
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pulmonary surfactant protein SP-D opsonises carbon nanotubes and augments their phagocytosis and subsequent pro-inflammatory immune response†
Kirsten M.
Pondman‡
ab,
Basudev
Paudyal‡
ac,
Robert B.
Sim
 d,
Anuvinder
Kaur
a,
Lubna
Kouser
a,
Anthony G.
Tsolaki
a,
Lucy A.
Jones
c,
Carolina
Salvador-Morales
e,
Haseeb A.
Khan
f,
Bennie
ten Haken
b,
Gudrun
Stenbeck
a and
Uday
Kishore
*a
d,
Anuvinder
Kaur
a,
Lubna
Kouser
a,
Anthony G.
Tsolaki
a,
Lucy A.
Jones
c,
Carolina
Salvador-Morales
e,
Haseeb A.
Khan
f,
Bennie
ten Haken
b,
Gudrun
Stenbeck
a and
Uday
Kishore
*a
aDepartment of Life Sciences, College of Health and Life Sciences, Brunel University London, Uxbridge UB8 3PH, UK. E-mail: uday.kishore@brunel.ac.uk; ukishore@hotmail.com; Fax: +44 (0)1895 269873
bNeuro Imaging, MIRA Institute, University of Twente, Enschede, The Netherlands
cFaculty of Science, Engineering and Computing, Kingston University Penrhyn Road, Kingston upon Thames, Surrey, KT1 2EE, UK
dDepartment of Pharmacology, University of Oxford, Mansfield Road, Oxford, OX1 3QT, UK
eBioengineering Department and Krasnow Institute for Advanced Study, George Mason University, Fairfax, 22030 Virginia, USA
fDepartment of Biochemistry, College of Science, King Saud University, Riyadh, Saudi Arabia
First published on 30th November 2016
Abstract
Carbon nanotubes (CNTs) are increasingly being developed for use in biomedical applications, including drug delivery. One of the most promising applications under evaluation is in treating pulmonary diseases such as tuberculosis. Once inhaled or administered, the nanoparticles are likely to be recognised by innate immune molecules in the lungs such as hydrophilic pulmonary surfactant proteins. Here, we set out to examine the interaction between surfactant protein D (SP-D), a key lung pattern recognition molecule and CNTs, and possible downstream effects on the immune response via macrophages. We show here that a recombinant form of human SP-D (rhSP-D) bound to oxidised and carboxymethyl cellulose (CMC) coated CNTs via its C-type lectin domain and enhanced phagocytosis by U937 and THP-1 macrophages/monocytic cell lines, together with an increased pro-inflammatory response, suggesting that sequestration of SP-D by CNTs in the lungs can trigger an unwanted and damaging immune response. We also observed that functionalised CNTs, opsonised with rhSP-D, continued to activate complement via the classical pathway, suggesting that C1q, which is the recognition sub-component of the classical pathway, and SP-D have distinct pattern recognition sites on the CNTs. Consistent with our earlier reports, complement deposition on the rhSP-D opsonised CNTs led to dampening of the pro-inflammatory immune response by THP-1 macrophages, as evident from qPCR, cytokine array and NF-κB nuclear translocation analyses. This study highlights the importance of understanding the interplay between innate immune humoral factors including complement in devising nanoparticle based drug delivery strategies.
Introduction
In recent years, carbon nanotubes (CNTs) have been considered as vehicles for vaccination and drug delivery.1,2 However, being “non-self”, these delivery platforms encounter the host's immune system which can alter their intended translational application. We have recently assessed how the innate immune system is able to recognise and process these nanoparticles, with the goal of extrapolating this knowledge for the development of a new generation of nanotherapeutics.3,4 One of the anatomical sites of targeted therapy using nanotherapeutics is the lungs and a number of studies have examined the challenges in delivering nanoparticles for treating a number of pulmonary diseases.5,6 Thus, understanding the mechanism and consequences of interaction between nanoparticles and the pulmonary innate immune system is of great importance.4Pulmonary toxicity of CNTs is one of the most discussed subjects in nanoparticle research. Lam et al. first showed that single-walled carbon nanotubes (SWNTs) caused dose- and time-dependent inflammation and fibrosis in mice,7 whilst another study showed minimal inflammation in the lungs.8 Granulomas were also shown to be induced by CNTs with subsequent fibrogenesis, possibly by alveolar macrophage-secreted chemokines causing fibroblast proliferation.9 While CNTs can cause respiratory deficiencies in mice,10 interstitial fibrotic lesions without infiltration have been shown around CNT clusters in rat lungs.9 These variable in vivo results can be attributed to the variations in CNT types (multi-, double- and single-walled), diameter and length, aggregation states, contamination with other materials, administration method and route, and surface coating.11 Studies comparing well-dispersed versus aggregated SWNTs found that poorly dispersed SWNTs formed clumps of 5 to 20 μm in diameter in the lungs, which triggered granuloma formation, whereas highly dispersed SWNTs that did not form any clumps and were found free in the tissue, gave rise to interstitial fibrosis, but no granulomatous lesions.12–14 Similar results were found in the case of MWNTs, where well dispersed samples were evenly distributed in cells of the lung parenchyma, to which the tissues responded by interstitial fibrosis of the alveolar wall, but with very limited granuloma formation.15
Surfactant proteins (SP) constitute up to 10% of the dry weight of pulmonary surfactant, with the rest composed of surfactant lipids.16 Of the four SPs, SP-B and SP-C are small hydrophobic peptides, which interact with surfactant lipids and maintain surfactant homeostasis, whilst SP-A and SP-D are large hydrophilic proteins, which perform a range of immunological functions.17 Both SP-A and SP-D can opsonise pathogens and foreign particles that may be deposited deep in the lungs and then interact with the alveolar macrophages via putative receptors, leading to their uptake and downstream immune response,17 suggesting that they can have a critical role to play in the processing of inhaled nanoparticles.
Oxidized (Ox) double-walled CNTs (DWNTs) bind efficiently with SP-A and SP-D via their homotrimeric C-type lectin domain or carbohydrate recognition domain (CRD), as shown by transmission electron microscopy.18,19 Since the physiological concentrations of SP-A and SP-D are relatively low in lung surfactant, their binding to nanoparticles can potentially cause significant depletion of these proteins, which can have a detrimental effect on the lung innate immune defence.19 Coating Ox-CNTs with SP-A results in an increased uptake of the particles by alveolar macrophages, while no inflammatory response is evoked, as determined by nitric oxide levels.20 Adsorption of lung surfactant corona, consisting of SP-A, B and D, enhances the uptake of SWNTs by RAW murine macrophages.21 Furthermore, pre-coating CNTs (pristine, oxidised or aminated) with porcine pulmonary surfactant stabilizes the suspensions of CNTs, but induces their clustering inside monocyte derived-macrophages.22 Coating SP-A as well as phosphatidylserine on CNTs has been shown to increase the efficiency of phagocytosis.23,24 Recently, SP-D was found to enhance uptake of polystyrene, carbon black and silica nanoparticles by alveolar macrophages and dendritic cells in mice.25 In murine alveolar macrophages, SP-A was found to inhibit agglomeration and uptake of amine-modified polystyrene nanoparticles (A-PS), whilst it promoted uptake of unmodified polystyrene nanoparticles (U-PS).26 U-PS and A-PS nanoparticles with bound SP-A or SP-D were found to neutralise influenza A viral cellular infection in vitro.27
We have recently examined the interaction of CNTs and other nanoparticles with innate immune humoral factors including complement. Classical pathway-activating CNTs get phagocytosed more effectively by macrophages, resulting in a down-regulation of the pro-inflammatory cytokine response,28 whereas Au–Ni nanowires, which activate complement weakly, are poorly phagocytosed, with an increased pro-inflammatory cytokine response by immune cells.4 Surface modifications of hydrophobic CNTs are of great importance in order for CNTs to be well-dispersed in aqueous media and this can be achieved by covalent or non-covalent surface coatings. We dispersed pristine multi-walled CNT (MWNTs), covalently by oxidation (Ox-CNT), or non-covalently with carboxymethyl-cellulose (CMC-CNT), each having carboxyl functional groups. Soluble complement factors, C1q and factor H, interacted differentially with functionalised CNTs and altered the immune response even without complement activation, suggesting that the recognition subcomponents of the complement pathways can recognise charge patterns due to CNT surface modifications.3,4
In the present study, we set out to examine the interaction of SP-D with Ox-CNT and CMC-CNT and its subsequent effect on complement activation, phagocytosis and immune response by macrophage cell lines.
Methods
Dispersion and functionalization of CNTs
CNTs (diameters 10–20 nm, length 5–20 μm) were purchased from Arry Nano (Frechen Königsdorf, Germany). Covalent functionalization or oxidation was achieved by a modified procedure, as previously described.29 CNTs were dispersed in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio mixture of sulphuric acid (H2SO4, 10 M) and nitric acid (HNO3, 10 M) by water bath sonication for 1 h (Transsonic t460 water bath sonicator, 85 Watt, 35 kHz), followed by probe sonication using QSonica sonicator (30 effective minutes with 50% on–50% off cycles, amplitude 100%, 25 watts) on ice. For non-covalent functionalization, the CNTs were dispersed in a 1 mg ml−1 solution of carboxymethyl cellulose (CMC) (Sigma) in a 1:2 mass ratio by probe sonication. Aggregates and clusters were removed by centrifugation at 8000g. Excess CMC was washed away using a 0.2 μm pore size polycarbonate track-etched membrane filter (Whatman) (Fig. 1a). For covalent functionalization, the strongly acidic mixture was refluxed at 120 °C for 48 h. The oxidised CNTs (Ox-CNTs) were washed extensively (3 cycle of centrifugation, 11000g, 30 min) and re-dispersed in water. Amorphous carbon residues were removed by overnight stirring of the Ox-CNTs in 10 mM NaOH30 and filtration washings with water using a 0.1 μm pore size polycarbonate track-etched membrane filter (Whatman) (Fig. 1b).
