
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time monitoring of plasmon induced dissociative electron transfer to the potential DNA radiosensitizer 8-bromoadenine†
R.
Schürmann
ab and
I.
Bald
 *ab
*ab
aInstitute of Chemistry, Physical Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14776 Potsdam, Germany. E-mail: bald@uni-potsdam.de
bBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany
First published on 27th December 2016
Abstract
The excitation of localized surface plasmons in noble metal nanoparticles (NPs) results in different nanoscale effects such as electric field enhancement, the generation of hot electrons and a temperature increase close to the NP surface. These effects are typically exploited in diverse fields such as surface-enhanced Raman scattering (SERS), NP catalysis and photothermal therapy (PTT). Halogenated nucleobases are applied as radiosensitizers in conventional radiation cancer therapy due to their high reactivity towards secondary electrons. Here, we use SERS to study the transformation of 8-bromoadenine (8BrA) into adenine on the surface of Au and AgNPs upon irradiation with a low-power continuous wave laser at 532, 633 and 785 nm, respectively. The dissociation of 8BrA is ascribed to a hot-electron transfer reaction and the underlying kinetics are carefully explored. The reaction proceeds within seconds or even milliseconds. Similar dissociation reactions might also occur with other electrophilic molecules, which must be considered in the interpretation of respective SERS spectra. Furthermore, we suggest that hot-electron transfer induced dissociation of radiosensitizers such as 8BrA can be applied in the future in PTT to enhance the damage of tumor tissue upon irradiation.
Introduction
Illumination of noble metal nanoparticles (NPs) with visible light leads to resonant oscillations of the electron gas, which are referred to as localized surface plasmons (LSP). In close proximity to the metal surface the electric field of the incoming and scattered light is highly enhanced by the LSP, which is exploited in surface enhanced Raman scattering (SERS), as the Raman scattering cross sections increase by several orders of magnitude.1,2 LSP can relax inelastically via reemitting a photon or through a non-radiative channel via electron hole pair production.3,4 These so called “hot electrons” can tunnel into unoccupied molecular orbitals of chemisorbed or physisorbed molecules and induce chemical reactions5–9 that can be monitored in real time using SERS.10,11Besides the direct electron transfer that is exploited in plasmonic catalysis “hot electrons” lose their energy very quickly via electron–electron scattering and subsequently via electron phonon scattering, which increases the temperature of the nanoparticles and their surroundings.12 In photothermal therapy (PTT) the increased temperature around hollow AuNPs13 or nanorods14 is used to kill cancer cells under illumination with NIR-lasers.15,16 Based on the work presented here we suggest that the combination of NPs with molecules incorporated in the DNA that strongly react with plasmon induced electrons might improve the efficiency of this therapy. Such potential radiosensitizers are halogenated nucleobases like 8-bromoadenie (8BrA) that strongly interact with low energy electrons17–20via dissociative electron attachment (DEA)21 whereby a transient negative ion decays into anionic and neutral fragments. This mechanism allows the damage of DNA below the ionization threshold.22 Brominated nucleobases that can be easily incorporated into the DNA possess a high electron affinity and the C–Br-bond is easily cleaved by attachment of electrons close to 0 eV.23,24 The formation of a radical inside the DNA leads to a strand break in a second step25 and thus increases the single strand break cross section.26
Here we demonstrate the dissociative hot electron transfer from plasmonically excited Au and AgNPs to 8BrA in a dry and aqueous environment monitored in real time using SERS. The reaction follows a fractal like kinetics and shows a fast reaction rate, as under typical SERS settings the majority of the analyte molecules are decomposed in a few hundred milliseconds.
