yield at 1–2% strain. The similarity in stiffness is as expected from simple short fibre models (such as Krenchel's)54 given the similar initial alignment (Fig. 5g), filler loading (Fig. 5f), and high aspect ratio of the CNTs in all cases; the absolute values are low, likely limited by the poor absolute alignment, nanotube bending, and potential microvoids. Given the low absolute stress at yield and consistency between samples, it can be assumed that the behaviour at this point is associated with the interface or matrix. In the post-yield regime, the SG-CNT/PVC composites stain harden and exhibit a significantly higher strain-to-failure (20.8 ± 1.26%, Fig. 5d) versus the shorter SWCNTs (3.9–9.3%), leading to a dramatic increase in the composite toughness (30.8 MJ m−3, Fig. 5e). This toughness represents an increase of 8.6 times and 2.4 times the Elicarb and Tuball SWCNT composite fibres, respectively, and is higher than even the pure PVC (27.1 MJ m−3). The increase in strain-to-failure for the SG-CNT composites is associated with the greatest increase in nanotube alignment (Fig. 5f), implying that the plastic deformation is associated with nanotube reorientation along the fibre axis of the entangled, long SG-CNTs. Conversely, the shorter CNTs produce composite fibres that fail soon after yield, leading to lower toughnesses and strain-to-failures; presumably the shorter CNTs are less entangled and readily pull-out of the yielding matrix. The slightly lower strength of the Elicarb versus both Tuball and SG-CNT composites is thought to be due to the presence of adsorbed amorphous carbons23 which may react reductively55 to covalently couple to the PVC but remain only weakly bound to the CNTs, reducing the load needed for pull-out and failure.
4. Conclusions
Supergrowth nanotubes can be dissolved by reductive charging in NaNp/DMAc to form stable, individualised solutions, more effectively than by other methods. Simple stirring of the nanotubide was sufficient to cut the nanotubes to ∼10 μm, still an order of magnitude higher than in typical SWCNT dispersions. The cutting was necessary to accelerate the very slow kinetics of dissolution of longer nanotubes; however, this more gentle process caused less damage and provided better dispersion than conventional ultrasonication. SWCNT/PVC composite fibres were created using a new reactive-coagulation fibre spinning route, through which the nanotubide is directly cross-linked with matrix polymer as the fibre forms. The polymer grafting produced surprisingly insulating, highly loaded SWCNT composites, with interesting dielectric properties, including a dielectric constant more than double that of PVC. The fibres formed using SG-CNTs are the strongest and toughest reported nanotubide-based fibres to date; however, the absolute performance was limited by the intrinsic performance of PVC, the intermediate SWCNT loadings (∼20 wt%) and relatively poor alignment. Control fibres were also prepared from typical, commercial SWCNTs, using the same process. The initial response to mechanical load was similar, however, the SG-CNTs showed a marked improvement, with a greatly extended plastic elongation after yield, whilst retaining high strength. The increased plastic strain resulted in a substantial improvement in toughness; an increase of 2.4 times when compared to the toughest short-SWCNT reinforced PVC composite fibre tested. The system provides further evidence that high aspect ratio SWCNTs are desirable for mechanical applications; the development of nanotubide routes as a method to process long SWCNTs, potentially in bulk quantities, is therefore crucial. The SWCNT/PVC composite fibres could be improved by refining the spinning conditions and introducing suitable post-processing conditions to maximise SWCNT content and alignment, and improve fibre macrostructure. The new reactive-wet-spinning process could also be applied to a variety of other, higher performance matrices through incorporation of nanotubide-reactive functional groups, or solutions of alternative charged nanomaterials.56 The reactive composite processing concept is also relevant to the preparation of interesting dielectric materials.Acknowledgements
The authors would like to acknowledge the EPSRC for the funding of this project (Doctoral Training Partnership, EP/M507878/1 and High Performance Ductile Composite Technology, EP/I02946X/1) and EU (MATFLEXEND Project #604093). In addition we extend our thanks and appreciation to Prof. Kenji Hata and his research group, The National Institute of Advanced Industrial Science and Technology in Japan (AIST), for providing the Supergrowth SWCNTs, and Thomas Swan Ltd for providing Elicarb SWCNTs. Supporting data can be requested from the corresponding author, but may be subject to confidentiality obligations. The authors declare no competing financial interest.References
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- S. A. Hodge, M. K. Bayazit, K. S. Coleman and M. S. P. Shaffer, Chem. Soc. Rev., 2012, 41, 4409–4429 RSC.
- L. X. Zheng, M. J. O'Connell, S. K. Doorn, X. Z. Liao, Y. H. Zhao, E. A. Akhadov, M. A. Hoffbauer, B. J. Roop, Q. X. Jia, R. C. Dye, D. E. Peterson, S. M. Huang, J. Liu and Y. T. Zhu, Nat. Mater., 2004, 3, 673–676 CrossRef CAS PubMed.
- A. N. Parra-Vasquez, N. Behabtu, M. J. Green, C. L. Pint, C. C. Young, J. Schmidt, E. Kesselman, A. Goyal, P. M. Ajayan, Y. Cohen, Y. Talmon, R. H. Hauge and M. Pasquali, ACS Nano, 2010, 4, 3969–3978 CrossRef CAS PubMed.
- J. Li, P. C. Ma, W. S. Chow, C. K. To, B. Z. Tang and J. K. Kim, Adv. Funct. Mater., 2007, 17, 3207–3215 CrossRef CAS.
- S. Ata, K. Kobashi, M. Yumura and K. Hata, Nano Lett., 2012, 12, 2710–2716 CrossRef CAS PubMed.
- C. L. Pint, S. T. Pheasant, M. Pasquali, K. E. Coulter, H. K. Schmidt and R. H. Hauge, Nano Lett., 2008, 8, 1879–1883 CrossRef CAS PubMed.
- K. Hata, D. N. Futaba, K. Mizuno, T. Namai, M. Yumura and S. Iijima, Science, 2004, 306, 1362–1364 CrossRef CAS PubMed.
- H. Kimura, D. N. Futaba, M. Yumura and K. Hata, J. Am. Chem. Soc., 2012, 134, 9219–9224 CrossRef CAS PubMed.
- T. Yamada, T. Namai, K. Hata, D. N. Futaba, K. Mizuno, J. Fan, M. Yudasaka, M. Yumura and S. Iijima, Nat. Nanotechnol., 2006, 1, 131–136 CrossRef CAS PubMed.
- J. A. Fagan, C. Y. Khripin, C. A. Silvera Batista, J. R. Simpson, E. H. Hároz, A. R. Hight Walker and M. Zheng, Adv. Mater., 2014, 26, 2800–2804 CrossRef CAS PubMed.
- S. D. Bergin, Z. Sun, P. Streich, J. Hamilton and J. N. Coleman, J. Phys. Chem. C, 2009, 114, 231–237 Search PubMed.
- A. Lucas, C. Zakri, M. Maugey, M. Pasquali, P. Schoot and P. Poulin, J. Phys. Chem. C, 2009, 113, 20599–20605 CAS.
- K. Kobashi, S. Ata, T. Yamada, D. N. Futaba, M. Yumura and K. Hata, Chem. Sci., 2012, 4, 727–733 RSC.
- H. Yoon, M. Yamashita, S. Ata, D. N. Futaba, T. Yamada and K. Hata, Sci. Rep., 2014, 4, 3907 CrossRef PubMed.
- K. Kazufumi, N. Hidekazu, Y. Takeo, N. Don, Y. Motoo and H. Kenji, Carbon, 2011, 49, 5090–5098 CrossRef.
- V. Davis, L. Ericson, A. Parra-Vasquez, H. Fan, Y. Wang, R. Smalley and M. Pasquali, Macromol., 2004, 37, 154–160 CrossRef CAS.