:1 volume ratio mixture of sulphuric acid (H2SO4, 10 M) and nitric acid (HNO3, 10 M) by water bath sonication for 1 h (Transsonic t460 water bath sonicator, 85 Watt, 35 kHz), followed by probe sonication using QSonica sonicator (30 effective minutes with 50% on–50% off cycles, amplitude 100%, 25 watts) on ice. For non-covalent functionalization, the CNTs were dispersed in a 1 mg ml−1 solution of carboxymethyl cellulose (CMC) (Sigma) in a 1:2 mass ratio by probe sonication. Aggregates and clusters were removed by centrifugation at 8000g. Excess CMC was washed away using a 0.2 μm pore size polycarbonate track-etched membrane filter (Whatman) (Fig. 1a). For covalent functionalization, the strongly acidic mixture was refluxed at 120 °C for 48 h. The oxidised CNTs (Ox-CNTs) were washed extensively (3 cycle of centrifugation, 11000g, 30 min) and re-dispersed in water. Amorphous carbon residues were removed by overnight stirring of the Ox-CNTs in 10 mM NaOH30 and filtration washings with water using a 0.1 μm pore size polycarbonate track-etched membrane filter (Whatman) (Fig. 1b).
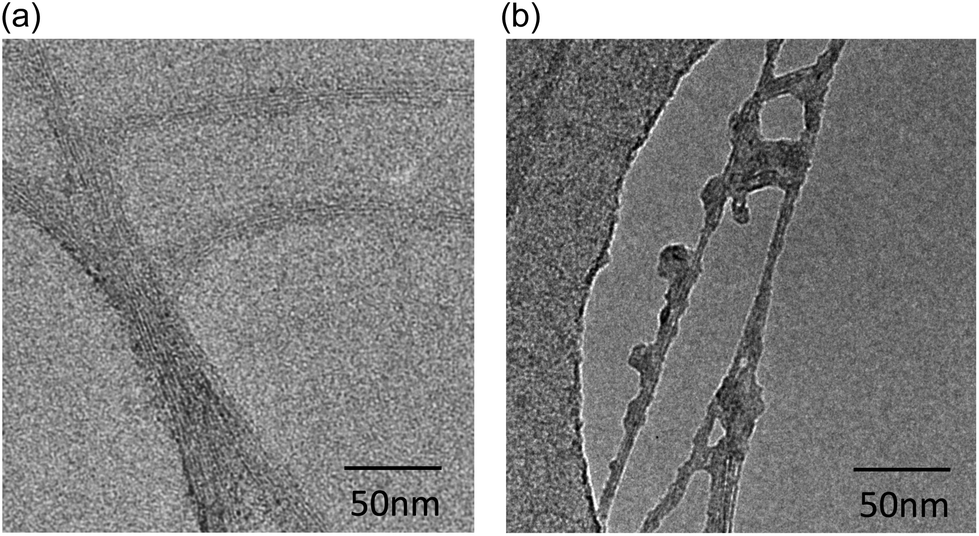 | ||
| Fig. 1 High resolution Transmission Electron Microscopy (TEM) of CNTs. (a) The pristine CMC-CNTs consist of a high number of parallel-undamaged carbon walls. The external diameter is approximately 22 nm while the inner diameter is 5 nm. (b) After acid treatment at high temperature, the outer walls of the oxidised CNTs (Ox-CNTs) show a high number of defects. In addition, the CNTs are shortened in the process (not shown). | ||
Production of a recombinant form of truncated lung surfactant protein D (rhSP-D)
A recombinant form of human lung surfactant protein D (rhSP-D) composed of eight N-terminal Gly-Xaa-Yaa triplets, α-helical neck region and the globular CRD region (rhSP-D), was expressed in E. coli BL21(λDE3) pLysS strain (Invitrogen) at 37 °C as described earlier.31 Briefly, transformants were grown in 500 ml LB containing ampicillin (100 μg ml−1) and chloramphenicol (50 μg ml−1) to an OD600 of 0.6. Protein expression was then induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3 h, the cells were centrifuged and then lysed by sonication.32 The sonicate was centrifuged at high speed (20000 rpm, 15 min) and the pellet containing rhSP-D as inclusion bodies was solubilized in 50 ml buffer A (50 mM Tris-HCl pH 7.5 + 100 mM NaCl + 5 mM CaCl2) with 10 mM β-mercaptoethanol and 8 M urea. The re-solubilized material was then dialyzed successively against buffer A containing 4 M urea, 2 M urea, 1 M urea and no urea, each for 2 h. The dialysate was further purified on a Q-Sepharose anion exchange resin (Pharmacia). The peak fractions, which eluted at 400 mM NaCl, were loaded onto a maltose–agarose column (Sigma) in buffer A + 5 mM CaCl2 and eluted with 5 mM EDTA pH 7.5. SDS-PAGE was used to examine each step for purity of rhSP-D (Fig. 2a). For endotoxin removal, 5 ml of polymyxin B agarose gel (Sigma) was packed in a 20 ml column and washed with 50 ml of 1% sodium deoxycholate and then further rinsed with 50 ml of autoclaved distilled water to completely remove sodium deoxycholate. The affinity purified rhSP-D was then mixed with the gel and left for 3 h at 4 °C on a shaker. The level of endotoxin (∼4 pg μg−1 of rhSP-D) in the rhSP-D protein, collected as unbound fraction, was determined using QCL-1000 Limulus amebocyte lysate system (BioWhittaker, Walkersville, MD, USA).
 | ||
| Fig. 2 Binding of rhSP-D to Ox-CNT and CMC-CNT. (a) SDS-PAGE (12% w/v acrylamide) analysis of recombinant SP-D. Recombinant human SP-D containing the homotrimeric neck and CRD regions (rhSP-D) was expressed in E. coli BL21(λDE3) pLysS. 3 h after induction with 0.4 mM IPTG, rhSP-D accumulated as an over-expressed protein of ∼20 kDa (lane 3) compared to uninduced cells (lane 2) and was found in inclusion bodies (lane 4). Following denaturation–renaturation in urea, the affinity-purified rhSP-D (Lane 5) was made LPS-free prior to use. Lane 1, protein molecular weight standard. (b) SDS-PAGE (12% w/v acrylamide) analysis of rhSP-D bound to Ox-CNTs and CMC-CNTs. CNTs were incubated overnight with rhSP-D in 1:2 weight ratio and washed extensively via centrifugation to remove unbound SP-D. Lane 1: molecular weight marker; lane 2: rhSP-D coated Ox-CNTs; lane 3: rhSP-D-coated CMC-CNTs. | ||
Binding of rhSP-D to Ox-CNT and CMC-CNT
rhSP-D was incubated overnight with CMC-CNT or Ox-CNT in a buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl and 5 mM CaCl2. Unbound protein was removed by repeated centrifugation at 17000g for 10 min and the CNTs were re-dispersed. Protein binding was analysed by SDS-PAGE. A range of protein: CNTs mass ratios was analysed (1:2, 1:1, 2:1, 4:1 and 8:1). No increase in binding was found above a 2:1 ratio; therefore, this ratio was chosen for subsequent experiments.
Complement consumption assays for the classical pathway
Complement consumption in normal human serum by CNTs was analysed using a complement hemolytic assay.33 CNT suspensions (100 μl of 100 μg ml−1 stock) in PBS were added to 100 μl of fresh human serum diluted 1:1 in dextrose gelatin veronal buffer with Mg2+ and Ca2+ (DGVB++). Zymosan (0.2 mg in 100 μl PBS; Sigma) served as a positive control, while serum alone was used as a negative control. To avoid possible interference by the CNTs with the complement assay, the samples were centrifuged (13000g, 10 min) after 1 h incubation at 37 °C, removing all CNTs from the sera. The remaining complement activity of the supernatants of each sample was determined. The supernatants were serially diluted 2-fold (1/10–1/5120 in DGVB++) using a 96 microtitre well plate. Then, 100 μl of each dilution was incubated with 100 μl of antibody (hemolysin)-sensitized sheep erythrocytes (EA) (TCS, Buckingham, UK), as described previously33 at the concentration of 108 cells per ml in DGVB++ for 1 h at 37 °C. Although the CNTs were removed from the sera, control experiments were performed by incubating the CNTs with EA for 2 h at 37 °C. No hemolysis was observed in these control experiments. After incubation, cells were spun down (700g, 10 min, room temperature), and released hemoglobin in the supernatant was measured at 541 nm. Total hemolysis (100%) was measured by lysing EA with water. Background spontaneous hemolysis (0%) was determined by incubating EA with DGVB++ only. Hemolytic complement (CH50) values, the serum dilution required for 50% cell lysis, were calculated and compared. The experiments were carried out in triplicate.
Phagocytosis assay
Uptake of CNTs by U937 cells (a monocytic cell line derived from histiocytic lymphoma) was measured using an ELISA type assay as previously described.28 First, CMC-CNTs and Ox-CNTs were biotinylated, as follows: 200 μg CNTs in 1 ml 0.1 M MES buffer [2-(N-morpholino)ethanesulfonic acid, pH 5] were stirred for 2 h with 1 mg pentylamine biotin (Pierce, Thermo Fisher Scientific) in the presence of 4 μg EDC [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide]. The reaction was stopped by adding 50 μl of 0.1 M ethanolamine (Sigma). The resulting biotin-CMC-CNTs and biotin-Ox-CNTs were dialyzed extensively against PBS (pH 7.4) in order to remove remaining reactants and MES.To confirm the efficiency of biotinylation, biotinylated CNTs were incubated with Alexa Fluor 488-conjugated streptavidin for 30 min and then washed via centrifugation at 17000g for 10 min three times and re-dispersed. Dispersed biotinylated CNTs were visualized with a Leica microscope using LAS software (ESI†).
U937 cells were cultured in complete RPMI 1640 medium (Gibco) containing 10% fetal calf serum (FCS), 2 mM L-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 1 mM sodium pyruvate, passaged, and then washed in AIM-V AlbuMAX serum free medium (GIBCO). In each well of a 24 well tissue culture plate, 5 × 105 cells were incubated in AIM-V AlbuMAX with the above-mentioned supplements (but no FCS). To each well, 20 μg of biotin-CMC-CNTs or biotin-Ox-CNTs were added and incubated for 6 h; a negative control was incubated with PBS only. Cells were harvested and washed five times in PBS using centrifugation at 300g and stored at −80 °C until further use. All experiments were performed in duplicate.
For quantification, microtitre wells (NUNC, Polysorb) were coated with 100 μl Avidin (Pierce) at 50 μg ml−1 in 0.1 M Na2CO3, pH 9.0 for 1 h at room temperature, followed by blocking with 1 mg ml−1 horse IgG in PBS for 1 h at room temperature. The pelleted U937 cells were lysed using 25 μl lysis buffer (10 mM HEPES, 20 mM NaCl, 0.5 mM EDTA, pH 7.5, 1% Triton X 100), following which 25 μl 0.1 mg ml−1 horse IgG in PBS (as blocking agent) was added. The lysate or biotinylated CNTs (to generate a standard curve) was incubated for 1 h at room temperature in the wells. The plate was washed 7 times with 0.1 mg ml−1 horse IgG in PBS and incubated with 1:2000 dilution of streptavidin–horseradish peroxidase (HRP) conjugate (Sigma) for 1 h at room temperature. 3,3′,5,5′-Tetramethylbenzidine (TMB) (Biolegend, London, UK) was used as a substrate for the HRP and the yellow product obtained after stopping the reaction with 2 N H2SO4 was read at 450 nm in a plate reader (BioRad).
Fluorescence microscopy
Qualitative analysis of biotinylated CMC-CNTs uptake was carried out using THP-1 cells. For these experiments, 1 × 105 THP-1 cells (per coverslip) were plated on 13 mm glass coverslips and treated for 24 h with 100 nM phorbol myristate acetate (PMA, Sigma) in complete RPMI 1640 containing 10% v/v FCS, 2 mM L-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 1 mM sodium pyruvate. Differentiated THP-1 cells were washed three times with PBS at 37 °C to remove excess PMA. Biotinylated CMC-CNTs coated with rhSP-D were incubated for 1 h at 37 °C in normal human serum in 1:1 ratio with DGVB++ buffer for complement deposition. CNTs were washed via centrifugation at 7000g for 10 min three times and re-dispersed via water bath sonication for 10 min before incubating with THP-1 cells. Cells were exposed to 4 μg ml−1 biotinylated CMC-CNTs coated with rhSP-D (rhSP-D + CMC-CNT), biotinylated CMC-CNT coated with rhSP-D and then complement deposited (rhSP-D + serum + CMC-CNT), complement deposited biotinylated CMC-CNTs (serum + CMC-CNTs) or biotinylated CMC-CNTs alone in 500 μl of serum-free RPMI 1640 medium for 2 h and 4 h. Cells were washed twice with PBS, fixed with 4% paraformaldehyde for 10 min, washed and processed for immunofluorescence. The coverslips were permeabilized using a buffer containing 20 mM HEPES–NaOH pH 7.4, 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100 and 10% sodium azide for 5 min on ice. The cells were stained for 30 min with 1.6 μM Hoechst 33342 (Life technologies) to visualize the nucleus, 2 μg ml−1 Alexa-Fluor546-conjugated wheat germ agglutinin (Invitrogen) to reveal the plasma membrane, and Alexa Fluor 488-conjugated streptavidin (Biolegend) to reveal biotinylated CMC-CNTs. Cells were washed twice, mounted using Citifluor anti-fade (Citifluor, UK), and observed under a Nikon Eclipse TE2000-S confocal microscope with 60× oil lens.
To observe nuclear translocation of NF-κB, 1 × 105 THP-1 cells per 13 mm coverslip were differentiated with PMA, as described above, and incubated with 4 μg ml−1 of rhSP-D bound CMC-CNTs (rhSP-D + CMC-CNT), rhSP-D bound and complement deposited CMC-CNT (rhSP-D + serum + CMC-CNT), complement deposited CMC-CNTs (serum + CMC-CNTs) or CMC-CNTs alone in 500 μl of serum-free RPMI 1640 medium for 2 h. Following fixation and permeabilization as described above, the cells were incubated with rabbit anti-NF-κB p65 polyclonal antibodies (1:500, Santa Cruz Biotech), followed by Alexa Fluor 488-goat anti-rabbit antibody (1:500, Abcam), and observed with a Leica Fluorescent microscope using LAS software.
Quantitative RT-PCR analysis
In a 24 well cell culture plate (Nunc), 10 μg of different forms of CNTs as described above, in PBS were added to each well containing 5 × 105 cell U937 cells in AIM-V AlbuMAX serum free medium and incubated for 15, 30, 45, 60, 120 or 360 min, and for 30, 60, 120 or 360 min in the case of differentiated THP-1 cells. Control samples included THP-1 or U937 cells incubated with PBS only for 30 min. Cells were harvested, spun down (3000g, 5 min), and stored at −80 °C. Total RNA was extracted from frozen cell pellets using the GenElute Mammalian Total RNA Purification Kit (Sigma-Aldrich). Samples were treated with DNase I to remove any contaminating DNA. To inactivate DNase I, samples were heated at 70 °C for 10 min, and then kept on ice. A Nano Drop 2000/2000c spectrophotometer (Thermo-Fisher Scientific) was used to determine the amount and purity (260/280 nm ratio) of RNA. The cDNA was synthesized using High Capacity RNA to cDNA Kit (Applied Biosystems).Primers (Table 1) were designed using Primer-BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
| Targets | Forward primer | Reverse primer |
|---|---|---|
| 18S | ATGGCCGTTCTTAGTTGGTG | CGCTGAGCCAGTCAGTGTAG |
| IL-1β | GGACAAGCTGAGGAAGATGC | TCGTTATCCCATGTGTCGAA |
| IL-6 | GAAAGCAGCAAAGAGGCACT | TTTCACCAGGCAAGTCTCCT |
| IL-10 | TTACCTGGAGGAGGTGATGC | GGCCTTGCTCTTGTTTTCAC |
| IL-12 | AACTTGCAGCTGAAGCCATT | GACCTGAACGCAGAATGTCA |
| TGF-β | GTACCTGAACCCGTGTTGCT | GTATCGCCAGGAATTGTTGC |
| TNF-α | AGCCCATGTTGTAGCAAACC | TGAGGTACAGGCCCTCTGAT |
| NF-κB | GTATTTCAACCACAGATGGCACT | AACCTTTGCTGGTCCCACAT |
| NLRP3 | GCCATTCCCTGACCAGACTC | GCAGGTAAAGGTGCGTGAGA |
The qPCR reaction consisted of 5 μl Power SYBR Green MasterMix (Applied Biosystems), 75 nM forward and reverse primer, and 500 ng template cDNA in a 10 μl reaction volume. PCR was performed using a Step One Plus Real-Time PCR System (Applied Biosystems). After initiation steps of 2 min at 50 °C and 10 min at 95 °C, the template was amplified for 40 cycles, each cycle comprised of 15 s at 95 °C and 1 min at 60 °C. Samples were normalized using the expression of human 18S rRNA. Data was analyzed using the Step One software v2.3 (Applied Biosystems). Ct (cycle threshold) values for each target gene were calculated. Relative expression of each target gene was calculated using the Relative Quantification (RQ) value, using the equation: RQ = 2−ΔΔCt for each cytokine target gene, and comparing relative expression with that of the 18S rRNA constitutive gene product. Assays were conducted in triplicate.
Multiplex cytokine array analysis
Secreted cytokines [interleukin (IL)-6, IL-10, IL-12p40, IL-12p70, IL-1α, IL-1β, tumor necrosis factor (TNF)-α, IL-15, IL-17A, IL-9, TNF-β, interferon (IFN)-α2], chemokines [monocyte chemoattractant protein (MCP)-3, macrophage-derived chemokine (MDC), eotaxin, fractalkine, growth regulated oncogene (GRO), IL-8, interferon gamma-induced protein (IP)-10, MCP-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, monokine induced by gamma interferon (MIG), interferon-inducible T-cell alpha chemoattractant (I-TAC), monokine induced by gamma interferon (MIG)], growth factors [IL-9, IL-2, epidermal growth factor (EGF), fibroblast growth factor (FGF)-2, granulocyte colony stimulating factor (G-CSF), granulocyte macrophage (GM)-CSF, IL-3, IL-7, vascular endothelial growth factor (VEGF)], and ligand and receptors [FMA-like tyrosine kinase 3 ligand (FLT-3L), interleukin 1 receptor antagonist (IL-1RA), sCD40L] were measured by MagPix Milliplex kit (EMD Millipore). Briefly, 25 μl of assay buffer was added to each well of a 96-well plate, followed by addition of 25 μl of standard, controls or supernatants of cells (treated with CNTs). 25 μl of analyte-specific antibody immobilised beads were added to each well, followed by incubation for 18 h at 4 °C. After washing the plate with the assay buffer, 25 μl of detection antibodies were incubated with the beads for 1 h at room temperature. 25 μl of streptavidin–phycoerythrin was then added and incubated for 30 minutes. Following a washing step, 150 μl of sheath fluid (BD Biosciences) was added to each well and the plate was read using the Luminex Magpix instrument (Bio-Rad).Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software). An unpaired 2-side t-test or a 2-way ANOVA was used on the data for any significant difference in expression. P values were computed and graphs compiled and analysed.Results
rhSP-D binds to Ox-CNT and CMC-CNT stably
Pristine CNTs of 5–20 μm in length and 10–20 nm diameters were well dispersed by either non-covalent coating with CMC or covalent functionalisation (oxidation); the dispersions were found highly stable for months. Transmission Electron Microscopy (TEM) imaging clearly revealed irregularities in the structure of the oxidised Ox-CNTs (Fig. 1b) which were not found on pristine CMC-CNTs (Fig. 1a). The Ox-CNTs were also significantly shortened (length 100–500 nm) (not shown). The NaOH treatment removed all amorphous carbon from the surface of the nanotubes (not shown). After incubation with rhSP-D and extensive washing, we found that purified rhSP-D (Fig. 2a) stably bound to both CMC-CNTs and Ox-CNTs, as evident from SDS-PAGE analysis (Fig. 2b).Coating of CNTs with recombinant human SP-D enhances complement consumption
In view of our earlier observation that Ox-CNTs and CMC-CNTs can activate the complement classical pathway,3,4,28 Ox-CNTs and CMC-CNTs coated with rhSP-D were investigated for complement activation via the classical pathway. Both rhSP-D coated CNTs showed a very significant (p < 0.0001, n = 3) enhancement (∼2 fold) in complement consumption compared to uncoated CNTs (Fig. 3). Complement activation, therefore, appears to be facilitated by rhSP-D coating on CNTs, suggesting that rhSP-D and C1q do not compete for binding to the same sites on the CNTs, i.e. rhSP-D does not inhibit C1q binding. Bound rhSP-D could also enhance complement activation by other mechanisms, e.g. by providing more sites for the covalent binding of C3b or C4b, thus enhancing formation of the convertase enzymes, C3bBb and C4b2a, which would increase consumption (activation) of complement. In a previous paper,28 it was shown that coating DWNT with other proteins, e.g. BSA and fibrinogen, can also enhance complement consumption, indicating that the effect arises due to the availability of more C4b/C3b binding sites. | ||
| Fig. 3 Complement consumption by rhSP-D coated CNTs. Hemolytic assay was carried out to determine complement consumption by rhSP-D coated and uncoated CMC-CNTs and Ox-CNTs. A significant increase in complement consumption by rhSP-D coated CNTs compared to uncoated counterparts was observed. Serum and zymosan were used as negative and positive control, respectively. All experiments were carried out in triplicate; error bars represent ±standard deviation. A 2-side unpaired t-test (with Welch's correction) was performed on the data to determine significant differences in complement consumption between each uncoated (control) and coated CNTs. All these comparisons were significant (p < 0.01). | ||
rhSP-D enhances uptake of CNTs by U937 and THP-1 macrophage cell lines
For the CMC-CNTs, binding of rhSP-D increased cellular uptake by about 30% (Fig. 4a), similar to previously reported increase in uptake of CMC-CNTs coated with serum and C1q.3,4,28 In contrast, there was a 30% decrease in cellular uptake of rhSP-D bound Ox-CNTs (Fig. 4a); this also fits with a similar pattern of cellular uptake seen with C1q and factor H.3,4 These opposing effects in uptake between the two different types of CNTs are intriguing. It is possible that the mode or density of binding of soluble pattern recognition receptors to CNTs can alter their ability to interact with their cognate receptors. There is also a large difference in length between the two types of coated CNTs, which may be relevant.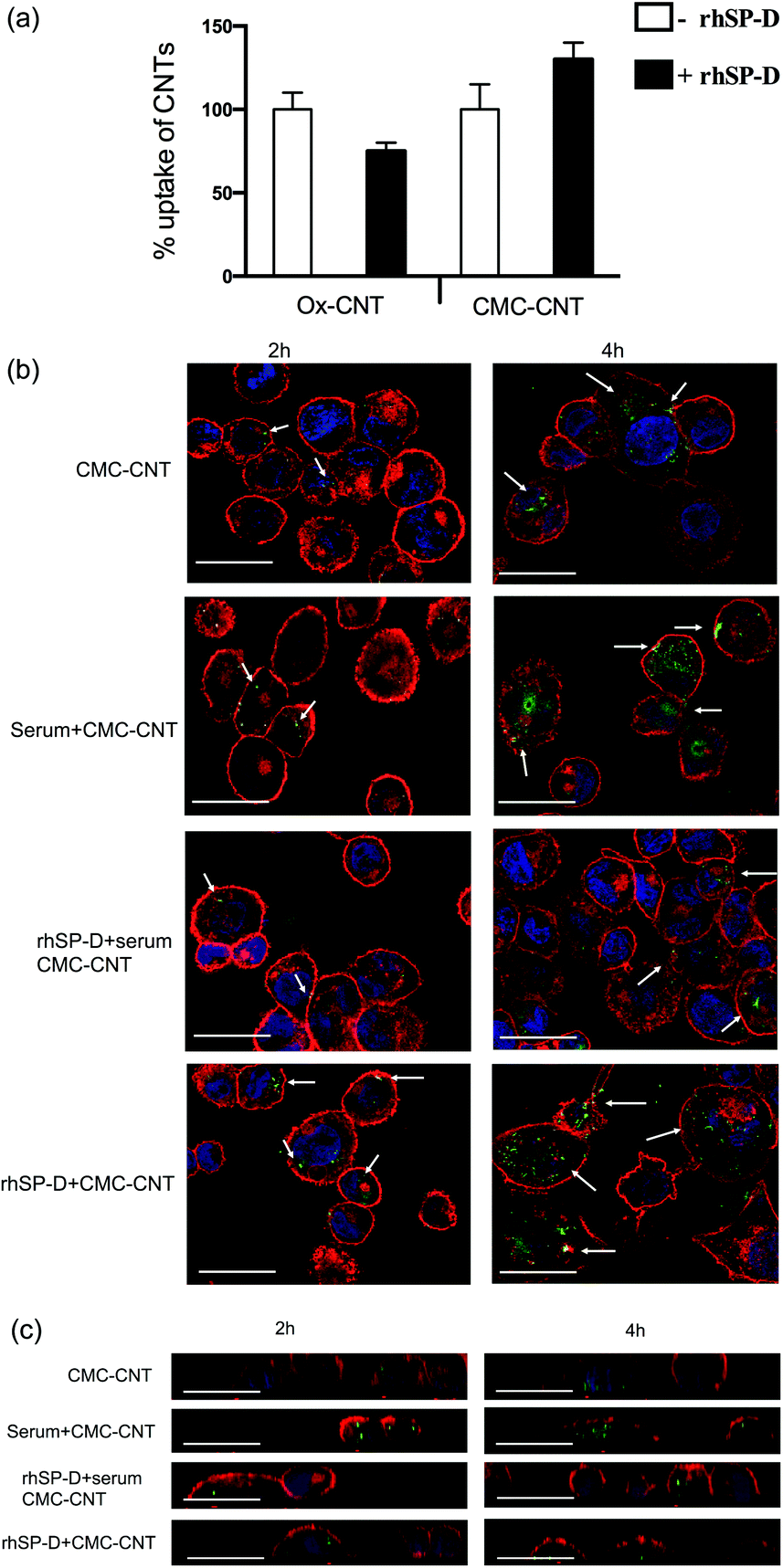 | ||
| Fig. 4 Differential uptake of CNTs by macrophage-like cell lines after coating with rhSP-D, and with or without serum treatment (i.e. complement deposition). (a) Biotinylated CMC-CNTs and biotinylated Ox-CNTs were incubated with rhSP-D in 1:2 w/w ratio (CNT:protein). After removal of unincorporated rhSP-D, U937 cells were incubated with 20 μg of rhSP-D bound biotinylated CMC-CNTs or biotinylated CMC-CNTs alone for 6 h. Following extensive washing in PBS, the cells were lysed and the amount of internalised CNTs was quantified by an ELISA type assay as described.28 All experiments were carried out in triplicate; error bars represent ±standard deviation; p = 0.0316 for Ox-CNTs and p = 0.053 for CMC-CNTs. (b) To confirm internalisation of CNTs, confocal microscopy was used. Differentiated THP-1 cells were exposed to rhSP-D bound biotinylated CMC-CNTs (rhSP-D + CMC-CNT), rhSP-D bound biotinylated CMC-CNT plus complement deposition (rhSP-D + serum + CMC-CNT), complement deposited biotinylated CMC-CNTs (serum + CMC-CNT), or biotinylated CMC-CNTs alone for 2 h and 4 h. Cells were washed, fixed, permeabilised and stained with Alexafluor-488 labelled streptavidin to reveal internalised biotinylated CMC-CNTs. Alexafluor 546-conjugated wheat germ agglutinin was used to reveal plasma membrane (red) and the nucleus was stained with Hoechst 33342 (Blue). Images shown are single sections taken with a Nikon confocal microscope; scale bar 20 μm. Arrows highlight endocytosed biotinylated CMC-CNTs. (c) Magnified orthogonal views of the same confocal images demonstrate uptake of biotinylated CMC-CNTs (green), plasma membrane in red, scale bar 20 μm. | ||
Confocal microscopy using differentiated THP-1 cells confirmed that rhSP-D, like serum treatment of CNTs, enhanced the uptake of CMC-CNTs (Fig. 4b). Uptake of CMC-CNTs did not seem to take place at all parts of the cells, as shown in orthogonal section of the THP-1 cells (Fig. 4c). Interestingly, complement deposition on rhSP-D coated CMC-CNTs (rhSP-D + serum) reduced uptake compared to rhSP-D coating alone or serum treatment alone at both early (2 hours) and late (4 hours) time points (Fig. 4b). The reduction in uptake of rhSP-D deposited CMC-CNTs with additional complement deposition could be due to steric hindrance or competition for a receptor.