Results and discussion
LSP that are created via laser illumination of AgNPs can decay non-radiatively by the formation of an electron hole pair, whereby the electron is excited from the sp-conduction band and has an energy between Ef and Ef + hν (Fig. 1):| hν + AgNP → e− + h+ | (1) |
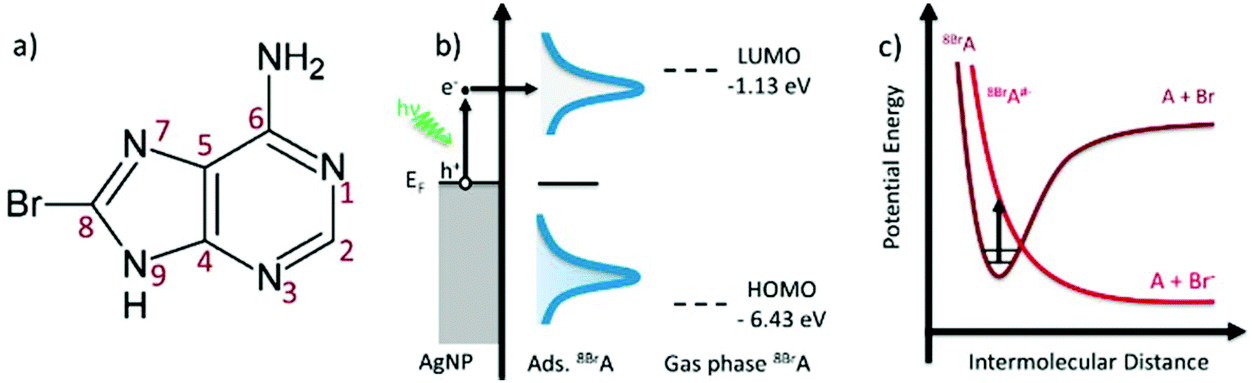 | ||
| Fig. 1 (a) Molecular structure of 8BrA; (b) schematic “hot electron” excitation in the silver nanoparticle with subsequent tunnelling into the LUMO of the adsorbed 8BrA; (c) schematic potential energy diagram illustrating DEA to 8BrA. | ||
On a time scale below 10 fs the excited electron loses its energy via electron–electron scattering until the electron gas approaches a Fermi–Dirac distribution.3 These so called “hot electrons” can tunnel into the lowest unoccupied molecular orbital (LUMO) of adsorbed 8BrA molecules and form a transient negative ion (TNI).21 Beyond that the electron can be directly injected into the LUMO via a coherent tunneling process before the electron interacts with the electron gas.27,28 In both cases a relatively unstable 8BrA anion is formed that can relax through the cleavage of the C8–Br bond.17,18
| 8BrA + e− → 8BrA#− → A˙ + Br− | (2) |
In the present experiment a AgNP solution with adsorbed 8BrA was dried on a Si wafer and SERS spectra were recorded as a function of the illumination time using a 532 nm laser with 250 μW focused on a 10 μm diameter spot. For a relatively short time of around 1 s Raman spectra were recorded with a band structure that is clearly assigned to 8BrA (Fig. 2a). The most remarkable bands are the ring breathing mode at 767 cm−1 and the C–Br bending- and stretching-mode at 299 cm−1 and 575 cm−1, respectively. With ongoing illumination, these bands are significantly decreasing in their intensity while simultaneously new bands arise at 336 cm−1, 629 cm−1, 735 cm−1, 967 cm−1, 1267 cm−1, 1329 cm−1, 1409 cm−1, 1465 cm−1 and 1575 cm−1 (indicated by a grey background in Fig. 2) that can be assigned to the SERS-spectrum of adenine (A).29 This indicates that the C–Br bond of the adsorbed 8BrA ruptures during laser irradiation most likely due to the dissociative attachment of an additional electron according to eqn (2). By capture of an H radical from the environment A is formed on the nanoparticle surface. A similar reaction has previously been demonstrated already with uracil.24 The transformation from 8BrA to A can be tracked by the intensity of the ring breathing mode at 735 cm−1 and 767 cm−1 (Fig. 2c).
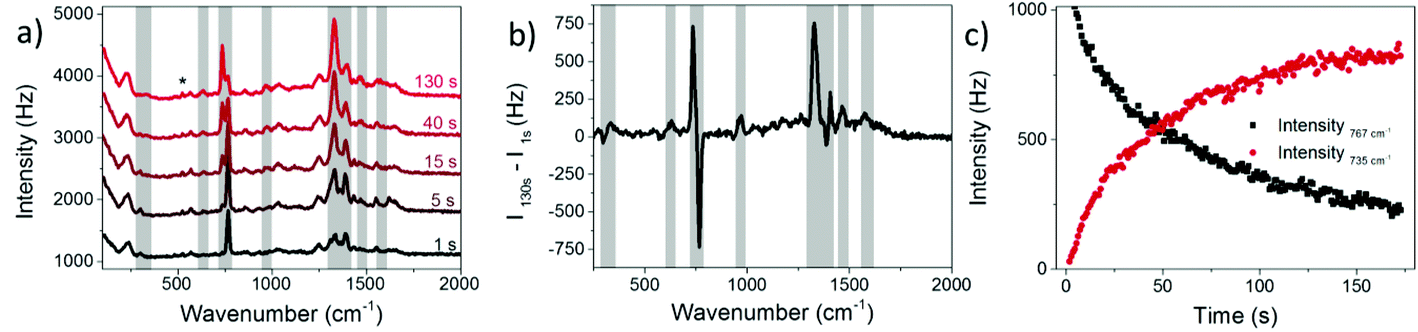 | ||
| Fig. 2 (a) SERS spectra of 8BrA on AgNPs dried on a Si-substrate for different illumination times. The Si band from the substrate at 521 cm−1 is marked with a *. (b) Difference spectra of t = 1 s and t = 130 s (same data set like in a). (c) Intensity of ring-breathing mode of 8BrA at 767 cm−1vs. ring breathing mode of A at 735 cm−1. | ||
The rate of the reaction depends on the concentration of 8BrA and the concentration of “hot electrons” in the illuminated area:
| Rate = −k [8BrA] [e−] | (3) |
However, it is difficult to monitor both concentrations simultaneously, in particular the number of hot electrons remains unknown. During continuous laser irradiation, a constant equilibrium concentration of “hot electrons” can be assumed as they are frequently reproduced and their timescales for excitation and relaxation are several orders of magnitude shorter than the timescales of the DEA reaction. Hence pseudo first-order kinetics is assumed to determine the observed rate coefficient kobs of the dissociation of 8BrA:
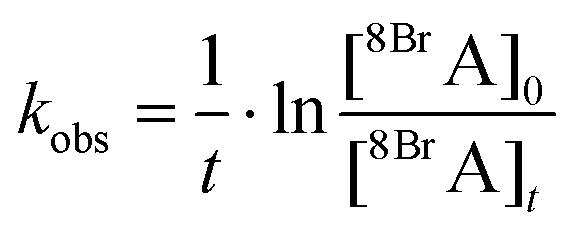 | (4) |
In Fig. 3akobs is plotted against time t, which demonstrates that the rate coefficient is not constant as expected for a (pseudo-) first order reaction, but decreases in time. This is attributed to the inhomogeneous distribution of reaction sites on the surface giving rise to a broad range of signal intensities and thus rate constants. In close proximity to the noble metal surface the electric field of the incoming and scattered light is strongly increased due to its interaction with the LSP, which strongly depends on the arrangement and structure of the nanoparticles. This electromagnetic enhancement increases the intensity of the Raman signal (Ij) by some orders of magnitude:1
| Ij = α2·gj4·I0 | (5) |
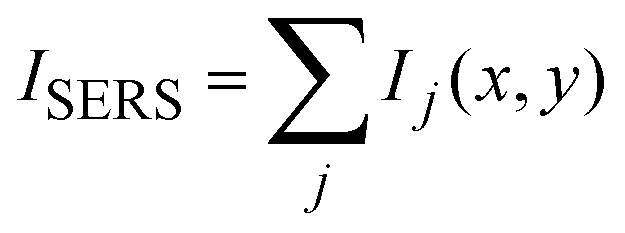 | (6) |
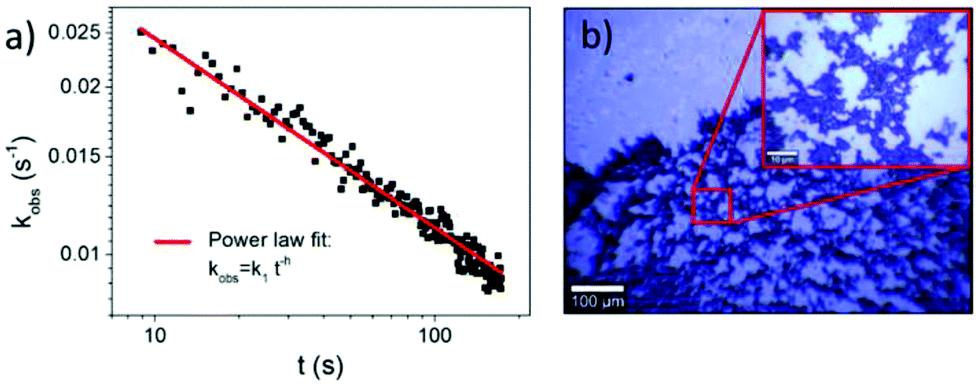 | ||
| Fig. 3 (a) Observed rate coefficient plotted against illumination time shows a power law dependence with h = 0.34 ± 0.01. (b) Light microscopy image of a dried AgNP sample on Si reveals a fractal structure of aggregated AgNPs. | ||
When assuming that the concentration of hot electrons at a certain position is in coherence with the magnitude of LSP, the local reaction rate correlates with the electromagnetic enhancement in the “hotspot”. Hence the 8BrA molecules that experience the strongest signal enhancement have the highest decomposition probability. Thus, the reaction centers with the highest rates only contribute in the beginning to the overall reaction, and consequently the observed reaction rate decreases with time.
This time dependence can be characterized by fractal-like kinetics,30 which describes heterogeneous reactions with geometrical constraints like hot electron catalyzed reactions,31 with a time dependent rate coefficient:
| kobs = k1·t−h, 0 ≤ h ≤ 1, t ≥ 1, | (7) |
According to eqn (4)kobs strongly depends on the starting intensity of the 8BrA signal that rapidly decreases during the first integration time. Assuming that the Raman enhancement factors of 8BrA and A are equivalent the sum of the intensities at 735 cm−1 and 767 cm−1 was used to determine the starting intensity of the 767 cm−1 peak.
20 measurements on the same sample at different positions have been performed and h was determined to be h = 0.36 ± 0.09, which indicates a 2-dimensional fractal lattice, for which h = 0.33.30Fig. 3a shows one of these measurements. The determined fractal dimension is in accordance with the fractal shape of the nanoparticle aggregates, which were observed in the bright field images (Fig. 3b) and described previously.32,33 It has to be mentioned that the standard deviation of h is larger than the error of h determined from the power law fit, as the composition of aggregates may vary on different positions of the sample.
Since k1 and h are not independent fitting parameters, h was set to 0.33 to avoid artifacts in the subsequent analysis. The rate constant k1 was determined as a function of the incident laser power P and fitted with a power function (see Fig. 4a):
| k1 = a·Pb | (8) |
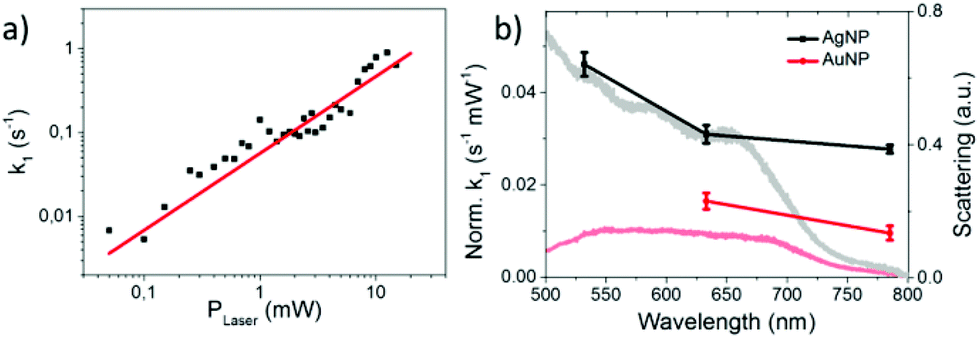 | ||
| Fig. 4 (a) Rate constant k1 as a function of the incident laser power fitted by a power law function (k1 = a·PLaserb with b = 0.92 ± 0.09). (b) Rate constant normalized by laser intensity for 8BrA dissociation on AgNPs (black) and AuNPs (red) for different laser wavelength and typical dark field spectra of aggregated NPs (grey and light red). | ||
The linear correlation between k1 and P with b = 0.92 ± 0.09 indicates a one photon process that is characteristic for hot electron induced reactions. For (partly) thermally induced reactions a super-linear power law dependence5 would be expected due to the exponential dependence of the reaction rate on temperature according to the Arrhenius equation. This conclusion is valid as long as the temperature on the AgNPs rises linearly with the incident laser power.12 Beyond that a direct photoexcitation is also unlikely for low photon fluences (102–104 W cm−2), because the HOMO–LUMO gap is about 5.3 eV according to ab initio calculations performed at the MP2/aug-cc-pVDZ level of theory.34 Therefore at least 3 photons with 2.33 eV (532 nm) are required to overcome the HOMO–LUMO gap.
We have also studied the transformation from 8BrA to A on a nanoparticle surface for 633 nm and 785 nm laser wavelength as well as on gold nanoparticles (AuNPs). In all cases a linear dependence on the laser power was observed (Fig. S3 in the ESI† and Fig. 4a). This is remarkable as the work functions of AgNPs (4.3 eV) and AuNPs (5.1 eV) are well above the photon energy of 1.61 eV (785 nm). This supports the model of dissociative electron transfer into the low lying LUMO of 8BrA. Normalizing k1 by the laser power reveals that the reaction rate increases with photon energy and is lower on AuNPs compared to AgNPs (see Fig. 4b) most probably due to the increased plasmonic enhancement on the AgNPs. We have recorded dark field scattering spectra on several spots of the sample, which show a broad surface plasmon resonance (SPR) ranging from around 400 nm for AgNPs and 500 nm for AuNPs to 700 nm. In particular for the near infrared photons with 785 nm wavelength, where the plasmonic enhancement is relatively weak, the SPR does not directly correspond to the normalized k1, even though the major trends are reflected. This might be explained by a resonant direct electron transfer into the adsorbed 8BrA. Nevertheless, on the basis of the present data it is not possible to say whether the reaction is dominated by a coherent tunnelling process or by Fermi–Dirac distributed electrons scattering into the molecular orbitals of the 8BrA molecule. In order to study the influence of the environment additional measurements have been performed in water using a 785 nm laser with 5.1 mW. Already after less than 400 ms more than half of the 8BrA in the relevant hotspots are dissociated (see Fig. 5). In solution it is difficult to determine accurate rate constants as the photoproduct is diffusing out of the focused area, hence the observed reaction rates represent only an infimum of the real reaction rate. In an aqueous environment the electron tunneling is much more favorable as the tunneling barrier is lowered by 0.8 eV.35 Therefore, an increased speed of reaction is expected as compared to dry samples.
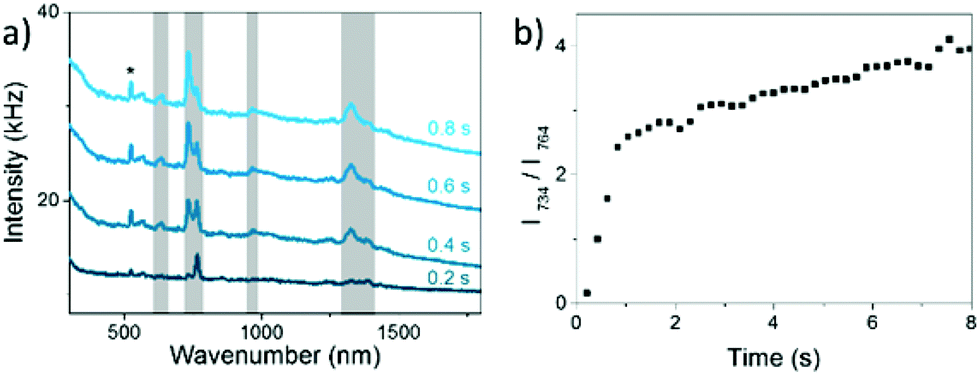 | ||
| Fig. 5 (a) SERS spectra of 8BrA on AgNPs in aqueous solution taken after different illumination times, irradiated with 785 nm with a power of 5.1 mW. The Si-band is marked with a *. (b) Ratio between 8BrA ring breathing mode at 764 cm−1 and A ring breathing mode at 734 cm−1 as a function of the illumination time (same data as in a). | ||
Conclusions
We demonstrate a dissociative electron transfer from laser illuminated noble metal nanoparticles to the potential DNA radiosensitizer 8BrA dried on a Si substrate as well as in solution. The reaction is described with fractal like kinetics. The observed interaction between the nanoparticles and 8BrA might help to improve photothermal cancer therapy. As the utilized substrate as well as the applied laser settings in this experiment were typical of SERS measurements, the plasmonically catalysed reaction can cause problems in the analysis of the Raman spectra, because the probe laser for the spectral analysis is the same that induces the decomposition of the analyte. As the time scale of the reaction is equivalent or faster than the typical accumulation time, one has to distinguish carefully between the SERS spectrum of the analyte molecule and its photoproducts. This should be done especially for molecules containing electrophilic groups such as halogens36,37 or nitro groups,38 which are known to decompose easily as a consequence of their interaction with “hot electrons”.Experimental section
Chemicals
8BrA was purchased from Carbosynth Ltd (UK) and A, silver nitrate and sodium citrate were purchased from Sigma Aldrich (Germany). All chemicals were diluted in Millipore water and used without further purifications. Gold nanoparticles with a diameter of 40 nm were purchased from BBI solutions.Silver nanoparticle preparation
AgNPs were prepared by the well-known procedure of Lee and Meisel.39 Briefly, 50 ml of 1 mM AgNO3 solution were brought to a rolling boil and 5 ml 38.8 mM trisodiumcitrate were added while the boiling was continued for 1 h under rigorous stirring. The produced AgNPs are 36 ± 10 nm in diameter and show an extinction maximum at 413 nm. The size distribution was determined by atomic force microscopy (AFM) using a Nanosurf Flex AFM equipped with a Tap150-Al-G cantilever in the tapping mode and image processing was performed with Gwyddeon 2.39 software. The UV-Vis-spectra were recorded using a Nanodrop 2000 of Thermo scientific.Surface enhanced Raman spectroscopy
In order to prepare the samples for the Raman measurements 100 μl of a 50 μM 8BrA solution were incubated for 2 h in 400 μl of the AgNP solution. Subsequently the dispersion was centrifuged and the residue was diluted in Millipore water two times. After a final centrifugation step a 2 μl droplet of the nanoparticle solution was dried on an oxygen plasma cleaned Si wafer. For the measurements in a liquid environment a 4 μl droplet was placed on a Si wafer that was surrounded by a bath of Millipore water to reduce the evaporation of the droplet. The Raman spectra were recorded using a Witec alpha 300 Raman-microscope with a 532 nm and a 785 nm laser and a Horiba Labram Raman-microscope with a 633 nm laser. The laser power was varied in the range from 50 μW to 50 mW and focused with a 10× objective on the sample. Scattering spectra of the sample were obtained using the Witec alpha 300 dark field unit with a 50× HD objective from Zeiss. Data analysis and processing were performed with Origin 9.1 software.Acknowledgements
This research was supported by the Federal Institute for Materials Research (BAM), a Marie Curie FP7 Integration Grant within the 7th European Union Framework Programme, by the Deutsche Forschungsgemeinschaft (DFG) and the University of Potsdam. We thank Dr Iwona Dąbkowska, University of Gdańsk, for providing results from ab initio calculations.References
- M. Moskovits, J. Raman Spectrosc., 2005, 36, 485–496 CrossRef CAS.
- J. Prinz, C. Heck, L. Ellerik, V. Merk and I. Bald, Nanoscale, 2016, 8, 5612–5620 RSC.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898 RSC.
- J. Y. Park, L. R. Baker and G. A. Somorjai, Chem. Rev., 2015, 115, 2781–2817 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CAS.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- T. Wadayama and M. Yokawa, Chem. Phys. Lett., 2006, 428, 348–351 CrossRef CAS.
- S. M. Kim, S. J. Lee, S. H. Kim, S. Kwon, K. J. Yee, H. Song, G. A. Somorjai and J. Y. Park, Nano Lett., 2013, 13, 1352–1358 CrossRef CAS PubMed.
- Z. Zhang, T. Deckert-Gaudig and V. Deckert, Analyst, 2015, 140, 4325–4335 RSC.
- W. Xie and S. Schlücker, Nat. Commun., 2015, 6, 7570 CrossRef PubMed.
- G. Baffou and R. Quidant, Laser Photonics Rev., 2013, 7, 171–187 CrossRef CAS.
- A. M. Gobin, M. H. Lee, N. J. Halas, W. D. James, R. A. Drezek and J. L. West, Nano Lett., 2007, 7, 1929–1934 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Laser. Med. Sci., 2008, 23, 217–228 CrossRef PubMed.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494 RSC.
- J. A. Webb and R. Bardhan, Nanoscale, 2014, 6, 2502 RSC.
- L. Chomicz, J. Leszczynski and J. Rak, J. Phys. Chem. B, 2013, 117, 8681–8688 CrossRef CAS PubMed.
- M. Wieczór, P. Wityk, J. Czub, L. Chomicz and J. Rak, Chem. Phys. Lett., 2014, 595–596, 133–137 CrossRef.
- Y. Park, K. Polska, J. Rak, J. R. Wagner and L. Sanche, J. Phys. Chem. B, 2012, 116, 9676–9682 CrossRef CAS PubMed.
- J. Rackwitz, J. Kopyra, I. Dąbkowska, K. Ebel, M. L. Ranković, A. R. Milosavljević and I. Bald, Angew. Chem., Int. Ed., 2016, 55, 10248–10252 CrossRef CAS PubMed.
- I. Bald, J. Langer, P. Tegeder and O. Ingólfsson, Int. J. Mass Spectrom., 2008, 277, 4–25 CrossRef CAS.
- B. Boudaïffa, Science, 2000, 287, 1658–1660 CrossRef.
- H. Abdoul-Carime, M. A. Huels, F. Brüning, E. Illenberger and L. Sanche, J. Chem. Phys., 2000, 113, 2517 CrossRef CAS.
- R. Schürmann and I. Bald, J. Phys. Chem. C, 2016, 120, 3001–3009 Search PubMed.
- S. Cecchini, S. Girouard, M. A. Huels, L. Sanche and D. J. Hunting, Radiat. Res., 2004, 162, 604–615 CrossRef CAS PubMed.
- A. Keller, J. Rackwitz, E. Cauët, J. Liévin, T. Körzdörfer, A. Rotaru, K. V. Gothelf, F. Besenbacher and I. Bald, Sci. Rep., 2014, 4, 7391 CrossRef CAS PubMed.
- K. Watanabe, D. Menzel, N. Nilius and H.-J. Freund, Chem. Rev., 2006, 106, 4301–4320 CrossRef CAS PubMed.
- C. Boerigter, U. Aslam and S. Linic, ACS Nano, 2016, 10, 6108–6115 CrossRef CAS PubMed.
- M. Pagliai, S. Caporali, M. Muniz-Miranda, G. Pratesi and V. Schettino, J. Phys. Chem. Lett., 2012, 3, 242–245 CrossRef CAS PubMed.
- R. Kopelman, Science, 1988, 241, 1620–1626 CAS.
- T. L. Thompson and J. T. Yates, J. Phys. Chem. B, 2006, 110, 7431–7435 CrossRef CAS PubMed.
- T. Kim, C.-H. Lee, S.-W. Joo and K. Lee, J. Colloid Interface Sci., 2008, 318, 238–243 CrossRef CAS PubMed.
- D. A. Weitz and M. Oliveria, Phys. Rev. Lett., 1984, 52, 1433–1436 CrossRef CAS.
- Private communication with Dr Iwona Dąbkowska.
- P. C. do Couto, B. J. Costa Cabral and S. Canuto, Chem. Phys. Lett., 2006, 429, 129–135 CrossRef CAS.
- N. Camillone, K. A. Khan, P. J. Lasky, L. Wu, J. E. Moryl and R. M. Osgood, J. Chem. Phys., 1998, 109, 8045 CrossRef CAS.
- E. P. Marsh, T. L. Gilton, W. Meier, M. R. Schneider and J. P. Cowin, Phys. Rev. Lett., 1988, 61, 2725–2725 CrossRef CAS PubMed.
- Z. Zhang, T. Deckert-Gaudig, P. Singh and V. Deckert, Chem. Commun., 2015, 51, 3069–3072 RSC.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr08695k |
| This journal is © The Royal Society of Chemistry 2017 |