- A. Pénicaud, P. Poulin, A. Derré, E. Anglaret and P. Petit, J. Am. Chem. Soc., 2005, 127, 8–9 CrossRef PubMed.
- S. Fogden, C. A. Howard, R. K. Heenan, N. T. Skipper and M. S. P. Shaffer, ACS Nano, 2012, 6, 54–62 CrossRef CAS PubMed.
- F. Hof, S. Bosch, S. Eigler, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2013, 135, 18385–18395 CrossRef CAS PubMed.
- S. Hodge, S. Fogden, C. Howard, N. Skipper and M. S. P. Shaffer, ACS Nano, 2013, 7, 1769–1778 CrossRef CAS PubMed.
- D. D. Tune, A. J. Blanch, C. J. Shearer, K. E. Moore, M. Pfohl, J. G. Shapter and B. S. Flavel, ACS Appl. Mater. Interfaces, 2015, 7, 25857–25864 CAS.
- A. J. Clancy, J. Melbourne and M. S. P. Shaffer, J. Mater. Chem. A, 2015, 3, 16708–16715 CAS.
- J. Chattopadhyay, A. K. Sadana, F. Liang, J. M. Beach, Y. Xiao, R. H. Hauge and W. E. Billups, Org. Lett., 2005, 7, 4067–4069 CrossRef CAS PubMed.
- H. Qian, E. S. Greenhalgh, M. S. P. Shaffer and A. Bismarck, J. Mater. Chem., 2010, 20, 4751–4762 RSC.
- Y. Martinez-Rubi, B. Ashrafi, J. Guan, C. Kingston, A. Johnston, B. Simard, V. Mirjalili, P. Hubert, L. Deng and R. Young, ACS Appl. Mater. Interfaces, 2011, 3, 2309–2317 CAS.
- J. N. Coleman, M. Cadek, R. Blake, V. Nicolosi, K. P. Ryan, C. Belton, A. Fonseca, J. B. Nagy, Y. K. Gun'ko and W. J. Blau, Adv. Funct. Mater., 2004, 14, 791–798 CrossRef CAS.
- Y. Liu and S. Kumar, ACS Appl. Mater. Interfaces, 2014, 6, 6069–6087 CAS.
- C. Mercader, A. Lucas, A. Derré, C. Zakri, S. Moisan, M. Maugey and P. Poulin, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18331–18335 CrossRef CAS PubMed.
- C. Jiang, A. Saha, C. C. Young, D. P. Hashim, C. E. Ramirez, P. M. Ajayan, M. Pasquali and A. A. Martí, ACS Nano, 2014, 8, 9107–9112 CrossRef CAS PubMed.
- J. E. Mark, Physical Properties of Polymers Handbook, Springer Science, 2nd edn, 2007 Search PubMed.
- A. M. Hezma, I. S. Elashmawi, A. Rajeh and M. Kamal, Phys. B, 2016, 495, 4–10 CrossRef CAS.
- A. A. Aljaafari, S. S. Ibrahim and T. A. El-Brolossy, Composites, Part A, 2011, 42, 394–399 CrossRef.
- T. Sterzyński, J. Tomaszewska, K. Piszczek and K. Skórczewska, Compos. Sci. Technol., 2010, 70, 966–969 CrossRef.
- V. J. Mkhabela, A. K. Mishra and X. Y. Mbianda, Carbon, 2011, 49, 610–617 CrossRef CAS.
- N. Ahmad, A. Kausar and B. Muhammad, Polym. – Plast. Technol. Eng., 2016, 55, 1076–1098 CrossRef CAS.
- H. J. Salavagione, G. Martínez and C. Ballesteros, Macromol., 2010, 43, 9754–9760 CrossRef CAS.
- X. L. Wu, eXPRESS Polym. Lett., 2010, 4, 723–728 CAS.
- A. J. Clancy, E. R. White, H. H. Tay, H. C. Yau and M. S. P. Shaffer, Carbon, 2016, 108, 423–432 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- N. Puech, C. Blanc, E. Grelet, C. Zamora-Ledezma, M. Maugey, C. Zakri, E. Anglaret and P. Poulin, J. Phys. Chem. C, 2011, 115, 3272–3278 CAS.
- S. Bergin, Z. Sun, D. Rickard, P. Streich, J. Hamilton and J. Coleman, ACS Nano, 2009, 3, 2340–2350 CrossRef CAS PubMed.
- D. Voiry, C. Drummond and A. Pénicaud, Soft Matter, 2011, 7, 7998–8001 RSC.
- D. N. Futaba, K. Hata, T. Namai, T. Yamada, K. Mizuno, Y. Hayamizu, M. Yumura and S. Iijima, J. Phys. Chem. B, 2006, 110, 8035–8038 CrossRef CAS PubMed.
- E. R. Meshot, M. Bedewy, K. M. Lyons, A. R. Woll, A. K. Juggernauth, S. Tawfick and J. A. Hart, Nanoscale, 2010, 2, 896–900 RSC.
- Y. Y. Huang, T. P. J. Knowles and E. M. Terentjev, Adv. Mater., 2009, 21, 3945–3948 CrossRef CAS.
- J. A. Fagan, J. R. Simpson, B. J. Bauer, S. H. Lacerda, M. L. Becker, J. Chun, K. B. Migler, A. R. Walker and E. K. Hobbie, J. Am. Chem. Soc., 2007, 129, 10607–10612 CrossRef CAS PubMed.
- N. Fakhri, F. C. MacKintosh, B. Lounis, L. Cognet and M. Pasquali, Science, 2010, 330, 1804–1807 CrossRef CAS PubMed.
- M. J. Green, N. Behabtu, M. Pasquali and W. W. Adams, Polymer, 2009, 50, 4979–4997 CrossRef CAS.
- A. Penicaud, L. Valat, A. Derre, P. Poulin, C. Zakri, O. Roubeau, M. Maugey, P. Miaudet, E. Anglaret, P. Petit, A. Loiseau and S. Enouz, Compos. Sci. Technol., 2007, 67, 795–797 CrossRef CAS.
- R. Blake, Y. K. Gun'ko, J. Coleman, M. Cadek, A. Fonseca, J. B. Nagy and W. J. Blau, J. Am. Chem. Soc., 2004, 126, 10226–10227 CrossRef CAS PubMed.
- M. B. Bryning, M. F. Islam, J. M. Kikkawa and A. G. Yodh, Adv. Mater., 2005, 17, 1186–1191 CrossRef CAS.
- B. Wu, D. Geng and Y. Liu, Nanoscale, 2011, 3, 2074–2085 RSC.
- H. Krenchel, Fibre reinforcement : theoretical and practical investigations of the elasticity and strength of fibre-reinforced materials, Akademisk Forlag, Copenhagen, 1964 Search PubMed.
- H. S. Leese, L. Govada, E. Saridakis, S. Khurshid, R. Menzel, T. Morishita, A. J. Clancy, E. R. White, N. E. Chayen and M. S. P. Shaffer, Chem. Sci., 2016, 7, 2916–2923 RSC.
- P. L. Cullen, K. M. Cox, M. K. Subhan, L. Picco, O. D. Payton, D. J. Buckley, T. S. Miller, S. A. Hodge, N. T. Skipper, V. Tileli and C. A. Howard, Nat. Chem., 2016, 9, 244–249 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data (SEM, AFM line profiles, Raman spectra, TGA), schematic of spinning equipment, full tabulation of mechanical data, discussions and calculations of critical length reinforcement and dielectric properties. See DOI: 10.1039/c7nr00734e |
| This journal is © The Royal Society of Chemistry 2017 |