Cytokine and transcription factor mRNA expression by U937 and THP-1 treated with rhSP-D bound CNTs
Cytokine and transcription factor gene expression profiles of U937 cells were measured at six time-points up to 6 h, following treatment with CNTs, with and without rhSP-D coatings. In U937 cells, mRNA levels for the pro-inflammatory cytokines (IL-1β, TNF-α and IL-12) were up regulated by Ox-CNTs, when compared to CMC-CNTs (Fig. 5a). For IL-1β, this observation is particularly dramatic. The effect of rhSP-D coating on CNTs was subtle, with a slight up-regulation in mRNA expression by Ox-CNTs for IL-1β and TNF-α. In the case of IL-12, for CMC-CNTs coated with rhSP-D, there was a slight down-regulation in mRNA levels. For the anti-inflammatory cytokines, IL-10 and TGF-β, there was a slight down-regulation of mRNA levels for Ox-CNTs and CMC-CNTs when bound to rhSP-D. The mobilisation of the pro-inflammatory response was also indicated in the up-regulation of NF-κB mRNA for Ox-CNTs coated with rhSP-D, although the effect seemed less pronounced in CMC-CNTs with rhSP-D. No significant changes were observed for NLRP3, suggesting that NLRP3 inflammasome activation was limited,34 as borne out by the 30% difference in uptake between the CNTs with and without rhSP-D coating (Fig. 4a).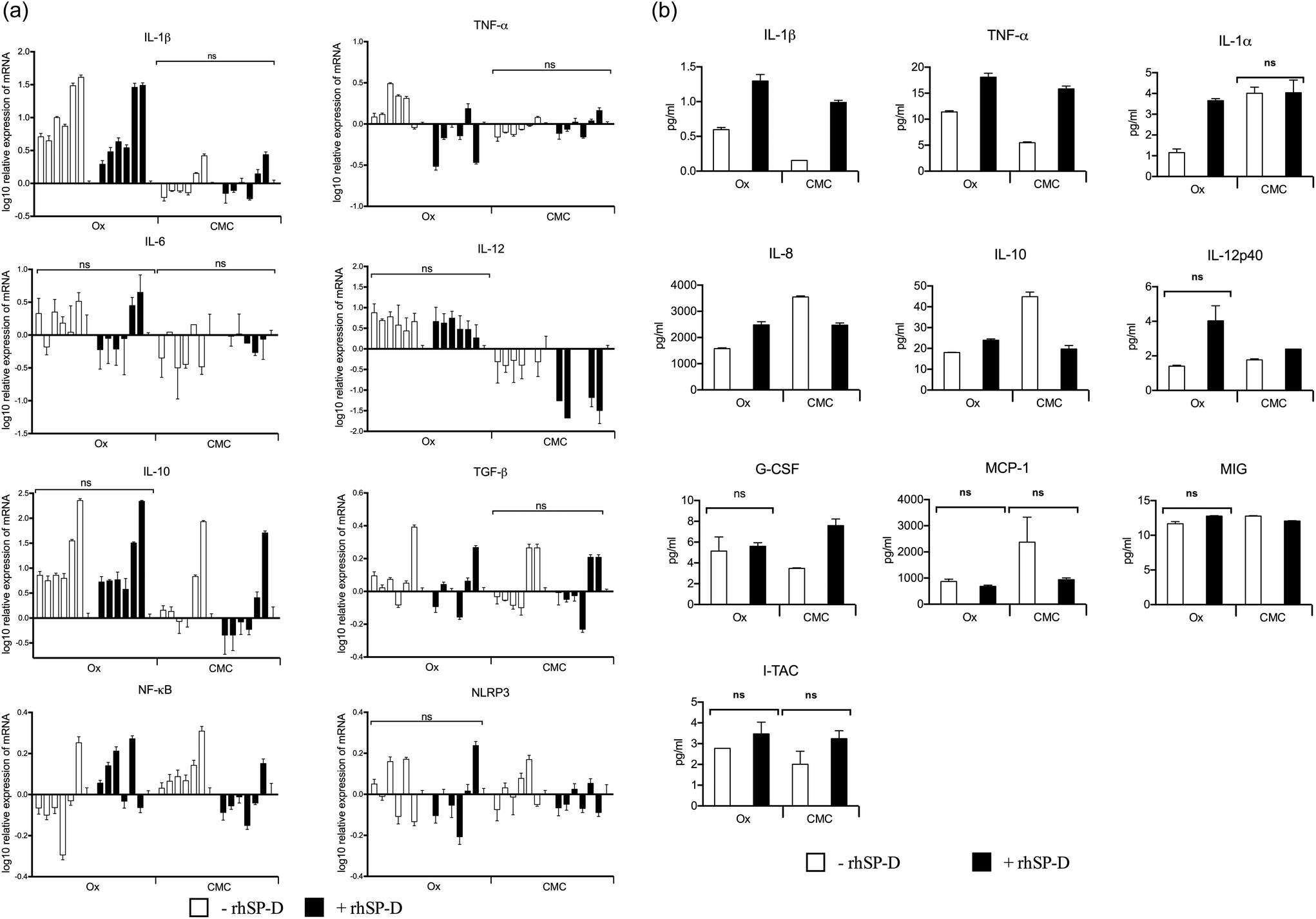 | ||
| Fig. 5 (a) Cytokine gene expression profile of U937 cells challenged with CNTs and rhSP-D bound CNTs. For the measurement of transcripts of pro- and anti-inflammatory cytokines as well as NF-κB and NLRP3, cells were incubated with each CNTs, for the following time points: 15, 30, 45, 60, 120 and 360 minutes (X-axis). In control experiments, cells were incubated with PBS only for 30 minutes. The expression of cytokines was measured using real-time qPCR and the data normalized to 18S rRNA gene expression as an endogenous control. Assays were conducted in triplicate. Error bars represent ±standard deviation. A 2-way ANOVA was performed on the data to determine significant differences in expression between each un-coated and coated nanoparticle. All these comparisons were significant (p < 0.05), except where shown (ns: not significant, p > 0.05). (b) Multiplex cytokine array analysis of the supernatants of U937 cells treated with CNTs and rhSP-D bound CNTs. Secreted cytokines, chemokines and growth factors (IL-8, IL-10, IL-12p40, IL-1α, IL-1β, TNF-α, MIG, I-TAC, MCP-1 and G-CSF) levels were measured by MagPix Milliplex kit (EMD Millipore). Supernatants were taken after 48 h treatment, except for MCP-1 (12 h). Error bars represent ±standard deviation. A 2-side unpaired t-test (with Welch's correction) was performed on the data to determine significant differences in protein levels between each un-coated and coated CNTs. All these comparisons were significant (p < 0.05), except where shown (ns: not significant, p > 0.05). | ||
Cytokine gene expressions of THP-1 cells were studied following treatment with rhSP-D coated CNTs, with or without complement deposition. rhSP-D coated CMC-CNTs up-regulated the pro-inflammatory cytokines (IL-1β and TNF-α) in comparison to complement deposited rhSP-D coated CMC-CNTs. However, we observed the opposite trend in the case of IL-12 mRNA level (Fig. 6a). For anti-inflammatory cytokines, unlike IL-10, TGF-β was slightly upregulated by complement-deposited and rhSP-D coated CMC-CNTs. NLRP3 was upregulated but NF-κB mRNA level was downregulated by complement deposition on the rhSP-D coated CMC-CNTs. Thus, complement deposition on CNTs dampened the pro-inflammatory effects of rhSP-D.
 | ||
| Fig. 6 (a) Cytokine gene expression profile of differentiated THP-1 cells challenged with rhSP-D coated CMC-CNT with or without serum treatment. rhSP-D bound CMC-CNTs were treated with or without serum. For the measurement of transcripts of pro-and anti-inflammatory cytokines as well as NF-κB and NLRP3, cells were incubated with each CNTs, at the following four time-points: 30, 60, 120 and 360 minutes (X-axis). In control experiments, cells were incubated with PBS only for 60 minutes. The expression of cytokines was measured using real-time qPCR and the data were normalized to 18S rRNA gene expression as an endogenous control. Assays were conducted in triplicate. Error bars represent ±standard deviation. A 2-way ANOVA was performed on the data to determine significant differences in expression between rhSP-D coated CMC-CNT with or without serum treatment. All these comparisons were significant (p < 0.01). (b). Multiplex cytokine array analysis of supernatants of THP-1 cells treated with rhSP-D bound CMC-CNT with or without serum. rhSP-D bound CMC-CNTs were incubated with differentiated THP-1 cells for 30 min, 1 h, 2 h, 6 h, 12 h, 24 h and 48 h. Cells from early time points (30 min, 1 h, 2 h, 6 h) were used for quantitative expression of different cytokines at transcriptional level. Culture supernatants from late time points (24 h and 48 h) were taken for measurement of protein level of secreted cytokines (IL-6, IL-10, IL-12p40, IL-12p70, IL-1α, IL-1β, TNF-α, IL-13, IL-15, IL-17A, IL-9, TNF-β), chemokines (MCP-3, MDC, eotaxin, fractalkine, GRO, IL-8, IP-10, MCP-1, MIP-1α and MIP-1β), growth factors (IL-2, EGF, FGF-2, G-CSF, GM-CSF, IL-3, IL-4, IL-5, IL-7 and VEGF), and other ligands and receptors (IFN-α2, IFN-γ, FLT-3L, IL-1RA and sCD40L) by using a commercially available MagPix Milliplex kit (EMD Millipore). The X-axis shows the incubation periods; 24 h and 48 h. Error bars represent ±standard deviation. A 2-way ANOVA was performed on the data to determine significant differences in expression between rhSP-D coated CNTs with or without serum treatment. All these comparisons were significant (p < 0.05), except where shown (ns: not significant, p > 0.05). | ||
Complement deposition negates the pro-inflammatory immune response induced by rhSP-D-coated CNTs
Since mRNA expression at early time points does not always give all the clues about immune response over an extended period, cytokine array analysis was performed on the supernatants obtained at 48 h after treatment of U937 and at 24 h and 48 h after treatment of THP-1 cells with CNTs (Fig. 5b and 6b). The data revealed a significant increase in the levels of pro-inflammatory cytokines for CNTs coated with rhSP-D, as indicated by IL-1α, IL-1β, TNF-α and IL-12p40. A similar profile of chemoattractant was seen in IL-8, I-TAC, MIG and MCP-1, indicating potential of rhSP-D to enhance inflammation. There was also an increase in G-CSF levels, particularly in CMC-CNTs coated with rhSP-D, suggesting a dampening of fibrotic responses consistent with an increase in the inflammatory response.35 IL-10 levels were down-regulated by the presence of rhSP-D coated on CMC-CNTs, whereas this was slightly enhanced for rhSP-D coated Ox-CNTs. The differences in protein levels between the un-coated CNT types were minor except for IL-8, IL-1α, IL-10 and MCP-1, where there was a marked enhancement of cytokine levels in CMC-CNTs compared to Ox-CNTs. The opposite pattern of protein secretion was observed for IL-1β and TNF-α, where they were suppressed in CMC-CNTs compared to Ox-CNTs.Like the U937 cells, the THP-1 cells produced an increased pro-inflammatory response when challenged with rhSP-D-coated CMC-CNTs (Fig. 6b). However, this effect was considerably reduced by complement deposition. Serum treatment of rhSP-D-bound CMC-CNTs led to a dramatic downregulation of IL-1β, TNF-α, IL-1α, IL-6, IL-8, MCP-1, MCP-3, MDC, GRO, IP-10, MIP-1β, C-CSF, GM-CSF and VEGF. There was slight, but significant down regulation in the levels of TNF-β, IL-17A, IL-12, IL-9 and IL-15. Other targets in the analyte arrays such as IL-10, IL-12p40, IFN-α2, eotaxin, fractalkine, MIP-1α, EGF, FGF-2, IL-3, IL-7, FLT3L and soluble CD40L were not significantly altered (Fig. 6b).
