
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dependence of the Ce(III)/Ce(IV) ratio on intracellular localization in ceria nanoparticles internalized by human cells†
Daniela
Ferraro
a,
Ilenia G.
Tredici
b,
Paolo
Ghigna
b,
Hiram
Castillio-Michel
c,
Andrea
Falqui
 d,
Cristiano
Di Benedetto
d,
Giancarla
Alberti
b,
Vittorio
Ricci‡
a,
Umberto
Anselmi-Tamburini‡
b and
Patrizia
Sommi‡
*a
d,
Cristiano
Di Benedetto
d,
Giancarla
Alberti
b,
Vittorio
Ricci‡
a,
Umberto
Anselmi-Tamburini‡
b and
Patrizia
Sommi‡
*a
aDepartment of Molecular Medicine, Human Physiology Unit, University of Pavia, 27100 Pavia, Italy. E-mail: patrizia.sommi@unipv.it; Fax: +39-0382-987664; Tel: +39-0382-987665
bDepartment of Chemistry, University of Pavia, 27100 Pavia, Italy
cEuropean Synchrotron Radiation Facility, 38000 Grenoble, France
dBiological and Environmental Sciences and Engineering Division, King Adullah University of Science and Technology, 23955-6900 Thuwal, Saudi Arabia
First published on 9th January 2017
Abstract
CeO2 nanoparticles (CNPs) have been investigated as promising antioxidant agents with significant activity in the therapy of diseases involving free radicals or oxidative stress. However, the exact mechanism responsible for CNP activity has not been completely elucidated. In particular, in situ evidence of modification of the oxidative state of CNPs in human cells and their evolution during cell internalization and subsequent intracellular distribution has never been presented. In this study we investigated modification of the Ce(III)/Ce(IV) ratio following internalization in human cells by X-ray absorption near edge spectroscopy (XANES). From this analysis on cell pellets, we observed that CNPs incubated for 24 h showed a significant increase in Ce(III). By coupling on individual cells synchrotron micro-X-ray fluorescence (μXRF) with micro-XANES (μXANES) we demonstrated that the Ce(III)/Ce(IV) ratio is also dependent on CNP intracellular localization. The regions with the highest CNP concentrations, suggested to be endolysosomes by transmission electron microscopy, were characterized by Ce atoms in the Ce(IV) oxidation state, while a higher Ce(III) content was observed in regions surrounding these areas. These observations suggest that the interaction of CNPs with cells involves a complex mechanism in which different cellular areas play different roles.
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) derive from free radicals originating from metabolic reactions. The extent and severity of the cellular damage exerted by ROS/RNS largely depends on the ability of the cells to neutralize these reactive species through physiological free radical scavengers, such as superoxide dismutase (SOD), catalase and glutathione peroxidase. When free radical concentrations exceed the antioxidant capacity of the cells, normal cell physiology is altered, causing the condition known as oxidative stress. Oxidative stress and generation of free radicals are associated with neurodegenerative conditions, such as Alzheimer's and Parkinson's diseases. High levels of ROS can also alter the immune response, causing chronic inflammation and immunological disorder, induce DNA damage, activate oncogenes and inactivate suppressor genes.1
CeO2 nanoparticles (CNPs) are under active investigation as promising agents in the therapy of several diseases involving free radicals or oxidative stress.2–8 CNPs are effective free radical scavengers with almost no toxicity. CNPs reduce inflammation caused by ROS or RNS,9 prevent radiation-induced damage,10 and have selective anticancer activity.11 Moreover, CNPs seem to exert a neuroprotective effect on neuronal cells and are effective in the treatment of Alzheimer's disease,5,8 as well as cerebral ischemia or hypoxia caused by stroke.4,12 Besides that, the free radical scavenging mechanism exerted by CNPs is not yet fully understood.
It has been suggested that the CNP scavenging properties are associated with the equilibrium between the two oxidation states of cerium, Ce(III) and Ce(IV), which is strongly dependent on the external environment/stimuli. According to this model, during oxidative stress, CNPs can react with excess superoxide and H2O2 and serve as cellular antioxidant agents, exerting a SOD/catalase mimetic activity via a self-regenerating mechanism.13 However, to date, this regenerative redox mechanism has been supported mostly by abiotic studies13,14 and cannot be considered completely proven. In fact, Cafun and colleagues15 have recently excluded, by an abiotic experiment under controlled conditions, the formation of a significant fraction of Ce(III) during catalase mimetic activity, showing instead that Ce(IV) is the prevalent oxidation state. Abiotic experiments are typically performed under different conditions from those expected in a real, more complex biological environment, and CNP activity in vivo, especially at the cellular level, is far from being clarified. Several questions are still open: how the evolution of the oxidative state of the CNP occurs during cell internalization and intracellular distribution; whether the catalytic activity changes are based on the intracellular localization; how such activity is influenced by the initial Ce(III)/Ce(IV) ratio; and how the presence of a protein corona and surface functionalization can modify the response of the CNP. Coating and surface charge have been shown to greatly affect CNP physicochemical characteristics and influence their antioxidant ability and biological activity.16,17 Asati et al.,16 for example, showed that modifying the surface charge by varying the type of coating could influence the intracellular distribution of the CNPs, directing them mostly to the lysosomes or favoring their cytoplasmic localization.
Investigation of some of these aspects has started, mainly in nematodes and plants.18,19 In an in vivo study on Caenorhabditis elegans, Collin and colleagues18 showed the influence of the superficial properties of NPs on cell internalization/localization, observing that initial surface chemistry favors specific sites of accumulation in tissues and organs. They also observed that the surface chemistry of CNPs has a profound impact on their toxicity and how the environment can modify it. In an investigation of the biodistribution and transformation of CNPs, the macroinvertebrate Planorbarius corneus was administered CNPs that initially only had Ce(IV).20 Inside the digestive gland of the snail, the Ce(IV) was mostly reduced to Ce(III) (∼78%). The authors speculated that the reduction occurred after CNP ingestion/internalization due to the interaction with the enzymes and metalloproteins present in the digestive gland, where digestion and detoxification take place. Ma and colleagues21 investigated the Ce speciation in cucumber plants at the cellular level, finding that the short time incubations favored Ce(IV) in all of the plant tissues, while prolonged exposures increased Ce(III) in the above-ground tissues. They suggested that the physicochemical interaction of the root exudates and CNPs at the nanobio interface is the necessary condition for the transformation of CNPs.21
However, a complete description of the mechanism of action of CNPs in human cells is still lacking. The present study aimed to bridge this gap, by investigating, in human cells, the modification of the Ce(III)/Ce(IV) ratio following internalization and its dependence on intracellular localization.
Experimental
CNP synthesis and characterization
CNPs were produced by direct precipitation from aqueous solution and stabilized by polyacrylic acid (PAA). Ce(NO3)3·6H2O (1.085 g, puriss. p.a. ≥99.0%) was dissolved in distilled water (43.65 mL). An aqueous suspension of PAA was prepared by diluting 600 mg commercial PAA suspension (63% w/w) with distilled water (31 mL). The diluted PAA solution (20 mL) was mixed with the Ce(NO3)3·6H2O solution. After mixing, concentrated NH4OH solution (5 mL, 28.0–30.0% NH3 basis) was added dropwise with stirring. Immediate quantitative precipitation occurred and the suspension was left under stirring. After 4 h, the suspension turned to a transparent brown solution, while after 1 day, the solution turned yellow. The obtained product had a CeO2 concentration of ∼6 mg mL−1. For cell treatment, CNPs were centrifuged and resuspended in order to remove debris, large agglomerates, and PAA in excess.CNPs were characterized by high-resolution transmission electron microscopy (HR-TEM), X-ray diffraction (XRD) and dynamic light scattering (DLS). For HR-TEM, a small drop of the aqueous suspension of CNPs was placed on an ultrathin carbon membrane 400-mesh copper grid and left for 5 min to dry. HR-TEM imaging was performed using an FEI Titan 80-300 Cube transmission electron microscope (Hillsboro, OR, USA), operating at an acceleration voltage of 300 kV, equipped with an S-Twin objective lens, an FEI X-FEG Schottky electron source, a 2k × 2k US1000 Gatan CCD Camera, and an EDS Si(Li) EDAX detector. The CNP dimension obtained from HR-TEM images was determined measuring more than hundred nanoparticles using ImageJ software.
XRD analysis was performed on films obtained by evaporating 500 μL CNP suspensions (concentration 6 mg mL−1) on glass microscope slides with an area of ∼2.2 cm2. The XRD patterns were acquired using a Bruker D8 Advance diffractometer (Bruker Corp., Billerica, MA, USA) with a Cu anticathode (λ-Cu-Kα = 1.541838 Å) operating at 40 kV and 40 mA. Diffractograms were acquired in θ–θ mode, with a step of 0.03° 2θ and an acquisition time of 20 s per step. Phase indexing, determination of lattice constants, and average grain sizes have been performed using the HighScore Plus software from PANalytical.
The hydrodynamic diameter of the CNPs was evaluated by DLS with Nano ZS90 DLS apparatus (Malvern Instruments, Malvern, UK). Three measurements were performed on diluted solutions (∼1 mg mL−1) for each sample, providing average sizes, distribution widths, polydispersion index and associated standard deviations (SDs).
Cells and cell treatments
HeLa cells (a gift from T. L. Cover, Vanderbilt University, Nashville, TN) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mM L-glutamine (all from Lonza, Basel, Switzerland) at 37 °C under a humidified atmosphere of 5% CO2 in air. Cells were seeded at 1.2 × 107 in 75 cm2 flasks. After 48 h, the cell monolayers were washed and incubated with fresh medium containing the CNP at a concentration of 200 μg mL−1 for 15 min to 24 h, as specified case by case. The CNPs formed a stable suspension in the culture medium and no sedimentation was observed, even at the end of the longest incubation time.ICP-OES analysis
The content of CeO2 in the cell culture pellet was determined by inductively coupled plasma optical emission spectrometry (ICP-OES), after mineral digestion of the samples. The samples were subjected to acid mineralization in a Parr acid digestion bomb with a Teflon liner, with diluted HNO3 [0.4 ml HNO3 for trace analysis (≥69.0%, Fluka) + 0.2 ml Milli-Q water] at 120 °C for 3 h (these experimental conditions were optimized according to a previously reported procedure).22 After cooling at room temperature, the solution was diluted to 100 mL with Milli-Q water (specific resistance: 18.2 MΩ cm−1) obtained from a Milli-Q Academic system (Millipore, Temecula, CA, USA); the total Ce content in this solution was determined by ICP-OES analysis (ICP-OES Optima 3300 D; Perkin Elmer, Santa Clara, CA, USA). The external calibration method was used for the determination of Ce in the Ce standards ranging from 0 to 10 mg L−1 (source: 1 g L−1 Ce Fluka standard solution in 1 M HNO3). The limit of detection (3× SD of the blank) and limit of quantification (10× SD of the blank) were 0.5 and 1.7 mg L−1, respectively, for Ce at 456.263 nm.MTT cytotoxicity assay
After 24 h of exposure, 0.9 mM MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich, St Louis, MO, USA) was added to the cells and, after 2 h of incubation at 37 °C, the medium was removed and 100 μL DMSO was added to dissolve the blue formazan that was formed. Cell viability was quantified by measuring absorbance at 540 nm using a microplate reader (Bio-Rad, Hercules, CA, USA) and expressed as a percentage of the control group (that was set to 100%).Comet assay
DNA single-strand breaks were detected as described by Ferraro et al.23 Cells treated with CNPs for 24 h were harvested and pelleted by centrifugation at 400g for 3 min at 4 °C. For evaluation of DNA damage, each duplicated sample was analyzed in a random manner, and for each sample, 100 nucleoids were scored and classified into five classes, according to the visual method described by Collins.24 The DNA damage was expressed in arbitrary units, where the minimum value was 0 (Class 0) and the maximum was 400 (Class 4). For each experimental condition, we carried out at least three independent experiments, each run in duplicate. The results are given as the means ± standard error of the mean (S.E.M.) of three independent experiments (n = 3).Evaluation of CNP pro-oxidant and antioxidant activities
The CNP pro-oxidant activity was evaluated by means of ROS-sensitive fluoroprobe CM-H2DCFDA25 (5(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate-acetyl ester; Molecular Probes). HeLa cells (7.5 × 104 mL−1) were seeded in ibiTreat μ-Slide 8 Wells (IBIDI, Martinsried, Germany) and cultured overnight. Cells previously treated for 24 h with or without CNPs were incubated with CM-H2DCFDA solution (in DMSO) at a final concentration of 10 μM in PBS for 30 min at 37 °C. The cells were carefully washed and immediately analyzed by fluorescence microscopy.To evaluate the CNP antioxidant activity, ROS were induced by exposing the cells to 100 μM tert-butyl hydroperoxide (Sigma-Aldrich) at the end of CNP incubation. After 30 min of induction, the ROS levels were evaluated as described above.
Samples were observed using an Olympus IX83 microscope (Olympus Inc., Tokyo, Japan) equipped with an APO 20/0.7 NA oil-immersion objective. ROS production was considered directly proportional to the fluorescence intensity and quantified using CellSens software (Olympus).
Immunofluorescence and light microscopy
Cells previously incubated with CNPs for 1, 6 and 24 h were processed for immunofluorescence as previously described.23 For more details see the ESI.†TEM and SEM of biological samples
After 24 h of incubation with CNPs, whole cells were processed for TEM or SEM.For TEM, cell monolayers, in 35 mm Petri dishes, were fixed by a mixture of 2% glutaraldehyde in 0.1 M cacodylate buffer (pH = 7.3) for 20 min at room temperature. To remove the fixative completely, cells were washed three times for 10 min in 0.1 M cacodylate buffer. Secondary fixation was performed by exposing cells to 1% aqueous OsO4 for 20 min at room temperature, followed by two washes of 5 min in pure distilled water. Cell monolayers were exposed to 1% uranyl acetate aqueous solution, for 20 min at room temperature. Specimens were washed twice with pure distilled water for 5 min, and then dehydrated through a graded series of ethanol (25, 50, 70, 90, 95 and 100%; 5 min each). Infiltration was carried out by placing cell monolayers in an increasing concentration of Durcupan ACM resin in 100% ethanol solvent (25% resin + 75% ethanol; 50% resin + 50% ethanol; 75% resin + 25% ethanol) for 90 min each, and 100% resin overnight at room temperature. On the next day, monolayers were embedded with a new resin, left for 2 h at room temperature, and then for 48 h at 60 °C.
Ultrathin sections (∼70 nm) were obtained with a Leica EM UC7 ultramicrotome (Leica, Wetzlar, Germany), using an ULTRA 45° diamond knife (DIATOME, Diatome, Hatfield, PA, USA). Sections were placed on 300-mesh copper grids, stained with lead citrate and washed with pure distilled water. Samples were then observed with a FEI TECNAI SPIRIT electron microscope, operating at an accelerating voltage of 120 kV, equipped with a Bio-Twin objective lens, a LaB6 electron source, a 4k × 4k FEI Eagle CCD Camera, and an EDS Si(Li) EDAX detector.
For SEM, HeLa cells grown on Si3N4 membranes (SPI, West Chester, PA, USA) and treated with CNPs were freeze-dried as described in detail in “In situ μXRF Imaging and Ce LIII-edge μXANES Analysis”. The samples were coated with a carbon film and observed with a field emission gun scanning electron microscope (Mira3 XMU; Tescan, Kohoutovice, Czech Republic) operating at 20 kV and equipped with an EDAX EDS microprobe and a BSE detector.
XANES
The X-ray absorption near edge spectroscopy (XANES) spectra at the Ce–LIII edge were measured at 100 K at the BM08 LISA beamline26 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France), in fluorescence mode, using a 13-element Ge detector for the fluorescence signal, and a double crystal Si(311) monochromator. The harmonic rejection was realized by means of palladium mirrors. The reproducibility in energy was checked periodically, typically after four or five scans, by measuring the absorption spectrum of a CeO2 standard, and every time it was found to be better than 0.1 eV. The spectra of CeO2 and Ce(NO3)3·6H2O standards were acquired in transmission mode at room temperature. For the measurements, an appropriate amount of sample was weighed to give a unit step jump at the Ce–LIII edge, and then thoroughly mixed with cellulose in a mortar and pestle.For the measurements, cells treated and processed as previously described were pelleted (∼5 × 107 cells) and washed with fresh culture medium. The wet pellet was transferred to a filter paper and centrifuged to remove any excess liquid, then the filter carrying the cells was flash frozen in LN2 and subsequently lyophilized.
The purified organelles were prepared according to Holden and Horton27 with slight modifications. At the end of 24 h treatment, HeLa cells were collected by trypsinization and centrifugation at 100g for 5 min at 4 °C. All the steps were performed at 4 °C. The cell pellet was washed with ice-cold PBS, incubated for 10 min in Digitonin Buffer (150 mM NaCl, 50 mM HEPES pH 7.4, 25 μg mL−1 digitonin), and centrifuged at 2000g for 10 min. The supernatant, corresponding to the cytosol-enriched fraction, was maintained in ice while the cell pellet was washed again, incubated in NP40 Buffer (150 mM NaCl, 50 mM HEPES pH 7.4, 1% NP40) for 30 min and centrifuged at 7000g for 10 min. The supernatant and the previously isolated cytosol fraction were combined and centrifuged again to collect the organelles which were transferred to a filter paper and lyophilized.
The samples were stored at −80 °C and carefully handled during the sample transfer to prevent temperature change. Before the measurements, the metallic sample holder was cooled with LN2, and the sample was rapidly mounted and immediately transferred into the cryostat where the temperature was lowered to 100 K. The whole mounting procedure lasted for <1 min to ensure that the sample temperature was always kept below −80 °C. Athena software was used for background removal and normalization of the acquired spectra:28 the pre-edge background was fitted by a straight line and subtracted. The spectra were normalized at the unit absorption coefficient at 400 eV above the edge, where the extended X-ray absorption fine structure (EXAFS) oscillations were negligible. The normalized spectra were then fitted by an arctan function and an appropriate number of Gaussian peaks to simulate all the channels for the electronic transitions at the Ce–LIII edge.29
In situ synchrotron radiation micro-X-ray fluorescence (μXRF) imaging and Ce LIII-edge micro-XANES (μXANES) analysis
HeLa cells were grown on Si3N4 membranes and treated with CNPs as described above. At the end of 24 h of incubation, samples were freeze-dried using the immersion freezing procedure with an LN2 container surrounding the liquid propane as a secondary cryogen, as described previously.30 Si3N4 membranes were blotted on a filter paper and the samples were immersed face up in liquid propane. Each membrane was sealed in a vial under LN2 vapor to avoid ice-crystal formation, and maintained at −80 °C. For X-ray synchrotron analysis, the samples were mounted in the pre-cooled sample holder under LN2 vapor and immediately inserted into the cryostat. The entire transfer process took <2 min.μXRF and Ce LIII-edge μXANES measurements were performed using a scanning X-ray microscope of beamline ID21 at ESRF. The beamline was equipped with a cryostage, which was passively cooled in a LN2 Dewar flask, keeping the sample stable at −170 ± 5 °C.31 Detectors included a Si3N4 diode for I0 and an 80 mm active area Silicon Drift Detector (Bruker) for the emitted fluorescence. Focus was achieved using fixed-curvature Kirkpatrick–Baez mirror optics. The photon flux was 5.7 × 1010 ph s−1 at 5.8 keV with a beam size of 1.0 × 0.5 μm2 (H × V). The μXRF maps were acquired with an incoming energy of 5.8 keV with 0.5 μm2 steps and an integration time of 100–150 ms. The μXRF acquisition was done in hyperspectral mode for which the XRF spectrum for each pixel in the image was registered. All maps were fitted using PyMCA software to obtain the intensity distribution of the elements.32 The μXANES spectra were acquired in fluorescence mapping mode by scanning the beam with a 0.6 × 0.6 μm2 step size and a 100 ms dwell time per pixel, with 5 eV energy steps in the region from 5.68 to 5.72 keV, 0.5 eV from 5.72 to 5.77 keV, and 2 eV from 5.77 to 5.84 keV. This resulted in 129 images recorded using a region of interest selective for Ce L3M4 and L3M5 emission lines, corrected for the detector dead time and normalized by I0. The stack of images was aligned using elastix33 and saved to an hdf5 file containing intensities and the energy values for each map to be processed using PyMCA for XANES spectra extraction. The Athena software was used for background removal and normalization: the spectra were handled as described above in the XANES section.
Results and discussion
The main focus of this study was to investigate whether and how CNPs undergo modification of their oxidation state once internalized by mammalian cells. This characterization, performed by XANES, is experimentally challenging. To allow meaningful XANES analysis and good biological compatibility, the CNPs must be properly designed. The Ce(III)/Ce(IV) equilibrium mostly involves the atoms on the surface of the CNPs,34 so CNPs with a high surface/volume ratio are required to ensure adequate XANES sensitivity, because the technique does not allow a distinction between the surface and core atoms. The diameter required to fulfil this requirement is estimated to be below 10 nm. The CNPs must also be well dispersed and form stable suspensions, showing minimal or no signs of agglomeration in an aqueous medium characterized by pH and composition compatible with the biological interaction. To ensure that the CNPs that we produced fulfilled all these requirements, we performed a wide spectrum of physicochemical and biological characterization, aiming to clarify the chemical and morphological properties of the CNPs and to reveal any indication of cellular damage and toxicity induced by their interaction with the cells.The experimental procedure must maintain intact the characteristics that the CNPs acquire after interaction with the biological system, avoiding any modification of the Ce(III)/Ce(IV) ratio during sample preparation and analysis. This requirement is particularly stringent. Water strongly absorbs X-rays at the energy of the Ce–LIII edge, making impossible the analysis of biological material in their culture medium. The oxidation state of the cerium atoms present on the surface of the nanoparticles is sensitive to the environment and can be drastically modified by interaction with oxidizing or reducing chemicals and even by unprotected exposure to air for several hours. For this reason, the use of any fixative and chemical dehydration process was avoided. To preserve the status of the CNPs at the end of the interaction with the cells, we froze the samples in liquid nitrogen (LN2) and maintained them under cryogenic conditions at all times, including during XANES analysis, while water was removed by freeze drying.
Physicochemical characterization and biological activity of CNPs
The XRD pattern of the synthetized CNPs is shown in Fig. 1A. All the diffraction peaks were indexed on the basis of the fluoritic CeO2 crystal structure. No additional peaks were observed. The peak position and integral breadth have been determined by fitting with Voight functions. The lattice parameter was determined to be 0.54262 nm, corresponding to a cell volume of 0.15973 nm3. The average grain size, evaluated using the Sherrer equation,35 was about 3.9 nm. | ||
| Fig. 1 Physicochemical characterization of the synthetized CNPs. (A) XRD pattern. (B) HR-TEM images of representative CNPs. Magnified regions, shown in insets, highlight the single-crystalline structure of CNPs, confirmed by the 2D-FFT diffractograms. Only the main lattice planes are indicated in the insets, to preserve readability, while full structural characterization is provided in each respective diffractogram. (C) CNP diameter distribution fitted with the log-normal function. | ||
The crystal structure of the nanoparticles was confirmed by HR-TEM analysis (Fig. 1B). This analysis evidenced also that the CNPs were monocrystalline with no evidence of internal defects and of significant agglomeration (Fig. 1B). From these images, the particle size was estimated to be 2.9 ± 0.94 nm (Fig. 1C). The hydrodynamic diameter, as detected from the DLS measurements, was significantly larger (13.9 ± 0.7 nm), accounting for the polymeric shell surrounding the CNPs.
To characterize the CNP–cell interaction we investigated the influence of CNPs on genotoxicity, cell viability, free radical scavenger properties, internalization and intracellular distribution.
To evaluate the extent of CNP internalization the amount of CeO2 present in the cells was quantified by ICP-OES after 1, 6 and 24 h of incubation. As expected, the amount of the internalized CNPs increased with time, going from ∼0.2 μg per 106 cells after 1 h of incubation to >1 μg per 106 cells after 24 h (Fig. 2A). A saturation effect was evident, as the incorporation of the CNPs appeared to slow down after the first few hours. Based on the results of this quantification, we focused our attention on 24 h incubation, because only at that time was the amount of CeO2 high enough to allow reliable XANES characterization.
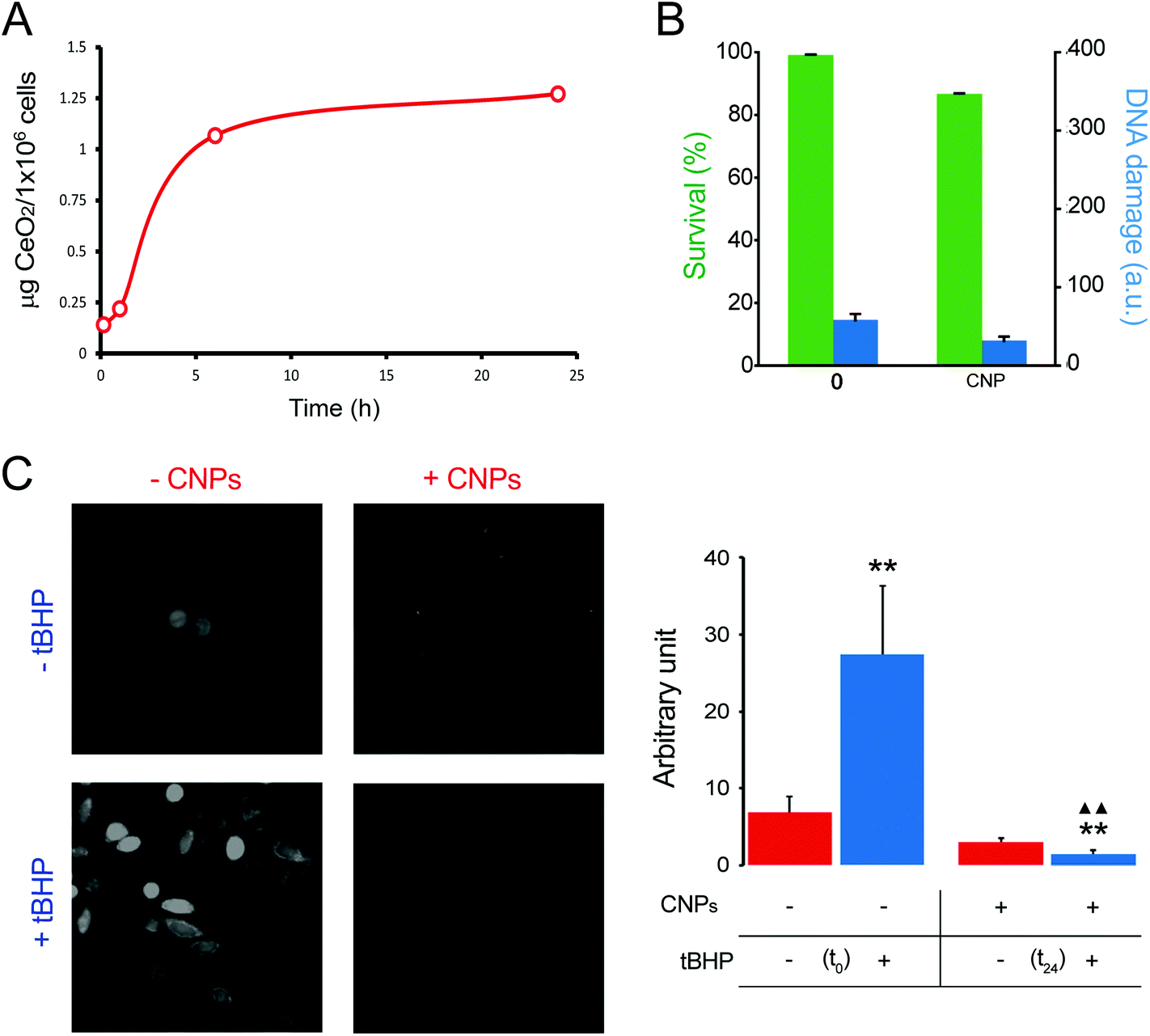 | ||
| Fig. 2 Biological characterization of CNPs. (A) Determination by ICP-OES of the cellular CNP content after 15 min, 1 h, 6 h and 24 h of incubation. (B) Effects of CNPs on cell viability (by MTT assay; green histograms) and DNA damage (by Comet assay; blue histograms) evaluated after 24 h of incubation. (C) Analysis of pro-oxidant and antioxidant activities of CNPs by CM-H2DCFDA after 24 h of incubation. Error bars represent S.E.M.; one-way ANOVA. **p < 0.01 versus each respective red column. ΔΔp < 0.01 versus the blue column. | ||
The effect of CNPs on cell viability was evaluated together with genotoxicity. Testing both aspects allowed us to obtain a more complete picture of the influence of CNPs on cell physiology. Cells exposed to CNPs for 24 h did not show evident signs of cytotoxic effects (evaluated by MTT analysis), because the cell viability was >85% if compared with that of the unexposed cells (Fig. 2B). Genotoxicity was investigated by means of the comet assay. We did not observe any DNA damage, and the level of damage was comparable with that obtained with control cells; both being damage Class 1 (in a 0–5 ranking) (Fig. 2B). These results confirmed that the CNPs used in this study were not detrimental to cell physiology, and that the procedure used for their synthesis did not introduce any factor that altered cell viability or induced genotoxicity. More importantly, these findings ensured that XANES characterization was performed on cells that remained healthy even after significant CNP internalization.
The level of oxidative stress induced by the CNPs represents another relevant point, because the literature is somehow discordant on this point.36,37 In general, CNPs possess antioxidant activity, although some studies have claimed that they also have a pro-oxidant effect.36,38 After 24 h of incubation, our CNPs did not show any pro-oxidant activity when monitored by using CM-H2DCFDA (Fig. 2C). Free radical scavenger activity of CNPs was evaluated by induction of ROS production by the exogenous oxidative stress inducer (tert-butyl hydroperoxide) and the resulting ROS level was monitored in the presence and absence of CNPs. We observed that CNPs clearly reduced the level of ROS (Fig. 2C).
The impact of CNPs on intracellular processing was evaluated by monitoring the modifications in lysosomes and autophagic vesicles in relation to the time of incubation (Fig. 3A and S1†). We observed that lysosomes (identifiable by Lamp1 antibody), which have a key role in processing exogenous and endogenous material, increased in number with incubation time (Fig. S1†). We could not follow directly CNP localization by light microscopy because these particles were not fluorescent. However, on the basis of previous experience with other types of NPs,23 the number of lysosomes could be related to internalization of the CNPs. We also investigated whether CNPs activated autophagy. By considering p62 as a marker, we observed that the autophagic vesicles were affected by the CNPs, showing a progressive increase with incubation time (1, 6 and 24 h) (Fig. 3A and S1†). CNP accumulation in endolysosomes was in agreement with the distribution observed by other authors, especially in the case of CNPs coated with PAA.16,17
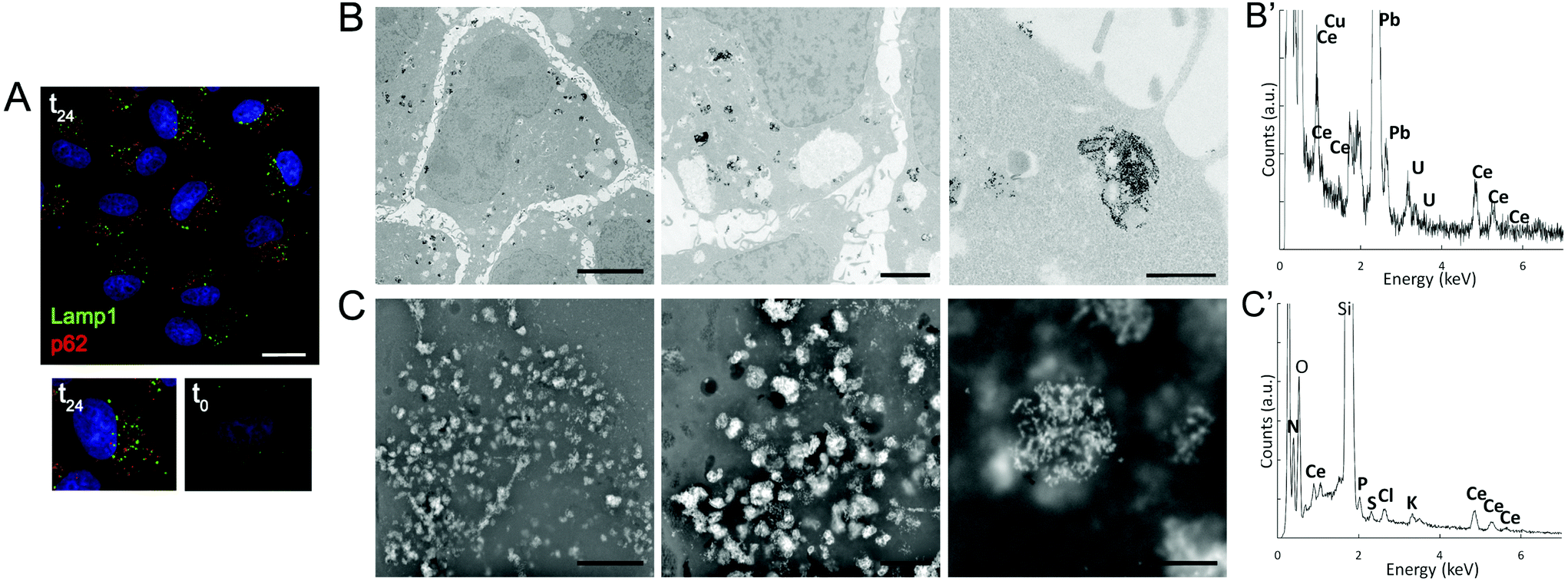 | ||
| Fig. 3 (A) Immunofluorescence of lysosomes (Lamp1) and autophagic vesicles (p62) of cells incubated for 24 h with CNPs (t24). Below, a comparison between single cells incubated with (t24) or without (t0) CNPs. Images are from one single confocal section. Bar, 50 μm. (B, C) Ultrastructural analysis of cells incubated for 24 h with CNPs. (B) Accumulation of CNP electron-dense aggregates mostly present inside the endosomal–lysosomal vesicles observed by TEM. Bars, 5, 1 and 0.5 μm for left, middle and right images. (B′) Representative TEM-EDS analysis of the CNP aggregates. Signals other than Ce are due to the sample holder and fixative. (C) Similar accumulation of CNPs observed by SEM using the BSE detector. Bars, 5, 1 and 0.5 μm for left, middle and right images. (C′) Representative SEM-EDS analysis of the CNP aggregates. Signals other than Ce are due to the sample holder and cell inorganic components. | ||
To confirm these light microscopy observations, the distribution of the internalized CNPs was analyzed by TEM, SEM, and also by μXRF. TEM showed accumulation of electron-dense aggregates of CNPs inside endosomal–lysosomal vesicles (Fig. 3B), as confirmed by in situ energy dispersive X-ray spectroscopy (EDS) analysis (Fig. 3B′). Similar CNP aggregates, identified by EDS analysis, were observed within the cells by SEM when the backscattered electron (BSE) detector was used (Fig. 3C and 3C′). μXRF allowed us to obtain an additional direct indication of the CNP distribution within cells incubated for 24 h (Fig. 4). The cell position and contours were identified using the distribution of K and Ca elements. The Ce distribution within the cells looked uneven. Most of the material was concentrated in a few “hot spots”, although some Ce appeared distributed, at a much lower concentration, in the rest of the cells.
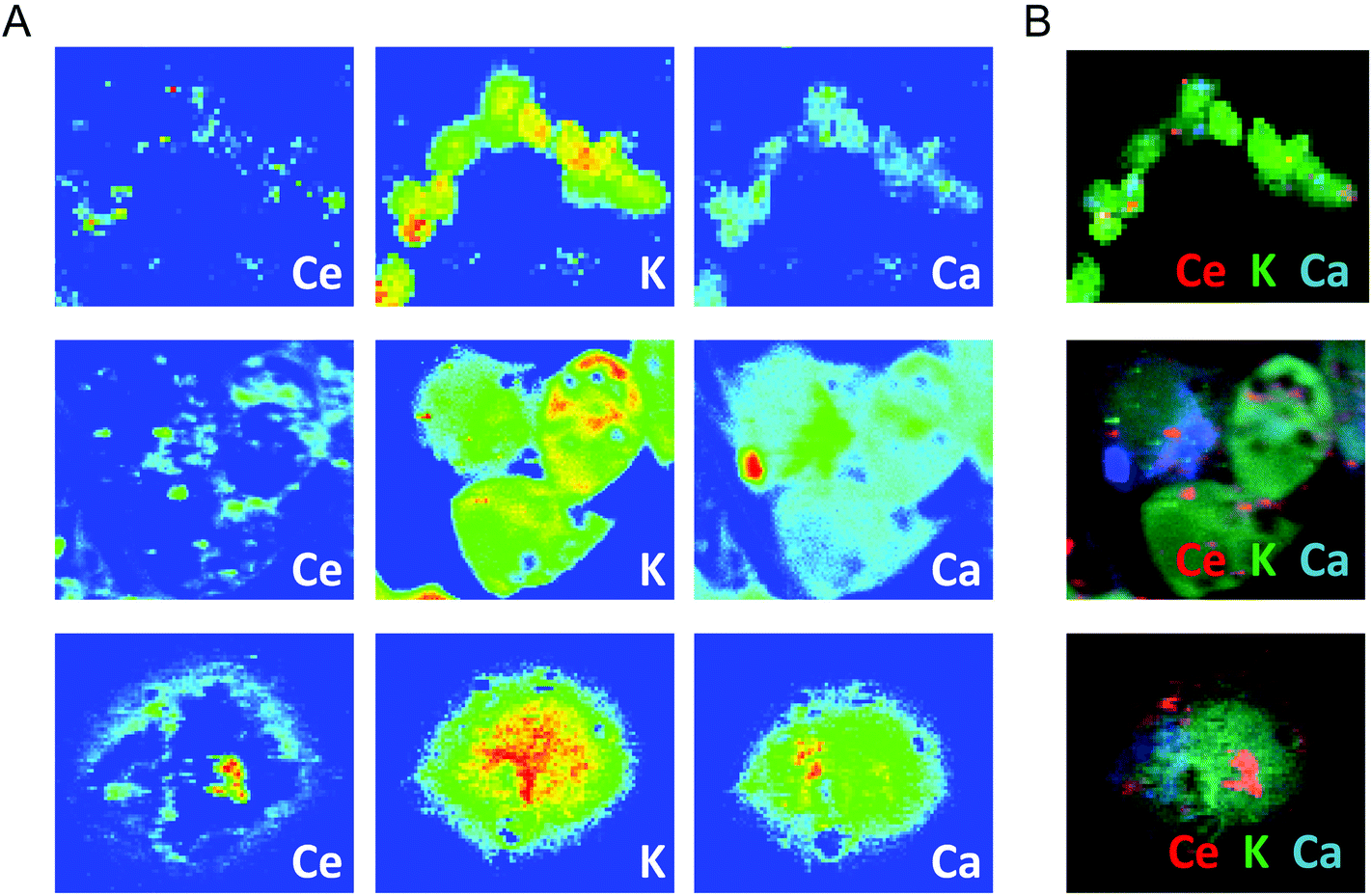 | ||
| Fig. 4 μXRF of the Ce distribution observed at three different magnifications in cells incubated with CNPs for 24 h. K and Ca distribution maps define the cell position and mark the cell contours. False-color maps show elemental concentrations within each individual image [from red (high) to blue (none)]. (B) Merged images of the elements reported in (A). | ||
XANES
The oxidation state of the incorporated CNPs was evaluated using X-ray absorption spectroscopy. This analysis was performed on cell pellets, to gain information on the average oxidation state of Ce, as well as on individual cells, using an μXANES setup. This last approach allowed us to obtain the distribution maps of the Ce oxidation state within each individual cell.The results relative to the XANES analysis of cell pellets are shown in Fig. 5. To discuss better the effect of the CNP–cell interaction, the spectra relative to the CNPs incubated with the cells for 24 h (Fig. 5D) are herein compared with those of three reference materials (Fig. 5A–C). It is important to notice that the cell culture medium does not contribute to modify the Ce(IV) initial CNP oxidation state, even in the case of a prolonged incubation time (Fig. S2†). Fig. 5A shows the Ce–LIII XANES spectra relative to bulk CeO2. The spectra can be discussed in terms of electronic transitions to empty states; the initial states being 2p3/2 in character.39 The transition to continuum states with the expulsion of a photoelectron gives rise to the step with an arctan shape (black line in Fig. 5A). All the other peaks present in Fig. 5 originated from transitions to discrete empty states. In particular, the peaks labeled a1 and a2 are dipole-allowed transitions to empty d states of the electronic configuration f0, split by crystal field effects. The peak labeled b is a dipole-allowed transition to empty d states of the electronic configuration f1![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) , with being a hole in the oxygen (ligand) 2p states. Thus, peaks a1, a2 and b represent different channels for electron excitation of different electronic configurations of Ce(IV), with the configuration f1 originating from the (partial) covalence of the Ce–O bond.40 The peak labeled c is mixed in nature, being due to dipole-allowed transitions to d states of the configuration f1, split by crystal field effects, and to dipole-allowed transitions to empty d states of the f1 configuration. As a result, the amplitude of peak c can be used to assess the presence of Ce(III) in the sample. Finally, the peak labeled d results from quadrupole-allowed/dipole-forbidden excitations to the empty 4f states.39,40
, with being a hole in the oxygen (ligand) 2p states. Thus, peaks a1, a2 and b represent different channels for electron excitation of different electronic configurations of Ce(IV), with the configuration f1 originating from the (partial) covalence of the Ce–O bond.40 The peak labeled c is mixed in nature, being due to dipole-allowed transitions to d states of the configuration f1, split by crystal field effects, and to dipole-allowed transitions to empty d states of the f1 configuration. As a result, the amplitude of peak c can be used to assess the presence of Ce(III) in the sample. Finally, the peak labeled d results from quadrupole-allowed/dipole-forbidden excitations to the empty 4f states.39,40
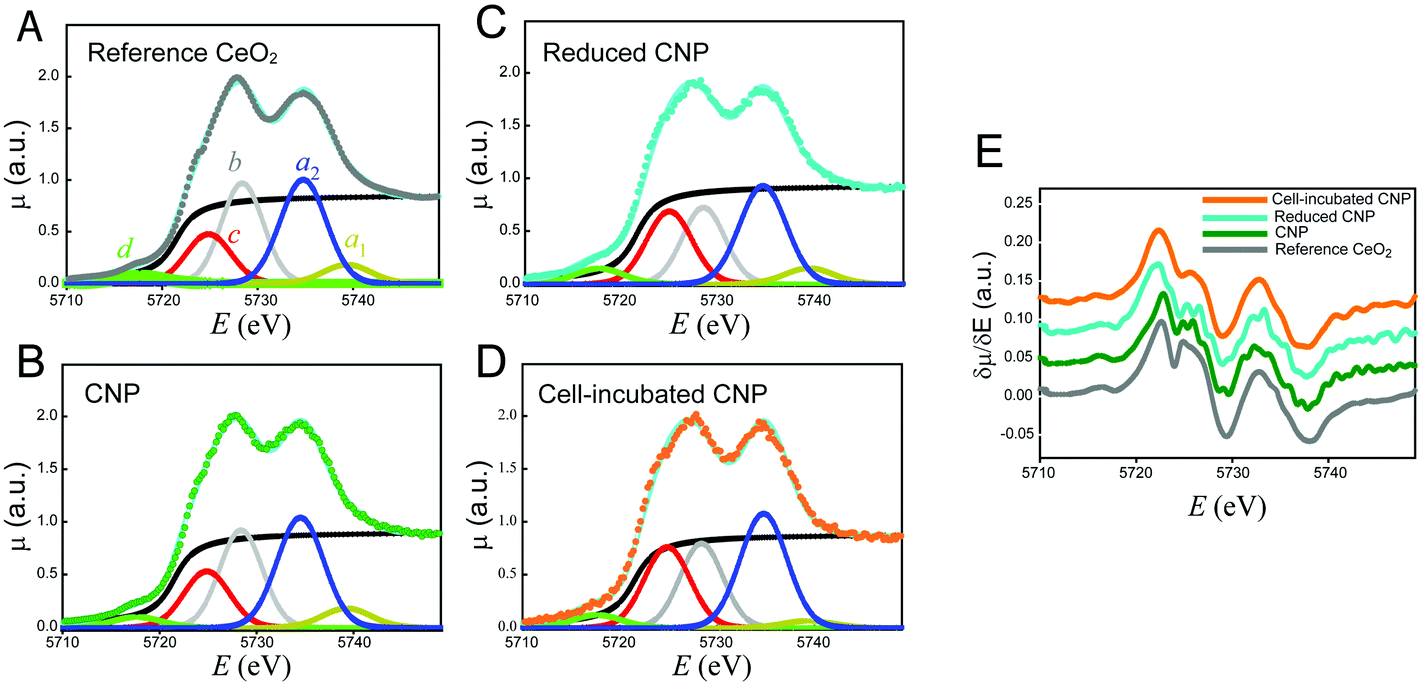 | ||
| Fig. 5 Ce–LIII XANES spectra of the reference CeO2 (A), CNPs (B), reduced CNPs (C) and CNPs incubated with cells for 24 h (D). These panels also show the fits relative to the peak components as described in the text. Panel (E) reports the corresponding derivatives. | ||
Fig. 5B shows the spectra of the CNPs before their interaction with the biological material. The spectra are similar to those of bulk CeO2 (Fig. 5A). A small decrease in intensity of the peaks was observed, particularly in the region of peaks c and b. This may have resulted from the difference in the surface to volume ratio of Ce atoms.41 In contrast, Fig. 5C shows the spectra relative to CNPs that were partially reduced chemically during chemical synthesis. These spectra showed an increase in the amount of Ce(III). In these CNPs, in addition to the above-mentioned effect, we detected a clear increase in the amplitude of peak c, together with a decrease in the amplitude of peak b. This effect may have been associated with an increase in the amount of Ce(III) and might be better appreciated in the derivative spectra reported in Fig. 5E. The XANES spectra were affected by the so-called chemical shift. The valence electrons screened the coulombic potential experienced by the core electrons involved in the XANES transitions. The lesser the valence electrons (the greater the oxidation state), the lesser the screening and thus the greater the binding energy of the core initial level. The net effect was a shift in energy of the spectral features (especially the energy position of the absorption edge) to a lower energy with the decreasing oxidation state. Fig. 5E clearly shows that the edge energy position of the reduced CNPs, represented by the first maximum in the derivative spectra, shifted towards a lower energy with respect to the regular CNPs and bulk CeO2.
Fig. 5D shows the Ce–LIII XANES of CNPs incubated for 24 h with the cells. The striking evidence is represented by the increase in the amount of Ce(III) resulting from this interaction. In addition to a marked increase in the relative intensity of peak C, a net shift in the energy position of the edge was observed in the derivative spectra of Fig. 5E. The spectra became completely equivalent to those of the CNPs that were chemically reduced. We conclude that the 24 h interaction between the CNPs and cells resulted in net localization of extra electrons to Ce states to produce Ce(III).
To the best of our knowledge, this result has not been reported previously in human cells, and supports the existence of a physiological mechanism that can modify the oxidation state of the CNPs. Similar results have been reported before in other, different biological models.18–20 Direct evidence of similar behavior in human cells after a significantly long incubation time (24 h) is promising in view of potential therapeutic application of CNPs, and is also relevant for clarifying the CNP mechanism of action. Since the content of Ce(III) in our starting CNPs was negligible, the formation and maintenance of a significant fraction of Ce atoms in this unstable oxidation state implies the existence of mechanisms that can reduce Ce(IV) and maintain the reduced form for a relevant time. This probably involves the presence of recycling mechanisms. The existence of such a mechanism is essential in view of the possible antioxidant activity of CNPs. Although Ce(III) is the form that is active towards some types of ROS, because it is easily oxidized, its activity would be short-lived without a parallel mechanism of recycling that can restore the reduced state of Ce. By combining the previous observations of SOD- and catalase-like activity of CNPs, Celardo et al.13 have recently proposed the existence of cycles involving parallel pathways of Ce oxidation and reduction. In these cycles, Ce(III) would always be responsible for the reduction of superoxides, while the formed Ce(IV) would be recycled through reactions involving oxidation of H2O2.
To clarify if the change in the oxidation state of CNPs was associated with specific localization within the cell, we conducted in situ analysis using a μXANES setup that allowed submicrometer resolution.42Fig. 6A shows an example of the Ce concentration map similar to those already reported in Fig. 4. CeO2 appeared to be mostly concentrated in small areas (hot spots), but a non-negligible amount outside the hot spots was observed in large regions within the cell limits. This complex distribution is better evidenced in Fig. 6B that shows also the intrinsic resolution of the μXRF/μXANES setup (2 μm). Full XANES spectra were acquired from different regions of this map (Fig. 6C and D). In particular, in Fig. 6C, spectrum 1 refers to the regions of the highest Ce content (red in the map of Fig. 6B); spectrum 2 is related to the regions of the intermediate Ce concentration (yellow in the map of Fig. 6B); and spectrum 3 is related to the remaining regions (i.e. an average of the full map after exclusion of the regions used for spectra 1 and 2). Reference spectra relative to bulk CeO2, for Ce(IV), and Ce(NO3)3, for Ce(III), are also reported. Significant differences were observed between these spectra. Spectrum 1 was similar to that of bulk CeO2. We conclude that the CNPs located in the hot spots presented Ce atoms in the Ce(IV) oxidation state. An increase in the spectral intensity at ∼5723 eV was apparent in spectrum 2 and even more evident in spectrum 3. In this same energy region, Ce(III) presented an intense emission line (Fig. 6C and D). It follows that, in the region of the cells away from the hot spots, a higher concentration of Ce(III) can be observed. To quantify the Ce(III)/Ce(IV) ratio, spectra 2 and 3 were fitted as linear combinations of spectrum 1 (used as a reference for Ce(IV)) and the spectrum of Ce(NO3)3·6H2O. These fittings were performed using the derivatives (Fig. S3†) to demonstrate better the shift in energy of the absorption edges. Ce(III) concentrations of 12% for spectrum 2 and 17% for spectrum 3 were obtained from this analysis. The different spectra and their derivatives are overlapped in Fig. 6B and C.
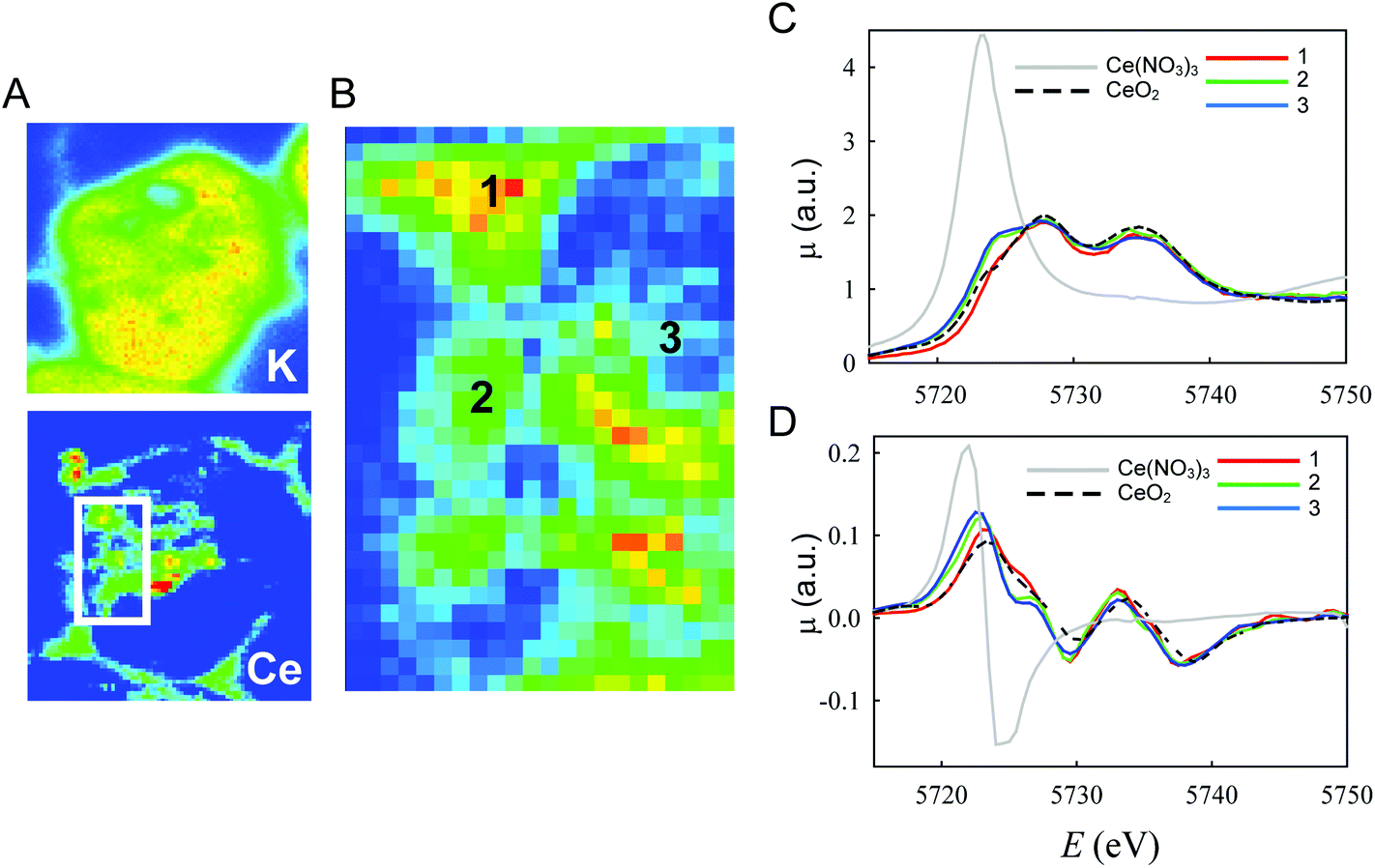 | ||
| Fig. 6 In situ μXANES analysis of the Ce oxidation state in cells incubated for 24 h with CNPs. (A) Representative μXRF on a single cell showing color-coded maps of Ce and K. Ce was mainly present in well-defined hot spots (red), although concentration gradients (from yellow to green) were also visible around them. (B) Enlarged boxed area from (A). Numbers indicate the regions in the cell where XANES analysis was performed. The acquired spectra (C) and the corresponding derivative (D) are shown with reference to CeO2 (Ce(IV)) and Ce(NO3)3 (Ce(III)). | ||
These findings were confirmed by the analysis of the Ce oxidation state in isolated subcellular compartments (e.g. endolysosomes and autophagosomes) obtained from cells incubated for 24 h with CNPs (Fig. 7). Analogous to the previous in situ analysis, only the Ce(IV) oxidation state was observed in those compartments.
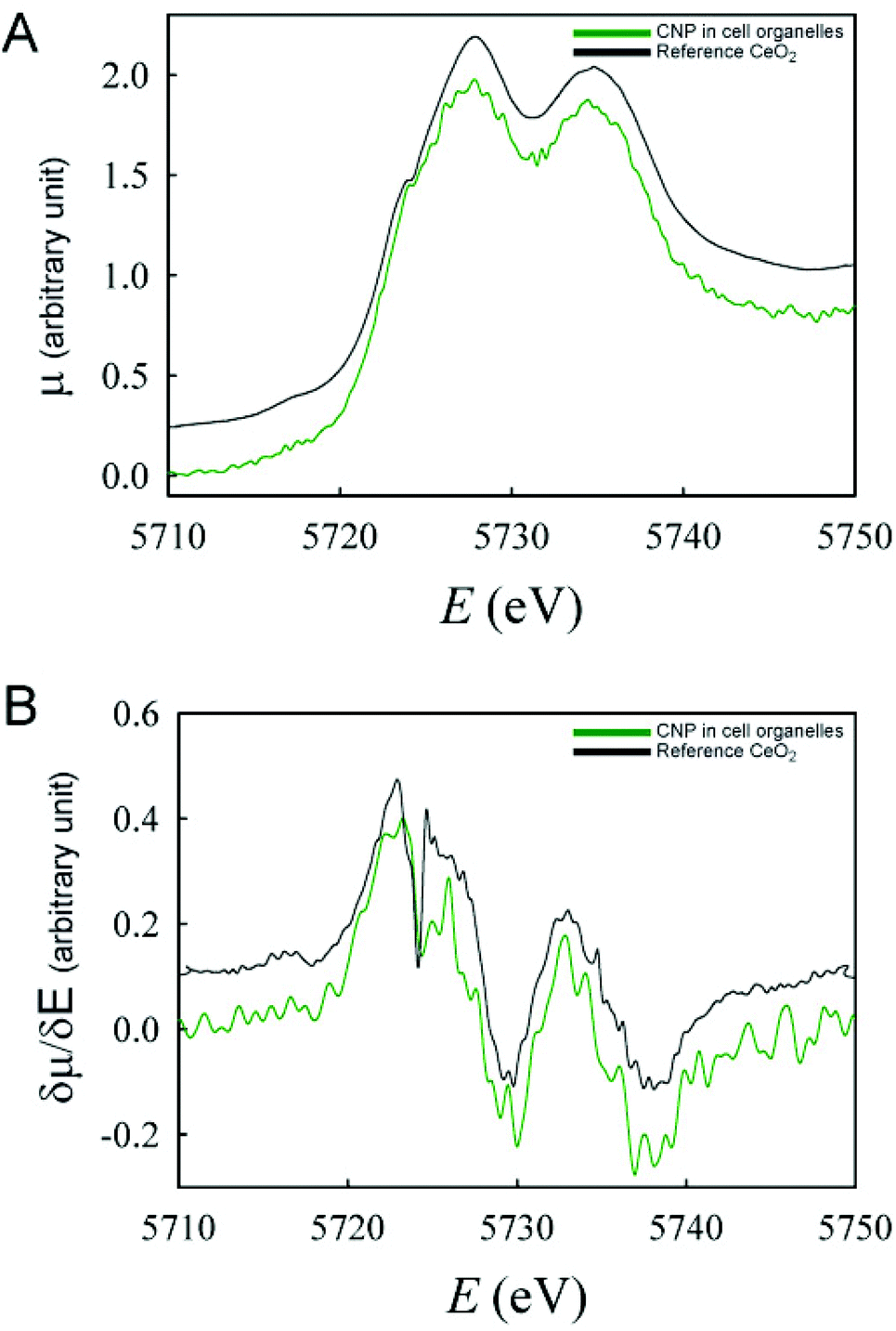 | ||
| Fig. 7 XANES spectra (A) and their respective derivatives (B) of CNPs inside the intracellular organelles isolated from cells incubated with CNPs for 24 h. | ||
Conclusions
Our findings represent a relevant and unprecedented result for human cells, suggesting that the interaction of CNPs with cells involves a complex mechanism, with different areas of the cell playing different roles. In particular, the results indicate that CNPs accumulated in the lysosomes are markedly different from those in the other regions of the cell. This suggests that the effectiveness of the CNPs and their ability to interact with ROS might depend on their localization. Although lysosomes are the main areas for CNP accumulation, additional CNPs can be found in other cell regions, although in smaller amounts. The two locations have different chemical environments, with lysosomes being characterized by an aggressive and acidic environment. It is not clear at this stage whether the dependence of the oxidation state on the localization represents different stages of a unique metabolic pathway, or whether it indicates the existence of parallel CNP processing mechanisms. In the first case, lysosomes might act as a storage location for CNPs, which may still be involved in ROS scavenging through a possible mechanism of exchange with the surrounding environment. In the other case, it is suggested that the two groups of CNPs are the result of different incorporation mechanisms and might have different biological activities.However, these results obtained in vivo in human cells, represent the first direct evidence of the CNP behavior inside human cells and a step toward the understanding of what makes ceria such a promising agent in the therapy of several diseases involving free radicals and/or oxidative stress.
Acknowledgements
We thank A. Casu (King Adullah University of Science and Technology, Thuwal) for the single-crystal structure analysis and E. Solcia (University of Pavia, Department of Molecular Medicine) for the ultrastructural cell analysis. This work was supported by Fondazione Cariplo (Grants No. 2011-2095).References
- L. Raj, T. Ide, A. U. Gurkar, M. Foley, M. Schenone and X. Li, et al., Selective killing of cancer cells by a small molecule targeting the stress response to ROS, Nature, 2011, 475(7355), 231–234 CrossRef CAS PubMed.
- R. W. Tarnuzzer, J. Colon, S. Patil and S. Seal, Vacancy engineered ceria nanostructures for protection from radiation-induced cellular damage, Nano Lett., 2005, 5(12), 2573–2577 CrossRef CAS PubMed.
- D. Schubert, R. Dargusch, J. Raitano and S.-W. Chan, Cerium and yttrium oxide nanoparticles are neuroprotective, Biochem. Biophys. Res. Commun., 2006, 342(1), 86–91 CrossRef CAS PubMed.
- A. Y. Estevez, S. Pritchard, K. Harper, J. W. Aston, A. Lynch and J. J. Lucky, et al., Neuroprotective mechanisms of cerium oxide nanoparticles in a mouse hippocampal brain slice model of ischemia, Free Radicals Biol. Med., 2011, 51(6), 1155–1163 CrossRef CAS PubMed.
- J. M. Dowding, W. Song, K. Bossy, A. Karakoti, A. Kumar and A. Kim, et al., Cerium oxide nanoparticles protect against Aβ-induced mitochondrial fragmentation and neuronal cell death, Cell Death Differ., 2014, 21(10), 1622–1632 CrossRef CAS PubMed.
- M. S. Wason and J. Zhao, Cerium oxide nanoparticles: potential applications for cancer and other diseases, Am. J. Transl. Res., 2013, 5(2), 126–131 CAS.
- L. Fiorani, M. Passacantando, S. Santucci, S. Di Marco, S. Bisti and R. Maccarone, Cerium Oxide Nanoparticles Reduce Microglial Activation and Neurodegenerative Events in Light Damaged Retina, PLoS One, 2015, 10(10), e0140387 Search PubMed.
- H. J. Kwon, M.-Y. Cha, D. Kim, D. K. Kim, M. Soh and K. Shin, et al., Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer's Disease, ACS Nano, 2016, 10(2), 2860–2870 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert and H. Shi, et al., Comparison of the Mechanism of Toxicity of Zinc Oxide and Cerium Oxide Nanoparticles Based on Dissolution and Oxidative Stress Properties, ACS Nano, 2008, 2(10), 2121–2134 CrossRef CAS PubMed.
- J. Colon, L. Herrera, J. Smith, S. Patil, C. Komanski and P. Kupelian, et al., Protection from radiation-induced pneumonitis using cerium oxide nanoparticles, Nanomed. Nanotechnol. Biol. Med., 2009, 5(2), 225–231 CrossRef CAS PubMed.
- L. Alili, M. Sack, A. S. Karakoti, S. Teuber, K. Puschmann and S. M. Hirst, et al., Combined cytotoxic and anti-invasive properties of redox-active nanoparticles in tumor-stroma interactions, Biomaterials, 2011, 32(11), 2918–2929 CrossRef CAS PubMed.
- P. K. Kim, D. W. Hailey, R. T. Mullen and J. Lippincott-Schwartz, Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(52), 20567–20574 CrossRef CAS PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Pharmacological potential of cerium oxide nanoparticles, Nanoscale, 2011, 3(4), 1411 RSC.
- Y.-J. Wang, H. Dong, G.-M. Lyu, H.-Y. Zhang, J. Ke and L.-Q. Kang, et al., Engineering the defect state and reducibility of ceria based nanoparticles for improved anti-oxidation performance, Nanoscale, 2015, 7(33), 13981–13990 RSC.
- J.-D. Cafun, K. O. Kvashnina, E. Casals, V. F. Puntes and P. Glatzel, Absence of Ce3+ Sites in Chemically Active Colloidal Ceria Nanoparticles, ACS Nano, 2013, 7(12), 10726–10732 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, Surface-charge-dependent cell localization and cytotoxicity of cerium oxide nanoparticles, ACS Nano, 2010, 4(9), 5321–5331 CrossRef CAS PubMed.
- N. Ould-Moussa, M. Safi, M.-A. Guedeau-Boudeville, D. Montero, H. Conjeaud and J.-F. Berret, In vitro toxicity of nanoceria: effect of coating and stability in biofluids, Nanotoxicology, 2014, 8(7), 799–811 CAS.
- B. Collin, E. Oostveen, O. V. Tsyusko and J. M. Unrine, Influence of Natural Organic Matter and Surface Charge on the Toxicity and Bioaccumulation of Functionalized Ceria Nanoparticles in Caenorhabditis elegans, Environ. Sci. Technol., 2014, 48(2), 1280–1289 CrossRef CAS PubMed.
- P. Zhang, Y. Ma, Z. Zhang, X. He, J. Zhang and Z. Guo, et al., Biotransformation of ceria nanoparticles in cucumber plants, ACS Nano, 2012, 6(11), 9943–9950 CrossRef CAS PubMed.
- T. Marie, A. Mélanie, B. Lenka, I. Julien, K. Isabelle and P. Christine, et al., Transfer, transformation, and impacts of ceria nanomaterials in aquatic mesocosms simulating a pond ecosystem, Environ. Sci. Technol., 2014, 48(16), 9004–9013 CrossRef CAS PubMed.
- Y. Ma, P. Zhang, Z. Zhang, X. He, J. Zhang and Y. Ding, et al., Where Does the Transformation of Precipitated Ceria Nanoparticles in Hydroponic Plants Take Place?, Environ. Sci. Technol., 2015, 49(17), 10667–10674 CrossRef CAS PubMed.
- M. G. Persico, P. Chiari, R. Biesuz and G. Alberti, Determination of 10B in lymphoma human cells after boron carrier treatment: comparison of 10BPA and immuno-nanoparticles, Chem. Pap., 2014, 68(2), 253–259 CAS.
- D. Ferraro, U. Anselmi-Tamburini, I. G. Tredici, V. Ricci and P. Sommi, Overestimation of nanoparticles-induced DNA damage determined by the comet assay, Nanotoxicology, 2016, 10(7), 861–870 CrossRef CAS PubMed.
- A. R. Collins, The comet assay for DNA damage and repair: principles, applications, and limitations, Mol. Biotechnol., 2004, 26(3), 249–261 CrossRef CAS PubMed.
- E. Eruslanov and S. Kusmartsev, Identification of ROS using oxidized DCFDA and flow-cytometry, Methods Mol. Biol., 2010, 594, 57–72 CAS.
- F. D'Acapito, S. Colonna, S. Pascarelli, G. Antonioli, A. Balerna and A. Bazzini, et al., GILDA (Italian beamline) on BM8, ESRF Newsl., 1998, 30, 42–44 Search PubMed.
- P. Holden and W. A. Horton, Crude subcellular fractionation of cultured mammalian cell lines, BMC Res. Notes, 2009, 2, 243 CrossRef PubMed.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541 CrossRef CAS PubMed.
- P. Ghigna, G. Spinolo, M. Scavini, U. A. Tamburini and A. V. Chadwick, The atomic and electronic structure of cerium substitutional defects in Nd 2- x Ce x CuO 4+ δ An XAS study, Phys. C, 1995, 253(1), 147–155 CrossRef CAS.
- S. De Carlo, Plunge Freezing (Holey Carbon Method), in Handbook of cryo-preparation methods for electron microscopy, ed. A. Cavalier, et al., CRC Press, Boca Raton, Florida, USA, 2009 Search PubMed.
- M. Salomé, M. Cotte, R. Baker, R. Barrett, N. Benseny-Cases and G. Berruyer, et al., The ID21 Scanning X-ray Microscope at ESRF, J. Phys.: Conf. Ser., 2013, 425(18), 182004 CrossRef.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra, Spectrochim. Acta, Part A, 2007, 62(1), 63–68 CrossRef.
- K. Marstal, F. Berendsen, M. Staring and S. Klein, SimpleElastix: A user-friendly, multi-lingual library for medical image registration, in IEEE Conference on Computer Vision and Pattern Recognition Workshops [Internet], 2016 [cited 2016 Aug 5]. p. 574–582. Available from: http://elastix.isi.uu.nl/marius/downloads/2016_c_WBIRa.pdf Search PubMed.
- B. Goris, S. Turner, S. Bals and G. Van Tendeloo, Three-dimensional valency mapping in ceria nanocrystals, ACS Nano, 2014, 8(10), 10878–10884 CrossRef CAS PubMed.
- E. K. Goharshadi, S. Samiee and P. Nancarrow, Fabrication of cerium oxide nanoparticles: characterization and optical properties, J. Colloid Interface Sci., 2011, 356(2), 473–480 CrossRef CAS PubMed.
- A. S. Karakoti, P. Munusamy, K. Hostetler, V. Kodali, S. Kuchibhatla and G. Orr, et al., Preparation and characterization challenges to understanding environmental and biological impacts of ceria nanoparticles, Surf. Interface Anal., 2012, 44(8), 882–889 CrossRef CAS PubMed.
- L. Rubio, B. Annangi, L. Vila, A. Hernández and R. Marcos, Antioxidant and anti-genotoxic properties of cerium oxide nanoparticles in a pulmonary-like cell system, Arch. Toxicol., 2015, 9(2), 269–278 Search PubMed.
- D. Ali, S. Alarifi, S. Alkahtani, A. A. AlKahtane and A. Almalik, Cerium Oxide Nanoparticles Induce Oxidative Stress and Genotoxicity in Human Skin Melanoma Cells, Cell Biochem. Biophys., 2015, 71(3), 1643–1651 CrossRef CAS PubMed.
- A. V. Soldatov, T. S. Ivanchenko, S. Della Longa, A. Kotani, Y. Iwamoto and A. Bianconi, Crystal-structure effects in the Ce L3-edge x-ray-absorption spectrum of CeO2: Multiple-scattering resonances and many-body final states, Phys. Rev. B: Condens. Matter, 1994, 50(8), 5074–5080 CrossRef CAS.
- J. A. Hernandez-Viezcas, H. Castillo-Michel, J. C. Andrews, M. Cotte, C. Rico and J. R. Peralta-Videa, et al., In Situ Synchrotron X-ray Fluorescence Mapping and Speciation of CeO2 and ZnO Nanoparticles in Soil Cultivated Soybean (Glycine max), ACS Nano, 2013, 7(2), 1415–1423 CrossRef CAS PubMed.
- J. Roggenbuck, T. Waitz, T. Wagner, A. Lotz, M. Froba and M. Tiemann, Ce LIII-XANES Investigation of Nanoporous CeO2, in HASYLAB-Jahresbericht HASYLAB/DESY, Hamburg, 2007, pp. 461–462 Search PubMed.
- J. Susini, M. Salomé, U. Neuhaeusler, O. Dhez, D. Eichert and B. Fayard, et al., The X-ray Microscopy and Micro-spectroscopy facility at the ESRF, Synchrotron Radiat. News, 2003, 16(3), 35–43 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr07701c |
| ‡ P. S., A.-T. U., and V. R. share last authorship. |
| This journal is © The Royal Society of Chemistry 2017 |