
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time in situ analysis of biocorona formation and evolution on silica nanoparticles in defined and complex biological environments†
Rickard
Frost
 *ab,
Christoph
Langhammer
b and
Tommy
Cedervall
c
*ab,
Christoph
Langhammer
b and
Tommy
Cedervall
c
aDepartment of Energy and Environment, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden. E-mail: rickard.frost@chalmers.se
bDepartment of Physics, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden
cBiochemistry and Structural Biology and NanoLund, Lund University, Box 124, SE-221 00 Lund, Sweden
First published on 23rd February 2017
Abstract
Biomolecules such as proteins immediately adsorb on the surface of nanoparticles upon their exposure to a biological environment. The formed adlayer is commonly referred to as biomolecule corona (biocorona) and defines the biological activity and toxicity of the nanoparticle. Therefore, it is essential to understand in detail the biocorona formation process, and how it is governed by parameters like composition of the biological environment, and nanoparticle size, shape and faceting. Here we present a detailed equilibrium and real time in situ study of biocorona formation at SiO2-nanoparticle surfaces upon exposure to defined (BSA, IgG) and complex (bovine serum, IgG depleted bovine serum) biological samples. We use both nanofabricated surface-associated Au core–SiO2 shell nanoparticles (faceted, d = 92–167 nm) with integrated nanoplasmonic sensing function and dispersed SiO2 nanoparticles (using DLS and SDS-PAGE). The results show that preadsorbed BSA or IgG are exchanged for other proteins when exposed to bovine serum. In addition, the results show that IgG forms a biocorona with different properties at curved (edge) and flat (facet) SiO2-nanoparticle surfaces. Our study paves the way for further real time in situ investigations of the biocorona formation and evolution kinetics, as well as the role of molecular orientation in biocorona formation, on nanoparticles with surface faceting.
Introduction
When nanomaterials are exposed to a biological environment, e.g. blood, serum, cytoplasm etc., constituents thereof inevitably adsorb to their surface.1,2 The formed adlayer is commonly referred to as a biomolecule corona (biocorona).3 During the biocorona formation process the nanomaterials gain new surface properties that may alter the fate of that material.4–6 Thus, the biological activity, e.g. cellular response and toxicity, of a nanomaterial is dependent on the environment that the material is/has been exposed to.7,8 For this reason, it is important to not only characterize nanomaterials as synthesized (synthetic identity) but also in situ when introduced in a biological environment (biological identity).The formation of the biocorona (hereafter referred to as corona) is typically a dynamic process in which different biomolecules compete for the available surface.9–11 Proteins with high concentration will likely bind first but with time be replaced by proteins with high affinity for the surface.12 The on/off rates and the concentration of each biomolecule will determine the contents in the corona over time until equilibrium is reached. For example, there are a number of studies describing the multiple protein binding to silica nanoparticles in complex protein mixtures,5,10,11,13–15 whereas another recent study reports that in human blood plasma a single protein is dominating when the particle surface area is scarce.11 An alternative view of the formation of the corona is that biomolecules stochastically adsorb irreversibly to the nanoparticle surface.16 It is likely that the nature of the nanoparticle surface determines which process dominates and that on some surfaces a combination of different processes occurs. However, very little is known about correlations between simultaneously occurring molecular adsorption processes and specific nanoparticle descriptors such as size and shape because real time corona formation studies are experimentally very difficult.
Commonly, when the corona formed in blood serum or plasma is studied, nanomaterials (often in the form of particles) are incubated in biological media. The formed complexes of the nanomaterial with adsorbed proteins, lipids or other biomolecules, are separated from unbound biomolecules (e.g. by centrifugation), and characterized (e.g. by mass spectrometry or gel electrophoresis). In this way, it is possible to identify the constituents of the corona. However, the described methodology has three main drawbacks: (1) the nanomaterials may aggregate upon incubation in biological media, (2) the formed corona may change during the purification process, and (3) only the equilibrium state is probed. Thus, the result of the final protein characterization may be affected in the sense that it not fully represents the initially formed corona. Another approach is to measure the size of the biomolecule/nanomaterial complex over time by dynamic light scattering (DLS), as the size of monodisperse complexes is related to the thickness of the corona. DLS analysis may be performed during incubation in biological media, thus purification of the nanoparticles may not be necessary if it is possible to distinguish between the response from the nanoparticles and the biological medium. However, it can be difficult to separate aggregation from the increase in hydrodynamic radius, which means that the result often is largely biased towards bigger entities. An alternative method is nanoparticle tracking analysis (NTA). However, in NTA there is often a need to dilute the samples before measurement, which again may change the equilibrium composition of the corona. The third method often used to measure the nanoparticle size, and to follow changes thereof is differential sedimentation centrifugation (DSC). The method is robust, but in mixed samples (of e.g. biomolecules and nanoparticles), the observed size of protein/nanoparticle complexes is difficult to assess, as the density of the protein/nanoparticle complexes is unknown.
As a complementary technology, to address some of the aforementioned shortcomings of existing techniques, we recently introduced a nanoplasmonic sensor (NPS) surface.17 This technique enables real-time in situ analysis of corona formation occurring on surface-associated nanofabricated metal core–dielectric shell nanostructures. In this arrangement, the nanostructures act as mimics of “free” dielectric nanoparticles in suspension with built-in sensing function. For convenience the used NPS surfaces are described in some detail in the ESI.† The sensor makes it possible to probe molecular adsorption events (kinetics) in situ and in real-time without the risk of sample aggregation or the need for purification procedures. In this way it effectively eliminates some of the key complicating factors of existing corona characterization techniques discussed above. In particular, it enables real-time in situ experiments of both initial corona formation and subsequent evolution. Moreover, this sensing platform is versatile due to the possibility to quite freely tailor the shell material and to fabricate nanostructures of different sizes. In the present work, we use nanoparticles that are comprised of a gold (Au) plasmonic core for molecular detection through localized surface plasmon resonance.18,19 A homogeneous 10 nm SiO2 dielectric layer is grown on the sensor chip surface to encapsulate the Au nanoparticles and in this way form a mimic of silica nanoparticles in solution20,21 (Fig. 1). We also note that the core–shell structures are not perfect spheres but faceted particles. Here, we capitalize on this effect by quantitatively analyzing the role of flat versus curved surfaces on a faceted nanoparticle, in corona formation. Core–shell nanostructures with different flat/curved ratios, applied to probe differences in protein adsorption behavior to the two surface regions, are obtained by fabricating facetted nanostructures of different size. The NPS experiment is different from most previous protein corona studies as the proteins are exposed to the sensor surface under a constant flow. The flow rate may change the binding kinetics and there is some evidence that there is an increased protein adsorption to the surface in a flow compared to a static situation.22–24 Although the applied NPS technique indeed provides relevant new information regarding the corona formation process, the use of multiple, complementary, analytical techniques is of key importance to a thorough characterization of the nanoparticle corona.
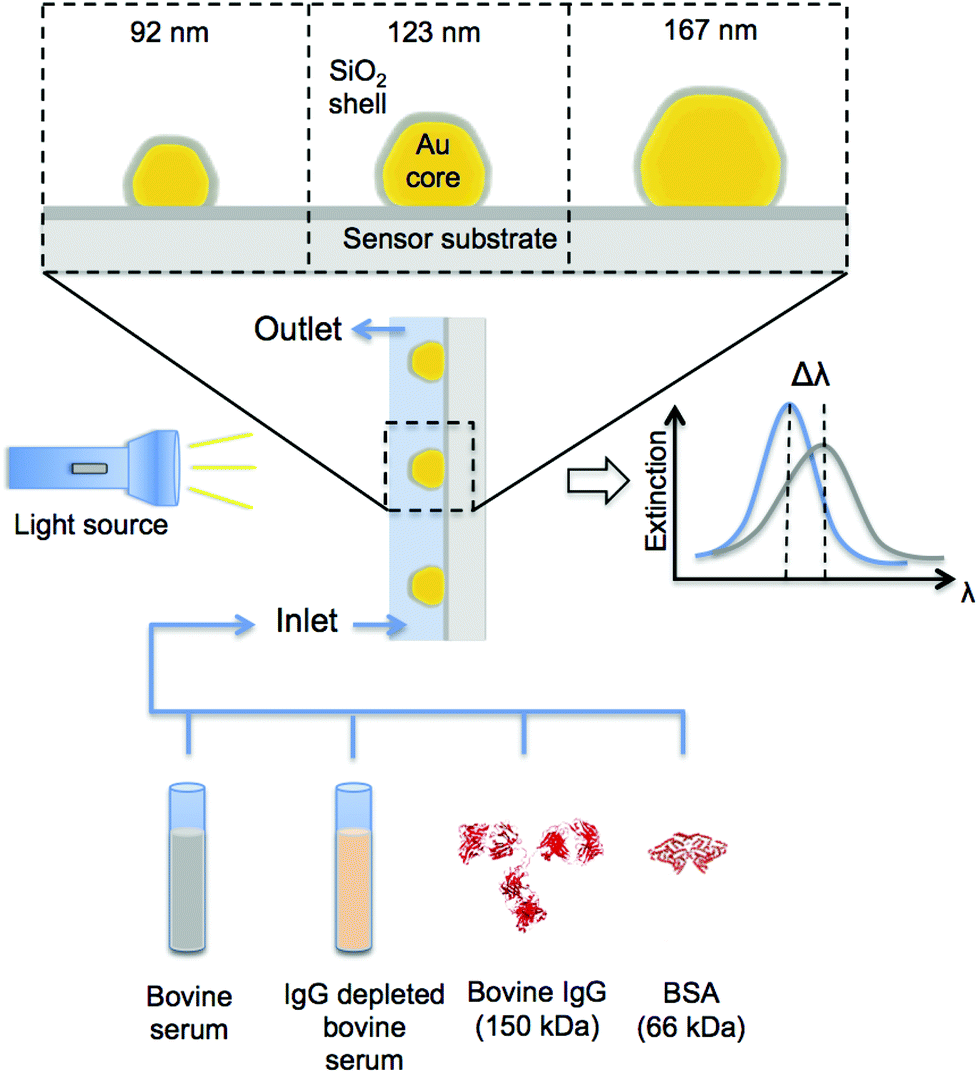 | ||
| Fig. 1 Schematic description of the used experimental setup for real-time nanoplasmonic sensing of corona formation and evolution. Three sizes of faceted Au-core/SiO2-shell nanoplasmonic sensors have been used, that is amorphous arrays of core–shell structures (coverage ∼10%) with average diameters of 92, 123 and 167 nm, respectively. Adsorption of molecules to the SiO2-shell generates a peak shift in the extinction spectra of the nanoplasmonic sensor, enabling real-time in situ analysis of the formed corona. Specifically, in our experiments the sensors have been exposed to bovine serum, IgG depleted bovine serum, IgG from bovine serum and bovine serum albumin (BSA). Sequential additions of these samples have also been performed to assess corona evolution under different conditions. | ||
Results and discussion
NPS of corona formation on differently sized SiO2 nanoparticle mimics
In this first part of our study, we use the nanoplasmonic sensing platform depicted in Fig. 1 to study the corona formation upon adsorption of BSA, IgG, bovine serum and IgG depleted bovine serum at the surface of differently sized Au core/SiO2 shell nanoparticles in situ. To this end, it is well established that particle size influences the protein content in the corona qualitatively and quantitatively.6,13 Furthermore, the particle size controls structural changes, and thus the function, of proteins bound to silica nanoparticles,25–27 as well as how proteins are ordered and oriented on the silica nanoparticle surface.26,28,29 The biological response of silica nanoparticles may also be dependent the structure (e.g. porosity) of the surface/shell.30 The biological samples were chosen for the purpose of elucidating the role of BSA and IgG, which are the two most abundant proteins in serum,31 during corona formation at nanoparticle surfaces. From the results presented in Fig. 2, it is evident by the observed peak shifts of the plasmonic sensor (Fig. 2A–C) that BSA generates a corona with less protein mass compared to IgG, which in turn has less mass compared to coronas formed in the more complex serum samples (see ESI† for experimental details). This data interpretation assumes a similar refractive index of the adsorbed protein layers since the true refractive indices are unknown. It has previously been observed that small proteins give more optically dense layers when adsorbed to a planar TiO2 surface compared to larger proteins. This trend implies that the layer of adsorbed BSA may have a slightly larger refractive index than the layer of IgG.32 As our results thus indicate, IgG does not have a major impact on the corona formation at SiO2 particles in bovine serum, as the response of the two serum samples (that is, with and without IgG) is seemingly similar. However, there is a slight difference between the two samples since IgG depleted bovine serum consistently generates a slightly smaller response compared to bovine serum. The reason for this observed difference could be a small change in the refractive index of the formed corona, depending on the presence or absence of IgG. | ||
| Fig. 2 (A–C) Nanoplasmonic sensing data of the adsorption of bovine serum, IgG depleted bovine serum (bovine serum–IgG), IgG and BSA to the three different types of sensors, that is, 92, 123 and 167 nm average particle size. (D–F) The thicknesses of the formed coronas (after 60 min adsorption) calculated as a function of their refractive indices based on eqn (1). The insets show TEM-images (top view) of the differently sized core/shell nanostructures where the surface facets of the Au-cores are clearly visible (all scale bars equal 20 nm). | ||
The magnitude of the NPS peak shift is related to both the thickness and the refractive index of the formed corona, according to the well established relation:33
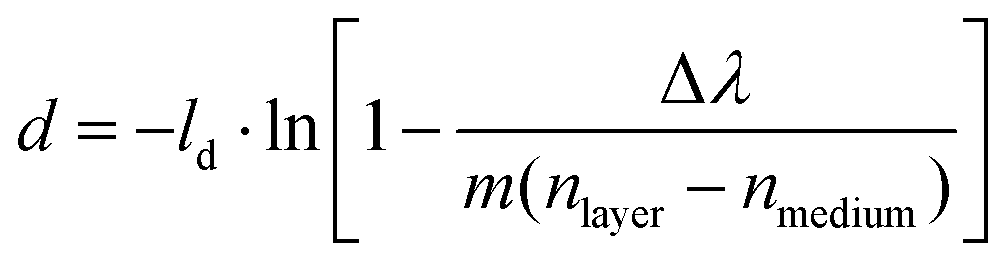 | (1) |
From eqn (1) it becomes clear that it is not possible using nanoplasmonic sensing alone to separate the film thickness of an adsorbed layer and its refractive index, and thus single out their absolute contributions to the experimentally measured peak shift. For this reason, we calculate the thickness of the corona (d) for each sample, using the recorded peak shift after 60 min incubation (Δλ), for a range of refractive indices (nlayer) according to eqn (1). The data are presented in Fig. 2D–F and show that the thicknesses of the acquired coronas vary and that dBSA < dIgG < dbovine serum-IgG ≤ dbovine serum over the entire range of considered refractive indices, assuming a similar refractive index32 of the formed protein coronas.
When comparing the data of the corona formation at the surface of the 92, 123 and 167 nm core/shell nanostructures some interesting differences can be seen. To understand them it is important to note the surface faceting of the nanostructures as shown in Fig. 2. Specifically, this means that it is the ratio between flat and curved areas at the nanoparticle surface that increases with the particle size and not the surface curvature that decreases. In fact the latter remains constant.17
We start our discussion with BSA since we have previously evaluated its response to the size – and thus the flat-to-curved surface area ratio – of the nanostructure,17 using the same experimental approach. From our previous study, in brief, we know that BSA gives rise to a larger plasmon resonance peak shift for the largest nanostructures upon corona formation. In the framework of the model we have developed in that study, this indicates that a denser protein layer is formed at the planar facets compared to the curved regions at the edges between the facets, likely due to increased surface interactions. Interestingly, applying the same concept to the data obtained in the present study, it turns out that the observed effect for BSA is small compared to the other samples, that is, IgG and the two serum samples (Table 1). Specifically, our data show that the plasmonic sensor response for IgG almost doubles relative to the response for BSA when the size of the nanostructures increases from 92 to 167 nm. Similarly, the response for the serum samples relative to the response for BSA also increases with the size of the nanostructures, although not to the same extent as for IgG. From these observations we draw the first conclusion that the corona formation indeed is different for the different systems under study. Specifically, following the same reasoning as developed for BSA in our earlier work,17 the larger fraction of planar areas on the facets of the 167 nm nanostructures compared to the smaller ones, allows for overall increased surface interactions, which in turn generates a globally denser protein corona and thereby a larger plasmonic sensor response. An alternative scenario also explaining the observed size-dependent differences of the plasmonic response to corona formation is that the proteins (especially IgG) adsorb with different orientations depending on where the adsorption occurs, i.e. at flat or curved regions. The importance of surface curvature on the orientation of protein binding to silica nanoparticles has previously been demonstrated for several proteins.28,29 In line with this scenario, IgG forms a thicker corona (e.g. protein adsorbed head on) at the planar facets and a thinner corona (e.g. protein adsorbed side on) at the curved regions. In the complex serum samples, which contain a large variety of different components, a combination of both described scenarios is likely to occur. Thus, due to the significantly larger response obtained for the serum samples, the results indicate that neither BSA nor IgG is a dominating component in the corona formed at the surface of SiO2 nanoparticles in bovine serum.
| Sample | 92 nm | 123 nm | 167 nm | |||
|---|---|---|---|---|---|---|
|
|
Max. rate (nm mg−1) |
|
Max. rate (nm mg−1) |
|
Max. rate (nm mg−1) | |
| BSA | 1.0 | 0.7 | 1.0 | 0.6 | 1.0 | 0.7 |
| IgG | 1.7 | 1.8 | 2.2 | 2.9 | 3.3 | 6.7 |
| Bovine serum–IgG | 4.4 | 3.1 | 4.6 | 3.8 | 5.8 | 9.0 |
| Bovine serum | 4.6 | 3.8 | 4.9 | 5.7 | 6.1 | 11.3 |



Kinetics of corona formation
To further evaluate the recorded data, we analyze the kinetics of the corona formation in detail. To this end we highlight that the applied protein concentrations are low compared to native protein concentrations in blood. For this reason, the kinetics of corona formation is likely significantly accelerated in in vivo situations. We also note that, due to an increased sensitivity factor, a larger NPS peak shift is expected upon corona formation when the size of the Au core increases. Thus, the observed adsorption rates during corona formation at the surface of the differently sized nanostructures are not easily quantitatively compared. To account for the different sensitivity of the sensor structures, we have normalized the data using experimentally determined calibration factors derived from the measured peak shift induced by atomic layer deposition of 5 nm Al2O3, i.e. a thin homogenous oxide layer (see ESI† for details). In Fig. 3 we show the sensitivity-factor-normalized peak shift signal induced by corona formation plotted versus the accumulated mass of protein the sensor has been exposed to. The flow rate is the same for all experiments but the protein concentration differs between the samples (see the Experimental section in ESI† for details). For this reason, it is convenient to use the exposure (accumulated mass of protein) to compare the kinetics for the different samples. Here, we have assumed an initial protein content of 80 mg mL−1 in the two serum samples. Due to the applied sensitivity-factor normalization, the obtained data from the three different nanoparticle sizes are now directly and quantitatively comparable. The data show that the initial rate of the corona formation is lower for BSA compared to the two serum samples, independent of the size of the core/shell nanostructures. This implies that BSA may, despite its high concentration, not be a dominant protein in the early events of the corona formation as in competition with other serum proteins many other proteins, including IgG with a relative high concentration, will bind faster. However, the kinetics of IgG adsorption varies significantly with the size. For the smallest nanostructures, IgG exhibits a fairly low rate of adsorption, although higher than for BSA. For larger nanostructures the rate of IgG adsorption increases and reaches a similar initial rate of adsorption as for the two serum samples for the largest structures. | ||
| Fig. 3 Kinetics of the corona formation upon addition of bovine serum, IgG depleted bovine serum (bovine serum–IgG), IgG and BSA using (A) 92 nm, (B) 123 nm and (C) 167 nm nanoplasmonic sensors. To account for the different sensitivity of the differently sized Au cores the measured peak shifts during corona formation were normalized using the experimentally determined sensitivity factors of the respective sensor (see ESI† for details). Hence, the shown data for the three different nanoparticle sizes can be directly and quantitatively compared. The insets show the normalized rate of adsorption during exposure to the first 0.6 mg of added protein. All experiments were performed under constant flow. For comparison between samples, the accumulated mass of added protein (exposure) is used in the present figure. | ||
Interestingly, the kinetics for the two serum samples also varies with the size of the nanostructures. For the largest nanostructures the initial rate of adsorption is markedly higher compared to the smallest structures. This result may be explained by (i) a more rapid adsorption to the planar areas compared to the curved regions (higher ratio of flat/curved areas at larger nanostructures), (ii) that flat and curved regions of the faceted nanostructures causes IgG and serum components adsorb with different conformation and/or orientations, or (iii) combinations of (i) and (ii).
Sequential corona formation: effect of BSA pre-adsorption
One of the main benefits with the NPS methodology is the possibility to monitor the corona formation process in real-time. This feature enables in situ analysis of corona evolution, e.g. induced changes in corona properties upon changes in the surrounding environment. Here, we investigate the effect of BSA preadsorption on the corona formation in bovine serum, IgG depleted bovine serum, and IgG at the surface of 123 nm core/shell nanostructures. Subsequently, we investigate the effect of BSA on the corona formed in the two serum samples and the effect of IgG depleted bovine serum on the corona formed by sequential exposures to BSA and IgG. The results, presented in Fig. 4A–C, show that BSA preadsorption does not prevent further adsorption of serum components or IgG. However, the preadsorbed BSA somewhat reduces the amount of serum components and IgG that adsorb to the surface. The dashed lines in Fig. 4A–C represent the response obtained during adsorption to bare surfaces, without preadsorbed BSA. Additionally, the results show that the corona formed in the two serum samples, on nanostructures with preadsorbed BSA, are not significantly affected by a second BSA exposure although the other serum proteins are no longer present in the solution (Fig. 4A and B). A corona formed by sequential exposures to BSA and IgG allows further adsorption of serum components during a subsequent exposure to IgG depleted bovine serum (Fig. 4C). The obtained response after exposure to IgG depleted bovine serum is similar on both pre-coated surfaces, independent on the IgG adsorption.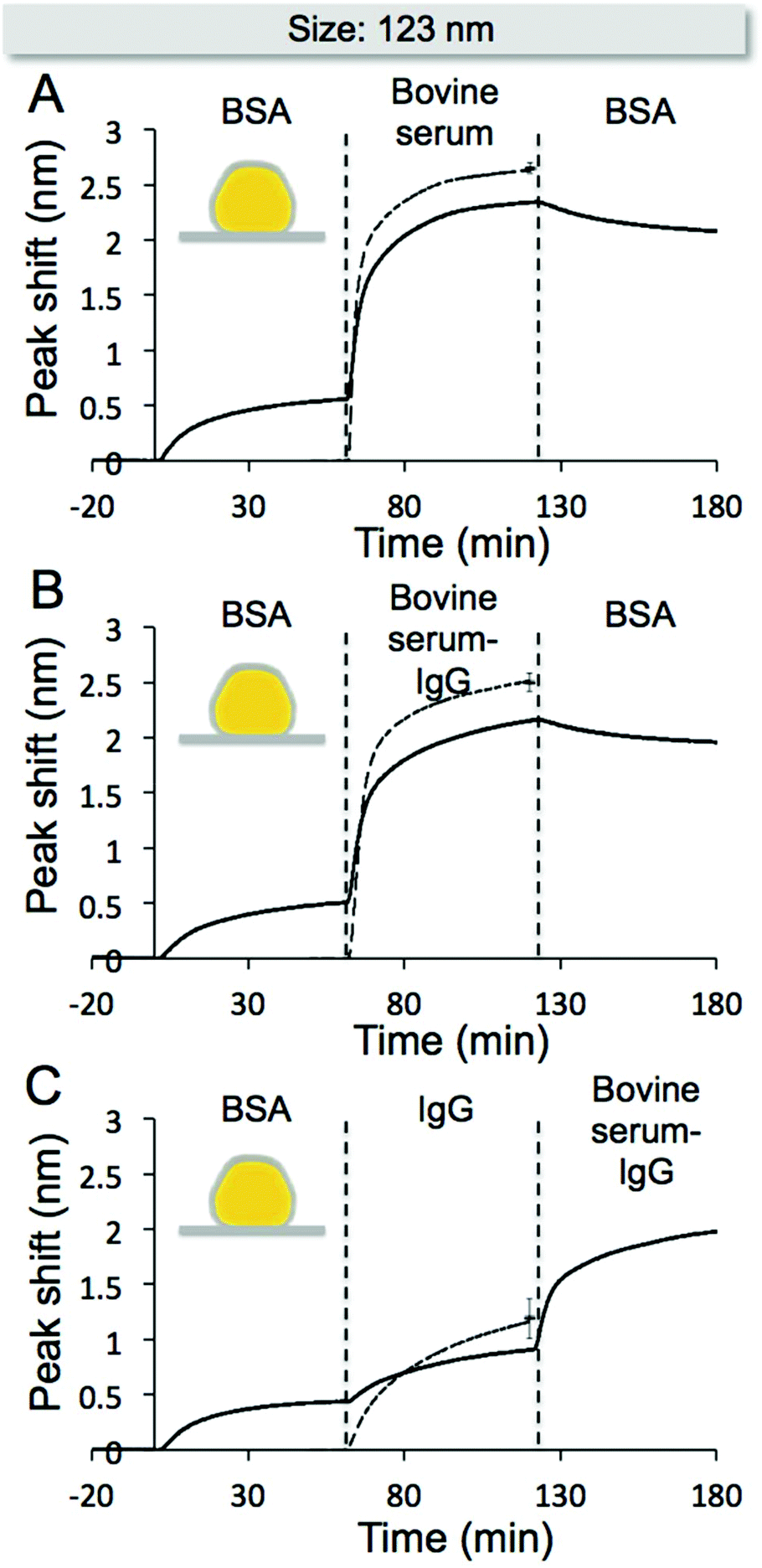 | ||
| Fig. 4 NPS data of sequential sample additions using 123 nm sensors (solid line). After initial adsorption of BSA the nanostructures were exposed to (A) bovine serum and thereafter BSA, (B) IgG depleted bovine serum and thereafter BSA, and (C) IgG and thereafter IgG depleted bovine serum. For comparison, the obtained response of bovine serum, IgG depleted bovine serum and IgG to the bare 123 nm sensors are included in the second step (dashed line). | ||
From the results in Fig. 4 it is clear that BSA does not form a complete, irreversibly bound monolayer at the surface of the nanostructures, as further adsorption of serum proteins and IgG is possible. These data may be explained by the following two scenarios: (i) BSA adsorption forms an incomplete monolayer, that is, IgG and serum proteins may directly adsorb to the SiO2-surface; (ii) BSA adsorbs reversibly allowing IgG and serum proteins to replace the preadsorbed BSA. Which scenario that is the most likely to occur is discussed in more detail below together with additional data obtained by DLS and SDS-PAGE.
Corona formation on silica nanoparticles in dispersions
The NPS data strongly indicate that BSA, IgG, and bovine serum form distinct coronas on silica nanoparticles and that exposure to bovine serum changes a preformed corona of BSA or IgG. However, we cannot unambiguously conclude from the NPS data whether there are still empty surface sites for the serum proteins to bind or if an exchange of proteins occurs (i.e. according to scenario (i) and (ii) mentioned above). To distinguish between these two possible explanations of the NPS data we set up experiments with silica nanoparticles in dispersion aiming to determine the size and the identity of adsorbed proteins. The size of the particles (d = 56 ± 9 nm by TEM) was chosen such that the curvature should be close to that of the curved region on the nanofabricated core–shell structures (corresponding to a diameter of about 52 nm, see ESI†). The somewhat larger size determined by DLS indicates that some nanoparticle aggregates are present.One important difference between the NPS experiments and the experiments in dispersion is that in dispersion we are studying the formed corona in equilibrium, instead of following the corona formation in real time. Four BSA concentrations and three silica particle concentrations were studied in detail. Table 2 shows the radii obtained by DLS for silica particles alone or together with BSA. There is a distinct increase in the size of the BSA incubated silica particles, which corresponds well with a monolayer of BSA. The size of the BSA/silica particle complex is not increasing with increasing concentrations indicating that the particle surface is saturated. However, it should be noted that DLS measures the hydrodynamic radius and that the formation of a BSA layer with low surface coverage cannot be excluded. The corresponding NPS results (Fig. 2) show that the effective refractive index of the BSA corona needs to be small (RI of about 1.36) to obtain a similar thickness of the protein layer, indicating a large degree of hydration.
| C (mg ml−1) | Radiusa (nm) | |||||
|---|---|---|---|---|---|---|
| BSA | 4.9% silica | %Pdb | 3.3% silica | %Pdb | 2.2% silica | %Pdb |
| a Radius calculated using a cumulative fit. b % polydispersity. | ||||||
| 2.1 | 43.9 ± 0.2 | 16 ± 2 | 44.2 ± 0.3 | 13 ± 2 | 43.4 ± 0.6 | 15 ± 1 |
| 1.4 | 44.3 ± 1.2 | 17 ± 4 | 43.5 ± 0.2 | 15 ± 1 | 43.2 ± 0.2 | 14 ± 2 |
| 0.9 | 45.0 ± 0 | 16 ± 1 | 43.2 ± 0.3 | 17 ± 1 | 43.8 ± 0.6 | 14 ± 1 |
| 0.62 | 44.8 ± 0.3 | 16 ± 1 | 42.9 ± 0.1 | 16 ± 1 | 43.4 ± 0.6 | 14 ± 1 |
| PBS | 38.3 ± 0.8 | 16 ± 2 | 37.9 ± 0.5 | 14 ± 1 | 38.5 ± 0.7 | 15 ± 2 |
| H2O | 37.4 ± 0.2 | 19 ± 1 | 37.8 ± 0.4 | 17 ± 2 | 39.3 ± 0.3 | 17 ± 1 |
Next, we mimicked the sequential corona formation that was performed in the NPS experiments in the DLS analysis. Thus, bare or BSA pre-incubated silica nanoparticles were mixed with bovine serum and after incubation for one hour the size of the protein/particle complexes was measured by DLS. The results show that the radii of the formed protein/particle complexes are similar, regardless if the silica particles were pre-incubated with BSA or not, Table 3. The peak corresponding to the smallest entities likely represent free proteins in the solution. In serum other structures, such as low- and high-density lipoprotein particles, are in the same size range (10 to 100 nm) as the nanoparticles and we cannot distinguish between if the multimodal size distribution arises from only the particles or is a result from the complex environment. However, the size distribution is clearly different from the size distribution of bovine serum alone.34 If the BSA forms an irreversible corona with high coverage the size of the BSA pre-coated particles is not expected to change after mixing with bovine serum. On the other hand, if the coverage is low and/or the adsorbed BSA is exchangeable other serum proteins may still bind, forming a thicker protein layer.
To further investigate whether BSA is irreversibly bound to the nanoparticles SDS-PAGE analysis of the BSA and the sequentially formed coronas was performed. After incubation in BSA or serum the particles were centrifuged and the pellets carefully washed with PBS. The bound proteins were desorbed with SDS and separated by SDS-PAGE. BSA pre-coated particles contain BSA also after the centrifugation and washing steps, Fig. 5A, indicating a strong binding to the silica particles. However, after incubation in bovine serum no BSA is bound to the silica particles, regardless if they were preincubated with BSA or not, see Fig. 5A. The lack of BSA is more easily seen in Fig. 5B where proteins around BSA are more separated. Actually, the protein profile is identical for bare and precoated silica particles, clearly suggesting that adsorbed BSA is exchanged by other serum proteins. This thus corroborates the interpretation of the corresponding NPS data discussed above. The low amount of BSA in the corona was expected, despite its high concentration in serum, as BSA binds with much slower kinetics than other serum proteins as shown in the NPS experiments summarized in Fig. 3 and because the affinity for the surface is likely low compared to other serum proteins. The reversibility of the BSA adsorption to the silica nanoparticles is also demonstrated when BSA precoated particles are incubated with IgG, Fig. 5C. The amount of BSA on the particles is clearly reduced as IgG is binding to the surfaces of the BSA precoated particles. Our results on the effect preadsorption of BSA correspond well with previous results using human albumin and human plasma.35
 | ||
| Fig. 5 Proteins adsorbed to silica particles visualized by SDS-PAGE. Particles and adsorbed proteins were separated from free proteins by centrifugation and adsorbed proteins were desorbed from the particles using SDS. (A) 12% SDS-PAGE (1) Particles incubated in 2.1 mg ml−1 BSA, (2) Particles incubated in 1.4 mg ml−1 BSA, (3) No particles and 2.1 mg ml−1 BSA, (4) No particles and 1.4 mg ml−1 BSA, (5) Particles preincubated in 2.1 mg ml−1 BSA and thereafter incubated in 75% bovine serum, (6) Particles preincubated in 1.4 mg ml−1 BSA and thereafter incubated in 75% bovine serum, (7) Particles preincubated in PBS and thereafter in 75% bovine serum, (8) 75% bovine serum without particles. (B) 10% SDS-PAGE (1) Particles incubated in 2.1 mg ml−1 BSA, 2. Particles preincubated in 2.1 mg ml−1 BSA and thereafter in 75% bovine serum, (3) Particles preincubated in PBS and thereafter in 75% bovine serum. (C) BSA and IgG adsorbed to silica particles visualized by SDS-PAGE. Particles preincubated with BSA and thereafter in (1) PBS, (2) 4 mg ml−1 IgG, (3) 2 mg ml−1 IgG. | ||
Conclusion
In conclusion, this article presents a systematic analysis of the corona formation in both defined (BSA and IgG) and complex (bovine serum and IgG depleted bovine serum) biological samples. We have applied three complementary analytical techniques (NPS, DLS and SDS-PAGE) to study the corona formation, in real time by measuring the corresponding kinetics, and at equilibrium. We also measured the subsequent evolution of the corona by sequential exposures to different biological samples by again measuring the corresponding kinetics and the equilibrium state. As one of the main results, we found that the NPS methodology, which is a new technique in the present context, is able to provide data that are not possible to obtain using standard methodologies in the field. Specifically, at conditions promoting aggregation of nanoparticles in suspension (e.g. during addition of IgG to the SiO2 nanoparticles in the present experiments), the use of surface associated core–shell nanoparticle mimics turned out to be beneficial since aggregation of the nanostructures at the sensor surface is prohibited. Furthermore, we have shown that the initial rate of adsorption of IgG and the serum samples increased with the size of the nanostructures (increasing ratio of flat/curved areas), in contrast to the initial rate of adsorption of BSA. Thus, the former samples either adsorbed at higher rate to the planar facets compared to the curved regions, or the proteins (especially IgG) adsorbed with different orientations depending on if the surface is curved or not. The significantly larger NPS response for the serum samples compared to the response for BSA and IgG furthermore showed that these proteins do not have a major impact on the corona formed in bovine serum.Finally, using SDS-PAGE, we also demonstrated that a formed BSA corona is largely replaced when the nanoparticle–BSA complex is exposed to bovine serum or IgG, in line with the NPS data. Thus, BSA adsorbed to SiO2 nanoparticles does not prevent further protein adsorption. Similarly, other serum proteins replace IgG when the nanoparticle–IgG complex is exposed to bovine serum. A more detailed summary of the main results, together with a schematic figure thereof, is given as ESI.† In essence, the presented data show the complementarity between the three different analytical techniques applied in this study. Specifically, together they generate time-resolved data of the adsorption processes (kinetics – NPS), data on corona thickness (DLS) and the size/identity (SDS-PAGE) of the adsorbed proteins. Thus, our approach clearly shows that, to better understand processes occurring at the nano–bio interface like corona formation, the use of complementary techniques is essential, and our results open up for further investigations regarding nanoparticle–protein interactions and the process of corona formation in complex biological environments.
Acknowledgements
This work was financially supported by Chalmers Area of Advance in Nanoscience and Nanotechnology, NanoLund, the Swedish Foundation for Strategic Environmental Research (MISTRA Environmental Nanosafety) and the Swedish Research Council (grant 2014-4956). Michael Persson at AkzoNobel is gratefully acknowledged for providing silica nanoparticles that were used for the DLS and SDS-PAGE analysis.References
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, Chem. Soc. Rev., 2015, 44, 6094–6121 RSC.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- D. Dell'Orco, M. Lundqvist, C. Oslakovic, T. Cedervall and S. Linse, PLoS One, 2010, 5, e10949 Search PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- C. Fedeli, D. Segat, R. Tavano, L. Bubacco, G. De Franceschi, P. P. de Laureto, E. Lubian, F. Selvestrel, F. Mancin and E. Papini, Nanoscale, 2015, 7, 17710–17728 RSC.
- L. Vroman, A. L. Adams, G. C. Fischer and P. C. Munoz, Blood, 1980, 55, 156–159 CAS.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- N. P. Mortensen, G. B. Hurst, W. Wang, C. M. Foster, P. D. Nallathamby and S. T. Retterer, Nanoscale, 2013, 5, 6372–6380 RSC.
- A. M. Clemments, P. Botella and C. C. Landry, ACS Appl. Mater. Interfaces, 2015, 7, 21682–21689 CAS.
- P. M. Kelly, C. Aberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Z. Krpetic and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- R. Frost, C. Wadell, A. Hellman, S. Molander, S. Svedhem, M. Persson and C. Langhammer, ACS Sens., 2016, 1, 798–806 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- C. Langhammer, E. M. Larsson, B. Kasemo and I. Zoric, Nano Lett., 2010, 10, 3529–3538 CrossRef CAS PubMed.
- E. M. Larsson, C. Langhammer, I. Zoric and B. Kasemo, Science, 2009, 326, 1091–1094 CrossRef CAS PubMed.
- F. A. A. Nugroho, C. Xu, N. Hedin and C. Langhammer, Anal. Chem., 2015, 87, 10161–10165 CrossRef CAS PubMed.
- J. L. Ortega-Vinuesa, P. Tengvall, B. Walivaara and I. Lundstrom, Biomaterials, 1998, 19, 251–262 CrossRef CAS PubMed.
- E. J. Choi, M. D. Foster, S. Daly, R. Tilton, T. Przybycien, C. F. Majkrzak, P. Witte and H. Menzel, Langmuir, 2003, 19, 5464–5474 CrossRef CAS.
- M. Salim, B. O'Sullivan, S. L. McArthur and P. C. Wright, Lab Chip, 2007, 7, 64–70 RSC.
- M. Lundqvist, I. Sethson and B. H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2006, 128, 3939–3945 CrossRef CAS PubMed.
- W. Shang, J. H. Nuffer, V. A. Muniz-Papandrea, W. Colon, R. W. Siegel and J. S. Dordick, Small, 2009, 5, 470–476 CrossRef CAS PubMed.
- S. Shrivastava, J. H. Nuffer, R. W. Siegel and J. S. Dordick, Nano Lett., 2012, 12, 1583–1587 CrossRef CAS PubMed.
- S. Shrivastava, S. A. McCallum, J. H. Nuffer, X. Qian, R. W. Siegel and J. S. Dordick, Langmuir, 2013, 29, 10841–10849 CrossRef CAS PubMed.
- A. B. Davila-Ibanez, R. Marino-Fernandez, M. Maceira-Campos, A. Garcia-Lorenzo, V. Martinez-Zorzano and V. Salgueirino, J. Phys. Chem. C, 2014, 118, 28266–28273 CAS.
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CAS.
- J. Voros, Biophys. J., 2004, 87, 553–561 CrossRef PubMed.
- O. Kedem, A. B. Tesler, A. Vaskevich and I. Rubinstein, ACS Nano, 2011, 5, 748–760 CrossRef CAS PubMed.
- C. R. Svensson, M. E. Messing, M. Lundqvist, A. Schollin, K. Deppert, J. H. Pagels, J. Rissler and T. Cedervall, PLoS One, 2013, 8, e74702 CAS.
- V. Mirshafiee, R. Kim, S. Park, M. Mahmoudi and M. L. Kraft, Biomaterials, 2016, 75, 295–304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06399c |
| This journal is © The Royal Society of Chemistry 2017 |