NF-κB nuclear translocation in response to rhSP-D coated CMC-CNTs can be halted by complement deposition
THP-1 cells were used to investigate nuclear translocation of NF-κB after CMC-CNT treatment using fluorescent staining techniques. NF-κB, a transcription factor, is an important regulator of expression of various pro-inflammatory cytokines induced in response to external stimuli. Fig. 7 shows fluorescence in cells exposed to CMC-CNTs for 2 h and stained with an antibody against the p65 subunit of NF-κB. As shown in Fig. 7 in the merged images, CMC-CNT exposure induced NF-κB nuclear translocation compared to the control (cells only). This nuclear translocation was more pronounced in the case of rhSPD-coated CMC-CNTs. However, no translocation was observed for complement deposited, rhSP-D-coated CMC-CNTs. This suggests that the pro-inflammatory effect observed for rhSP-D coated CMC-CNTs was negated by serum deposition on rhSP-D coated CMC-CNTs. This effect is further evident in the downregulation of TNF-α and IL-1β mRNA levels (Fig. 6a), and the secretion of pro-inflammatory cytokines (TNF-α, IL-1β, IL-2 and IL-6), and chemokines (IL-8, MCP-1, IP-10) (Fig. 6b).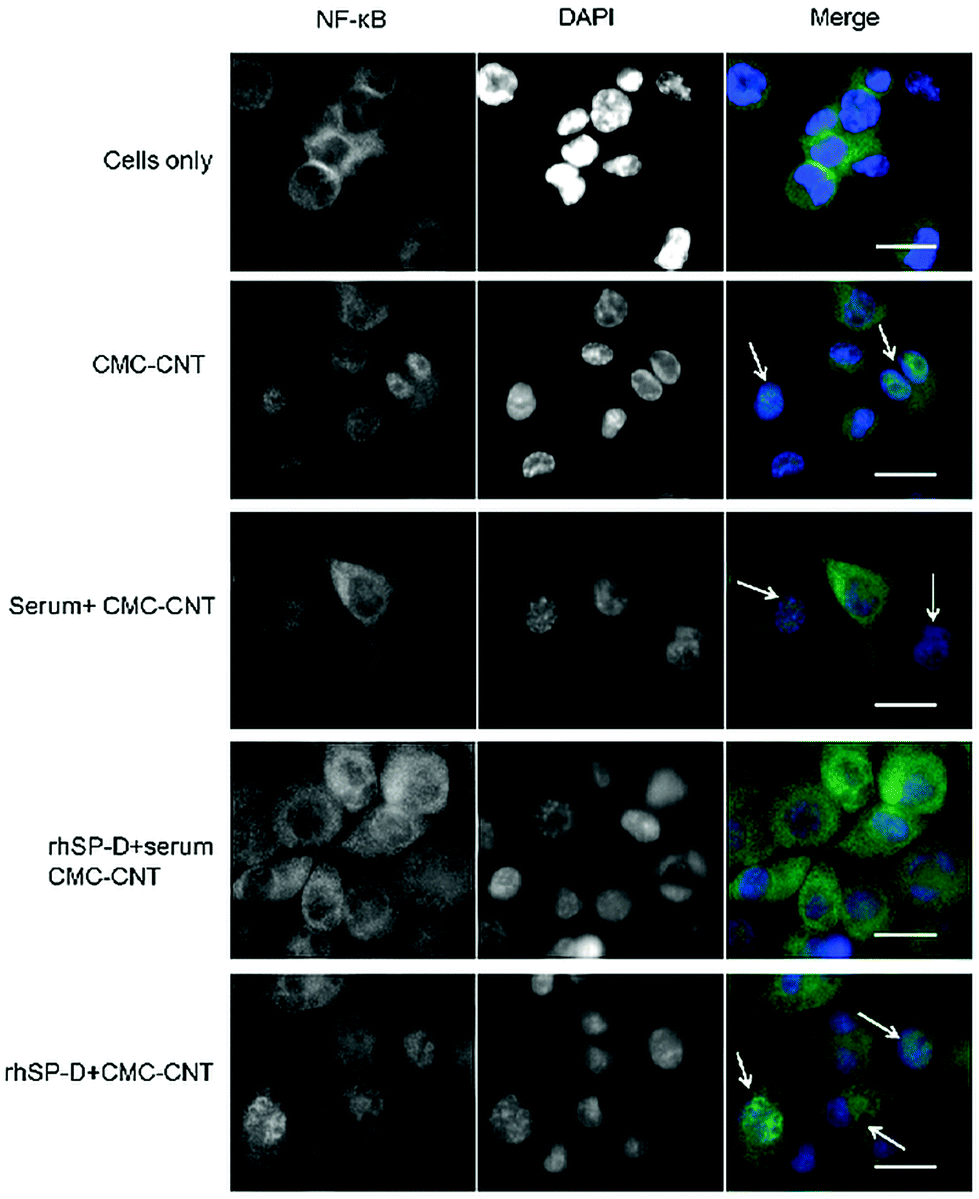 | ||
| Fig. 7 Effect of rhSP-D and/or complement deposition on CMC-CNT on NF-κB cytoplasm to nucleus translocation. Differentiated THP-1 cells were exposed to rhSP-D bound CMC-CNTs (rhSP-D + CMC-CNT), rhSP-D bound CMC-CNT with complement deposition (rhSP-D + serum + CMC-CNT), complement deposited CMC-CNTs (serum + CMC-CNTs), or CMC-CNT alone for 2 h. Cells were washed, fixed, permeabilised and incubated with rabbit anti-NF-κB p65 antibodies, followed by Alexa Fluor 488-goat anti rabbit antibody (green). The nucleus was stained with Hoechst 33342 (blue). Scale bar 20 μm. Arrows highlight nuclear NF-κB in the merged images. | ||
Discussion
Carbon nanotubes (CNTs) have exceptional physical properties that make them attractive and amenable for their exciting use as therapeutic vehicles in the lungs.5,6,36,37 Thus, it is of great importance to understand the biological interactions these CNTs will have in the lungs, especially with innate immune components, which are likely to be the first host defence system to recognise these nanoparticles.3,38 Inevitably, use of CNTs in the lungs will bring them into contact with pulmonary surfactant, including hydrophilic surfactant proteins, SP-A and SP-D, which are Ca2+-dependent pattern recognition innate immune molecules.17Of the two hydrophilic surfactant proteins, SP-D has been shown to mount a strong pro-inflammatory response when engaging with a variety of pathogens.39,40 Its structure includes a multimerising cysteine-containing N-terminal region, a triple-helical collagen-like region, a trimerising α-helical coiled-coil neck region, followed by a C-terminal C-type lectin or carbohydrate recognition domain (CRD). The primary subunit of three polypeptide chains is tetramerised, and can polymerise further to form fuzzy ball-like dodecamers.17 In this study, we have used recombinant human SP-D expressed in E. coli, which consists of three CRDs, a neck and eight gly-Xaa-Yaa collagen region repeats, where Xaa and Yaa could be any amino acid. The C-terminal homotrimeric CRD region is the principal ligand-binding domain of SP-D that recognises carbohydrate and/or charge patterns on pathogens, allergens, receptors/binding proteins, and apoptotic cells.41 This ligand binding causes opsonic effects, leading to enhanced phagocytosis and clearance mechanisms via superoxidative burst and mostly pro-inflammatory immune response by phagocytic cells.42In vivo models of allergy43–45 and SP-D knock-out mice studies have revealed that SP-D can modulate adaptive immune responses further downstream from its effector functions that can include helper T cell polarisation43 and apoptosis induction in immune-activated cells.31,46,47 Thus, interaction between SP-D and nanoparticles can have profound and wide-ranging impact on the pulmonary innate and adaptive immune mechanisms.
Both SP-A and SP-D have been shown to interact with nanoparticles and alter their phagocytosis. Metal oxide nanoparticles can bind SP-A purified from bronchoalveolar lavage, depending on their surface chemistry.48 Enhanced uptake of nanoparticles by alveolar macrophages has been reported by SP-A, which prefers hydrophobic coating, whilst SP-D prefers hydrophilic surface on the nanoparticles.49,50 Decreased uptake of nanoparticles has also been shown in alveolar macrophages and lung dendritic cells isolated from SP-D knockout mice, when compared to wild type mice.25
In the present study, we set out to examine the effect of rhSP-D binding to the surface of functionalised CNTs (Ox-CNT and CMC-CNTs) on their uptake by macrophage cell lines. THP-1 cells constitutively express complement receptor 3 (CR3) whose surface expression is further up-regulated by PMA treatment.51 The observed enhanced uptake of rhSP-D coated CMC-CNTs could be thus CR3 mediated, as observed for SP-A.52 We also tested if rhSP-D coated CNTs continue to activate the complement classical pathway, exploring the possibility of independent and multiple interactions of key innate immune molecules with CNTs. It has been shown previously that serum treatment (i.e. complement deposition) enhances CNT uptake by macrophages (U937), B cells (Raji),4 and human bronchial epithelial cells.52
We have previously shown that complement deposition occurs on CNTs, mainly via the classical pathway, which enhances their uptake by complement receptors on macrophages and B cells, whilst down-regulating the pro-inflammatory cytokine response via the up-regulation of IL-10.4,28 We have also shown that C1q, the first recognition subcomponent of the classical pathway, binds to CNTs via its ligand recognising globular (gC1q) domain, as revealed by the use of recombinant globular head modules (ghA, ghB and ghC).3,4 It is likely that the binding of C1q to CNTs is through the recognition of charge patterns.3,4,53,54 SP-D is of similar structure to C1q but is a mainly carbohydrate or charge pattern recognition molecule, perhaps recognising vicinal diol groups as does mannose-binding lectin.19 Thus, in addition to the modulatory properties of rhSP-D after binding to CNTs, we also examined the effect of rhSP-D bound to CNTs on complement activation. Interestingly, complement consumption was enhanced 2-fold by rhSP-D when it was bound to Ox-CNTs as well as CMC-CNTs. These observations indicate that SP-D binds different sites than C1q on CNTs, and thus does not inhibit binding of C1q to CNTs. Increased complement activation by rhSP-D coated CNTs is most likely caused by bound rhSP-D forming additional binding sites for C4b and C3b. In contrast, the recombinant ghA, ghB and ghC modules, bound to CNTs, have been shown to inhibit classical pathway activation.3,4 This is because they do compete with C1q for binding to the CNTs.
We also examined the downstream cytokine response from macrophage cell lines treated with CNTs with and without rhSP-D coating. rhSP-D bound CNTs up-regulated the pro-inflammatory response in U937 and THP-1 cells. It is also likely that the majority of rhSP-D coated CNTs interact with Toll-like receptor 4 (TLR4), and thus initiate the IL-1β pathway release via NLRP3 inflammasome activation.34 The up-regulation of IL-12 may indicate Th1 cell polarisation,55 suggesting that SP-D switches from eliciting a Th2 to a Th1 response during interaction with CNTs. These reports are consistent with an earlier report.4 rhSP-D coating on both types of CNTs was also found to up-regulate G-CSF and I-TAC, which are important for the mobilisation of neutrophils and T cells, respectively. This further illustrates the potential of rhSP-D to cause inflammatory response in conjunction with CNTs.
IL-1α and IL-1β are prototypical inflammatory cytokines, which can additionally induce endothelial cells to produce infiltration-causing MCP-1.56 In addition, IL-1 also induces IFN-γ production by Th1 and NK cells via IL-12 secretion by antigen presenting cells.57 In addition, IL-1 causes matrix metalloproteinases production by resident fibroblasts causing extracellular matrix degradation. Thus, suppression of these cytokines, whose generation involves inflammasomes and TLR4, is beneficial for pulmonary homeostasis. It is curious that IL-1 itself can upregulate the pyrogenic and acute phase protein TNF-α in addition to perhaps TLR4 mediated interaction involving SP-D and CNT.58
One crucial observation being reported here is the up-regulation of VEGF by rhSP-D bound CNTs. VEGF is known to increase edema (vascular permeability) in asthma, involving Th2 dependent allergen sensitisation and pulmonary inflammation.59 It is quite clear that CNT-bound rhSP-D does the opposite of soluble rhSP-D in murine models;43 however this effect can be overcome by complement deposition. A similar role can be attributed in Th2-dependent lung diseases. IL-8, as a crucial neutrophil chemoattractant, has been shown to be involved in acute lung injury.60 NF-κB nuclear translocation induced by nanoparticles has been previously shown in human kidney cells.61 Similarly, both in vivo and in vitro studies have shown activation of NF-κB and production of pro-inflammatory mediators such as TNF-α, IL-1β, IL-8 and IL-6 by metallic nanoparticles62 and single-walled CNTs.63
The results presented here raise several important issues. Functionalized CNTs, as a charge pattern recognition target, can be recognised by SP-D and C1q simultaneously. This study is one of few which we have published, in which the binding of several other proteins, e.g. human serum albumin (HSA), bovine serum albumin (BSA), fibrinogen, human C1q, recombinant fragments of C1q, and SP-A, have been studied.3,4,19,20,28,33 It has been shown that coating CNTs with other proteins such as BSA and fibrinogen can enhance complement activation.28 However, the binding of SP-D from the lung surfactant fluid to CNTs is highly selective, and therefore, SP-D binding to CNT in the lung will occur more avidly than the binding of other proteins. Thus, in the context of lungs, the effects of SP-D will dominate the effect of other proteins. In addition, we have also carried out control experiments using maltose-binding protein (MBP), which did not modify the effect of CNTs on immune cells.
SP-D opsonisation, similar to complement deposition, leads to enhanced uptake of CNTs by macrophages. However, both opsonins (SP-D and complement), when together, restrict the entry of the CNTs into macrophages. The ability of complement to suppress pro-inflammatory response by rhSP-D bound CNTs makes a good argument for coating of CNTs with small fragments of recombinant complement components for therapeutic applications.
In the context of the lungs, the use of therapeutic CNTs needs to be carefully considered, since their interaction with SP-D could cause harmful inflammation analogous to what has been observed with asbestos.64,65 Furthermore, it has been reported that carbon black particles have the ability to completely sequester SP-D from suspension.66 Sequestration of surfactant proteins aggravated ongoing Streptococcus penumoniae infection in rats exposed to airborne particles of sizes less than 10 μm.67 Furthermore, sequestration of SP-D and SP-A by nanoparticles was shown to alter the ability of these proteins to neutralise influenza A infection in vitro.27 Immobilization of these innate immune molecules by nanoparticles could thus make the host susceptible to viral and bacterial infection. Modified CNTs intended for administration to the lung should therefore be examined for their interaction with SP-D.
Authors’ contributions
KM and BP carried out most of the crucial experiments; RBS, AK, LK, AGT, LAJ, CSM, HAK and BTH provided crucial reagents and technical help; GS carried out most of microscopy work and helped with writing; UK led the project with ideas and the design of experiments, in addition to writing the manuscript.Acknowledgements
H. A. K. acknowledges the Deanship of Scientific Research at King Saud University for funding via Group no. RGP-009.Notes and references
- T. R. Fadel and T. M. Fahmy, Trends Biotechnol., 2014, 32, 198–209 CrossRef CAS PubMed.
- R. V. Mundra, X. Wu, J. Sauer, J. S. Dordick and R. S. Kane, Curr. Opin. Biotechnol., 2014, 28, 25–32 CrossRef CAS PubMed.
- K. M. Pondman, L. Pednekar, B. Paudyal, A. G. Tsolaki, L. Kouser, H. A. Khan, M. H. Shamji, B. Ten Haken, G. Stenbeck, R. B. Sim and U. Kishore, Nanomedicine, 2015, 11, 2109–2118 CAS.
- K. M. Pondman, A. G. Tsolaki, B. Paudyal, M. H. Shamji, A. Switzer, A. A. Pathan, S. M. Abozaid, B. Ten Haken, G. Stenbeck, R. B. Sim and U. Kishore, J. Biomed. Nanotechnol., 2016, 12, 197–216 CrossRef CAS PubMed.
- J. C. Bonner, Expert Rev. Respir. Med., 2011, 5, 779–787 CrossRef CAS PubMed.
- S. Choudhary and V. Kusum Devi, J. Controlled Release, 2015, 202, 65–75 CrossRef CAS PubMed.
- C. W. Lam, J. T. James, R. McCluskey and R. L. Hunter, Toxicol. Sci., 2004, 77, 126–134 CrossRef CAS PubMed.
- D. B. Warheit, B. R. Laurence, K. L. Reed, D. H. Roach, G. A. Reynolds and T. R. Webb, Toxicol. Sci., 2004, 77, 117–125 CrossRef CAS PubMed.
- J. B. Mangum, E. A. Turpin, A. Antao-Menezes, M. F. Cesta, E. Bermudez and J. C. Bonner, Part. Fibre Toxicol., 2006, 3, 15 CrossRef PubMed.
- X. Wang, P. Katwa, R. Podila, P. Chen, P. C. Ke, A. M. Rao, D. M. Walters, C. J. Wingard and J. M. Brown, Part. Fibre Toxicol., 2011, 8, 2–13 CrossRef PubMed.
- S. Y. Madani, A. Mandel and A. M. Seifalian, Nano Rev., 2013, 4, 21521 Search PubMed.
- A. A. Shvedova, J. P. Fabisiak, E. R. Kisin, A. R. Murray, J. R. Roberts, Y. Y. Tyurina, J. M. Antonini, W. H. Feng, C. Kommineni, J. Reynolds, A. Barchowsky, V. Castranova and V. E. Kagan, Am. J. Respir. Cell Mol. Biol., 2008, 38, 579–590 CrossRef CAS PubMed.
- A. A. Shvedova, E. Kisin, A. R. Murray, V. J. Johnson, O. Gorelik, S. Arepalli, A. F. Hubbs, R. R. Mercer, P. Keohavong, N. Sussman, J. Jin, J. Yin, S. Stone, B. T. Chen, G. Deye, A. Maynard, V. Castranova, P. A. Baron and V. E. Kagan, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2008, 295, L552–L565 CrossRef CAS PubMed.
- A. A. Shvedova, E. R. Kisin, A. R. Murray, C. Kommineni, V. Castranova, B. Fadeel and V. E. Kagan, Toxicol. Appl. Pharmacol., 2008, 231, 235–240 CrossRef CAS PubMed.
- D. W. Porter, A. F. Hubbs, R. R. Mercer, N. Wu, M. G. Wolfarth, K. Sriram, S. Leonard, L. Battelli, D. Schwegler-Berry, S. Friend, M. Andrew, B. T. Chen, S. Tsuruoka, M. Endo and V. Castranova, Toxicology, 2010, 269, 136–147 CrossRef CAS PubMed.
- J. Goerke, Biochim. Biophys. Acta, 1998, 1408, 79–89 CrossRef CAS.
- U. Kishore, T. J. Greenhough, P. Waters, A. K. Shrive, R. Ghai, M. F. Kamran, A. L. Bernal, K. B. Reid, T. Madan and T. Chakraborty, Mol. Immunol., 2006, 43, 1293–1315 CrossRef CAS PubMed.
- M. Rybak-Smith, K. M. Pondman, E. Flahaut, C. Salvador-Morales and R. B. Sim, in Carbon Nanotubes for Biomedical Applications, ed. R. Klingeler and B. R. Sim, Springer, Berlin Heidelberg, 2011, pp. 183–210 Search PubMed.
- C. Salvador-Morales, P. Townsend, E. Flahaut, C. Vénien-Bryan, A. Vlandas, M. L. H. Green and R. B. Sim, Carbon, 2007, 45, 607–617 CrossRef CAS.
- C. Salvador-Morales, Z. Khan, J. Zamory, V. Tran, A. Cedeno, J. Umanzor-Alvarez, U. Kishore and R. B. Sim, J. Adv. Microsc. Res., 2013, 8, 93–99 CrossRef CAS.
- A. A. Kapralov, W. H. Feng, A. A. Amoscato, N. Yanamala, K. Balasubramanian, D. E. Winnica, E. R. Kisin, G. P. Kotchey, P. Gou, L. J. Sparvero, P. Ray, R. K. Mallampalli, J. Klein-Seetharaman, B. Fadeel, A. Star, A. A. Shvedova and V. E. Kagan, ACS Nano, 2012, 6, 4147–4156 CrossRef CAS PubMed.
- M. Gasser, P. Wick, M. J. Clift, F. Blank, L. Diener, B. Yan, P. Gehr, H. F. Krug and B. Rothen-Rutishauser, Part. Fibre Toxicol., 2012, 9, 9–17 CrossRef PubMed.
- N. V. Konduru, Y. Y. Tyurina, W. Feng, L. V. Basova, N. A. Belikova, H. Bayir, K. Clark, M. Rubin, D. Stolz, H. Vallhov, A. Scheynius, E. Witasp, B. Fadeel, P. D. Kichambare, A. Star, E. R. Kisin, A. R. Murray, A. A. Shvedova and V. E. Kagan, PLoS One, 2009, 4, e4398 Search PubMed.
- B. Stringer and L. Kobzik, Am. J. Respir. Cell Mol. Biol., 1996, 14, 155–160 CrossRef CAS PubMed.
- M. Kendall, P. Ding, R. M. Mackay, R. Deb, Z. McKenzie, K. Kendall, J. Madsen and H. Clark, Nanotoxicology, 2013, 7, 963–973 CrossRef CAS PubMed.
- Z. McKenzie, M. Kendall, R. M. Mackay, H. Whitwell, C. Elgy, P. Ding, S. Mahajan, C. Morgan, M. Griffiths, H. Clark and J. Madsen, Nanotoxicology, 2015, 9, 952–962 CrossRef PubMed.
- Z. McKenzie, M. Kendall, R. M. Mackay, T. D. Tetley, C. Morgan, M. Griffiths, H. W. Clark and J. Madsen, Philos. Trans. R. Soc. London, Ser. B, 2015, 370, 20140049 CrossRef PubMed.
- K. M. Pondman, M. Sobik, A. Nayak, A. G. Tsolaki, A. Jakel, E. Flahaut, S. Hampel, B. Ten Haken, R. B. Sim and U. Kishore, Nanomedicine, 2014, 10, 1287–1299 CAS.
- Y. Li, X. Zhang, J. Luo, W. Huang, J. Cheng, Z. Luo, T. Li, F. Liu, G. Xu, X. Ke, L. Li and H. J. Geise, Nanotechnology, 2004, 15, 1645 CrossRef CAS.
- R. Verdejo, S. Lamoriniere, B. Cottam, A. Bismarck and M. Shaffer, Chem. Commun., 2007,(5), 513–515 RSC.
- L. Mahajan, T. Madan, N. Kamal, V. K. Singh, R. B. Sim, S. D. Telang, C. N. Ramchand, P. Waters, U. Kishore and P. U. Sarma, Int. Immunol., 2008, 20, 993–1007 CrossRef CAS PubMed.
- E. Dodagatta-Marri, A. S. Qaseem, N. Karbani, A. G. Tsolaki, P. Waters, T. Madan and U. Kishore, Methods Mol. Biol., 2014, 1100, 273–290 CAS.
- C. Salvador-Morales, E. Flahaut, E. Sim, J. Sloan, M. L. Green and R. B. Sim, Mol. Immunol., 2006, 43, 193–201 CrossRef CAS PubMed.
- E. Meunier, A. Coste, D. Olagnier, H. Authier, L. Lefevre, C. Dardenne, J. Bernad, M. Beraud, E. Flahaut and B. Pipy, Nanomedicine, 2012, 8, 987–995 CAS.
- F. Romero, D. Shah, M. Duong, R. B. Penn, M. B. Fessler, J. Madenspacher, W. Stafstrom, M. Kavuru, B. Lu, C. B. Kallen, K. Walsh and R. Summer, Am. J. Respir. Cell Mol. Biol., 2015, 53, 74–86 CrossRef CAS PubMed.
- J. G. Li, W. X. Li, J. Y. Xu, X. Q. Cai, R. L. Liu, Y. J. Li, Q. F. Zhao and Q. N. Li, Environ. Toxicol., 2007, 22, 415–421 CrossRef CAS PubMed.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed.
- K. M. Pondman, R. B. Sim and U. Kishore, in Encyclopedia of Nanotechnology, ed. B. Bhushan, Springer, Netherlands, Dordrecht, 2015, pp. 1–8 Search PubMed.
- U. Kishore, A. L. Bernal, M. F. Kamran, S. Saxena, M. Singh, P. U. Sarma, T. Madan and T. Chakraborty, Arch. Immunol. Ther. Exp., 2005, 53, 399–417 CAS.
- A. Nayak, E. Dodagatta-Marri, A. G. Tsolaki and U. Kishore, Front. Immunol., 2012, 3, 131 Search PubMed.
- P. Silveyra and J. Floros, Front. Biosci., 2012, 17, 407–429 CrossRef CAS.
- U. Kishore, T. Madan, P. U. Sarma, M. Singh, B. C. Urban and K. B. Reid, Immunobiology, 2002, 205, 610–618 CrossRef CAS.
- T. Madan, U. Kishore, M. Singh, P. Strong, H. Clark, E. M. Hussain, K. B. Reid and P. U. Sarma, J. Clin. Invest., 2001, 107, 467–475 CrossRef CAS PubMed.
- T. Madan, K. B. Reid, M. Singh, P. U. Sarma and U. Kishore, J. Immunol., 2005, 174, 6943–6954 CrossRef CAS.
- A. S. Qaseem, S. Sonar, L. Mahajan, T. Madan, G. L. Sorensen, M. H. Shamji and U. Kishore, Mol. Immunol., 2013, 54, 98–107 CrossRef CAS PubMed.
- L. Mahajan, P. Gautam, E. Dodagatta-Marri, T. Madan and U. Kishore, Expert Rev. Proteomics, 2014, 11(3), 355–369 CrossRef CAS PubMed.
- L. Mahajan, H. Pandit, T. Madan, P. Gautam, A. K. Yadav, H. Warke, C. S. Sundaram, R. Sirdeshmukh, P. U. Sarma, U. Kishore and A. Surolia, PLoS One, 2013, 8, e85046 Search PubMed.
- C. Schulze, U. F. Schaefer, C. A. Ruge, W. Wohlleben and C. M. Lehr, Eur. J. Pharm. Biopharm., 2011, 77, 376–383 CrossRef CAS PubMed.
- C. A. Ruge, U. F. Schaefer, J. Herrmann, J. Kirch, O. Canadas, M. Echaide, J. Perez-Gil, C. Casals, R. Muller and C. M. Lehr, PLoS One, 2012, 7, e40775 CAS.
- C. A. Ruge, J. Kirch, O. Canadas, M. Schneider, J. Perez-Gil, U. F. Schaefer, C. Casals and C. M. Lehr, Nanomedicine, 2011, 7, 690–693 CAS.
- J. Plescia and D. C. Altieri, Biochem. J., 1996, 319(Pt 3), 873–879 CrossRef CAS PubMed.
- H. Haniu, N. Saito, Y. Matsuda, T. Tsukahara, K. Maruyama, Y. Usui, K. Aoki, S. Takanashi, S. Kobayashi, H. Nomura, M. Okamoto, M. Shimizu and H. Kato, Toxicol. in Vitro, 2013, 27, 1679–1685 CrossRef CAS PubMed.
- U. Kishore and K. B. Reid, Immunopharmacology, 2000, 49, 159–170 CrossRef CAS PubMed.
- R. Ghai, P. Waters, L. T. Roumenina, M. Gadjeva, M. S. Kojouharova, K. B. Reid, R. B. Sim and U. Kishore, Immunobiology, 2007, 212, 253–266 CrossRef CAS PubMed.
- V. Athie-Morales, H. H. Smits, D. A. Cantrell and C. M. Hilkens, J. Immunol., 2004, 172, 61–69 CrossRef CAS.
- A. Sica, J. M. Wang, F. Colotta, E. Dejana, A. Mantovani, J. J. Oppenheim, C. G. Larsen, C. O. Zachariae and K. Matsushima, J. Immunol., 1990, 144, 3034–3038 CAS.
- A. Billiau, Adv. Immunol., 1996, 62, 61–130 CAS.
- M. Yamazoe, C. Nishitani, M. Takahashi, T. Katoh, S. Ariki, T. Shimizu, H. Mitsuzawa, K. Sawada, D. R. Voelker, H. Takahashi and Y. Kuroki, J. Biol. Chem., 2008, 283, 35878–35888 CrossRef CAS PubMed.
- C. G. Lee, H. Link, P. Baluk, R. J. Homer, S. Chapoval, V. Bhandari, M. J. Kang, L. Cohn, Y. K. Kim, D. M. McDonald and J. A. Elias, Nat. Med., 2004, 10, 1095–1103 CrossRef CAS PubMed.
- T. C. Allen and A. Kurdowska, Arch. Pathol. Lab. Med., 2014, 138, 266–269 CrossRef CAS PubMed.
- I. Pujalte, I. Passagne, B. Brouillaud, M. Treguer, E. Durand, C. Ohayon-Courtes and B. L'Azou, Part. Fibre Toxicol., 2011, 8, 1–10 CrossRef PubMed.
- C. Monteiller, L. Tran, W. MacNee, S. Faux, A. Jones, B. Miller and K. Donaldson, Occup. Environ. Med., 2007, 64, 609–615 CrossRef CAS PubMed.
- M. Pacurari, X. J. Yin, J. Zhao, M. Ding, S. S. Leonard, D. Schwegler-Berry, B. S. Ducatman, D. Sbarra, M. D. Hoover, V. Castranova and V. Vallyathan, Environ. Health Perspect., 2008, 116, 1211–1217 CrossRef CAS PubMed.
- S. L. Cassel, S. C. Eisenbarth, S. S. Iyer, J. J. Sadler, O. R. Colegio, L. A. Tephly, A. B. Carter, P. B. Rothman, R. A. Flavell and F. S. Sutterwala, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9035–9040 CrossRef CAS PubMed.
- C. Dostert, V. Petrilli, R. Van Bruggen, C. Steele, B. T. Mossman and J. Tschopp, Science, 2008, 320, 674–677 CrossRef CAS PubMed.
- M. Kendall, L. Brown and K. Trought, Inhalation Toxicol., 2004, 16(Suppl 1), 99–105 CrossRef CAS PubMed.
- J. T. Zelikoff, L. C. Chen, M. D. Cohen, K. Fang, T. Gordon, Y. Li, C. Nadziejko and R. B. Schlesinger, Inhalation Toxicol., 2003, 15, 131–150 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr08807d |
| ‡ These authors contributed equally to this study, thus, they are joint first authors. |
| This journal is © The Royal Society of Chemistry 2017 |