
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A tale of four kingdoms – isoxazolin-5-one- and 3-nitropropanoic acid-derived natural products
Tobias
Becker
a,
Jacques
Pasteels
b,
Christiane
Weigel
c,
Hans-Martin
Dahse
c,
Kerstin
Voigt
c and
Wilhelm
Boland
 *a
*a
aDepartment of Bioorganic Chemistry, Max Planck Institute for Chemical Ecology, Jena, Germany. E-mail: boland@ice.mpg.de
bDepartment of Biology, Université Libre de Bruxelles, Brussels, Belgium
cLeibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Jena, Germany
First published on 8th March 2017
Abstract
Covering up to September 2016
This review reports on natural compounds that derive from the isoxazolinone ring as well as the 3-nitropropanoic acid (3-NPA) moiety. These structural elements occur in compounds that have been identified in plants, insects, bacteria and fungi. In particular, plants belonging to the family of legumes produce such compounds. In the case of insects, isoxazolin-5-one and 3-NPA derivatives were found in leaf beetles of the subtribe Chrysomelina. A number of these natural products have been synthesized so far. In the case of the single compound 3-NPA, several synthetic strategies have been reported and some of the most efficient routes are reviewed. The toxicity of 3-NPA results from its ability to bind covalently to the catalytic center of succinate dehydrogenase causing irreversible inhibition of mitochondrial respiration. As a motif that is produced by many species of plants, leaf beetles and fungi, different detoxification mechanisms for 3-NPA have evolved in different species. These mechanisms are based on amide formation of 3-NPA with amino acids, reduction to β-alanine, ester formation or oxidation to malonic acid semialdehyde. The biosynthetic pathways of 3-NPA and isoxazolin-5-one moieties have been studied in fungi, plants and leaf beetles. In the case of fungi, 3-NPA derives from aspartate, while leaf beetles use essential amino acids such as valine as ultimate precursors. In the case of plants, it is supposed that malonate serves as a precursor of 3-NPA, as indicated by feeding of 14C-labeled precursors to Indigofera spicata. In other leguminous plants it is suggested that asparagine is incorporated into compounds that derive from isoxazolin-5-one, which was indicated by 14C-labeled compounds as well. In the case of leaf beetles it was demonstrated that detection of radioactivity after 14C-labeling from a few precursors is not sufficient to unravel biosynthetic pathways.
 Tobias Becker | Tobias Becker studied chemistry at the University of Bremen and received his Bachelor degree in 2010. Then he moved to the Friedrich Schiller University in Jena and received his Master degree in 2012. After finishing his studies Tobias joined the group of Prof. Wilhelm Boland at the Max Planck Institute for Chemical Ecology to start as a PhD student in the field of leaf beetle defensive chemistry. Tobias defended his PhD thesis in December 2016 and is currently working in the Department of Bioorganic Chemistry to study further aspects of the biosynthesis of defensive compounds in leaf beetles. |
 Jacques Pasteels | Jacques M. Pasteels studied Biology in the Université libre de Bruxelles (ULB), received his PhD in 1966, and was promoted to Assistant Professor in 1967. In 1968–1969 he went to the USA as Assistant Professor in Chico State, California, and postdoc in Athens Georgia, in the laboratory of Prof. M. S. Blum. Back in the ULB, he became full Professor in 1979 for Animal Behavior and Entomology until becoming Emeritus in 2004. He is member of the Belgian Royal Academy of Science. He was President of the International Society of Chemical Ecology in 1991, and received from this society the Silver Medal Award for his pioneering and outstanding contribution to the field. |
 Kerstin Voigt | Kerstin Voigt is the head of the Jena Microbial Resource Collection (JMRC), a joint strain collection of the University and Hans Knöll Institute (formerly: IMET) collections maintaining about 50 |
 Wilhelm Boland | Wilhelm Boland studied Chemistry and received his PhD from the University of Cologne in 1978. He was a Research Associate at the Institute of Biochemistry (University of Cologne) until 1987 and moved then to the University of Karlsruhe as a Professor for Organic Chemistry. In 1994 he became Full Professor for Bioorganic Chemistry at the University of Bonn. In 1997 he joined the Max Planck Society and became Director of the Department of Bioorganic Chemistry in the Max Planck Institute for Chemical Ecology in Jena. Professor Boland was the 1st Simeone-Silverstein lecturer (1995) of the International Society of Chemical Ecology and president of the society from 2008 to 2009. |
1. Introduction
Isoxazolin-5-one and 3-nitropropanoic acid (or 3-nitropropionic acid, 3-NPA) moieties occur in a number of natural products, and all share a remarkable structural diversity. In principle, three isomeric forms of the five-membered fully unsaturated isoxazol-derived heterocycles exist. These are the 2- and 3-isoxazolin-5-one (isoxazol-5(4H)-one and isoxazol-5(2H)-one, respectively) as well as the isoxazol-5-ol isomers (Fig. 1).1,2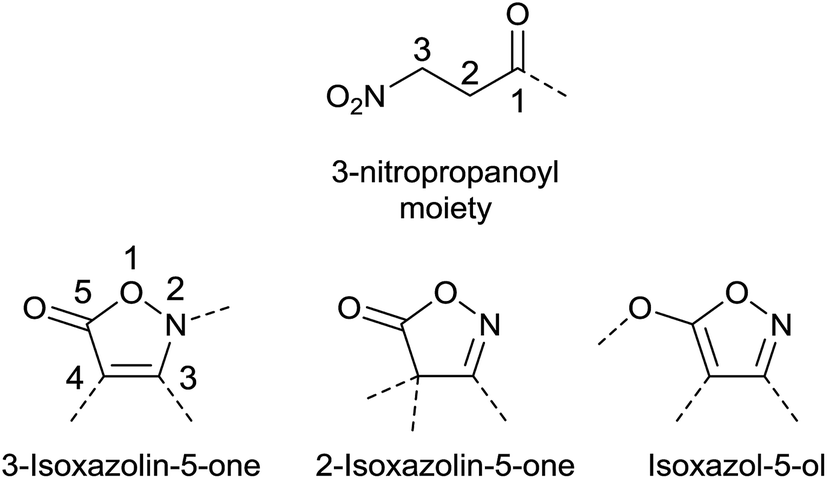 | ||
| Fig. 1 Structural motifs of isoxazolin-5-one and -5-ol isomers as well as 3-nitropropanoic acid. | ||
Both structural motifs are similar due the inherent O–C–C–C–N–O sequence, and mainly differ in the oxidation state of the nitrogen atom and the open-chain vs. ring structure.
In biological sources, a number of natural compounds derived from isoxazolin-5-one have so far been identified in several kingdoms. Such compounds occur across the plant family of Fabaceae (Leguminosae, Papilionaceae), which contains roughly 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 species within 751 genera.3 Many of these plants, e.g. Lathyrus sativus or Pisum sativum, are important food sources for livestock as well as humans. Furthermore, isoxazolin-5-ones are found in many species of leaf beetles (Chrysomelidae), belonging to the subtribe Chrysomelina. At the same time 3-NPA derived compounds occur in a number of leguminous plant and leaf beetle species. Pure 3-NPA is a toxin, causing neurodegeneration with symptoms and pathology very similar to those found in patients with Huntington’s disease.4 In some plants and beetles, 3-NPA and isoxazolin-5-one moieties are both produced by one organism. In addition, some studies revealed a fundamental relationship in the biosyntheses of the isoxazolinone heterocycle and 3-NPA in leaf beetles.5,6 Whether in plants such relationships occur, producing both moieties, is not yet known. Since the prolonged consumption of seeds of some species of the genus Lathyrus (Leguminosae), e.g. L. sativus, causes the neuronal disease of lathyrism, many of the secondary metabolites in these plants, those deriving from isoxazolin-5-one and from 3-NPA, have been characterized in terms of neurotoxicity. These studies revealed different relationships among natural products derived from isoxazolin-5-one and 3-NPA to the disease of lathyrism, either via direct toxicity or upon the involvement of these natural products in the biosynthesis of other toxins, especially the neurotoxic β-N-oxalylamino-L-alanine (β-ODAP). Hence, attempts to genetically engineer such plants to reduce the economic damage caused by these metabolites are promising.
500 species within 751 genera.3 Many of these plants, e.g. Lathyrus sativus or Pisum sativum, are important food sources for livestock as well as humans. Furthermore, isoxazolin-5-ones are found in many species of leaf beetles (Chrysomelidae), belonging to the subtribe Chrysomelina. At the same time 3-NPA derived compounds occur in a number of leguminous plant and leaf beetle species. Pure 3-NPA is a toxin, causing neurodegeneration with symptoms and pathology very similar to those found in patients with Huntington’s disease.4 In some plants and beetles, 3-NPA and isoxazolin-5-one moieties are both produced by one organism. In addition, some studies revealed a fundamental relationship in the biosyntheses of the isoxazolinone heterocycle and 3-NPA in leaf beetles.5,6 Whether in plants such relationships occur, producing both moieties, is not yet known. Since the prolonged consumption of seeds of some species of the genus Lathyrus (Leguminosae), e.g. L. sativus, causes the neuronal disease of lathyrism, many of the secondary metabolites in these plants, those deriving from isoxazolin-5-one and from 3-NPA, have been characterized in terms of neurotoxicity. These studies revealed different relationships among natural products derived from isoxazolin-5-one and 3-NPA to the disease of lathyrism, either via direct toxicity or upon the involvement of these natural products in the biosynthesis of other toxins, especially the neurotoxic β-N-oxalylamino-L-alanine (β-ODAP). Hence, attempts to genetically engineer such plants to reduce the economic damage caused by these metabolites are promising.
Within the biological kingdoms of fungi and of bacteria, natural products derived from 3-NPA and isoxazolin-5-one have also been detected. These findings are of special interest in terms of the health of human food, which is often contaminated by molds; these molds may produce the neurotoxin 3-NPA. Fermentation processes of food can also induce the production of 3-NPA by encouraging the time-dependent growth of corresponding fungi.7 In marine organisms, benzisoxazolinones with interesting antibiotic8 or antifouling9 properties have been detected. Similar bioactivities were also observed in terrestrial, plant-produced isoxazolin-5-one derivatives. The motif of small nitro-acids, such as 3-NPA, was found to exhibit some activity against tuberculosis-inducing bacteria.10
2. Occurrence and structural diversity
2.1. 3-NPA and derived compounds
3-Nitropropanoic acid (3-NPA) 1 was first isolated by Gorter11 in 1920 after acidic hydrolysis of hiptagin 2, a glucoside from the plant Hiptage madablota (Fig. 2).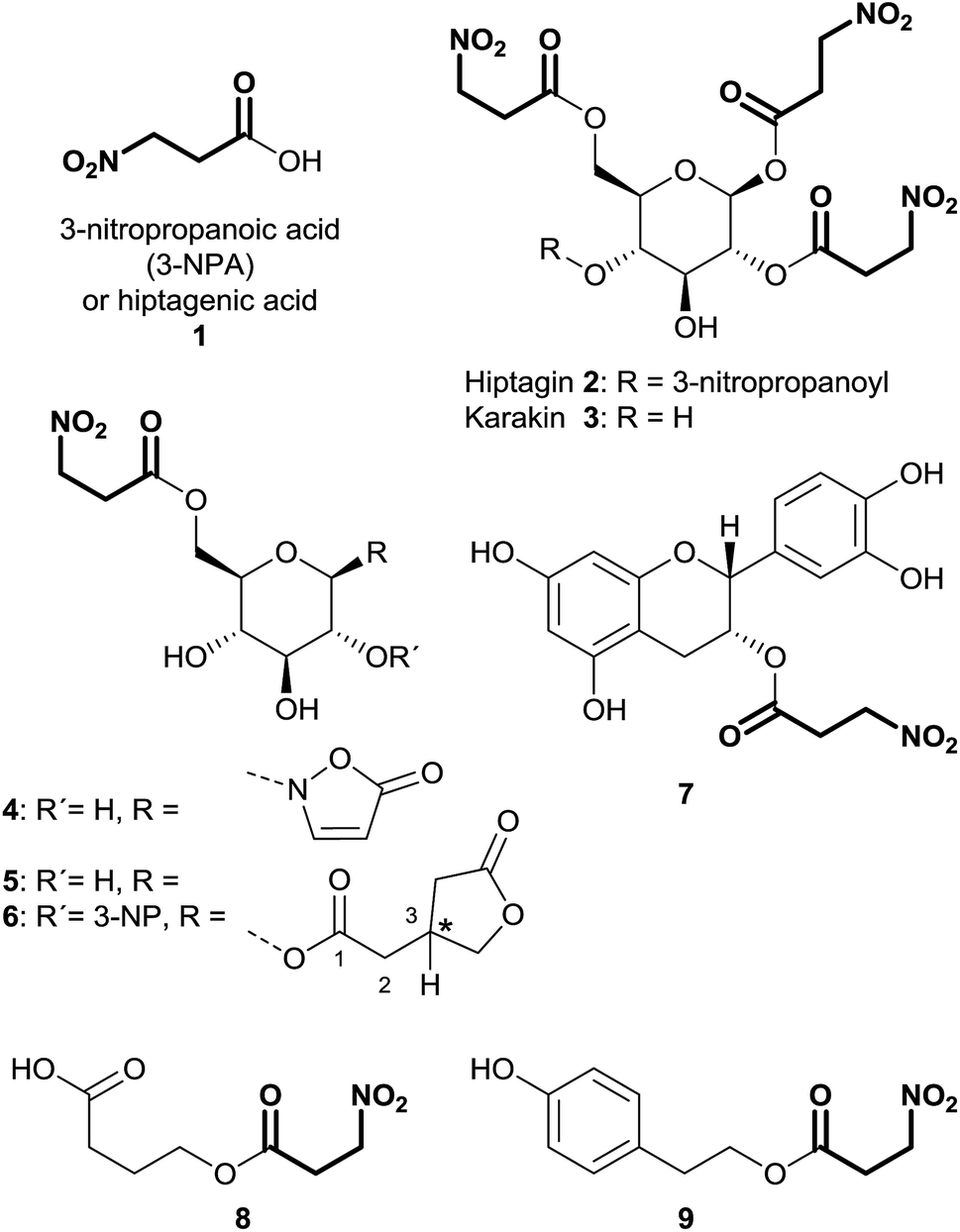 | ||
| Fig. 2 3-NPA and derived compounds from diverse organisms; * = configuration not yet determined. | ||
Since the molecular structure of this compound was not identified, 3-NPA was designated with the trivial name “hiptagenic acid”. Later, Carrie12 isolated a substance showing an identical mixed melting point to hiptagenic acid, after applying the same hydrolytic protocol to karakin 3 (Fig. 2), which is a glucoside occurring in Corynocarpus laevigata fruits. The identity of 3-NPA as hiptagenic acid was finally shown by Carter and McChesney13 as reviewed earlier by Raistrick and Stössl.14 Since these first discoveries of 3-NPA derivatives, an increasing number of species from different kingdoms have been identified as producers of natural compounds deriving from 3-NPA. In plants, 3-NPA and its compounds have been detected in the families of Fabaceae (Leguminosae or Papilionaceae), Malpighiaceae, Corynocarpaceae, and Violaceae.15 More than 500 species of leguminous plants belonging to at least seven genera, as shown in the cases of Astragalus, Coronilla, Hippocrepis, Indigofera, Lotus, Securigera and Scorpiurus, produce such compounds.16 In particular, the amount of 3-NPA per fresh weight is higher than 100 nmol mg−1, which is equivalent to more than 7% of the soluble fixed nitrogen in the shoots of some of these plants.16,17 The chemical diversity of natural products containing 3-NPA in plants that have been identified so far is represented by the free acid itself16,18,19 and O-acyl compounds of glucose and glucosides,11,19–30e.g. hiptagin 2,11,31 karakin 3,12,22 compound 4 (ref. 29 and 30) and epimers of 5 and 6,20 as well as from (−)-epicatechin32 (compound 7) and 4-hydroxybutanoic acid 8 (ref. 33) (Fig. 2). For an overview of the occurrence of glucosides derived from 3-NPA see R. Parry et al.34 It is important to note that the structures of compounds 5 and 6 were originally assigned as 1-O-acyl glucosides20 as shown in Fig. 2, while aliphatic O-glucosides were shown for these compounds in the corresponding reviews.34,35 In the case of ref. 35 no stereochemistry was addressed at all, and thus the structure of hiptagin 2 was not assigned correctly in this review either. The absolute stereochemistry of the epimers of 5 and 6 was not assigned, although there was no indication of diastereoisomers in these isolates.20 The aglycon, isolated from Astragalus canadensis samples, 3-hydroxymethyl glutaric acid, was optically inactive.36 A racemization of this free acid can proceed via hydrolysis, followed by re-lactonization or via direct intramolecular attack of the free carboxyl function (Fig. 3).
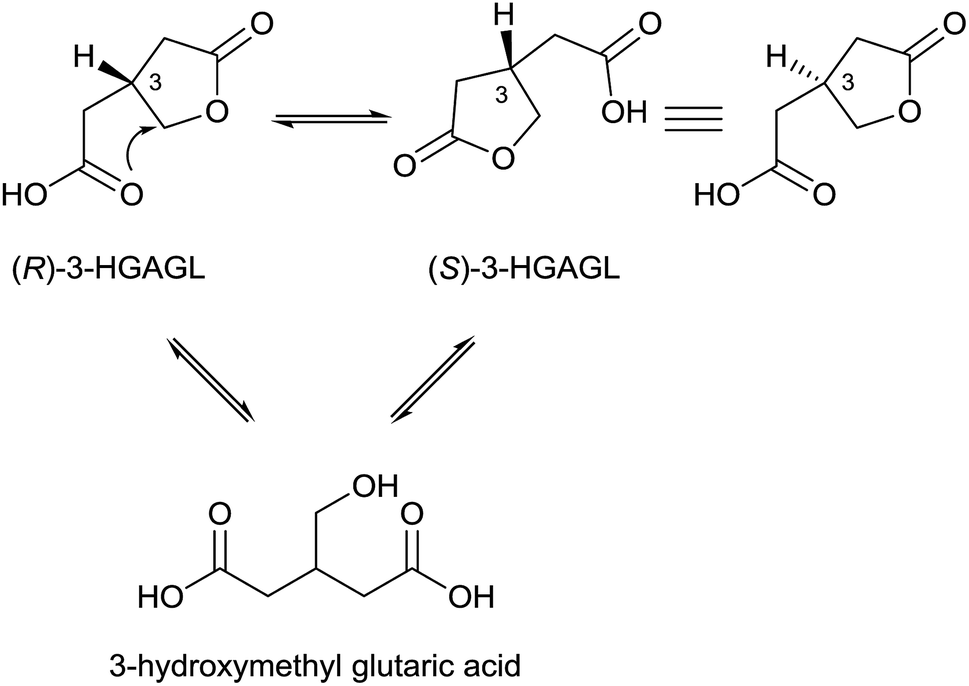 | ||
| Fig. 3 Suggested equilibria between (R)- and (S)-3-hydroxymethyl glutaric acid γ-lactone (3-HGAGL) in solution. | ||
Hence, even though the aglycones ((R)- and (S)-3-HGAGL) occur as racemates, selective O-acylation of a glucosyl donor with an activated form of one of the possible enantiomers of 3-hydroxymethyl glutaric acid γ-lactone can, via enzymatic catalysis, lead to pure diastereomers of compounds 5 and 6 at the 3-position of the lactone, as reported by Benn et al.20
In fungal species, free 3-NPA has been identified in Aspergillus, Arthrinium, Diaporthe and Penicillium genera.15,37–39 Furthermore, the ester compound 4-hydroxyphenethyl 3-nitropropanoate 8 was isolated from endophytic Phomopsis sp. PSU-D15 (Fig. 2).40
3-NPA derived compounds were also detected in leaf beetle species (Coleoptera: Chrysomelidae), belonging to the subtribe Chrysomelina (Chrysomelini), e.g. Phaedon cochleariae and Chrysomela populi (Fig. 2).41–46 In these organisms, compound 4 is present in all life stages of the insect, occurring as a major secondary metabolite in the eggs, the larval hemolymph, the pupae and the adult hemolymph and their defensive secretions.6,41–45,47–51 Additionally, in adult secretions of the four leaf beetle species Gastrophysa atrocyanea, Plagiodera versicolora distincta, Chrysomela vigintipunctata costella, and Gastrolina depressa, belonging to the subtribe Chrysomelina, diverse esters of glucose containing 3-NPA and isoxazolin-5-one motifs were indicated by TLC and their structures were determined by HPLC, MS and NMR analyses.46 These findings describe structures without the isoxazolin-5-one heterocycle, bearing up to three 3-NPA moieties in different positions on the sugar.46
In the eggs, concentrations around 10−2 M (M = mol L−1) of compound 4 were reported for Gastrophysa viridula, Chrysomela tremulae and C. populi.44 The same concentration ranges were determined for this compound in the larval hemolymph,41 and show increasing tendencies with respect to the larval body weight.5,41
To gain a comprehensive picture of the occurrence of 3-NPA and its derivatives in natural samples, it is of interest whether 3-NPA can be found only in its unaltered free form, or if it is found as a derivative, e.g. an ester as shown in many examples in Fig. 2. In leaf beetles, the content of free 3-NPA in the hemolymph was detected only in low amounts that were probably caused by the immediate hydrolysis of ester compounds, e.g.4, in fresh natural samples. In adult secretions, the free toxin was detected, probably occurring due to hydrolysis after transport of the corresponding glucosides from the hemolymph. In contrast, studies concerning the formation 3-NPA in microbiota often describe the production of the free toxin. In plants, as shown above, mainly the free acid and the O-acyl compounds of 3-NPA and glucosides or other alcohols have been described. Nevertheless, in many studies it remains unclear whether 3-NPA occurs as a derivative, the free toxin or in both forms.
In order to detect free 3-NPA, several screening methods have been established, mainly to analyze toxic contaminations (such as 3-NPA) in food sources, caused by microbiota.52–55 In MS analyses, nitro alkanes exhibit characteristic ions, e.g. NO2− (m/z 46), allowing the application of precursor ion scans and other established MS techniques.56,57 These analyses proceed under mild ionization conditions, such as atmospheric pressure chemical ionization (APCI), and can be coupled with previous LC separation, which is necessary for the detection of most of the 3-NPA derivatives from natural samples that have been described so far. In addition to their specific behavior in mass spectrometry, 3-NPA derivatives show characteristic NMR signals. The two methylene groups in the 3-NPA moiety show solvent dependent diagnostic signals around δH 3.02 (C-2-H2, triplet, 3J2,3 = 5.8 Hz), δC 31.7 (C-2) ppm and δH 4.71 (C-3-H2, triplet, 3J2,3 = 5.8 Hz), δC 70.7 (C-3) ppm in the case of ester 4 in D2O.41,45,46,58
2.2. Isoxazolin-5-one derivatives
Natural products of the described class mainly occur as derivatives of 3-isoxazolin-5-one, rather than the 2-isoxazolin-5-one or the isoxazolol isomers. So far, 3-isoxazolin-5-one derivatives have been identified in plants,59–73 leaf beetles (Chrysomelina),6,41–51 bacteria of the genus Streptomycetes74–76 as well as the cold-water sponge Geodia barretti,9 the fungus Fusarium larvarum77 and the marine bacterium Pseudoalteromonas flavipulchra JG1.8 The structural diversity of naturally occurring isoxazolin-5-one compounds varies from derivatives of amino acids (TAN-950A 10, BIA 11 and ACI 12),59,74–76 peptides (γ-glu-AEI 13 and γ-glu-BIA 14),59,61,62,64,78 ethylamine (AEI 15),79 carboxylates (CMI 16 and 17)59,70,79 and cyanoethane (BIP or CEI 18)62,68,71 to glucosides, e.g. compounds 4,6,29,30,41,42,44–5119 (ref. 41, 44, 45 and 67) and 20 (ref. 65 and 69) (Fig. 4). Furthermore, benz-3-isoxazolin-5-ones such as parnafungin B1 21,77 bromobenzisoxazolone barettin 22 (ref. 9) and N-hydroxybenzoisoxazolone 23 (ref. 8) have been isolated. Common abbreviations and names used for the mentioned compounds in the cited literature are presented in Fig. 4.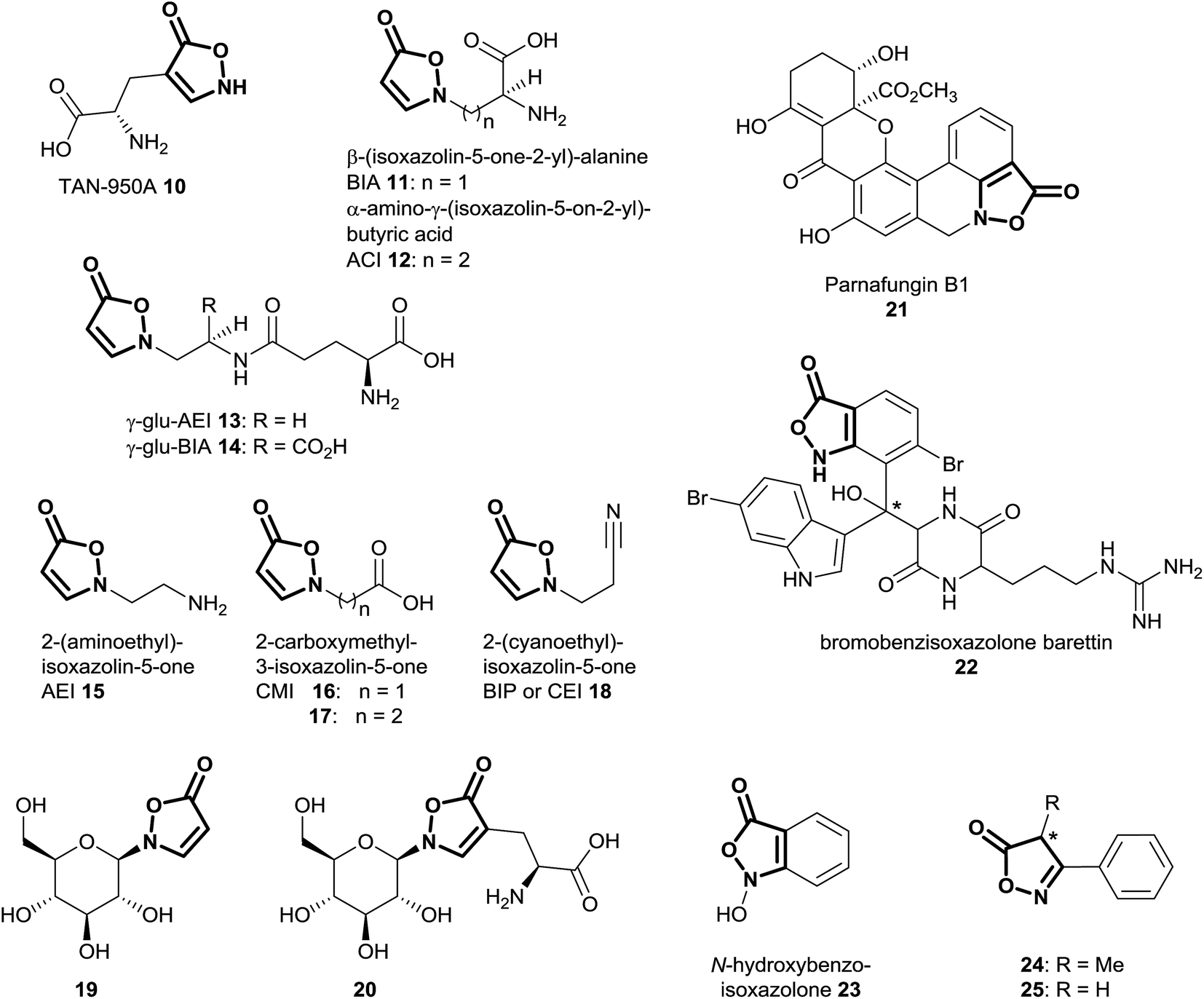 | ||
| Fig. 4 Structures of naturally occurring isoxazolin-5-one derivatives with highlighted isoxazolin-5-one structure; * = stereochemistry is under investigation; compounds 24 and 25 were indicated only by GC-MS (see text). | ||
In Lathyrus odoratus (Leguminosae), it was shown that single 3-isoxazolin-5-one derivatives 11–13, 15 and 18–20 occur in contents of up to >1% of the dry weight. Compound 19 is produced in amounts of up to 2.64 nmol mg−1 fresh weight in 7 day-old seedlings of L. odoratus.65 In general, as expected, the amounts of these compounds show spatial and temporal variations in the plant.65
In leaf beetles, similar trends were observed, as compounds 4 and 19 occur in amounts of 2 to 12 nmol mg−1 body weight in Phaedon cochleariae.5,41 Analogously, these compounds were detected in millimolar concentrations in the eggs of these insects.44
Within compound 4, both of the discussed moieties, 3-NPA and 3-isoxazolin-5-one, occur in the same molecule. This substance has been identified as a major component of secretions and hemolymph of leaf beetles,6,41,42,44,45,47,50,51 as well as of two species of Astragalus29,30 plants (Fabaceae). Furthermore, various plants from the Fabaceae family produce 3-isoxazolin-5-one and 3-NPA derivatives. Thus, it is of interest to know in which other species, especially in the legume family, both moieties can be detected at the same time. To study the occurrence of 3-isoxazolin-5-one derivatives, UV-absorption spectra were measured.59,80 These analyses revealed strong absorption of the 2-substituted derivatives of the heterocycle around 260 nm,59,80 with extinction coefficients above 104 L mol−1 cm−1.58,73 These properties indicate the occurrence of putative 3-isoxazolin-5-one compounds in natural samples, as shown by capillary zone electrophoresis62 or HPLC-UV81-based analyses.
If the isolated amounts or concentrations of the compounds in natural samples are sufficient, additional NMR data can be acquired.41 Typical NMR signals of 3-isoxazolin-5-one derivatives occur around δH 5.52 (C-4-H, doublet, 3J3,4 = 3.7 Hz), δC 91.7 (C-4) ppm and δH 8.47 (C-3-H, doublet, 3J3,4 = 3.7 Hz), δC 155.0 (C-3) ppm as well as δC 174.7 (C-5) ppm, in 2-substituted, 3,4-unsubstituted compound 4 in D2O.41,45,46,58 The benzisoxazolinone compound 22 shows carbon shifts at δC 170.3 (C-5) , 137.7 (C-3) and 138.7 (C-4) ppm in d6-DMSO.9 In compound 21, typical carbon shifts occurred at δC 167.3 (C-5), 155.6 (C-3) and 109.7 (C-4) ppm (d6-DMSO).77
There is some indication from GC-MS analyses of methanolic extracts of species belonging to the endophytic fungal genus Xylaria that 3-phenyl-4-methyl-2-isoxazolin-5-one 24 (Fig. 4) may be a component of the secondary metabolites.82 For this substance, no isolation and structure elucidation was carried out, and thus the absolute stereochemistry of the putative structure is unknown.82 The presence of the 2-isoxazolin-5-one derivative was indicated only by comparison with the NIST compound library (95% identity in the EI-mass spectra).82 Analogously, indications for the occurrence of 3-phenyl-2-isoxazolin-5-one 25 in the Chinese traditional medicine prescription “Xiaoxuming” decoction (aqueous boiling extraction of herbs) were obtained by similar GC-MS-based analyses.83 Since these compounds are used particularly as synthetic intermediates,84 the occurrence of such 2-isoxazolin-5-one derivatives in Xylaria and related species should be investigated by additional analytical techniques, e.g. HPLC-(HR)MS as well as NMR.
In analyses applying GC-MS methods using hydroxylamine- and trimethylsilyl (TMS)-derivatization of human urine samples, acetoacetic acid has been observed to form TMS derivatives of 3-methyl-isoxazol-5-ol and 3-methyl-3-isoxazolin-5-one.85 Hence, these compounds were identified as artefacts, formed by the application of hydroxylamine to the natural samples.85 In a further publication, a similar observation has been described including the isolation and characterization by 13C NMR of the untreated 3-methyl-2-isoxazolin-5-one after reaction of hydroxylamine and acetoacetic acid.86
3. Biosynthesis of 3-isoxazolin-5-one and 3-NPA derivatives
3.1. Microbiota
Early publications on the biosynthesis of 3-NPA from fungal origin describe the incorporation of aspartate into 3-nitropropanoic acid.87 By applying radioactive [4-14C]-DL-aspartic acid to Penicillium atrovenetum, it was demonstrated that 2% is incorporated, showing 96% of the labeling at the C-1 position in 3-NPA 1.87 Later, it was observed that the L-isomer of aspartic acid is the preferred precursor.88 Hence, it could be demonstrated that other compounds associated with the tricarboxylic acid cycle, e.g. pyruvate and acetate, are also precursors of 3-NPA in this fungus.89 In contrast to the findings in leaf beetles, β-alanine failed to be incorporated into compound 1 after the application of radioactive precursors.87 Additional experiments indicated that aspartate serves as a nitrogen source for the biosynthesis of 3-NPA in P. atrovenetum.88 Using [2-13C,15N]-DL-aspartic acid, Baxter et al. showed by NMR analyses that the C–N bond in aspartate is conserved upon 3-NPA biosynthesis in P. atrovenetum.90 Further experiments on the biosynthesis of 3-NPA in this fungus resulted in the confirmation of L-nitrosuccinate 26 as an intermediate within this metabolic pathway, which indicates that the pathway proceeds via direct oxidation of the nitrogen atom in the precursor L-aspartic acid (Fig. 5).37,91 This oxidation is probably a stepwise process, where the consumption of dioxygen produces N-hydroxy intermediates, as indicated by the application of 18O2 to the fungus.37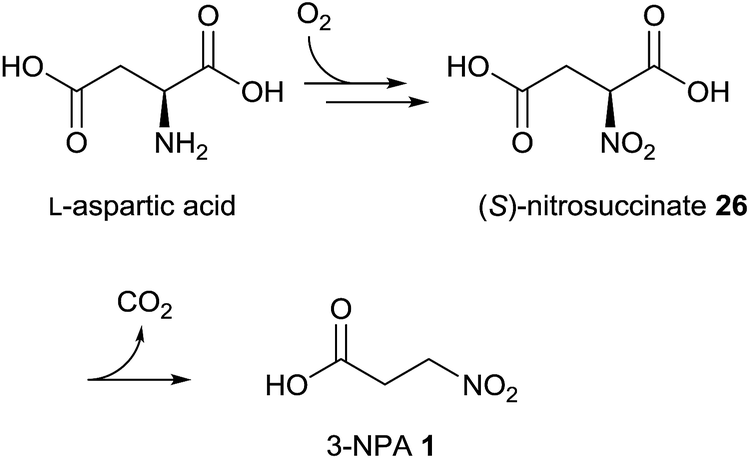 | ||
| Fig. 5 Biosynthetic pathway of 3-NPA in P. atrovenetum.37 | ||
Furthermore, it was indicated that compound 26 decarboxylates spontaneously under physiological conditions and hence an enzymatically catalyzed transformation to 3-NPA is probably not required.92
In vitro studies with enzymes from Streptomyces and other bacteria also showed that 3-NPA is formed via oxidation of L-aspartate.93 In this case the reaction is catalyzed by a flavin-dependent oxidoreductase FzmM, which is involved in the biosynthesis of fosfazinomycin.93 Thus, 3-NPA occurs as a putative intermediate within the biosynthesis of this natural product in bacteria.
3.2. Leaf beetles
First experiments on the biosynthesis of isoxazolin-5-one and 3-NPA derivatives in the case of leaf beetles were carried out with adults of the species Chrysomela tremulae (Chrysomelina).6,51 For this purpose, [14C4]-aspartate covered leaves were presented to adults of C. tremulae for one week.6 After this time, radioactivity was measured in 3-isoxazolin-5-one glucosides 4 and 19, which are the major components of the defensive adult secretions. In the 3-NPA ester 4, a higher specific activity was determined and after acidic hydrolysis, the resulting free glucose showed negligible radioactivity. As a consequence, it was concluded that both moieties, 3-NPA as well as the 3-isoxazolin-5-one heterocycle, derive from aspartate as a precursor, analogous to the biosynthesis of 3-NPA in fungi. Since the natural products 4 and 19 were not detected in the host plants of the insects, these results further indicated that these secondary metabolites are de novo produced by the insects. No information about the intactness of the incorporation or corresponding intermediates in the pathway was provided in this study. Furthermore, low relative incorporations of aspartate into compounds 4 and 19 of around 0.016% were determined. These adult defensive compounds were also detected in high amounts (mM concentrations) in the larval41 and adult hemolymph as well as the pupae. Consequently, the feeding experiments were repeated and expanded using leaf beetle larvae, which are easier to rear and treat, by applying stable isotope labeled compounds.5 These studies revealed an incorporation of randomly re-assembled fragments of aspartate into compounds 4 and 19. In contrast, the intact incorporation of the essential amino acid valine was observed. Further investigations revealed that the biosynthetic pathway proceeds via propanoic acid (in the propanoyl-CoA form) as well as β-alanine. This pathway has been described previously,94 proceeding via oxidation of propanoyl-CoA to form acrylyl-CoA,94,95 followed by addition of ammonia or further oxidation to malonate semialdehyde, which can be transaminated.Whether only one or both of these pathways occur in leaf beetles has not yet been investigated. Nevertheless, the last two reactions (addition of ammonia as well as transamination) could provide β-Ala. The latter compound is then step-wise oxidized at the nitrogen atom to form compound 27 and the corresponding oxime 29 (E/Z) or 3-NPA 1, probably via the dihydroxylated intermediate 28. Intermediates 27 and 29, as well as 3-NPA, were synthesized as [13C,15N]-labeled compounds and applied in vivo to the beetles, analogous to commercial multiple stable-isotope-labeled aspartate, valine, propanoic acid and β-alanine. The oxime 29 (or the respective CoA-ester) can be cyclized to produce the unsubstituted 3-isoxazolin-5-one 30 (and its tautomers), which was shown to be transformed into compound 19 by reaction with α-UDP-glucose in 1H NMR assays. As a last step, 3-NPA is activated under consumption of ATP to form the corresponding S-CoA ester, which is transesterified to form the nitroester 4 (Fig. 6).5
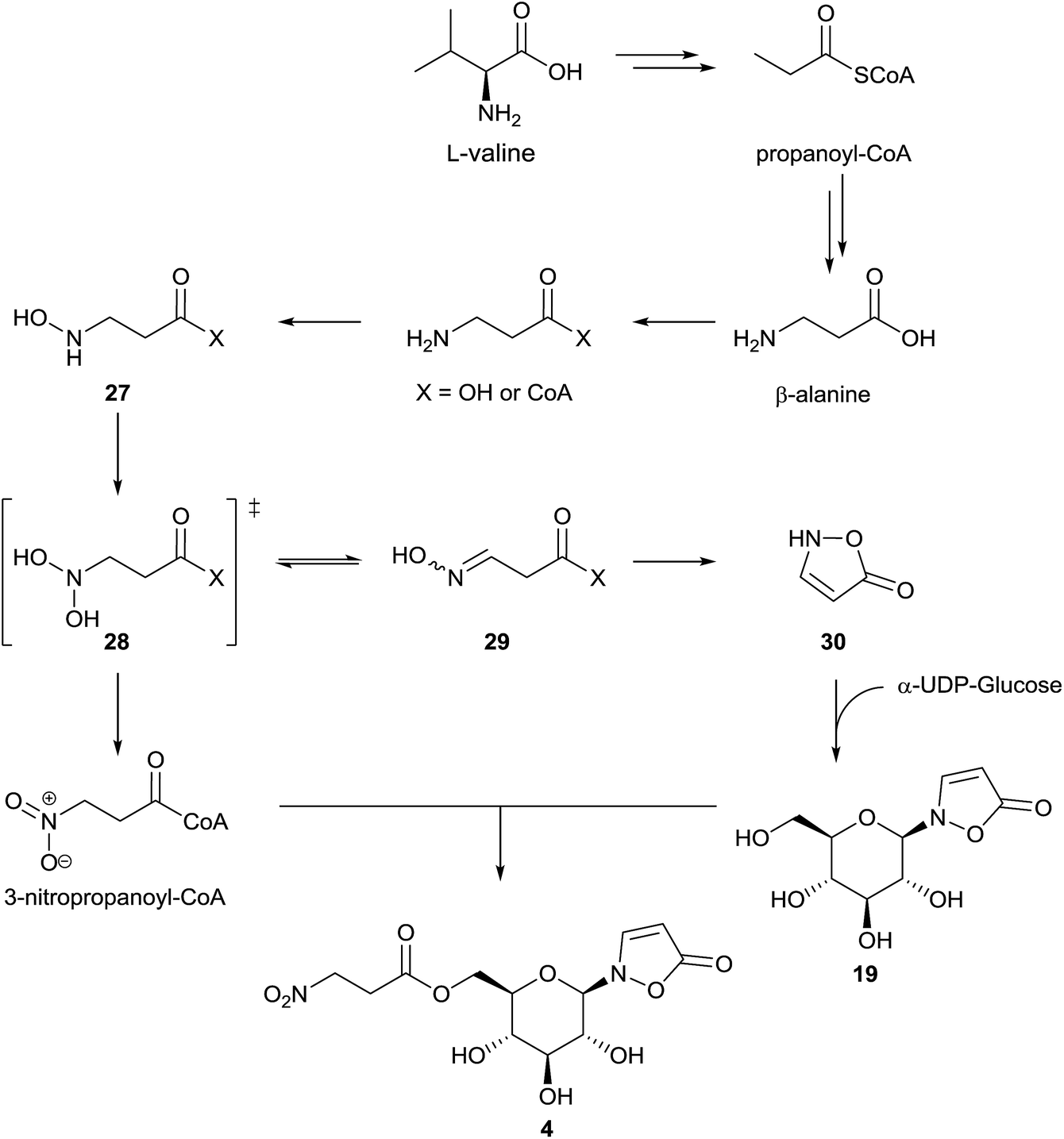 | ||
| Fig. 6 Proposed biosynthetic pathway of compounds 4 and 19 in Chrysomelina. | ||
In agreement with the previous results,6 the relative incorporation of aspartate was determined to be 0.019%,5 while the incorporation of valine was 8.1 fold higher. In the case of β-alanine and propanoic acid, the relative incorporations were 17.4 ± 11.2% and 2.3 ± 1.5%, respectively.5 These results5,6 reveal the fundamental relationship of the isoxazolin-5-one biosynthesis to the biosynthesis of 3-NPA in leaf beetles. In some species, the concentration of the 3-NPA moiety increases with the larval body weight, while the concentration of the glucoside 19 is more or less constant.5,41 These observations indicate tight control of different enzymes involved in the β-alanine N-oxidation, allowing selective oxidation of the amino group to form either the oxime (by enzyme group I) or the nitro group (by enzyme group II).
Recently published results5 on the biosynthesis of compounds 4 and 19 in Chrysomelina relate to juvenile beetles. The extent to which the biosynthesis in the adult beetles equals the biosynthesis of these compounds in the larval stage has not yet been investigated; it is likely that identical pathways exist, given the identical genotype in all life stages of one organism.
3.3. Plants
To study the biosynthesis of 3-NPA in the leguminous plant Indigofera spicata (creeping indigo), several putative radioactive labeled precursors were applied.96 In these experiments, a relative incorporation of [2-14C]-malonate of between 0.017 and 0.047% was detected, while aspartate failed as a precursor, shown by the absence of radioactivity in 3-NPA after the plants were fed on 14C-labeled aspartic acid.96 Consequently, [2-14C]-malonyl-monohydroxamic acid 32 as a putative malonate-derived intermediate was synthesized and applied to the plant, resulting in relative incorporation rates of between 0.018 and 0.081%.96 Therefore, a biosynthetic pathway for the 3-NPA production in I. spicata via malonate, malonyl monoamide 31, N-hydroxy-β-alanine 27 and malonyl monohydroxamate 32 was suggested (Fig. 7, right pathway).96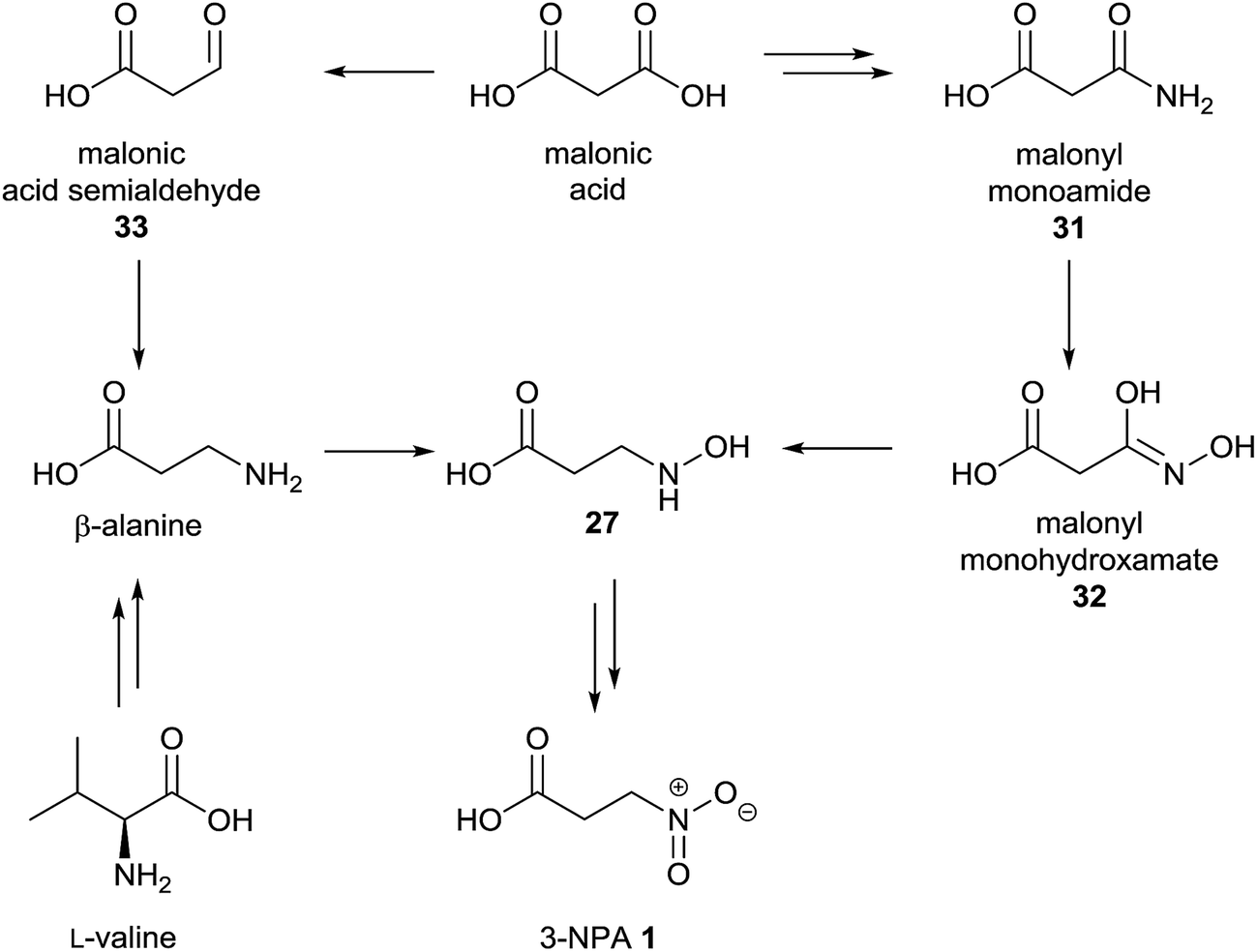 | ||
| Fig. 7 Proposed biosynthetic pathway of 3-NPA 1 in I. spicata, adapted from Candlish et al.96 (right) and alternative pathway (left). | ||
In addition to the described findings, the position of incorporation within the C3-unit of 3-NPA was determined with different results. These experiments indicate that incorporation occurs in the (expected) C-2 position as well as C-3, but not the C-1 position.96 A positive incorporation into positions other than C-2 indicate random fragmentation and the re-assembly of malonate due to diverse metabolic reactions within the plant, as exemplified in the biosynthesis of 3-NPA in leaf beetles via randomly re-assembled aspartate.5 In addition, the determined relative incorporations were rather small, being comparable to the (indirect) incorporation of aspartate into compounds 4 and 19 in leaf beetles.5,6 Thus, a direct biosynthetic pathway deriving from non-malonate-derived metabolites, e.g. valine and propanoate as well as β-alanine, may not be excluded from the presented results of this96 study. In bacterial97 as well as archaeal98 species, it was shown that malonate can be reduced to form malonate semialdehyde 33. As this substance is a precursor of β-alanine94 in diverse organisms, it is of interest whether the observed malonate incorporation is due to the subsequent formation of β-Ala via transamination of malonate semialdehyde 33 with e.g. glutamate (Fig. 7, left pathway).
Since aspartate was not a precursor of 3-NPA in creeping indigo, the involvement of symbiotic microbiota in the biosynthesis of this toxin from primary metabolites seems unlikely. Nevertheless, 3-NPA occurs in a range of different plant species that may show different metabolic pathways for 3-NPA biosynthesis, and thus some contribution of microorganisms in the 3-NPA production cannot be ruled out. Extracts of a number of endophytic fungi contained 3-NPA, indicating microbiota play a role in the total production of this compound.99
Investigations on the biosynthesis of the isoxazolin-5-one moiety of different natural products were carried out in some Lathyrus, Pisum, Leucaena (Leguminosae) and Citrullus (Cucurbitaceae) species (Fig. 8).
 | ||
| Fig. 8 Proposed biosynthetic pathways for the biosyntheses of isoxazolin-5-one derivatives 10 to 14, 19, 20 and 30 in different plant species, reconstructed from the cited literature. | ||
In vivo studies in L. odoratus plants using [14C4]-asparagine revealed the positive incorporation into compounds BIA 11, ACI 12, CEI 18 and 20.100 Photohydrolysis experiments indicated that the incorporation of [14C4]-asparagine occurs to different extents in the side-chain and the heterocycle.100 In L. sativus, it was demonstrated that the reaction of O-acetyl-L-serine (OAS) with isoxazolin-5-one 30 results in the formation of BIA 11.101 This reaction was supposed to be catalyzed by cysteine synthase isolated from the plant.102 Furthermore, it was shown that BIA 11 is a precursor in the biosynthesis of the plant toxins L-α,β-diaminopropanoic acid and β-N-oxalyl-L-α,β-diaminopropanoic acid as well as γ-glu-AEI 13 and γ-glu-BIA 14.64,103–105 Later, it was demonstrated that enzyme isolates of cysteine synthases from L. sativus as well as L. odoratus can also catalyze the reaction of isoxazolin-5-one and OAS to form TAN-950A 10, the 4-substituted isomer of BIA 11, which is a structural motif in the glucoside 20.106 The glucoside 19 is formed via the reaction of isoxazolin-5-one 30 and α-UDP-glucose. This transformation is catalyzed by enzyme extracts from L. odoratus as well as P. sativum.107 NMR experiments showed that this reaction also occurs in leaf beetles.5 After reaction of 19 with OAS, compound 20 is formed. This reaction is catalyzed by extracts from L. odoratus, P. sativum, Citrullus vulgaris and Leucaena leucocephala.108In vitro assays using enzyme extracts from L. odoratus indicated that the biosynthesis of ACI 12, the homologue of BIA 11, proceeds via isoxazolin-5-one as well as S-adenosyl-L-methionine as substrates.109,110
As described in previous chapters, the isoxazolin-5-one and 3-NPA moieties occur in Astragalus species in the same molecule, as found in compound 4. Whether there is a relationship between the metabolic pathways of both moieties, similar to the biosynthesis of these motifs in leaf beetles, is not yet known.
Additional analyses may shed light on related topics such as the incorporation of applied precursors into the isoxazolin-5-one and 3-NPA motifs of the natural products. Furthermore, the genetic, transcriptomic and proteomic basics of the 3-NPA biosynthesis should be addressed in important food plants such as L. sativus.111–113
4. Chemical synthesis and properties of 3-NPA and 3-isoxazolin-5-one derivatives
4.1. Syntheses of 3-NPA
An almost quantitative method to realize the synthesis of 3-NPA 1 was described based on the oxidative cleavage of the corresponding β-lactam 34 conducted by a dioxirane (Fig. 9a).114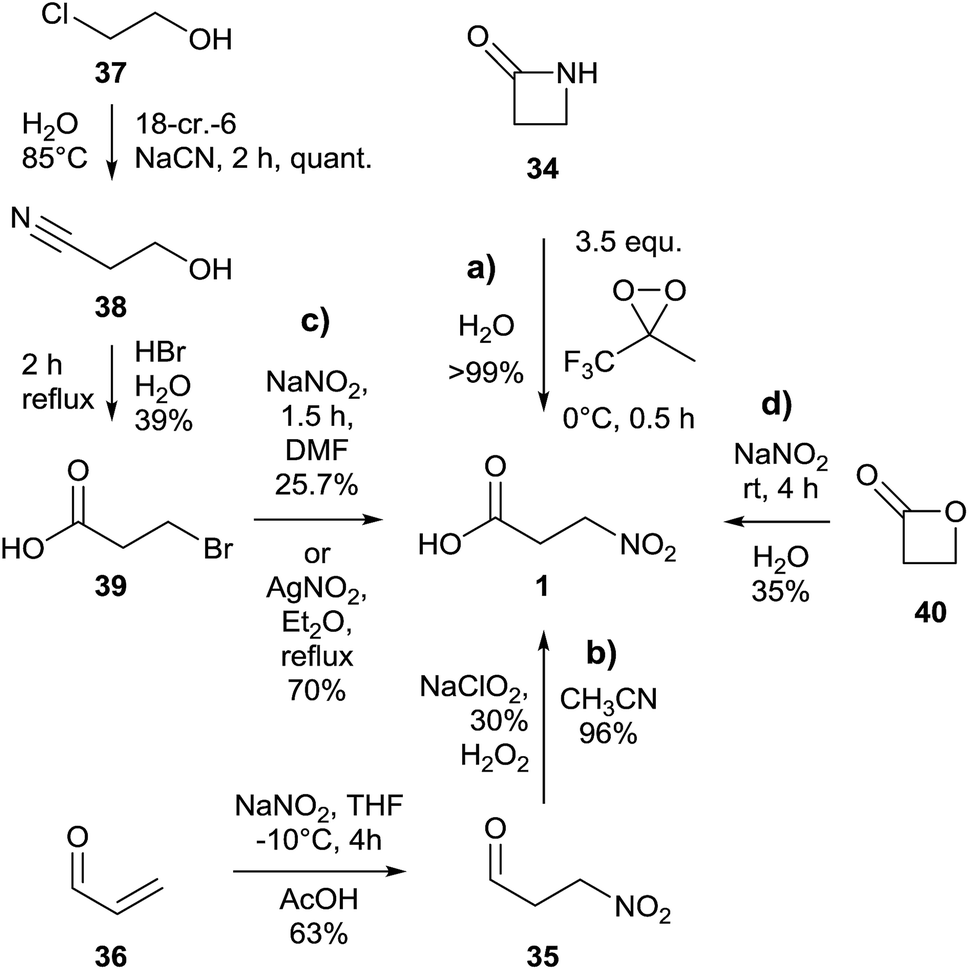 | ||
| Fig. 9 Synthetic procedures for the synthesis of 3-nitropropanoic acid. | ||
An alternative way to achieve the synthesis of 3-NPA 1 proceeds via oxidation of the analogous aldehyde 3-nitropropanal 35, starting from commercial acrolein 36 (Fig. 9b).115
In order to realize a tailored introduction of stable heavy isotopes into 3-NPA 1 to study biosynthetic aspects of this component, a three step synthesis in which 2-chloroethanol 37 was used as the starting material yielded intermediate 3-hydroxypropionitrile 38 as well as 3-bromopropanoic acid 39 (Fig. 9c).5,37,41 The yield of the last step of the latter method was further improved by using AgNO2 as a source for the nitro group instead of NaNO2.116 Previous attempts using 3-iodopropanoic acid and silver nitrite in water yielded only 14% of the product.117
Furthermore, the synthesis of 3-NPA 1 using β-propiolactone 40 and sodium nitrite in one step and giving a moderate yield was described (Fig. 9d).118
4.2. Syntheses of naturally occurring isoxazolin-5-one derivatives
Syntheses of N-substituted 3-isoxazolin-5-one derivatives were achieved using the sodium salt of the heterocycle 30.59,72,119,120 The latter compound was synthesized starting from ethyl propiolate 41via ethyl 3-(hydroxyimino)propanoate 42 (Fig. 10a).80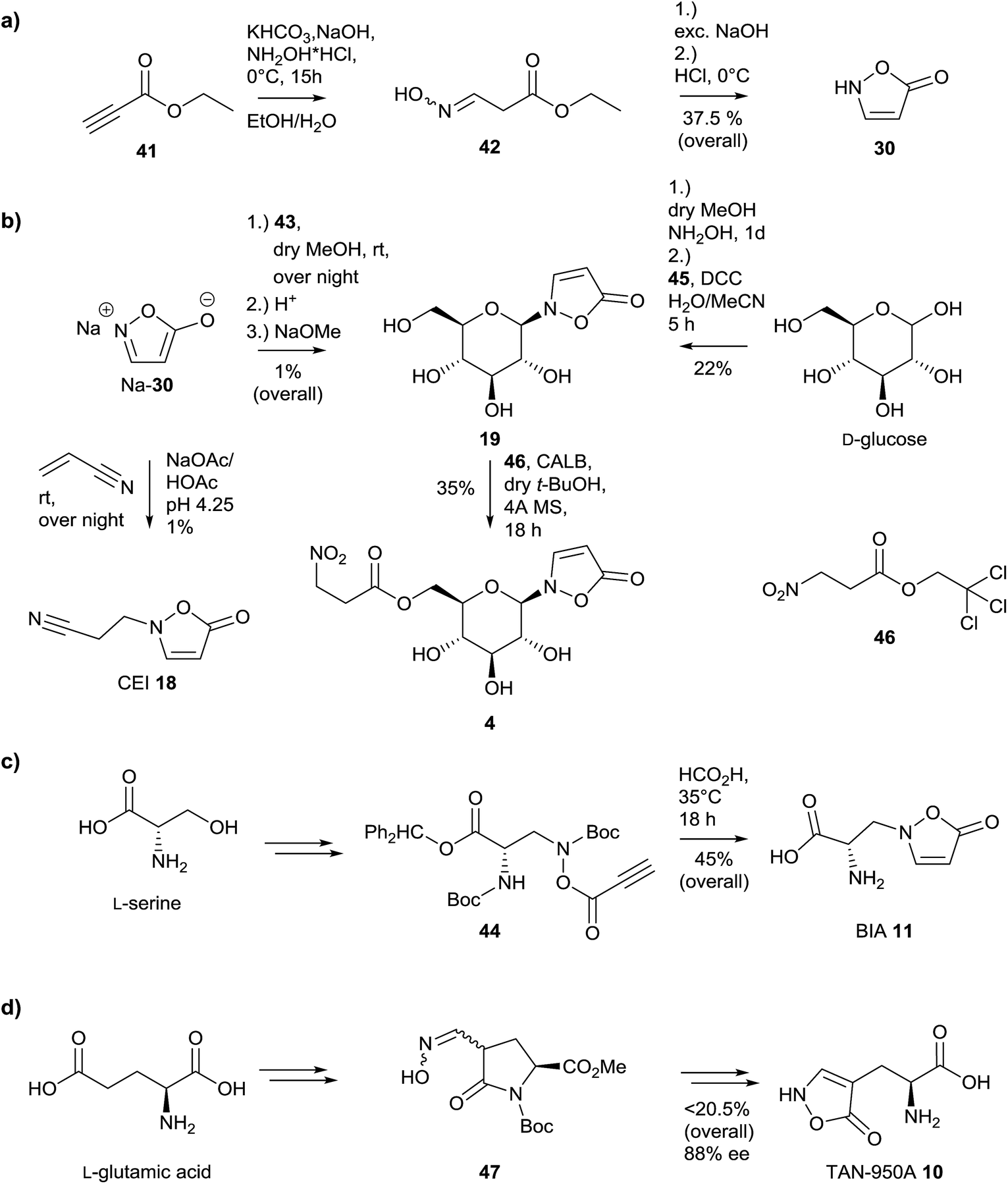 | ||
| Fig. 10 Synthetic strategies to provide naturally occurring 3-isoxazolin-5-one derivatives 4, 10, 11, 18, 19 and 30, adapted from the literature. | ||
Many attempts with alkyl bromides and chlorides failed or gave only low yields, as in the case of compound 19, which was synthesized via 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide 43 (Fig. 10b).59,119 This observation is in part due to the low selectivity of these reactions, which leads to the formation of 3-, 4- as well as O-substituted derivatives of 3-isoxazolin-5-one.119 A further attempt where O-acetylserine reacted with isoxazolin-5-one under catalysis by pyridoxal phosphate provided BIA 11 in a very low yield of 0.15%.121
Thus, an alternative method for the synthesis of 3,4-unsaturated 3-isoxazolin-5-one derivatives was developed by Baldwin and coworkers, based on a 5-endo-dig reaction.122,123 The construction of the heterocycle proceeds via O-acylation of an (N-boc-protected) N-hydroxy derivative with propynoic acid 45 to form compound 44, followed by an attack of the triple bond by the amino functional group, released upon formic acid-catalyzed deprotection (Fig. 10c).123 Upon application of this 5-endo-dig strategy, the naturally occurring compounds BIA 11 as well as the glucoside 19 could be synthesized (Fig. 10b and c).58,122–124 Compound 19 can be obtained directly from D-glucose in two steps and one pot.124 Upon transesterification using Candida antarctica lipase B (CALB) as well as an trichloro ester 46, compound 4 can be synthesized in moderate yields.58 This provides rapid access to 13C6-labeled 4 from [13C6]-19, which is easily accessible from commercial [13C6]-D-glucose. The yields of the obtained products are sufficient to provide the required amounts of authentic standards for qualitative and quantitative analyses.5,41,123 Using these standards, aspects of the biosynthesis of these natural products can be studied using synthetic isotopically labeled intermediates.5,104,123 Furthermore, quantifications can be simplified and are more reproducible if samples are spiked with defined amounts of stable-isotope-labeled internal standards (SIL-IS).5,125
A further synthesis was described for the construction of compound 10, starting from L-glutamic acid (Fig. 10d).76,126,127 The synthesis of TAN-950A proceeds over six steps from L-glutamic acid via oxime 47 in moderate yield and with a loss of 12% of the initial optical purity. The synthetic protocol has also been applied to D-glutamic acid resulting in an enantiomeric excess of 72%.
In the case of benzisoxazolinones, synthetic models for parnafungins were described.128,129 For further studies on the synthesis and properties of isoxazolones as well as -5-oles and related compounds see Beccalli130 and Sørensen131et al.
4.3. Properties of isoxazolin-5-one derivatives
Aqueous solutions of unsubstituted 3-isoxazolin-5-one 30 show rapid degradation, forming cyanoacetic acid as a main product.80 Under alkaline pH, compound 30 shows increased stability.80 In contrast, N-substituted 3-isoxazolin-5-one derivatives show rapid degradation in alkaline media upon hydrolysis and are stable under acidic conditions.59,124,132 The stability at low pH that was observed for many 2-substituted 3-isoxazolin-5-one derivatives, especially in the corresponding N-glucosides,124,132 was unexpected, due to the general instability of glycosides under acidic conditions. The stability of glucoside 19 persists even in the presence of β-glucosidase from almonds.124 In different solvents, the content of all three possible tautomers of compound 30 changes.80 Under alkaline conditions, the anionic form of the corresponding isoxazol-5-ol predominates, while in chloroform the 2-isoxazolin-5-one isomer is mainly observed.80 Under acidic conditions, the content of the 3-isoxazolin-5-one increases to 55%.80Upon irradiation at wavelengths around the absorption maximum (263 ± 3 nm) of 2-substituted 3-isoxazolin-5-one derivatives, the heterocycle decomposes efficiently, forming a range of different products such as corresponding amines as well as N-acetyl amines and CO2.133 The quantum yields of these photohydrolysis reactions were determined to be above 0.3 in naturally occurring 3-isoxazolin-5-one derivatives.59,124,133
5. Toxicology
5.1. Toxic mechanisms of 3-nitropropanoic acid
3-Nitropropanoic acid 1 is a mitochondrial toxin due to its ability to irreversibly inhibit the generally occurring enzyme succinate dehydrogenase.134 This property is due to the isoelectronic character of 3-NPA compared to succinic acid (Fig. 11), which is the natural substrate of mitochondrial complex II.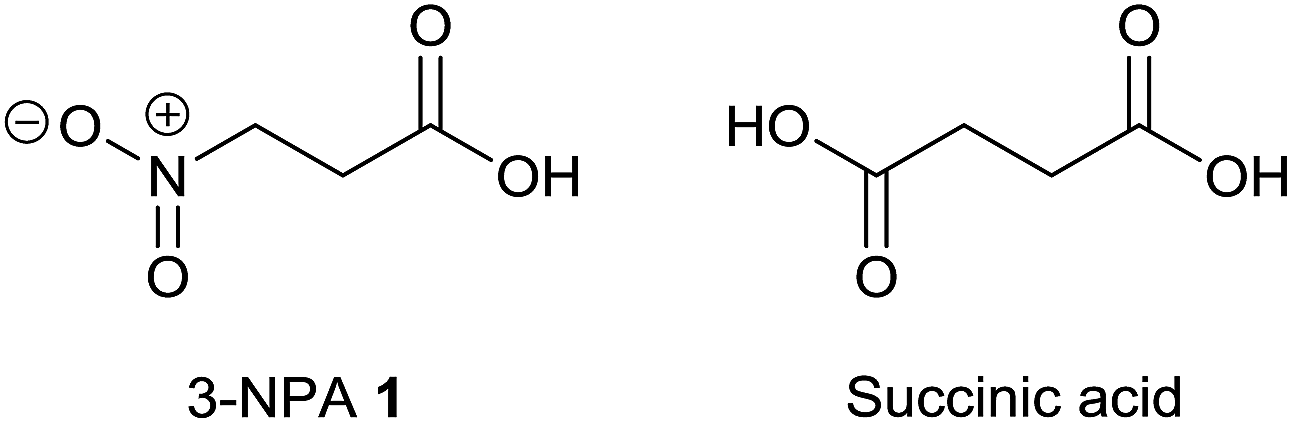 | ||
| Fig. 11 Structures of 3-NPA and succinic acid. | ||
Thus 3-NPA 1 is able to bind effectively to the catalytic center of succinate dehydrogenase.135 Several species deriving from 3-NPA that might deactivate succinate dehydrogenase have been suggested, e.g. 3-nitroacrylate,136 which could be formed due to oxidation of 3-NPA catalyzed by complex II, unaltered 3-NPA,135 the (dianionic) 3-nitronate isomer (P3N)35 or even its carbanion.134
X-ray as well as mass spectrometry analyses demonstrated that a covalent adduct of 3-NPA and the enzyme is formed, which supports the observation of succinate dehydrogenase as an irreversible inhibition mentioned.136 Furthermore, X-ray diffraction provides evidence for the formation of an unusual cyclic adduct between the guanidino group of the side-chain of Arg297 and 3-NPA within the catalytic center. This product is reconstructed from the original publication to bear most likely a 1,2,4-triazole moiety (Fig. 12).136
 | ||
| Fig. 12 Mass spectra and X-ray supported suggestion for the structure of the side-chain of Arg297 in 3-NPA-treated enzyme (bottom), compared with the Arg297 side-chain in the untreated complex II (top), adapted from Huang et al.136 | ||
The suggested structure is supported by MS analyses of tryptic peptides before and after treatment of complex II with 3-NPA, indicating a mass shift of 83 Da in the digestive fragments after addition of 3-NPA to solutions of complex II. In addition, the tryptic peptides show losses of 44 Da, which indicates decarboxylation, and thus the involvement of the nitro group instead of the carboxylic acid of 3-NPA in the formation of the covalent adduct.136 Alternative covalent addition products, e.g. with the contribution of the flavin cofactor35 or other functional groups in the enzyme,137e.g. mercapto groups, were not indicated by these136 data.
Whether the formation of this covalent addition product requires the complex II catalyzed oxidation of 3-NPA to form 3-nitroacrylate within the catalytic center, prior to inactivation, is still under investigation.136 The former experiments indicated a rapid inactivation of complex II upon application of 3-nitroacrylate within 15 s, but four to five equivalents of the inhibitor were required in the case of 3-nitroacrylate, while in the case of 3-NPA one equivalent was sufficient for inactivation.138
A previous study based on the crystal structure of complex II binding 3-NPA shows a non-covalent complex between substrate and enzyme.135 These findings were mainly supported by X-ray data of lower resolution and they do not explain the observed irreversible inhibition. Thus, these structures might show a pre-inactivation complex between the enzyme and 3-NPA.
Although the observed irreversible inhibition of complex II leads to mitochondrial energy impairment due to interference with the tricarboxylic acid cycle (Krebs cycle), the lethal dose in mice and rats indicates medium acute toxicity after oral administration, with LD50 values between 60 and 120 mg kg−1 body weight.139 Nevertheless, chronic 3-NPA administration causes neurodegeneration.140,141 The effects caused by 3-NPA are similar to the symptoms of Huntington’s disease,142,143 and thus compound 1 was used as a tool to study this illness, as it induced the symptoms in animal models.4,144,145 Typical symptoms of Huntington’s disease include, in particular, dysfunctions of movement such as dystonia, chorea, and hypokinesia.143 3-NPA intake was shown to poison cattle that fed on food that contained this toxin.142 Significant economic damage occurs due to poisoning by the ingestion of plants or microorganisms, e.g. from moldy sugarcane containing Arthrinium spp.15
Due to the obvious general toxicity of 3-NPA, the toxin was also characterized in terms of some antitumor toxicity using human cell lines.146
In addition to the toxicity against mammalian organisms, 3-NPA showed toxicity against many other cell types, e.g. against Mycobacterium tuberculosis H37Ra147 as well as H37Rv,10 due to the general toxicological mechanism of compound 1. Different minimum inhibition concentrations (MIC) have been published, in the range of 3.3 μM and 50 μg mL−1 (420 μM) for H37Ra10,147 and 12.5 μg mL−1 (105 μM) for H37Rv.10
For a more comprehensive overview of toxicological parameters and corresponding experiments see Burdock et al.139 as well as Madhusudan et al.142
5.2. Detoxification of 3-NPA
Since toxic 3-nitropropanoic acid 1 occurs in many plants and leaf beetles as well as in microbiota, different detoxification mechanisms have evolved in various organisms that are exposed to significant doses of 3-NPA. In insect generalists, e.g. Melanoplus species148 or Spodoptera littoralis,149 which feed on leguminous plants among many others, it was shown that 3-NPA detoxification is carried out by the formation of amino acid amides (Fig. 13).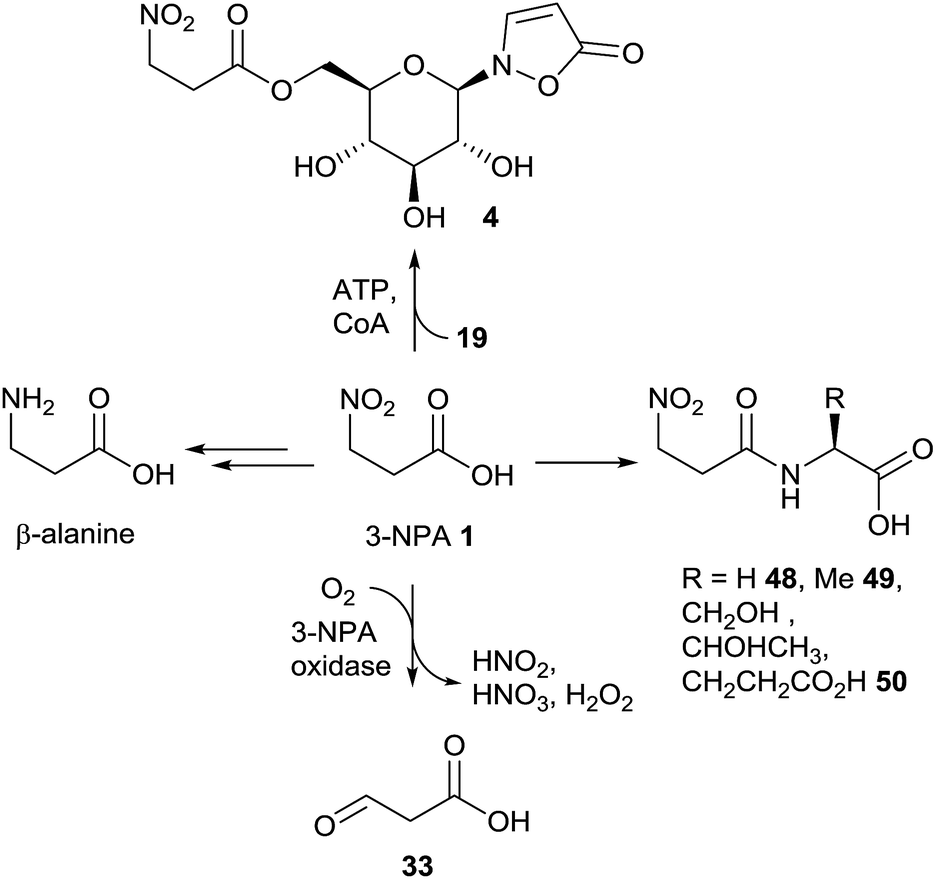 | ||
| Fig. 13 Detoxification pathways of 3-NPA in different organisms. | ||
After the formation of these amino acid conjugates, e.g. derived from glycine 48, alanine 49 and glutamic acid 50, the amides are excreted via the malphigian tubules over the hind gut of the insects.149
In (leguminous) plants, the ability to detoxify 3-NPA 1via oxidation probably evolved to protect sensitive tissue from intoxication.16,150 This oxidation is catalyzed by 3-NPA oxidase, which delivers malonate semialdehyde 33, nitrite, nitrate and hydrogen peroxide (Fig. 13).16,150 The activity of this enzyme in various leguminous plants correlates with the amount of 3-NPA that is produced by the organism.16
Similar reactions have been observed in P. atrovenetum, where a flavoprotein was identified as oxidizing 3-NPA (or its homologue propionate-3-nitronate) to produce compound 33 and a number of further compounds, e.g. superoxide.151 When E. coli cells were genetically modified to produce this oxidase, the bacteria were shown to be protected from 3-NPA mediated toxicity.152
Just as the consumption of propionate-3-nitronate151 was indicated by a decrease of oxygen concentration upon the addition of alkaline 3-NPA solution to the medium, the fungal protein was named as propionate-3-nitronate oxidase.151 In these studies, buffered solutions with pH values of around 8 were applied, yielding higher contents of the nitronate species, but no optimum pH range of the enzyme was determined.151 In contrast, the plant-produced 3-NPA oxidase showed maximum activity within a slightly acidic reaction medium (pH 5).150 Thus, in plants the occurrence of a nitronate dianion species, prior to oxidative cleavage of 3-NPA, is unlikely to be required.
It has been shown that mixed cultures of microbiota from the ruminal gut can metabolize 3-NPA to form β-alanine (Fig. 13).153 This pathway is of interest in so far as it explains the adaptation of ruminants to this toxin.154,155
In leaf beetles, the prevention from self-intoxication with 3-NPA 1 is realized by the formation of the ester compound 4 (Fig. 13).41 After the injection of [1-13C,15N]-3-nitropropanoic acid into larval hemolymph, isotopically labeled product 4 was detected, showing a mass shift of Δm/z = 2. This detoxification hypothesis is supported by sufficient concentrations of the glucoside precursor, compound 19, exceeding the amount of the corresponding nitro-ester 4 in early larval stages.5,41 The formation of the ester compound 4 equals the last step in the biosynthesis of this compounds in leaf beetles (Chrysomelina), as shown in the biosynthesis section in this article.
5.3. Toxicology of isoxazolin-5-one derivatives
Diverse non-natural isoxazolin-5-ones, isoxazol-5-oles and related compounds show promising biological activity, and thus are used as drugs for diverse targets.156–158 In naturally occurring isoxazolinones, various experiments have been carried out to explore the corresponding ecological relevance as well the application potential of these compounds. Since it was observed that the consumption of plants belonging to the genus Lathyrus, e.g. Lathyrus sativus (grass pea), causes the neuronal disease neurolathyrism,159 many studies concerning biological functions of natural isoxazolinones have focused on the involvement of such compounds in corresponding neuropathologies.In vitro investigations concerning the non-proteinogenic amino acid BIA 11 and its homologue ACI 12 showed that compound 11 has some excitotoxic potential.160 The excitotoxic potential of 11 was lower or in some cells comparable to the values of the well characterized neurotoxin β-N-oxalyl-L-α,β-diaminopropanoic acid (β-ODAP), which occurs together with BIA 11 in leguminous plants. The application of 6-cyano-7-nitroquinoxaline-2,3-dione indicated that the toxicity of 11 was mediated by non-N-methyl-D-aspartate type receptors.160 Compound 12 showed no significant excitotoxic effects in these experiments, but it was observed previously that neurolathyrism in young chicks was caused by this substance.161 Since these compounds were readily available from natural sources, further toxicological aspects have been investigated, e.g. antibiotic activity. In the case of BIA 11, antifungal activity, including mycelial as well as cell growth inhibition, has been observed in several strains of yeast species, without showing any antibacterial activity.162 A minimum inhibitory concentration (MIC) of 3 μM for BIA 11 was determined for Saccharomyces cerevisiae in minimal medium.163 Thus, compound 11 may play a significant role as a plant-protecting agent by influencing the rhizosphere, especially during early developmental stages in young legumes.162
Compound 10 (TAN-950A), which is a structural analogue of BIA 11, showed inhibitory effects on an L-glutamate/L-aspartate transporter (GLAST) and is significantly expressed in glial cells.164,165 In these experiments, no effects were observed for the corresponding glucoside 20.164 Furthermore and analogous to BIA 11, TAN-950A 10 was identified as an antifungal antibiotic agent, showing activity against some Candida strains as well as Saccharomyces cerevisiae with minimum inhibitory concentrations of 0.78 to 3.13 μg mL−1.166 It was demonstrated that activity persisted in response to the application to systemic Candida albicans TA infections in mice.166
In the case of the naturally occurring benzisoxazolin-5-one derivatives, promising bioactivities have been observed. It was demonstrated by using the Candida albicans Fitness Test that the parnafungins are antifungal agents,77,167,168 inhibiting mRNA polyadenylation (for the structure of parnafungin B1 21 see Fig. 4). It was observed by affinity selection/mass spectrometry that the “straight” isomer, assigned as parnafungin A, binds with higher affinity to polyadenosine polymerase (PAP) compared to the “bent” structures of other parnafungins.169
The marine sponge secondary metabolite bromobenzisoxazolone barettin 22 was identified as an antifouling agent, as it inhibits the settlement (EC50 = 15 nM) of barnacle larvae (Balanus improvisus).9 The efficiency of this inhibition is about 60 times more potent than that of isolated structural analogues, which lack the benzisoxazolinone moiety.9
The N-hydroxybenzisoxazolinone 23 showed antibacterial activity against V. anguillarum, V. harveyi VIB 286, A. hydrophila and S. aureus in TLC bioautography overlay assays.8
Since antifungal properties have been observed in a number of isoxazolin-5-one derivatives, glucoside 19 was tested against various cell lines of human and microbial cells. For this purpose, solutions of glucoside 19 (0.1 to 1 mg mL−1) were added to cell lines of phyto- and entomopathogens as well as to human pathogens.170 None of those tests resulted in any visible inhibition of growth of the cells or the mycelium of the treated cell lines. Analogously, neither increased mortality nor repulsion of Myrmica rubra (Formicidae) individuals feeding on 10−2 M solutions of compound 19 was observed.44 These observations indicate that some of the natural isoxazolinone derivatives may not cause (neuro-)toxicity. In the case of compound 19, a major ecological benefit for its producers may arise from derivatization with a 3-nitropropanoyl residue, which allows high amounts of this toxin to be stored, as shown in the case of leaf beetles.41 This hypothesis is further supported by a significant deterrent effect of compound 4 against M. rubra,44 which shows that storage of 3-NPA in the form of the corresponding ester in the leaf beetle hemolymph is non-toxic for the organism.
Although this hypothesis is plausible, many plants produce simple glucose esters of 3-NPA, without the presence of the isoxazolin-5-one moiety, as shown in Section 2. Furthermore, high amounts of the 3-NPA free glucoside 19 as well as many other isoxazolin-5-one derivatives of ambiguous biological function occur in various species of different kingdoms. These observations may indicate the as-yet undiscovered ecological importance of many natural isoxazolin-5-one derivatives, beyond a role within neurotoxicity or antibiotic activity.
6. Conclusions and perspectives
Natural compounds derived from 3-NPA and isoxazolin-5-one occur throughout four kingdoms of biology and show considerable structural diversity. In several species of Chrysomelina leaf beetles and leguminous plants of the genus Astragalus, both structural motifs occur at the same time an in the same molecule. In most cases, it is still unclear whether such a relationship occurs within 3-NPA and/or isoxazolin-5-one producers. Furthermore, whether 3-NPA is produced as a pure compound, or as a derivative, e.g. an ester, is not yet apparent.Biochemical fundamental research about the neurodegenerative toxicity of 3-NPA has been carried out, and results show that this nitro acid irreversibly inhibits the mitochondrial respiration via formation of a cyclic addition product with succinate dehydrogenase. Whether the unaltered compound 3-NPA is involved in the final deactivation, or its oxidized form 3-nitroacrylate, or the corresponding dianionic tautomers propionate-3-nitronate and the 3-deprotonated carbanion is under investigation.
These toxicological properties mainly contribute to the ecological function of 3-NPA as a general poison, one that is produced in significant amounts by plants and beetles as well as fungi. Nevertheless, it seems that there are no studies specifically aimed at investigating the evolution of the interactions between the organisms producing 3-NPA and isoxazolin-5-one derivatives and their predators, as have been carried out for producers of cardenolides,171 iridoids172 and nicotine.173 Evidence for a defensive function in the case of leaf beetles is indirect, deduced from the secretion of compounds in well-established defensive glands; further evidence is found in defensive toxins that are produced by species of other taxa, e.g. cardenolides174 and triterpene saponins,51 which release the secretion in response to disturbance, and from the obvious general toxicity of 3-NPA.
As widespread occurrence of 3-NPA and its derivatives shows, several fundamentally different detoxification mechanisms have evolved in various species of different kingdoms. The detoxification pathways include ester or amide formation with sugars and with amino acids. These reactions increase the storage potential of 3-NPA and protect the organism that produces it. Furthermore, it has been suggested that 3-NPA can be reduced to β-alanine by certain microorganisms or cleaved upon oxidation to form malonate semialdehyde, hydrogen peroxide, nitrate and nitrite, as well as other products, as shown in microbiota and (leguminous) plants.
In the case of isoxazolin-5-one derivatives, toxicological investigations were carried out to analyze their neurotoxicity as well as antibiotic and antifouling properties. Some promising results were found, although the biological function of many other isoxazolinone natural products, e.g. compound 19, still seems obscure.
Biosynthetic pathways have been studied in legumes and fungi, as well as leaf beetles. In fungi, MS, NMR and radioactivity experiments including degradation procedures to localize the incorporated label were conducted, resulting in the discovery of aspartate as the ultimate precursor. This concept was transferred to leaf beetles, resulting in positive radioactive labeling after aspartate feeding. MS investigations later showed that in Chrysomelina larvae, aspartate is incorporated through random re-assembly rather than direct incorporation. Instead of a metabolic route with aspartic acid as the ultimate precursor, a pathway starting from L-valine via propanoyl-CoA and β-alanine leads to the direct formation of isoxazolin-5-one and 3-NPA derived natural compounds. In the leguminous plant I. spicata, an alternative pathway for the biosynthesis of 3-NPA was suggested on the basis of radioactive labeling experiments. These studies resulted in the positive incorporation of malonate, while aspartate failed to be incorporated into 3-NPA. Whether alternative pathways, e.g. from valine and propanoyl-CoA, lead to a more direct production of 3-NPA in legumes has not yet been investigated. The formation of isoxazolin-5-one derivatives in plants has been studied independently from the biosynthesis of 3-NPA, resulting in the positive incorporation of isoxazoline-5-one as a general precursor for corresponding natural products. This compound can be transferred to various acceptors in plants, e.g. S-adenosyl-L-methionine, O-acetyl-L-serine or α-UDP glucose, leading to the biosynthesis of various natural compounds. Nevertheless, many questions regarding the biosynthesis of isoxazolin-5-one derivatives in plants remain unanswered, especially with respect to the exploration of corresponding biosynthetic intermediates and ultimate precursors. Whether a relationship between 3-NPA and isoxazolin-5-one production occurs in plants, as it does in leaf beetles,5 is unknown. Furthermore, it is of interest whether the biosyntheses are due to autogenous de novo production or to the involvement of (endo-)symbionts, or to both, especially in animals and plants producing 3-nitropropanoic acid or derived compounds.
The ecological importance of isoxazolin-5-one and of 3-NPA derived natural compounds for their producers can be better understood once the details of their biosynthetic pathways have been investigated; these details include related genes and enzymes, especially those supported by knock-out experiments which provide genetic information and enable studies of chemo-ecological aspects. Genetic information can also improve the health effects and safety of important food plants, e.g. L. sativus and P. sativum, by making use of tailored genetic engineering. Gene clusters showing in vitro activity for the production of 3-NPA from aspartate have recently been described in some Streptomycetes strains.93
In order to unravel important aspects of occurrence, amounts, biological properties as well as the biosyntheses, the chemical synthesis and subsequent qualitative and quantitative analyses of these natural products and their putative biosynthetic intermediates can be a useful tool. Some of the naturally occurring isoxazolinones have already been synthesized, but due to multiple synthetic steps, the tailored incorporation of isotope labels remains challenging. Nevertheless, some attempts using synthetic isotope labeled isoxazolinone glucosides have led to the improvement of quantitative analyses (SIL-IS).5,125
7. Acknowledgement
Open Access funding provided by the Max Planck Society.8. References
- D. Hellwinkel, Systematic Nomenclature of Organic Chemistry A Directory to Comprehension and Application of its Basic Principles, Springer Berlin Heidelberg, Berlin, 2001 Search PubMed.
- R. B. Fox and W. H. Powell, Nomenclature of Organic Compounds Principles and Practice, Oxford University Press, Oxford, 2001 Search PubMed.
- M. J. M. Christenhusz and J. W. Byng, Phytotaxa, 2016, 261, 17 CrossRef.
- I. Túnez, I. Tasset, V. Pérez-De La Cruz and A. Santamaría, Molecules, 2010, 15, 878 CrossRef PubMed.
- T. Becker, K. Ploss and W. Boland, Org. Biomol. Chem., 2016, 14, 6274–6280 CAS.
- T. Randoux, J. C. Braekman, D. Daloze and J. M. Pasteels, Naturwissenschaften, 1991, 78, 313–314 CrossRef CAS PubMed.
- X. Qiao, W. Fu, Y. Qiao, H. Huang, X. Kang, C. Wang and D. Li, Shipin Kexue, 2011, 32 Search PubMed.
- M. Yu, J. Wang, K. Tang, X. Shi, S. Wang, W.-M. Zhu and X.-H. Zhang, Microbiology, 2012, 158, 835–842 CrossRef CAS PubMed.
- E. Hedner, M. Sjögren, S. Hodzic, R. Andersson, U. Göransson, P. R. Jonsson and L. Bohlin, J. Nat. Prod., 2008, 71, 330–333 CrossRef CAS PubMed.
- D. U. Ganihigama, S. Sureram, S. Sangher, P. Hongmanee, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Eur. J. Med. Chem., 2015, 89, 1–12 CrossRef CAS PubMed.
- K. Gorter, Bull. Jard. bot. Buitenz., 1920, 2, 187–202 CAS.
- M. S. Carrie, J. Soc. Chem. Ind., London, 1934, 53, 288T–289T Search PubMed.
- C. L. Carter and W. J. McChesney, Nature, 1949, 164, 575–576 CrossRef CAS PubMed.
- H. Raistrick and A. Stössl, Biochem. J., 1958, 68, 647–653 CrossRef CAS PubMed.
- R. C. Anderson, W. Majak, M. A. Rassmussen, T. R. Callaway, R. C. Beier, D. J. Nisbet and M. J. Allison, J. Agric. Food Chem., 2005, 53, 2344–2350 CrossRef CAS PubMed.
- C. R. Hipkin, D. J. Simpson, S. J. Wainwright and M. A. Salem, Nature, 2004, 430, 98–101 CrossRef CAS PubMed.
- D. J. Simpson, S. J. Wainwright and C. R. Hipkin, Vet. Rec., 1999, 145, 169–171 CrossRef CAS PubMed.
- M. C. Williams and R. C. Barneby, Brittonia, 1977, 29, 310–326 CrossRef CAS.
- M. C. Williams, F. R. Stermitz and R. D. Thomas, Phytochemistry, 1975, 14, 2306–2308 CrossRef CAS.
- M. Benn, Y. Bai and W. Majak, Phytochemistry, 1995, 40, 1629–1631 CrossRef CAS.
- C. L. Carter, J. Sci. Food Agric., 1951, 2, 54–55 CrossRef CAS.
- W. Majak and M. Benn, Phytochemistry, 1994, 35, 901–903 CrossRef CAS.
- B. G. Moyer, P. E. Pfeffer, K. M. Valentine and D. L. Gustine, Phytochemistry, 1979, 18, 111–113 CrossRef CAS.
- W. S. Garcez, F. R. Garcez and A. Barison, Biochem. Syst. Ecol., 2003, 31, 207–209 CrossRef CAS.
- W. S. Garcez, F. R. Garcez, N. K. Honda and A. J. R. d. Silva, Phytochemistry, 1989, 28, 1251–1252 CrossRef CAS.
- W. Majak, M. Benn, D. McEwan and M. A. Pass, Phytochemistry, 1992, 31, 2393–2395 CrossRef CAS.
- W. Majak and R. J. Bose, Phytochemistry, 1976, 15, 415–417 CrossRef CAS.
- B. G. Moyer, P. E. Pfeffer, J. L. Moniot, M. Shamma and D. L. Gustine, Phytochemistry, 1977, 16, 375–377 CrossRef CAS.
- M. H. Benn, W. Majak and R. Aplin, Biochem. Syst. Ecol., 1997, 25, 467–468 CrossRef CAS.
- L. F. Pistelli, in Stud. Nat. Prod. Chem., ed. R. Atta ur, Elsevier, 2002, vol. 27, Part H, pp. 443–545 Search PubMed.
- C. Gnanasunderam and O. R. W. Sutherland, Phytochemistry, 1986, 25, 409–410 CrossRef CAS.
- L. Qiu, Y. Liang, G.-H. Tang, C.-M. Yuan, Y. Zhang, X.-Y. Hao, X.-J. Hao and H.-P. He, Phytochem. Lett., 2013, 6, 368–371 CrossRef CAS.
- M. A. Salem, J. M. Williams, S. J. Wainwright and C. R. Hipkin, Phytochemistry, 1995, 40, 89–91 CrossRef CAS.
- R. Parry, S. Nishino and J. Spain, Nat. Prod. Rep., 2011, 28, 152–167 RSC.
- K. Francis, C. Smitherman, S. F. Nishino, J. C. Spain and G. Gadda, IUBMB Life, 2013, 65, 759–768 CrossRef CAS PubMed.
- S. C. Gulati, H. J. Klosterman and L. J. Schermeister, Phytochemistry, 1972, 11, 1516 CrossRef.
- R. L. Baxter, A. B. Hanley, H. W. S. Chan, S. L. Greenwood, E. M. Abbot, I. J. Mcfarlane and K. Milne, J. Chem. Soc., Perkin Trans. 1, 1992, 2495–2502 RSC.
- A. Andolfi, A. Boari, M. Evidente, A. Cimmino, M. Vurro, G. Ash and A. Evidente, J. Nat. Prod., 2015, 78, 623–629 CrossRef CAS PubMed.
- J. C. Polonio, M. A. d. S. Ribeiro, S. A. Rhoden, M. H. Sarragiotto, J. L. Azevedo and J. A. Pamphile, Fungal Biol., 2016, 1600–1608 CrossRef CAS PubMed.
- V. Rukachaisirikul, U. Sommart, S. Phongpaichit, J. Sakayaroj and K. Kirtikara, Phytochemistry, 2008, 69, 783–787 CrossRef CAS PubMed.
- G. Pauls, T. Becker, P. Rahfeld, R. R. Gretscher, C. Paetz, J. M. Pasteels, S. H. von Reuss, A. Burse and W. Boland, J. Chem. Ecol., 2016, 42, 240–248 CrossRef CAS PubMed.
- J. M. Pasteels, M. Rowell-Rahier, J.-C. Braekman and D. Daloze, in Novel aspects of the biology of Chrysomelidae, ed. P. H. Jolivet, M. L. Cox and E. Petitpierre, Kluwer Academic Publisher, Dordrecht, The Netherlands, 1994, vol. 50, pp. 289–301 Search PubMed.
- J. M. Pasteels and M. Rowell-Rahier, Entomography, 1989, 6, 423–432 Search PubMed.
- J. M. Pasteels, D. Daloze and M. Rowell-Rahier, Physiol. Entomol., 1986, 11, 29–37 CrossRef CAS.
- J. M. Pasteels, J. C. Braekman, D. Daloze and R. Ottinger, Tetrahedron, 1982, 38, 1891–1897 CrossRef CAS.
- W. Sugeno and K. Matsuda, Appl. Entomol. Zool., 2002, 37, 191–197 CrossRef CAS.
- J. M. Pasteels, M. Rowell-Rahier, J. C. Braekman and D. Daloze, Biochem. Syst. Ecol., 1984, 12, 395–406 CrossRef CAS.
- M. Rowell-Rahier and J. M. Pasteels, J. Chem. Ecol., 1986, 12, 1189–1203 CrossRef CAS PubMed.
- J. M. Pasteels, Biochem. Syst. Ecol., 1993, 21, 135–142 CrossRef CAS.
- J. M. Pasteels, A. Termonia, D. Daloze and D. M. Windsor, in Proceedings of the Fifth International Symposium on the Chrysomelidae, Iguacu, 2000, ed. D. G. Furth, Pensoft Publishers, 2003, vol. 21, pp. 261–275 Search PubMed.
- P. Laurent, J.-C. Braekman, D. Daloze and J. Pasteels, Eur. J. Org. Chem., 2003, 2003, 2733–2743 CrossRef.
- B. Warth, A. Parich, J. Atehnkeng, R. Bandyopadhyay, R. Schuhmacher, M. Sulyok and R. Krska, J. Agric. Food Chem., 2012, 60, 9352–9363 CrossRef CAS PubMed.
- E. Varga, T. Glauner, F. Berthiller, R. Krska, R. Schuhmacher and M. Sulyok, Anal. Bioanal. Chem., 2013, 405, 5087–5104 CrossRef CAS PubMed.
- M. Peitzsch, M. Sulyok, M. Taubel, V. Vishwanath, E. Krop, A. Borras-Santos, A. Hyvarinen, A. Nevalainen, R. Krska and L. Larsson, J. Environ. Monit., 2012, 14, 2044–2053 RSC.
- K. F. Nielsen and J. Smedsgaard, J. Chromatogr. A, 2003, 1002, 111–136 CrossRef CAS PubMed.
- J. Dron, E. Abidi, I. E. Haddad, N. Marchand and H. Wortham, Anal. Chim. Acta, 2008, 618, 184–195 CrossRef CAS PubMed.
- O. S. Chizhov, V. I. Kadentsev, G. G. Palmbach, K. I. Burstein, S. A. Shevelev and A. A. Feinsilberg, Org. Mass Spectrom., 1978, 13, 611–617 CrossRef CAS.
- T. Becker, H. Görls, G. Pauls, R. Wedekind, M. Kai, S. H. von Reuss and W. Boland, J. Org. Chem., 2013, 78, 12779–12783 CrossRef CAS PubMed.
- F. Lambein, Y.-H. Kuo and R. V. Parijs, Heterocycles, 1976, 4, 567–593 CrossRef CAS.
- L. Rahbœk and C. Christophersen, in The Alkaloids: Chemistry and Biology, Academic Press, 2001, vol. 57, pp. 185–233 Search PubMed.
- P. Rozan, Y.-H. Kuo and F. Lambein, Phytochemistry, 2001, 58, 281–289 CrossRef CAS PubMed.
- B. Chowdhury, P. Rozan, Y.-H. Kuo, M. Sumino and F. Lambein, J. Chromatogr. A, 2001, 933, 129–136 CrossRef CAS PubMed.
- P. Rozan, Y.-H. Kuo and F. Lambein, J. Agric. Food Chem., 2000, 48, 716–723 CrossRef CAS PubMed.
- Y.-H. Kuo, F. Ikegami and F. Lambein, Phytochemistry, 1998, 49, 43–48 CrossRef CAS.
- F. Ikegami, F. Lambein, Y. H. Kuo and I. Murakoshi, Phytochemistry, 1984, 23, 1567–1569 CrossRef CAS.
- F. Lambein, J. K. Khan, C. Becu and A. D. Bruyn, Phytochemistry, 1992, 31, 887–892 CrossRef CAS.
- Y.-C. Lai and S.-S. Lee, Nat. Prod. Commun., 2013, 8, 363–365 CAS.
- K. Ichimura, H. Ono, A. Soga, H. Shimizu-Yumoto, K. Kohata and M. Nakayama, Hortic. J., 2016, 85, 161–168 CrossRef.
- Y.-H. Kuo, F. Lambein, F. Ikegami and R. Van Parijs, Plant Physiol., 1982, 70, 1283–1289 CrossRef CAS PubMed.
- F. Ikegami, Y. Tajima, S. Ohmiya, F. Lambein and I. Murakoshi, Chem. Pharm. Bull., 1984, 32, 2450–2451 CrossRef CAS.
- H. Shimizu-Yumoto, N. Hayashi, K. Ichimura and M. Nakayama, J. Chromatogr. A, 2012, 1245, 183–189 CrossRef CAS PubMed.
- F. Lambein, L. Van Rompuy, A. De Bruyn and R. Van Parijs, Arch. Int. Physiol. Biochim., 1974, 82, 187 CAS.
- L. Van Rompuy, F. Lambein, R. De Gussem and R. Van Parijs, Biochem. Biophys. Res. Commun., 1974, 56, 199–205 CrossRef CAS PubMed.
- T. Iwama, Y. Nagai, S. Harada, K. Itoh and A. Nagaoka, Eur. J. Pharmacol., 1990, 183, 471–472 CrossRef.
- T. Iwama, Y. Nagai, N. Tamura, S. Harada and A. Nagaoka, Eur. J. Pharmacol., 1991, 197, 187–192 CrossRef CAS PubMed.
- S. Tsubotani, Y. Funabashi, M. Takamoto, S. Hakoda and S. Harada, Tetrahedron, 1991, 47, 8079–8090 CrossRef CAS.
- C. A. Parish, S. K. Smith, K. Calati, D. Zink, K. Wilson, T. Roemer, B. Jiang, D. Xu, G. Bills, G. Platas, F. Peláez, M. T. Díez, N. Tsou, A. E. McKeown, R. G. Ball, M. A. Powles, L. Yeung, P. Liberator and G. Harris, J. Am. Chem. Soc., 2008, 130, 7060–7066 CrossRef CAS PubMed.
- P. Rozan, H. Y. Kuo and F. Lambein, Amino Acids, 2001, 20, 319–324 CrossRef CAS PubMed.
- A. De Bruyn, G. Verhegge and F. Lambein, Magn. Reson. Chem., 1991, 29, 641–643 CrossRef CAS.
- F. De Sarlo, G. Dini and P. Lacrimini, J. Chem. Soc. C, 1971, 86 RSC.
- J. K. Khan, Y.-H. Kuo, N. Kebede and F. Lambein, J. Chromatogr. A, 1994, 687, 113–119 CrossRef CAS.
- X. Liu, M. Dong, X. Chen, M. Jiang, X. Lv and G. Yan, Food Chem., 2007, 105, 548–554 CrossRef CAS.
- Z. Li, K. Ni, X. Liao and G. Du, Chin. J. Pharm. Anal., 2006, 26, 577–584 CAS.
- C. Ni and X. Tong, J. Am. Chem. Soc., 2016, 138, 7872–7875 CrossRef CAS PubMed.
- G. A. Mills, V. Walker and J. M. Mellor, Clin. Chim. Acta, 1989, 184, 93–97 CrossRef CAS.
- R. Libert, J. P. Draye, F. Van Hoof, A. Schanck, J. P. Soumillion and E. de Hoffmann, Biol. Mass Spectrom., 1991, 20, 75–86 CrossRef CAS PubMed.
- A. J. Birch, B. J. McLaughlin, H. Smith and J. Winter, Chem. Ind., 1960, 26, 840–841 Search PubMed.
- P. D. Shaw and J. A. McCloskey, Biochemistry, 1967, 6, 2247–2253 CrossRef CAS PubMed.
- J. H. Birkinshaw and A. M. L. Dryland, Biochem. J., 1964, 93, 478–487 CrossRef CAS PubMed.
- R. L. Baxter, E. M. Abbot, S. L. Greenwood and I. J. Mcfarlane, J. Chem. Soc., Chem. Commun., 1985, 564–566 RSC.
- R. L. Baxter, A. B. Hanley and H. W. S. Chan, J. Chem. Soc., Chem. Commun., 1988, 757–758 RSC.
- R. L. Baxter, S. L. Smith, J. R. Martin and A. B. Hanley, J. Chem. Soc., Perkin Trans. 1, 1994, 2297–2299 RSC.
- Z. Huang, K.-K. A. Wang and W. A. van der Donk, Chem. Sci., 2016, 7, 5219–5223 RSC.
- Y. Kaziro and S. Ochoa, in Adv. Enzymol. Relat. Areas Mol. Biol., John Wiley & Sons, Inc., 2006, pp. 283–378 Search PubMed.
- D. Peter, B. Vögeli, N. Cortina and T. Erb, Molecules, 2016, 21, 517 CrossRef PubMed.
- E. Candlish, L. J. La Croix and A. M. Unrau, Biochemistry, 1969, 8, 182–186 CrossRef CAS PubMed.
- I. A. Berg, D. Kockelkorn, W. Buckel and G. Fuchs, Science, 2007, 318, 1782–1786 CrossRef CAS PubMed.
- G. Strauss and G. Fuchs, Eur. J. Biochem., 1993, 215, 633–643 CrossRef CAS PubMed.
- P. Chomcheon, S. Wiyakrutta, N. Sriubolmas, N. Ngamrojanavanich, D. Isarangkul and P. Kittakoop, J. Nat. Prod., 2005, 68, 1103–1105 CrossRef CAS PubMed.
- F. Lambein, R. Van Vaerenbergh and Y. H. Kuo, Arch. Physiol. Biochem., 1984, 92, B38–B39 CrossRef.
- I. Murakoshi, F. Kato, J. Haginiwa and L. Fowden, Chem. Pharm. Bull., 1973, 21, 918–920 CrossRef CAS.
- F. Ikegami, G. Ongena, R. Sakai, S. Itagaki, M. Kobori, T. Ishikawa, Y.-H. Kuo, F. Lambein and I. Murakoshi, Phytochemistry, 1993, 33, 93–98 CrossRef CAS.
- F. Ikegami, A. Yamamoto, Y.-H. Kuo and F. Lambein, Biol. Pharm. Bull., 1999, 22, 770–771 CAS.
- Y.-H. Kuo, F. Lambein, L. C. Mellor, R. M. Adlington and J. E. Baldwin, Phytochemistry, 1994, 37, 713–715 CrossRef CAS.
- F. Lambein, O. Godelieve and K. Yu-Haey, Phytochemistry, 1990, 29, 3793–3796 CrossRef CAS.
- F. Ikegami, M. Kamiya, Y.-H. Kuo, F. Lambein and I. Murakoshi, Biol. Pharm. Bull., 1996, 19, 1214–1215 CAS.
- I. Murakoshi, F. Ikegami, F. Kato, J. Haginiwa, F. Lambein, L. Van Rompuy and R. Van Parijs, Phytochemistry, 1975, 14, 1269–1270 CrossRef CAS.
- I. Murakoshi, F. Ikegami, F. Kato, J. Haginawa, F. Lambein, L. V. Rompuy and R. V. Parijs, Phytochemistry, 1975, 14, 1515–1517 CrossRef CAS.
- F. Ikegami, R. Sakai, T. Ishikawa, Y.-H. Kuo, F. Lambein and l. Murakoshi, Biol. Pharm. Bull., 1993, 16, 732–734 CAS.
- A. Callebaut, F. Lambein and R. Van Parijs, Arch. Int. Physiol. Biochim., 1977, 85, 157–158 CAS.
- V. K. Yadav and S. L. Mehta, Curr. Sci., 1995, 68, 288–292 Search PubMed.
- S. Kumar, G. Bejiga, S. Ahmed, H. Nakkoul and A. Sarker, Food Chem. Toxicol., 2011, 49, 589–600 CrossRef CAS PubMed.
- M. C. V. Patto, R. Amarowicz, A. N. A. Aryee, J. I. Boye, H. J. Chung, M. A. Martin-Cabrejas and C. Domoney, Crit. Rev. Plant Sci., 2015, 34, 105–143 CrossRef.
- C. Annese, L. D’Accolti, R. Filardi, I. Tommasi and C. Fusco, Tetrahedron Lett., 2013, 54, 515–517 CrossRef CAS.
- P. Silva, J. Costa and V. Pereira, Synth. Commun., 2001, 31, 595 CrossRef CAS.
- J. K. Addo, P. Teesdale-Spittle and J. O. Hoberg, Synthesis, 2005, 12, 1923–1925 Search PubMed.
- M. T. Bush, O. Touster and J. E. Brockman, J. Biol. Chem., 1951, 188, 685–693 CAS.
- H. B. Hass, H. Feuer and S. M. Pier, J. Am. Chem. Soc., 1951, 73, 1858 CrossRef CAS.
- L. Van Rompuy, N. Schamp, N. De Kimpe and R. Van Parijs, J. Chem. Soc., Perkin Trans. 1, 1973, 2503–2506 RSC.
- L. Van Rompuy, N. Schamp and R. Van Parijs, Arch. Int. Physiol. Biochim., 1973, 81, 394–395 CAS.
- I. Murakoshi, F. Ikegami, Y. Yoneda, H. Ihara, K. Saicata and C. Koide, Chem. Pharm. Bull., 1986, 34, 1473–1478 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington and D. J. Birch, Tetrahedron Lett., 1985, 26, 5931–5934 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington and L. C. Mellor, Tetrahedron, 1994, 50, 5049–5066 CrossRef CAS.
- T. Becker, P. Kartikeya, C. Paetz, S. H. von Reuss and W. Boland, Org. Biomol. Chem., 2015, 13, 4025–4030 CAS.
- S. Arrivault, M. Guenther, S. C. Fry, M. M. F. F. Fuenfgeld, D. Veyel, T. Mettler-Altmann, M. Stitt and J. E. Lunn, Anal. Chem., 2015, 87, 6896–6904 CrossRef CAS PubMed.
- M. Itoh, D. Hagiwara and T. Kamiya, Tetrahedron Lett., 1975, 16, 4393–4394 CrossRef.
- E. Schröder and E. Klieger, Justus Liebigs Ann. Chem., 1964, 673, 196–207 CrossRef.
- Q. Zhou and B. B. Snider, J. Org. Chem., 2010, 75, 8224–8233 CrossRef CAS PubMed.
- Q. Zhou and B. B. Snider, Org. Lett., 2009, 11, 2936–2939 CrossRef CAS PubMed.
- E. M. Beccalli, D. Pocar and C. Zoni, Recent developments in the chemistry of isoxazol-5-ones, Società Chimica Italiana, Rome, Italy, 2003 Search PubMed.
- U. S. Sørensen and P. Krogsgaard-Larsen, Org. Prep. Proced. Int., 2001, 33, 515–564 CrossRef.
- F. Lambein and R. Van Parijs, Biochem. Biophys. Res. Commun., 1970, 40, 557–564 CrossRef CAS PubMed.
- A. De Bruyn, G. Verhegge and F. Lambein, Planta Med., 1992, 58, 159–162 CrossRef CAS PubMed.
- T. A. Alston, L. Mela and H. J. Bright, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3767–3771 CrossRef CAS.
- F. Sun, X. Huo, Y. Zhai, A. Wang, J. Xu, D. Su, M. Bartlam and Z. Rao, Cell, 2005, 121, 1043–1057 CrossRef CAS PubMed.
- L. S. Huang, G. Sun, D. Cobessi, A. C. Wang, J. T. Shen, E. Y. Tung, V. E. Anderson and E. A. Berry, J. Biol. Chem., 2006, 281, 5965–5972 CrossRef CAS PubMed.
- D. J. Smith and R. C. Anderson, J. Agric. Food Chem., 2013, 61, 763–779 CrossRef CAS PubMed.
- C. J. Coles, D. E. Edmondson and T. P. Singer, J. Biol. Chem., 1979, 254, 5161–5167 CAS.
- G. A. Burdock, I. G. Carabin and M. G. Soni, Food Chem., 2001, 75, 1–27 CrossRef CAS.
- E. Brouillet, P. Hantraye, R. J. Ferrante, R. Dolan, A. Leroy-Willig, N. W. Kowall and M. F. Beal, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7105–7109 CrossRef CAS.
- M. F. Beal, E. Brouillet, B. G. Jenkins, R. J. Ferrante, N. W. Kowall, J. M. Miller, E. Storey, R. Srivastava, B. R. Rosen and B. T. Hyman, J. Neurosci., 1993, 13, 4181–4192 CAS.
- G. S. Madhusudan, I. G. Carabin, J.C. Griffiths and G. A. Burdock, Reviews in Food and Nutrition Toxicity. Chapter 5: β-Nitropropionic acid in the Diet: Toxicity Aspects, CRC Press LLC, 2004 Search PubMed.
- T. Alexi, P. E. Hughes, R. L. M. Faull and C. E. Williams, NeuroReport, 1998, 9, R57–R64 CrossRef CAS PubMed.
- E. Brouillet, C. Jacquard, N. Bizat and D. Blum, J. Neurochem., 2005, 95, 1521–1540 CrossRef CAS PubMed.
- E. Brouillet, F. Condé, M. F. Beal and P. Hantraye, Prog. Neurobiol. (Oxford, U. K.), 1999, 59, 427–468 CrossRef CAS.
- F. T. Lu, D. C. Ma, W. Yan, J. Guo and L. H. Bai, Lett. Appl. Microbiol., 2015, 61, 165–170 CrossRef CAS PubMed.
- P. Chomcheon, S. Wiyakrutta, N. Sriubolmas, N. Ngamrojanavanich, D. Isarangkul and P. Kittakoop, J. Nat. Prod., 2005, 68, 1103–1105 CrossRef CAS PubMed.
- W. Majak, D. L. Johnson and M. H. Benn, Phytochemistry, 1998, 49, 419–422 CrossRef CAS.
- A. Novoselov, T. Becker, G. Pauls, S. H. von Reuss and W. Boland, Insect Biochem. Mol. Biol., 2015, 63, 97–103 CrossRef CAS PubMed.
- C. R. Hipkin, M. A. Salem, D. Simpson and S. J. Wainwright, Biochem. J., 1999, 340, 491–495 CrossRef CAS PubMed.
- D. J. T. Porter and H. J. Bright, J. Biol. Chem., 1987, 262, 14428–14434 CAS.
- K. Francis, S. F. Nishino, J. C. Spain and G. Gadda, Arch. Biochem. Biophys., 2012, 521, 84–89 CrossRef CAS PubMed.
- R. C. Anderson, M. A. Rasmussen and M. J. Allison, Appl. Environ. Microbiol., 1993, 59, 3056–3061 CAS.
- Y. Zhang, R. Long, C. M. Warzecha, J. A. Coverdale, E. A. Latham, M. E. Hume, T. R. Callaway, M. R. O’Neil, R. C. Beier, R. C. Anderson and D. J. Nisbet, Anaerobe, 2014, 26, 7–13 CrossRef CAS PubMed.
- R. C. Anderson, M. A. Rasmussen, N. S. Jensen and M. J. Allison, Int. J. Syst. Evol. Microbiol., 2000, 50, 633–638 CrossRef CAS PubMed.
- S. N. Maddila, S. Maddila, W. E. van Zyl and S. B. Jonnalagadda, Res. Chem. Intermed., 2016, 42, 2553–2566 CrossRef CAS.
- H. Kiyani and F. Ghorbani, Res. Chem. Intermed., 2016, 42, 6831–6844 CrossRef CAS.
- D. A. Bachovchin, S. J. Brown, H. Rosen and B. F. Cravatt, Nat. Biotechnol., 2009, 27, 387–394 CrossRef CAS PubMed.
- Y. W. Woldeamanuel, A. Hassan and G. Zenebe, J. Neurol., 2012, 259, 1263–1268 CrossRef PubMed.
- M. Riepe, P. S. Spencer, F. Lambein, A. C. Ludolph and C. N. Allen, Nat. Toxins, 1995, 3, 58–64 CrossRef CAS PubMed.
- F. Lambein and B. D. Vos, Arch. Int. Physiol. Biochim., 1981, 89, B66–B67 Search PubMed.
- S. U. Schenk, F. Lambein and D. Werner, Biol. Fertil. Soils, 1991, 11, 203–209 CrossRef CAS.
- S. U. Schenk and D. Werner, Phytochemistry, 1991, 30, 467–470 CrossRef CAS.
- K. Kusama-Eguchi, T. Kusama, F. Ikegami, F. Lambein and K. Watanabe, Mol. Brain Res., 1997, 52, 166–169 CrossRef CAS PubMed.
- N. Tamura, Y. Matsushita, T. Iwama, S. Harada, S. Kishimoto and K. Itoh, Chem. Pharm. Bull., 1991, 39, 1199–1212 CrossRef CAS PubMed.
- S. Hakoda, S. Tsubotani, T. Iwasa, M. Suzuki, M. Kondo and S. Harada, J. Antibiot., 1992, 45, 854–860 CrossRef CAS PubMed.
- G. F. Bills, G. Platas, D. P. Overy, J. Collado, A. Fillola, M. R. Jiménez, J. Martín, A. G. del Val, F. Vicente, J. R. Tormo, F. Peláez, K. Calati, G. Harris, C. Parish, D. Xu and T. Roemer, Mycologia, 2009, 101, 449–472 CrossRef CAS PubMed.
- B. Jiang, D. Xu, J. Allocco, C. Parish, J. Davison, K. Veillette, S. Sillaots, W. Hu, R. Rodriguez-Suarez, S. Trosok, L. Zhang, Y. Li, F. Rahkhoodaee, T. Ransom, N. Martel, H. Wang, D. Gauvin, J. Wiltsie, D. Wisniewski, S. Salowe, J. N. Kahn, M.-J. Hsu, R. Giacobbe, G. Abruzzo, A. Flattery, C. Gill, P. Youngman, K. Wilson, G. Bills, G. Platas, F. Pelaez, M. T. Diez, S. Kauffman, J. Becker, G. Harris, P. Liberator and T. Roemer, Chem. Biol., 2008, 15, 363–374 CrossRef CAS PubMed.
- G. C. Adam, C. A. Parish, D. Wisniewski, J. Meng, M. Liu, K. Calati, B. D. Stein, J. Athanasopoulos, P. Liberator, T. Roemer, G. Harris and K. T. Chapman, J. Am. Chem. Soc., 2008, 130, 16704–16710 CrossRef CAS PubMed.
- Microorganisms, Conidiobolus coronatus, Beauveria Bassiana, Alternaria alternata, Fusarium graminearum, Cladosporium herbarium, Glomerella cingulata, Bacillus subtilis, Staphilococcus aureus, Escherichia coli, Pseudomonas aeruginosa, Enterococcus faecalis, Mycobacterium vaccae, Sporobolomyces salmicolor, Candida albicans and Penicillium notatum.
- A. A. Agrawal, G. Petschenka, R. A. Bingham, M. G. Weber and S. Rasmann, New Phytol., 2012, 194, 28–45 CrossRef CAS PubMed.
- M. Wink, Phytochemistry, 2003, 64, 3–19 CrossRef CAS PubMed.
- X. W. Wang and J. L. Bennetzen, Mol. Genet. Genomics, 2015, 290, 11–21 CrossRef CAS PubMed.
- S. Dobler, D. Daloze and J. M. Pasteels, Chemoecology, 1998, 8, 111–118 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |