
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-performance thermoelectricity in edge-over-edge zinc-porphyrin molecular wires†
Mohammed
Noori‡
ab,
Hatef
Sadeghi
 a and
Colin J.
Lambert
*a
a and
Colin J.
Lambert
*a
aQuantum Technology Centre, Department of Physics, Lancaster University, Lancaster LA1 4YB, UK. E-mail: h.sadeghi@lancaster.ac.uk; c.lambert@lancaster.ac.uk
bDepartment of Physics, College of Science, Thi-Qar University, Thi-Qar, Iraq
First published on 31st March 2017
Abstract
If high efficiency organic thermoelectric materials could be identified, then these would open the way to a range of energy harvesting technologies and Peltier coolers using flexible and transparent thin-film materials. We have compared the thermoelectric properties of three zinc porphyrin (ZnP) dimers and a ZnP monomer and found that the “edge-over-edge” dimer formed from stacked ZnP rings possesses a high electrical conductance, negligible phonon thermal conductance and a high Seebeck coefficient of the order of 300 μV K−1. These combine to yield a predicted room-temperature figure of merit of ZT ≈ 4, which is the highest room-temperature ZT ever reported for a single organic molecule. This high value of ZT is a consequence of the low phonon thermal conductance arising from the stacked nature of the porphyrin rings, which hinders phonon transport through the edge-over-edge molecule and enhances the Seebeck coefficient.
Introduction
Thermoelectric materials, which convert heat to electrical energy, could have an enormous impact on global energy consumption, but at present their efficiency is too low and the most efficient materials are toxic and have limited global supply. Recently, in an effort to overcome these limitations, thermoelectric effects in low-dimensional structures and molecular-scale systems have begun to be investigated.1–14 Nanostructures are promising, because transport takes place through discrete energy levels and in molecular-scale junctions, this leads to room-temperature quantum interference, which opens further avenues for enhancing the conversion of heat into electrical energy.15The efficiency of a thermoelectric (TE) material or device is determined by the dimensionless thermoelectric figure of merit ZT = GS2T/κ, where G is the electrical conductance, T is temperature, S is the thermopower (Seebeck coefficient) and κ = κel + κph is the thermal conductance due to electrons (κel) and phonons (κph). The Seebeck coefficient characterizes the ability of a thermoelectric material to convert heat to electricity and is defined as S = −ΔV/ΔT, where ΔV is the voltage difference generated between the two ends of the junction when a temperature difference ΔT is established between them.7,16–19 Enhancing the efficiency of TE materials is not easy, because all parameters are correlated. For example at the fundamental level, the electronic properties G, S and κel are related, because they are all derived from the transmission coefficient Tel(E) describing electrons of energy E passing from one electrode to other through a molecule (see Methods). In particular the Seebeck coefficient S is approximately proportional to the slope of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Tel(E), evaluated at the Fermi energy EF, whereas the electrical conductance is proportional to Tel(EF). Therefore, if the Fermi energy lies in a region of high slope, close to a transmission resonance then both G and S are enhanced.20 On the other hand, to decrease the thermal conductance κ, which appears in the denominator of ZT, both electron and phonon transport must be engineered. Therefore, simultaneous consideration of both electron and phonon transport is needed to develop new materials for thermoelectricity.
Tel(E), evaluated at the Fermi energy EF, whereas the electrical conductance is proportional to Tel(EF). Therefore, if the Fermi energy lies in a region of high slope, close to a transmission resonance then both G and S are enhanced.20 On the other hand, to decrease the thermal conductance κ, which appears in the denominator of ZT, both electron and phonon transport must be engineered. Therefore, simultaneous consideration of both electron and phonon transport is needed to develop new materials for thermoelectricity.
Since only a few groups worldwide are able to measure the thermal conductance of single molecules, theoretical investigation is needed to identify new strategies to simultaneously suppress phonons and enhance S and G. Recent proposals to reduce phonon transport in molecular junctions include weakening the overlap between the continuum of vibrational states in the electrodes and discrete vibrational modes of the molecules,21 taking advantage of the weak interaction between different parts of the molecules, as in π–π stacked structures19 and using the low Debye frequency of electrodes to filter high-frequency phonons.20 The challenge is to identify new materials and device structures in which such strategies can be realized in the laboratory. In this paper, we present a comparative theoretical study of the thermoelectric properties of four different zinc porphyrin structures and elucidate a new strategy for simultaneously increasing their thermopower and reducing their thermal conductance leading to a high value of ZT.
Methods
The geometry of each structure consisting of gold electrodes and a single zinc porphyrin molecule was relaxed to a force tolerance of 20 meV Å−1 using the SIESTA28 implementation of density functional theory (DFT), with a double-ζ polarized basis set (DZP) and generalized gradient functional approximation (GGA-PBE) for the exchange and correlation functionals,30,31 which is applicable to arbitrary geometries. A real-space grid was defined with an equivalent energy cutoff of 150 Ry. From the relaxed xyz coordinate of the system, sets of xyz coordinates were generated by displacing each atom in positive and negative x, y, and z directions by δq′ = 0.01 Å. The forces in three directions qi = (xi, yi, zi) on each atom were then calculated by DFT without geometry relaxation. These values of force are combined with the method described in ref. 20 to calculate the dynamical matrix and thermal conductance due to phonons.To calculate the electronic properties of the molecules in the junction, from the converged DFT calculation, the underlying mean-field Hamiltonian H was combined with our quantum transport code, GOLLUM29 to calculate the transmission coefficient Tel(E) for electrons of energy E passing from the source to the drain. The electrical conductance Gel(T) = G0L0, the electronic contribution of the thermal conductance κel(T) = (L0L2 − L12)/hTL0 = and the thermopower S(T) = −L1/eTL0 of the junction are calculated from the electron transmission coefficient Tel(E) where  and f(E,T) is the Fermi–Dirac probability distribution function f(E,T) = (e(E−EF)/KBT + 1)−1, T is the temperature, EF is the Fermi energy, G0 = 2e2/h is the conductance quantum, e is electron charge, and h is the Planck's constant.
and f(E,T) is the Fermi–Dirac probability distribution function f(E,T) = (e(E−EF)/KBT + 1)−1, T is the temperature, EF is the Fermi energy, G0 = 2e2/h is the conductance quantum, e is electron charge, and h is the Planck's constant.
Results and discussion
Fig. 1 shows four different zinc porphyrin (ZnP) structures investigated below. The first 1 is a ZnP monomer.22 Structure 2 is an edge-over-edge ZnP dimer, in which two ZnPs are locked together by meso-position pyridines.23,24 Structure 3 comprises two ZnPs connected by an oligoyne linker,22,25,26 while 4 comprises two ZnPs connected through meso-position pyridines.27 In what follows, our aim is to demonstrate that of the above structures, the edge-over-edge ZnP dimer 2 is by far the most efficient thermoelectric energy converter. From a structural point of view, this arises because the pyridyl rings of 2 are locked and therefore ring rotation, which would otherwise reduce the electrical conductance, is eliminated. Secondly, the edge-over-edge rigid conformation of 2 increases its rigidity, which pushes the internal vibrational modes to higher frequencies. This reduces room temperature thermal conductance, because modes with frequencies greater than ∼25 meV do not contribute significantly. Thirdly, longitudinal modes entering one end of the edge-over-edge molecule must convert to flexural modes to pass from one porphyrin to the other, which creates extra phonon scattering and reduces thermal conductance. | ||
| Fig. 1 The device structures investigated consist of four different zinc porphyrin (ZnP) monomer structures. (1) Edge-over-edge ZnP, (2) a ZnP-dimer linked by an oligoyne chain, (3) a ZnP-dimer linked by two pyridyl rings (4). | ||
For the structures of Fig. 1, Fig. 2 show the transmission coefficients for electrons with energy E and phonons of energy ħω, passing through a molecule from the left electrode to the right electrode, calculated using the method described in ref. 20. We first carry out geometrical optimization of each molecule placed between two gold electrodes using the SIESTA28 implementation of density functional theory (DFT) to find the ground state optimized positions of the atoms relative to each other (see Methods). From the ground state geometry, we obtain the mean-field Hamiltonian of each system comprising both electrodes and molecules and use our transport code GOLLUM29 to calculate the transmission coefficients Tel(E) (see Methods). In each case the optimal angle between the porphyrins is zero, which corresponds to the maximum conductance that could be obtained.22 The electronic transport properties of 1 and 3 have been studied experimentally in the literature,22 so we used these to benchmark our calculations. As shown in Table S1 of the ESI,† our calculated conductances for these molecules are in good agreement with experiment. The electron transmission of 4 is much smaller than 1, 2 and 3, whereas the transmission of 2 is either equal to that of 3 near the HOMO resonance or lower in the vicinity of the middle of the HOMO–LUMO gap. As shown in Fig. (SI2†), this is reflected in the electrical conductance as a function of temperature.
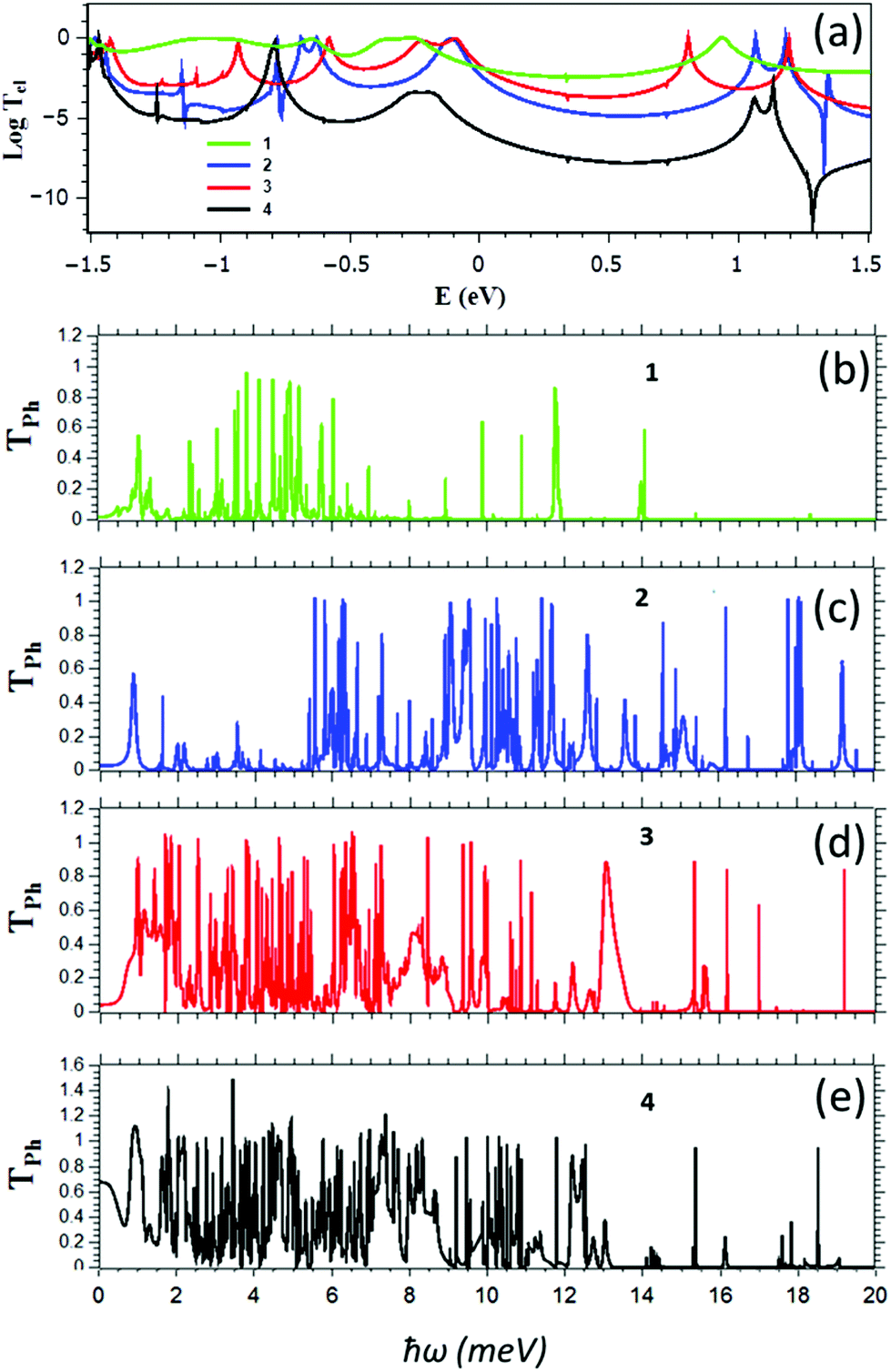 | ||
| Fig. 2 (a) Electron transmission coefficients as a function of energy and (b–e) phonon transmission coefficients as a function of ħω for the ZnP monomer 1, the edge-over-edge ZnP 2, the ZnP dimer connected via an oligoyne chain 3 and ZnP dimer connected through pyridyl rings 4. | ||
To calculate the vibrational properties of each structure, we use the harmonic approximation to construct the dynamical matrix D. Each atom is displaced from its ground-state equilibrium position by δq′ and −δq′ in the x, y, and z directions and the forces on all atoms calculated in each case. For 3n degrees of freedom (n = number of atoms), the 3n × 3n dynamical matrix Dij = (Fqi(δq′j) − Fqj(−δq′j))/2Mijδq′j is constructed, where F and M are the force and mass matrices (see ref. 20 for more details). For an isolated molecule, the square root of the given values of D determines the frequencies ω associated with the vibrational modes of the molecule in the junction (see the ESI†). For a molecule within a junction, the dynamical matrix describes an open system composed of the molecule and two semi-infinite electrodes and is used to calculate the transmission coefficient Tph(ω) for phonons with energy ħω passing through the molecule from the right to the left electrodes.20
Fig. 2b–e shows Tph(ω) for the four structures of Fig. 1. It is apparent that the widths of the resonances in the edge-over-edge ZnP-dimer 2 are smaller than those of the other structures and the low energy phonons (in the range 2–5 meV) are either suppressed or pushed to higher frequencies. This can be demonstrated using the participation ratio of the dimer molecular cores 2, 3 and 4 and comparing the integrated density of states N(ħω) of 2, 3 and 4. As shown in Fig. S1 of the ESI,† the participation ratio of the molecule core (ZnPs and linkers) connected to the gold surface is mostly due to the in-plane (PRy) and out of plane (PRx) transverse modes in structures 3 and 4, whereas out-of plane transverse modes are mainly suppressed or converted to in-plane transverse modes and moved to the higher frequency, reflecting the higher rigidity of the edge-over-edge structure. In addition, the integrated density of states is almost the same for 3 and 4, whereas for low frequencies, the integrated density of states of 2 is smaller than 3 and 4. This means the thermal conductance is reduced significantly in 2, because transmission of the low energy modes is suppressed due to the scattering from in-plane modes to cross-plane transverse modes. In addition, some modes are pushed to higher frequency, although this is smaller effect compared with the suppression of low frequency transmission. Overall, these two effects combine to yield a lower phonon thermal conductance in 2.
The thermal conductance of the junction (κ = κph + κel) is obtained by summing the contributions from both electrons (κel) and phonons (κph). The electronic (phonon) thermal conductances are calculated from the electron (phonon) transmission coefficients shown in Fig. 2a–e. Fig. 3a shows that the ZnP monomer 1 has the lowest value of κph while 4 has the highest. This is counter-intuitive, because one would expect a higher thermal conductance for shorter molecules. However, due to the more rigid nature of the monomer, its vibrational modes are pushed to higher frequencies and therefore their contribution to the room temperature conductance is suppressed. In addition, Fig. 3b and c show that the thermal conductance due to the electrons κel of the dimer ZnP 3 is higher than those of the edge-over-edge ZnP and structures 1 and 4 for a wide range of energy in the vicinity of DFT predicted Fermi energy. The crucial point is that almost for all Fermi energies, the electronic contribution to the thermal conductance is higher than the phonon contribution. This is significant, because to achieve a high-ZT material, one needs to only focus on engineering the electronic properties of structure 2.
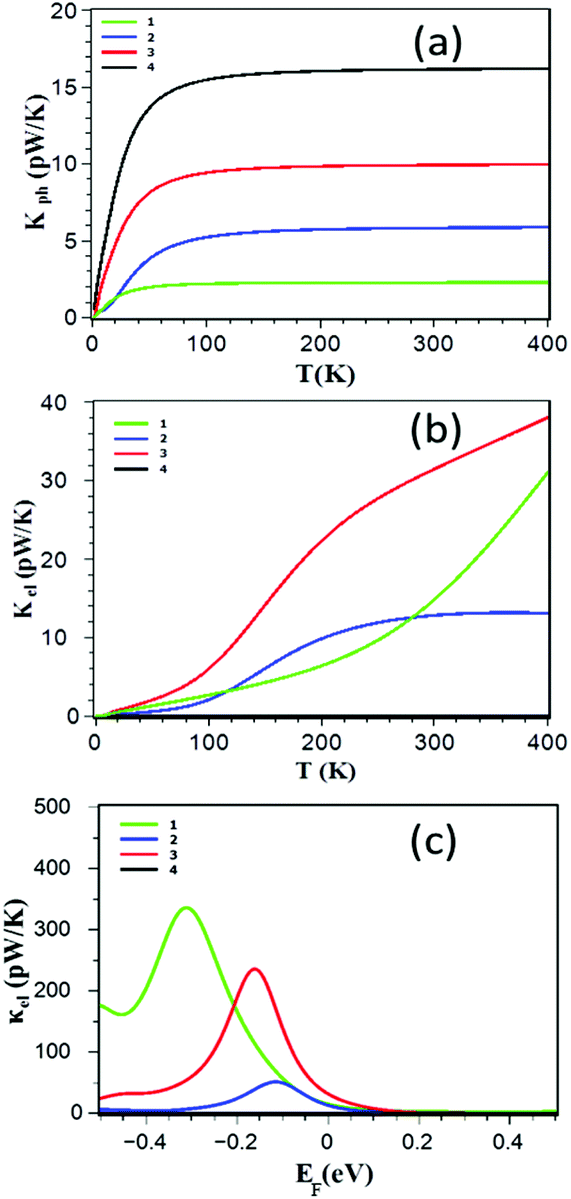 | ||
| Fig. 3 (a) Phonon thermal conductances (b) electronic thermal conductance as a function of temperature, (c) room-temperature electronic thermal conductance as a function of Fermi energy EF calculated using the DFT-predicted Fermi energy. The results are shown for the ZnP monomer 1, the edge-over-edge ZnP 2, the ZnP dimer connected through an oligoyne chain 3 and the ZnP-dimer connected through pyridyl rings 4. | ||
To examine the thermoelectric properties of 1–4, we obtained the Seebeck coefficient of all structures from the electron transmission coefficient Tel(E), as described in the Methods. Fig. 4a shows the Seebeck coefficients as a function of Fermi energy EF (and also as a function of temperature in Fig. S3 of the ESI†) and reveals that the edge-over-edge ZnP dimer 2 has a higher Seebeck coefficient than 1, 3 and 4 due to the higher slope of lnTel(EF) over a wide range of Fermi energies between the HOMO and LUMO. Since the electronic contribution to the thermal conductance is higher in 1, 2 and 3, the contribution of the phonons is negligible. Furthermore the electrical conductance is proportional to the electronic thermal conductance, so they cancel each other in ZT. Consequently as shown in Fig. 4b, due to the high Seebeck coefficient of the edge-over-edge dimer, a ZT as high as ≈4 is obtained when EF lies in a wide energy window in the vicinity of the DFT-predicted Fermi energy. Fig. 4b also shows that the less-rigid structure 4 is not promising for efficient conversion of the heat to electricity. Although all of these structures are made from ZnP, this study shows the importance of the molecular design. The more rigid edge-over-edge ZnP dimer 2 shows a very high ZT, whereas the less conductive structure 3 is unattractive for thermoelectricity.
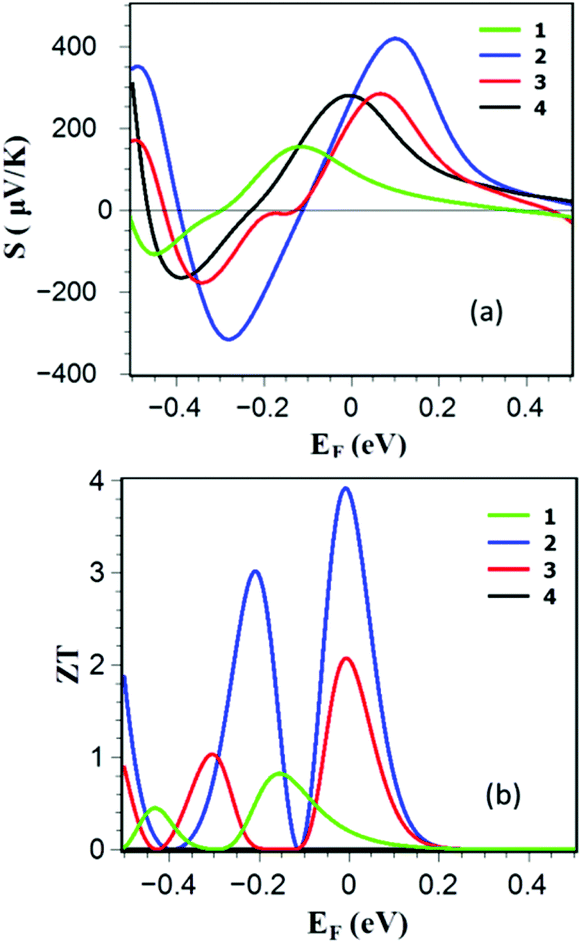 | ||
| Fig. 4 (a) Seebeck coefficient S and (b) full thermoelectric figure of merit ZT as a function of Fermi energy for the ZnP monomer 1, edge-over-edge ZnP 2, ZnP connected through an oligoyne chain 3 and ZnP-dimer connected through pyridyl rings 4. | ||
To our knowledge, there currently exist no measurements of the Seebeck coefficient of porphyrins and no measurements of their ZT. The Seebeck coefficient of n-alkanedithiol (ADT) (n = 2, 3, 4, 5, 6, 8) was found to range from 6.8 to 2.4 μV K−1, depending on the length n,32,33 whereas 1,4-benzenedithiol (BDT), 4,4′-dibenzenedithiol (DBDT) and 4,4′′-tribenzenedithiol (TBDT) attached to Au electrodes have been measured by several groups34–41 and found to increase with the number of phenyl rings, ranging from 7 μV K−1 to 16 μV K−1, by changing the terminal groups, measurements on 1,4-bis((trimethylstannyl)methyl)-n-phenyl (n = 1, 2, 3, 4) revealed that the Seebeck coefficient increased to 24 μV K−1 for the longest molecules. On the other hand 1,4-n-phenylenediamine (PDA) (n = 1, 2, 3) was found to have lower values in the range of 2.1–10.4 μV K−1.32,42,43 These values are comparable to the Seebeck coefficient of oligothiophenes, which can be as high as 14.8 μV K−1 for ethanethioate-terminated terthiophenes on gold.44
Changing the terminal groups to pyridine generally switches transport towards the LUMO and changes the sign of the Seebeck coefficient. For example the Seebeck coefficients of 4,4′-bipyridine and 1,2-di(4-pyridyl)ethylene attached to gold electrodes were found to be approximately −9 μV K−1 and −10 μV K−1 respectively.43,45
Fullerenes tend to have higher Seebeck coefficients, ranging from −10 to −30 μV K−1 for C60 with different metallic electrodes46 to −33 μV K−1 for C60 dimers47 and up to −31.6 μV K−1 C82 endohedral fullerenes.48 The sign of the endohedral fullerene Sc3N@C80 was shown to be sensitive to pressure, ranging from −25 μV K−1 to +25 μV K−1, depending on the orientation of the molecule on a gold substrate.49
The length dependent thermal conductance in Au-alkanethiol-SiO2 junctions by the scanning thermal microscopy (SThM) technique has been measured to vary from 25 pW K−1 for the shortest chains to 10 pW K−1 for chains of 18 carbon atoms.50 The thermal conductance of Au-alkanedithol (n = 8, 9, 10)-GaAs junctions at room temperature51 was measured to be approximately 27 MW m−2 K−1, assuming a 1 nm2 footprint per molecule, this equates to 27 pW K−1 per molecule. Values ranging from 35 to 65 MW m−2 K−1 were measured for different terminal groups.52
Single-molecule thermoelectricity is a rapidly expanding field of fundamental research and our paper is aimed primarily at further developing our understanding at the single-molecule level. Acquisition of the fundamental understanding of single-molecule thermoelectricity will underpin the design and synthesis of high-performance molecular films, but will not be sufficient, because a range of additional issues arise when single molecules are placed in parallel to form a molecular film. Ref. 53 discusses the question of how a statistical ensemble of single-molecule conductances and Seebeck coefficients combine in parallel to yield the thermoelectric properties of a dilute molecular film and concludes that a conductance-weighted single-molecule Seebeck coefficient is the relevant quantity. For more dense films, thermoelectric properties may be modified by intermolecular interactions, although as mentioned above, measurements of single-molecule thermal conductances of alkanes50 are comparable to the thermal conductance per molecule obtained from the measurements on molecular films51,52 and therefore at least for alkanes, such interactions do not appear to be significant.
Conclusions
In summary, we have compared the thermoelectric properties of three ZnP dimers and a ZnP monomer and found that the edge-over-edge-like dimer possesses a negligible phonon thermal conductance and a high Seebeck coefficient of the order of 300 μV K−1. These transport properties combine to yield a room-temperature figure of merit of ZT ≈ 4, which is the highest room-temperature ZT ever reported for an organic material. This high ZT value is a consequence of low phonon thermal conductance, which arises from the edge-over-edge stacking of the porphyrin rings and hinders phonon transport through the molecule.Acknowledgements
This work is supported by UK EPSRC grants EP N017188/1, EP/M014452/1, the European Union Marie-Curie Network MOLESCO 606728 and the Ministry of Higher Education and Scientific Research, Thi-Qar University, Iraq. We dedicate this paper to the memory of the late Thomas Wandlowski, a world-leading scientist and a true friend, whose premature passing is a great loss to the molecular-electronics community.References
- H. Sadeghi, S. Sangtarash and C. J. Lambert, Sci. Rep., 2015, 5, 9514–9520 CrossRef CAS PubMed.
- M. Christensen, A. B. Abrahamsen, N. B. Christensen, F. Juranyi, N. H. Andersen, K. Lefmann, J. Andreasson, C. R. Bahl and B. B. Iversen, Nat. Mater., 2008, 7, 811–815 CrossRef CAS PubMed.
- G. Joshi, H. Lee, Y. Lan, X. Wang, G. Zhu, D. Wang, R. W. Gould, D. C. Cuff, M. Y. Tang and M. S. Dresselhaus, Nano Lett., 2008, 8, 4670–4674 CrossRef CAS PubMed.
- W. Kim, S. L. Singer, A. Majumdar, D. Vashaee, Z. Bian, A. Shakouri, G. Zeng, J. E. Bowers, J. M. Zide and A. C. Gossard, Appl. Phys. Lett., 2006, 88, 242107 CrossRef.
- R. Venkatasubramanian, E. Siivola, T. Colpitts and B. O'quinn, Nature, 2001, 413, 597–602 CrossRef CAS PubMed.
- L. Hicks, T. Harman, X. Sun and M. Dresselhaus, Phys. Rev. B: Condens. Matter, 1996, 53, R10493 CrossRef CAS.
- P. Reddy, S.-Y. Jang, R. A. Segalman and A. Majumdar, Science, 2007, 315, 1568–1571 CrossRef CAS PubMed.
- A. I. Hochbaum, R. Chen, R. D. Delgado, W. Liang, E. C. Garnett, M. Najarian, A. Majumdar and P. Yang, Nature, 2008, 451, 163–167 CrossRef CAS PubMed.
- K. Baheti, J. A. Malen, P. Doak, P. Reddy, S.-Y. Jang, T. D. Tilley, A. Majumdar and R. A. Segalman, Nano Lett., 2008, 8, 715–719 CrossRef CAS PubMed.
- A. I. Boukai, Y. Bunimovich, J. Tahir-Kheli, J.-K. Yu, W. A. Goddard Iii and J. R. Heath, Nature, 2008, 451, 168–171 CrossRef CAS PubMed.
- K. Schwab, E. Henriksen, J. Worlock and M. L. Roukes, Nature, 2000, 404, 974–977 CrossRef CAS PubMed.
- K. Uchida, S. Takahashi, K. Harii, J. Ieda, W. Koshibae, K. Ando, S. Maekawa and E. Saitoh, Nature, 2008, 455, 778–781 CrossRef CAS PubMed.
- X. Zheng, W. Zheng, Y. Wei, Z. Zeng and J. Wang, J. Chem. Phys., 2004, 121, 8537–8541 CrossRef CAS PubMed.
- M. Paulsson and S. Datta, Phys. Rev. B: Condens. Matter, 2003, 67, 241403 CrossRef.
- L. A. Algharagholy, Q. Al-Galiby, H. A. Marhoon, H. Sadeghi, H. M. Abduljalil and C. J. Lambert, Nanotechnology, 2015, 26, 475401 CrossRef PubMed.
- M. Wierzbicki and R. Świrkowicz, J. Phys.: Condens. Matter, 2010, 22, 185302 CrossRef CAS PubMed.
- C. S. Lau, H. Sadeghi, G. Rogers, S. Sangtarash, P. Dallas, K. Porfyrakis, J. Warner, C. J. Lambert, G. A. D. Briggs and J. A. Mol, Nano Lett., 2015, 16, 170–176 CrossRef PubMed.
- N. A. Zimbovskaya, J. Phys.: Condens. Matter, 2014, 26, 275303 CrossRef PubMed.
- H. Sadeghi, S. Sangtarash and C. J. Lambert, Beilstein J. Nanotechnol., 2015, 6, 1176–1182 CrossRef CAS PubMed.
- H. Sadeghi, S. Sangtarash and C. J. Lambert, Nano Lett., 2015, 15, 7467–7472 CrossRef CAS PubMed.
- R. Y. Wang, R. A. Segalman and A. Majumdar, Appl. Phys. Lett., 2006, 89, 173113 CrossRef.
- G. Sedghi, V. M. García-Suárez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martín, D. Bethell, S. J. Higgins, M. Elliott and N. Bennett, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS PubMed.
- R. T. Stibrany, J. Vasudevan, S. Knapp, J. A. Potenza, T. Emge and H. J. Schugar, J. Am. Chem. Soc., 1996, 118, 3980–3981 CrossRef CAS.
- J. Vasudevan, R. T. Stibrany, J. Bumby, S. Knapp, J. A. Potenza, T. J. Emge and H. J. Schugar, J. Am. Chem. Soc., 1996, 118, 11676–11677 CrossRef CAS.
- A. K. Burrell, D. L. Officer, P. G. Plieger and D. C. Reid, Chem. Rev., 2001, 101, 2751–2796 CrossRef CAS PubMed.
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed.
- K. F. Cheng, C. M. Drain and K. Grohmann, Inorg. Chem., 2003, 42, 2075–2083 CrossRef CAS PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- J. Ferrer, C. J. Lambert, V. M. García-Suárez, D. Z. Manrique, D. Visontai, L. Oroszlany, R. Rodríguez-Ferradás, I. Grace, S. Bailey and K. Gillemot, New J. Phys., 2014, 16, 093029 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter, 1999, 59, 7413 CrossRef.
- J. A. Malen, et al. , Nano Lett., 2009, 9, 1164 CrossRef CAS PubMed.
- S. Y. Guo, G. Zhou and N. J. Tao, Nano Lett., 2013, 13, 4326 CrossRef CAS PubMed.
- A. Tan, S. Sadat and P. Reddy, Appl. Phys. Lett., 2010, 96, 013110 CrossRef.
- A. Tan, et al. , J. Am. Chem. Soc., 2011, 133, 8838 CrossRef CAS PubMed.
- S. K. Lee, et al. , Nano Lett., 2014, 14, 5276 CrossRef CAS PubMed.
- Y. Kim, et al. , Appl. Phys. Lett., 2016, 109, 033102 CrossRef.
- D. Z. Manrique, Q. Al-Galiby, W. Hong and C. J. Lambert, Nano Lett., 2016, 16, 1308–1316 CrossRef CAS PubMed.
- M. Tsutsui, M. Taniguchi and T. Kawai, Nano Lett., 2008, 8, 3293 CrossRef CAS PubMed.
- T. Morikawa, et al. , Nanoscale, 2014, 6, 8235 RSC.
- S. Kaneko, et al. , Appl. Phys. Express, 2015, 8, 095503 CrossRef.
- D. Kim, P. S. Yoo and T. Kim, J. Korean Phys. Soc., 2015, 66, 602 CrossRef CAS.
- T. Kim, et al. , Nano Lett., 2014, 14, 794 CrossRef CAS PubMed.
- W. B. Chang, et al. , Chem. Mater., 2014, 26, 7229 CrossRef CAS.
- J. R. Widawsky, et al. , Nano Lett., 2012, 12, 354 CrossRef CAS PubMed.
- S. K. Yee, et al. , Nano Lett., 2011, 11, 4089 CrossRef CAS PubMed.
- C. Evangeli, et al. , Nano Lett., 2013, 13, 2141 CrossRef CAS PubMed.
- S. K. Lee, et al. , Nanoscale, 2015, 7, 20497 RSC.
- L. Rincon-Garcia, et al. , Nat. Mater., 2016, 15, 289 CrossRef CAS PubMed.
- T. Meier, et al. , Phys. Rev. Lett., 2014, 113, 060801 CrossRef CAS PubMed.
- R. Y. Wang, R. A. Segalman and A. Majumdar, Appl. Phys. Lett., 2006, 89, 173113 CrossRef.
- M. D. Losego, et al. , Nat. Mater., 2012, 11, 502 CrossRef CAS PubMed.
- C. J. Lambert, H. Sadeghi and Q. Al-Galiby, C. R. Phys., 2016, 17(10), 1084 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr09598d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |