
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficiency of photoinduced electron transfer in mono- and di-nuclear iridium complexes: a comparative study
Sourav
Kanti Seth
,
Parna
Gupta
and
Pradipta
Purkayastha
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, WB, India. E-mail: ppurkayastha@iiserkol.ac.in
First published on 5th June 2017
Abstract
Iridium complexes have been recognized as the most widely used class of emitters because of their efficient spin–orbit coupling and hence relaxation of the spin selection rule. The strong phosphorescence of the Ir(III) complexes is induced by the triplet metal-to-ligand charge transfer (3MLCT). However, there are significant differences in the photophysical and electrochemical properties of the mono- and dinuclear Ir(III) complexes. Photoinduced energy and electron transfer (PET) is frequently observed in supramolecular Ir(III) cyclometalated complexes. Herein, we synthesized a mononuclear and a dinuclear Ir(III) complex ([Ir(ppy)2(fmp)][PF6] and [{Ir(ppy)2}2(H2bpib)][PF6]2, represented by 1 and 2 in the text, respectively) to compare their PET efficiencies. Spectroscopic and electrochemical studies reveal that 1 (consisting of a single iridium center) acts as a better electron donor as compared to 2 (consisting of two iridium centers) during PET.
Introduction
Cyclometalated iridium(III) complexes are well known for their rich photochemical and photophysical properties.1,2 These complexes show properties of organic light-emitting diodes,3–6 have applications in photovoltaic cells,7,8 and are used as biological labeling reagents9,10 because they possess high luminescence quantum yields and long excited state lifetimes. Iridium complexes have been recognized as the most widely used class of emitters because of their efficient spin–orbit coupling and hence relaxation of the spin selection rule. The strong phosphorescence of the Ir(III) complexes is induced by the triplet metal-to-ligand charge transfer (3MLCT), reflecting the character of both the metal centre and the organic ligand.11Some phosphorescent dinuclear Ir(III) complexes show high luminescent efficiencies and good electroluminescent abilities. Therefore, highly phosphorescent dinuclear Ir(III) complexes have been synthesized and fully characterized. However, significant differences in the photophysical and electrochemical properties of the mono- and dinuclear complexes have recently been reported. Yang et al. showed that the key factor in determining the photophysical and electrochemical properties of dinuclear complexes is their unique molecular orbital spatial distribution pattern.6 They also noted that the efficiency of the organic light-emitting diode (OLED) constructed from the dinuclear iridium(III) complex was much higher than of that constructed with the corresponding mononuclear counterpart.
Photoinduced energy and electron transfer processes are frequently observed in supramolecular Ir(III) cyclometalated complexes.12–14 It is also apparent that these polynuclear complexes possess enhanced extinction coefficients as compared to their mononuclear analogues.15,16 Spin–orbit coupling is facilitated by the presence of additional metal centers that, in turn, enhance the radiative rate constant and hence increase the efficiency of phosphorescence.4,17 In the semi-classical electron transfer (ET) theory, the reaction rates are governed by three principal parameters: (i) electronic coupling between the donor and acceptor, (ii) the change in the reaction free-energy (ΔG°), and (iii) the extent of solvation accompanying the ET reaction.18 Moreover, ET rates can be limited by the dynamics of nuclear motion through the frequency factor νN.
Through extensive literature search, we could hardly find any reports on Ru or Re complexes with the simultaneous presence of fmp/ppy or H2bpib/ppy ligands as used in the present study. However, reports on Ru complexes with fmp/bpy or fmp/phen and H2bpib/phen or H2bpib/bpy ligands are available.19–21 It is inappropriate to compare the reported Ir and the Ru complexes of the present case because ppy acts as a cyclometalating ligand in our Ir complexes, whereas bpy/phen has been used for the Ru complexes. However, an overall comparison of their properties can be made. The Ru complexes show absorption maxima at around 280 nm (ε ≈ 6 × 104 M−1 cm−1) and 370 nm (ε ≈ 4 × 104 M−1 cm−1) due to the π–π* transition and at around 460 nm (ε ≈ 2 × 104 M−1 cm−1) attributed to 1MLCT. The Ir complexes show similar absorption bands at around 280 nm due to π–π* transition, and 370 nm (ε ≈ 6 × 104 M−1 cm−1) and 470 nm (ε ≈ 6 × 103 M−1 cm−1) bands are considered to be due to 1LC/1MLCT (LC = ligand centred, MLCT = metal-to-ligand charge transfer) and MC (metal centred) transitions, respectively.22 Differences in the molar extinction coefficients (ε) in the visible region of the spectrum make the Ru analogues better visible light absorbers. Moreover, higher spin–orbit coupling constant of Ir (3909 cm−1) makes intersystem crossing easier as compared to that of Ru (1042 cm−1).22 This makes Ir a better candidate for a more populated triplet excited state than its Ru analogue. Despite comparable triplet excited state lifetimes for both types of complexes (1–2 μs), emission quantum yields for the Ir complexes are higher than those for the Ru complexes.23,24 Moreover, the more populated, long-lived triplet excited state is better for efficient electron transfer than the less populated triplet excited state. Cyclic voltammogram shows that the Ir centres (Ir3+/Ir4+) more easily oxidize (∼1.20–1.25 V) than the Ru centres (Ru2+/Ru3+ ∼1.30–1.40 V) in the ground state.25
As light harvesting building blocks, Ir(III) complexes can transform solar energy into electrical energy through ultrafast ET. Hence, these potential candidates for energy harvesting are worth investigating through physicochemical studies. We have chosen two Ir(III) complexes, one mononuclear and one dinuclear (1 and 2 in Scheme 1), to compare their photoinduced electron transfer (PET) efficiencies. Spectroscopic and electrochemical studies reveal that 1 (consisting of a single iridium centre) acts as a better electron donor as compared to 2 (consisting of two iridium centres) during PET.
 | ||
| Scheme 1 Structures of the 1 ([Ir(ppy)2(fmp)][PF6]) and 2 ([{Ir(ppy)2}2(H2bpib)][PF6]2) complexes. | ||
Results and discussion
The two Ir(III) complexes, a mononuclear and a dinuclear (1 and 2), were compared in terms of PET. Complex 1 was synthesized using a mixture of [Ir(ppy)2Cl]2, methanol, dichloromethane, and acetonitrile in appropriate proportions and refluxing it for 5 h at 90 °C, and then, a reddish orange solution was obtained. To synthesize complex 2, a mixture of [Ir(ppy)2Cl]2, 2 (0.1 mmol), and acetonitrile/dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was refluxed for 4 h at 90 °C to obtain an orange coloured mixture. The mixture was cooled down to room temperature and the solvent was evaporated using a rotary evaporator. Both complexes were precipitated by adding potassium hexafluorophosphate (KPF6) in dichloromethane–methanol 1:1 mixture at room temperature and purified by silica gel column chromatography.26
:1 v/v) was refluxed for 4 h at 90 °C to obtain an orange coloured mixture. The mixture was cooled down to room temperature and the solvent was evaporated using a rotary evaporator. Both complexes were precipitated by adding potassium hexafluorophosphate (KPF6) in dichloromethane–methanol 1:1 mixture at room temperature and purified by silica gel column chromatography.26
The systems in the present study consisted of TEOA as the sacrificial donor, complex 1 or 2 as the photosensitizer, and methyl viologen dichloride (MVCl2) as the electron acceptor. The absorption spectra of the complexes show two intense bands at around 280 nm and 360 nm and one weak broad band peaking at around 465 nm in 1:1 acetonitrile–water (Fig. 1). The band at 280 nm is attributed to the spin-allowed π–π* ligand-to-ligand charge transfer (1LLCT), whereas those at 360 nm and 465 nm are mainly due to 1MLCT and d–d transitions in the metal centre/s (MC).26 The molar extinction coefficient (ε371 = 6.6 × 104 M−1 cm−1) for 2 is almost twice that of 1 (ε340 = 3.1 × 104 M−1 cm−1) in acetonitrile.
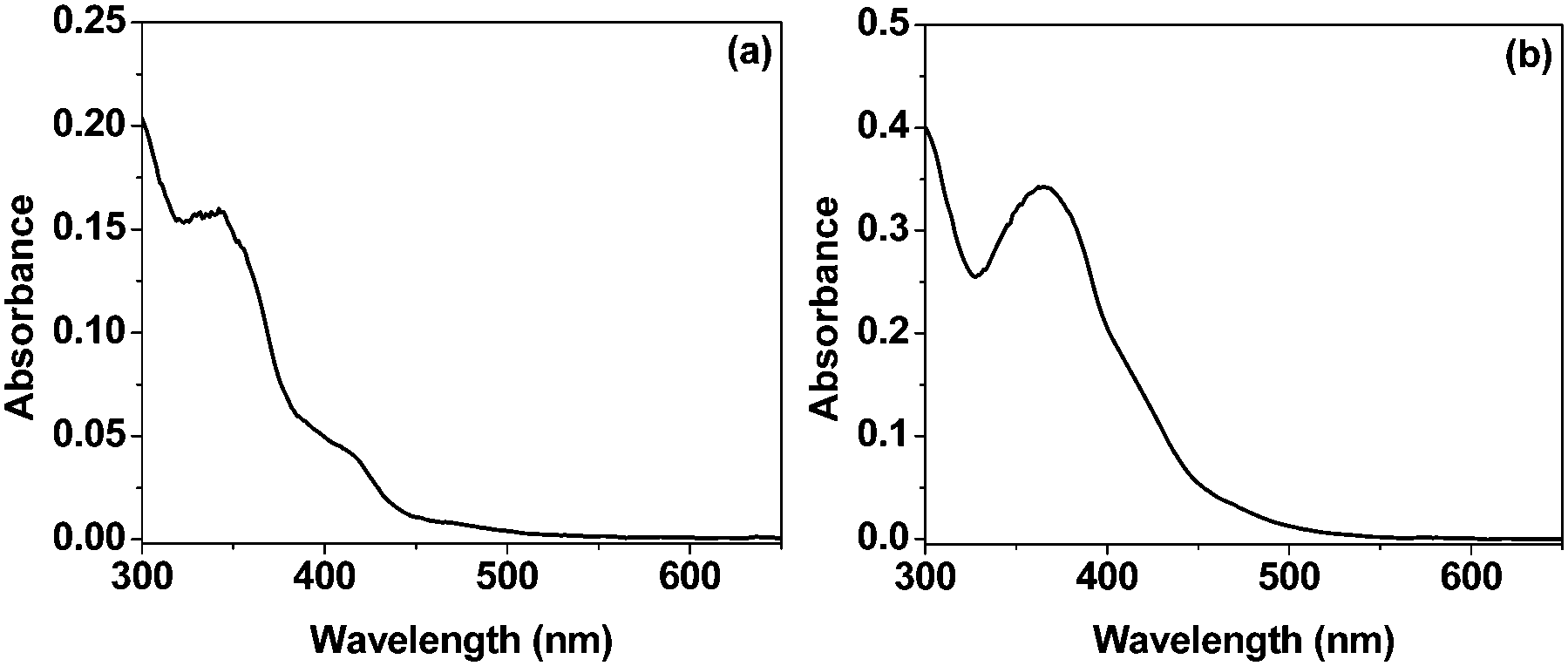 | ||
| Fig. 1 Absorption spectra of the 1:1 H2O–acetonitrile solutions of 1 and 2. The concentration of both complexes is 20 μM. | ||
Interestingly, irradiating the solutions of the individual complexes, added with the sacrificial electron donor and the acceptor, with the radiation of wavelength 370 nm turns the light yellow solution blue. This produces stable methyl viologen radical cations (MV˙+) (see later) that absorb at 605 nm (Fig. 2).27–29Fig. 2(c) shows that this phenomenon (formation of MV˙+) initially occurs faster in 2, but despite a slower start, the radical cation formation becomes faster in 1 over time and overtakes that observed in 2. The sacrificial electron donor TEOA partially neutralizes the overall molecular charges of 1 and 2 and helps the acceptor MV2+ approach them to facilitate electron transfer. The slower electron transfer in the bimetallic complex 2 can be presumed to be due to lesser approachability of the MV2+ cations.
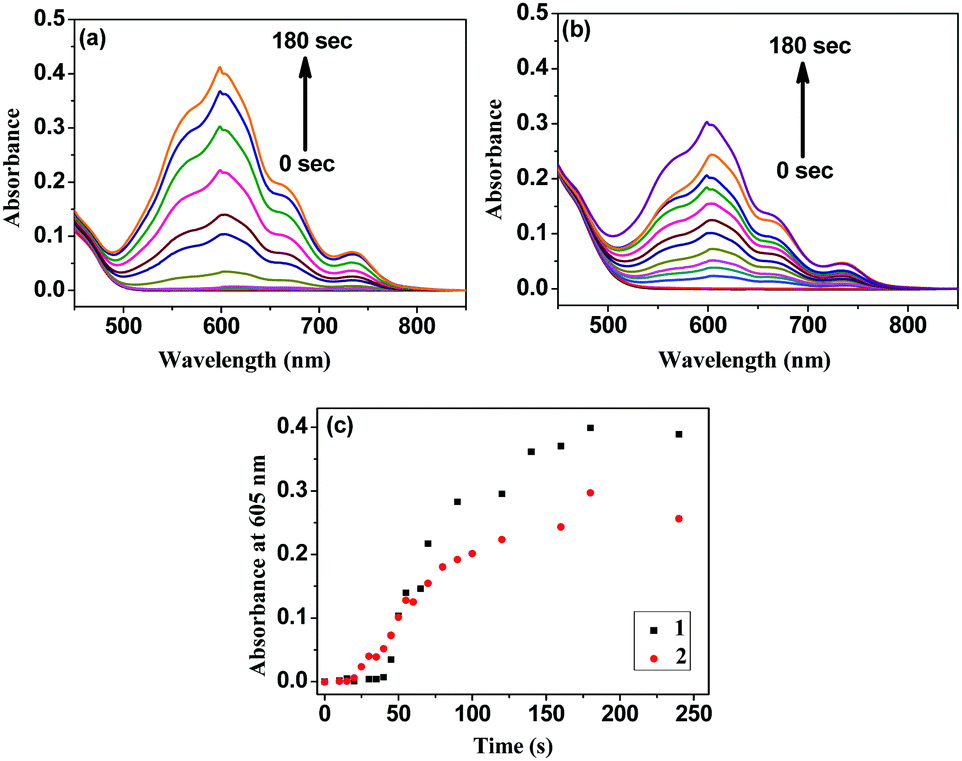 | ||
| Fig. 2 Absorption spectra of the solutions containing (a) 20 μM of 1, 625 μM of TEOA, and 100 μM of MV2+, and (b) 20 μM of 2, 625 μM of TEOA, and 100 μM of MV2+ with different light exposure times at wavelength 370 nm; (c) comparative plots of absorbance at 605 nm with time. | ||
A similar trend was observed for the quenching of the fluorescence of 1 and 2 in the presence of TEOA and MV2+ (Fig. 3). Both complexes show a broad emission peaking at 590 nm upon excitation at 370 nm in 1:1 acetonitrile–water. Previous studies showed that the presence of iridium in a compound eases intersystem crossing (ISC), rendering a populated triplet state (3MLCT and 3LC).29 Consequently, it is expected that the presence of two iridium centres in 2 would facilitate ISC more than the presence of a single iridium centre in 1. Emission mainly occurs from the triplet states (3MLCT and 3LC) for both the sensitizers. During the emission scans, N2 gas was purged through the sample solutions after each addition of MV2+ to minimize the effect of dissolved oxygen for obtaining maximum yields of the CT emissions from the triplet states.
 | ||
| Fig. 3 Quenching of fluorescence of 20 μM (a) 1 and (b) 2 added with 625 μM TEOA and 0–4.375 mM of MV2+; (c) Stern–Volmer quenching plots for the two complexes. | ||
Quenching of the emissions from two sensitizers upon the addition of MV2+ was compared via a Stern–Volmer plot using the equation, I0/I = 1 + K[Q], where I0 is the emission intensity in absence of a quencher, I is the emission intensity in presence of a quencher, K is the Stern–Volmer quenching constant, and [Q] is the quencher concentration. From the plot of I0/I vs. [MV2+] (Fig. 3c), K's for 1 and 2 were found to be 220.3 M−1 and 112.4 M−1, respectively. Despite the fact that 2 has more 3MLCT character in its excited state than 1, electron transfer to MV2+ is found to be less efficient in 2 than that in 1 mainly due to the less approachability of the electron acceptor cations in 2.
The time-resolved fluorescence decay measurements for the two sensitizers in presence of TEOA and different concentrations of MV2+ show a decrease in the average lifetime 〈τ〉 for both 1 and 2 (Fig. 4 and Tables 1 and 2). The average lifetime is calculated using 〈τ〉 = ∑αiτi/∑αi, where τi indicate the lifetimes for the individual decay components and αi are their respective contributions. The Stern–Volmer plots obtained using the equation, τ0/τ = 1 + K[Q], corroborate the steady state data, where τ0 is the lifetime of the sensitizer in the absence of MV2+, τ is the lifetime of the sensitizer in the presence of MV2+, K is the Stern–Volmer quenching constant, and [Q] is the concentration of MV2+. From the plot of τ0/τ vs. [MV2+] (Fig. 4c), K's for 1 and 2 were calculated to be 377.4 M−1 and 135.2 M−1, respectively. The third component τ3 appears in 1 because of the existence of the aldehyde functionality that may generate a CT state. 〈τ〉 decreases much faster in the case of 1 as compared to that in the case of 2 due to more favourable electron transfer in the first case.
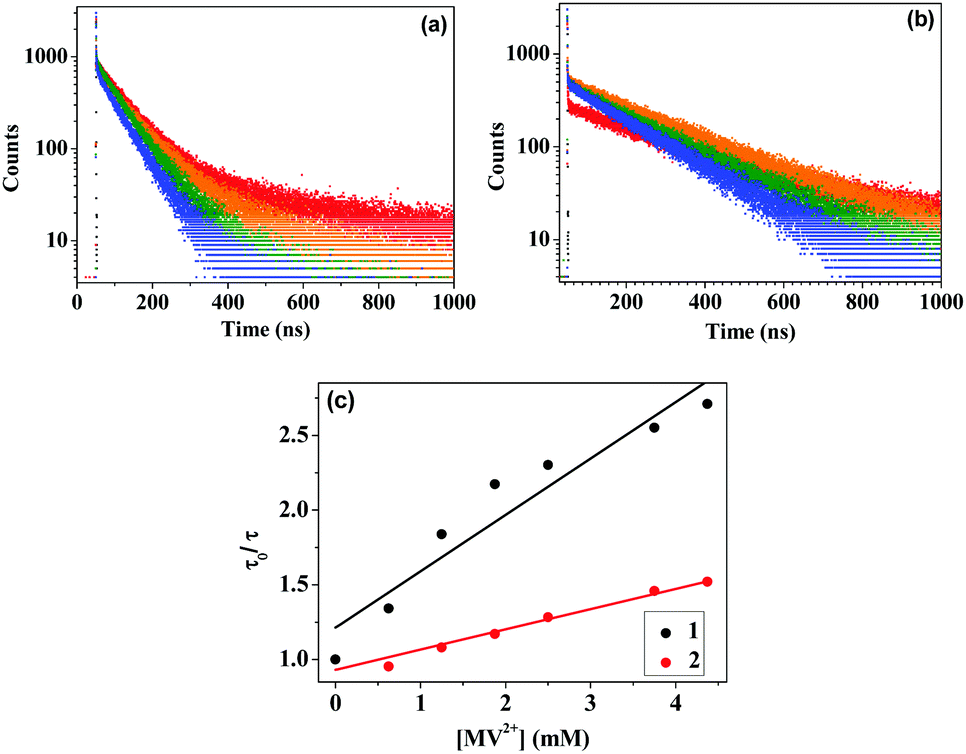 | ||
| Fig. 4 Time-resolved fluorescence data of 20 μM (a) 1, and (b) 2, added with 625 μM TEOA and 0–4.375 mM of MV2+; (c) Stern–Volmer quenching plots for the two complexes. | ||
| [MV2+] (mM) | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) | 〈τ〉 (ns) | χ 2 |
|---|---|---|---|---|---|
| 0.0 | 0.32 (1.59) | 372.1 (28.77) | 72.7 (69.64) | 157.7 | 1.05 |
| 0.625 | 0.33 (2.05) | 237.9 (28.31) | 72.2 (69.64) | 117.6 | 1.12 |
| 1.250 | 0.33 (2.00) | 142.1 (31.84) | 61.3 (66.16) | 85.8 | 1.08 |
| 1.875 | 0.31 (2.17) | 48.1 (35.96) | 89.3 (61.87) | 72.6 | 1.10 |
| 2.500 | 0.32 (3.56) | 40.4 (24.10) | 81.2 (72.85) | 68.5 | 1.13 |
| 3.750 | 0.33 (3.19) | 9.6 (3.96) | 66.2 (92.85) | 61.8 | 1.11 |
| 4.375 | 0.36 (2.26) | 8.4 (3.17) | 61.2 (94.57) | 58.2 | 1.09 |
| [MV2+] (mM) | τ 1 (ns) | τ 2 (ns) | 〈τ〉 (ns) | χ 2 |
|---|---|---|---|---|
| 0.0 | 0.33 (2.51) | 261.0 (97.49) | 254.4 | 1.07 |
| 0.625 | 0.18 (1.49) | 271.0 (98.51) | 266.9 | 1.01 |
| 1.250 | 0.19 (1.36) | 238.8 (98.64) | 235.6 | 1.04 |
| 1.875 | 0.19 (1.67) | 221.2 (98.33) | 217.5 | 1.05 |
| 2.500 | 0.20 (1.87) | 202.2 (98.13) | 198.4 | 1.06 |
| 3.750 | 0.19 (1.64) | 177.5 (98.36) | 174.6 | 1.09 |
| 4.375 | 0.19 (2.16) | 171.0 (97.84) | 167.3 | 1.11 |
Oxidative electron transfer in 1 and 2 was confirmed through electrochemical studies. A CV study allowed us to understand the details of the ground state electronic movements in the coordination complexes in the absence and presence of TEOA and MV2+ (Fig. 5). 1 and 2 show irreversible oxidation peaks at around 1.25 V and 1.32 V, respectively, which are attributed to the oxidation of the iridium centres, Ir(III) → Ir(IV). In the presence of TEOA, new oxidation peaks arise for both 1 and 2 at around 0.78 V and 0.81 V, respectively. Hence, 2 shows an oxidation peak 30 mV higher than that of 1. These low voltage oxidation peaks probably appear due to ligand oxidations. It is useful to note here that upon the addition of TEOA, the solution becomes alkaline (pH 8–10 depending upon the amount of TEOA added to the solution). Hence, the –NH on the ligand can get deprotonated by the base, leaving a net negative charge on the N-atom. This is the origin of the oxidation peaks for the ligand. The structure of 1 suggests more electron density on the N-atom as compared to that of 2 as the benzaldehyde moiety has an electron pushing effect and in 2, there is greater room for the delocalization of the electron density. This makes 1 more oxidizable as compared to 2, as suggested by the voltage.
 | ||
| Fig. 5 Cyclic voltammograms of 1 and 2 in the absence and presence of TEOA and MV2+. | ||
The solutions of 1 and 2 turn blue upon irradiation with 370 nm light, suggesting the formation of MV˙+. Note that in absence of TEOA, no blue coloration appears. This can be explained in terms of partial neutralization of the net positive charge of the complexes due to the addition of TEOA that, otherwise, restricts the approach of the electron acceptor MV2+. This effect is more pronounced in monopositive 1 as compared to the dipositive 2. Furthermore, the iridium oxidation peaks shift to 1.21 V and 1.26 V for 1 and 2, respectively. In the presence of TEOA, the electron density significantly increases on the ancillary ligands, and hence, oxidation becomes much easier.
The formation and existence of the MV˙+ radical in solution was established via flash photolysis experiments in the presence of the sacrificial donor TEOA (Fig. 6). The transient absorption spectrum for 1 shows a ground state bleach at around 350 nm, one sharp positive absorption band at 400 nm, and a broad band at around 550 nm (Fig. 6a). These are indicative of the excited 3MLCT species. The absorption decays with time at the abovementioned three wavelengths (Fig. 6b). MV˙+ shows characteristic absorption bands at 390 and 605 nm. In the presence of TEOA and MV2+, growth components are observed at 400 nm and 600 nm, respectively, which are indicative of MV˙+ (Fig. 6d).30,31 Similar observations for 2 also confirm the formation of MV˙+ in this case. Hence, conversion of MV2+ to MV˙+ under the present experimental conditions provides evidence for efficient PET in 1 and 2 in the presence of TEOA and MV2+, in concurrence with the steady state data.
 | ||
| Fig. 6 Flash photolysis data for 1 (a–d) and 2 (e–h) showing the corresponding transient absorption spectra and decays in the absence (b and f) and presence (d and h) of TEOA and MV2+. | ||
Conclusions
Herein, a comparative study was carried out to understand the effect on PET due to the presence of one or two iridium centres in a coordination complex. For this purpose, we synthesized Ir complexes and analyzed them spectroscopically. The high spin–orbit coupling constant for Ir (3909 cm−1) makes the non-radiative ISC feasible. Although the excited state lifetime of 2 is higher than that of 1 due to its more stable structure, the latter becomes a more efficient electron donor in the excited state due to other factors. The presence of a sacrificial electron donor (TEOA) and a potential electron acceptor, MV2+, makes PET feasible in the presence of 370 nm radiation. The absence of TEOA does not allow ready availability of the acceptor in the vicinity of the complexes due to electrostatic repulsion; hence, no MV˙+ formation is observed (no blue coloration of the solution). TEOA makes the solutions of 1 and 2 alkaline and the net positive charge on the complexes is significantly suppressed significantly to facilitate the approachability of MV2+ and hence favours PET. Electrochemical analysis shows that 1 is more easily oxidized than 2 in the presence of TEOA, making the monometal-centred 1 a better candidate for PET than the bimetallic 2.Experimental
Materials
The precursor materials IrCl3·3H2O, 2-phenylpyridine (ppy), 1,10-phenanthroline, and terephthalaldehyde were purchased from Sigma-Aldrich and used as received. Solvents used for synthetic purposes were of analytical grade. The spectroscopic measurements were conducted using spectroscopy grade solvents. The cyclometalated iridium(III) chloro-bridged dimer [Ir(ppy)2Cl]2, 1,10-phenanthroline-5,6-dione, 2-(4-formylphenyl)imidazo[4,5-f][1,10]-phenanthroline (fmp), and 2,2-p-phenylene(imidazo[4,5-f][1,10]-phenanthroline) (H2bpib) were synthesized according to literature procedures.19,32Syntheses
The compounds 1 and 2 were synthesized following previously reported procedures.26 Both complexes were precipitated by adding potassium hexafluorophosphate (KPF6) in 1:1 dichloromethane–methanol at room temperature and purified by silica gel column chromatography using a 1–10% methanol in dichloromethane mixture. Orange-yellow coloured complexes were obtained and the structures were confirmed by 1H-NMR spectroscopy and ESI-MS.
Instruments and methods
The UV-visible spectra were obtained using a Hitachi U-2900 double beam spectrophotometer. Herein, 400 μL of 20 μM 1 was taken in a micro quartz cuvette in 1:1 acetonitrile–water mixture for the experiments. To this, triethanolamine (TEOA), which is a sacrificial electron donor, was added ([TEOA] = 625 μM) followed by the addition of methyl viologen (MV2+), which acts as an electron acceptor ([MV2+] = 100 μM). N2 gas was purged through the solution for 15 minutes to remove dissolved oxygen, and absorbance was obtained by exposing the solution to a continuum 75 W xenon lamp with different exposure times from 0 to 180 seconds at 370 nm. A similar procedure was followed for 2. The corresponding emission spectra were obtained using a PTI QuantaMaster-40 spectrofluorometer.
The fluorescence lifetimes for the respective systems were obtained using a Horiba Jobin Yvon time-resolved spectrofluorimeter with picoseconds time resolution using the time-correlated single-photon counting (TCSPC) method. The instrument is furnished with a FluoroHub single photon counting controller and a FC-MCP-50SC MCP-PMT detection unit. The samples were excited by a 377 nm laser head and emission was monitored at 590 nm. The fluorescence decay was obtained with the gradual addition of MV2+ (0–4.375 mM) after purging the solutions with N2 gas for 15 minutes before each measurement. The same procedure was followed for 2.
Cyclic voltammetry (CV) was performed using a potentiostat/galvanostat (model 263A) from Princeton Applied Research equipped with a cell stand from BASi. A Ag/AgCl reference electrode, Pt-wire counter electrode, and Pt-disc working electrode were used in this experiment. Herein, 4 ml of 1 mM 1 solution in 1:1 acetonitrile–water was put into a glass container. An excess amount of tetrabutylammonium perchlorate (about 170 mg) was added to this solution, followed by purging with N2 for 15 minutes. The cyclic voltammogram was obtained for the system at a 50 mV s−1 scan rate. The same process was followed for the other solutions.
The nanosecond flash photolysis setup obtained from Applied Photophysics comprising a Nd:YAG laser (Lab series, Model Lab 150, Spectra Physics) was used for obtaining the transient absorption spectra. Samples were excited at 355 nm using the Nd:YAG laser. Then, 25 μM of 1 in 2 ml of 1:1 acetonitrile–water was placed in a cuvette, and the solution was degassed by purging with argon gas for 20 minutes. Transient absorption spectra were obtained using a continuum 150 W xenon lamp placed perpendicularly to the laser beam. Spectra for the solution containing 25 μM of 1, 250 μM of MV2+, and 500 μM of TEOA were similarly obtained. The same procedure was followed for 2.
Acknowledgements
SKS is thankful to the Council of Scientific and Industrial Research for a Senior Research Fellowship. This work is supported by the Department of Science and Technology through research grants ST/FT/CS-057/2009 (PG) and EMR/2015/000950 (PP). The authors thank Prof. Samita Basu of Saha Institute of Nuclear Physics for allowing the flash photolysis experiments.Notes and references
- I. M. Dixon, J. P. Collin, J. P. Sauvage, L. Flamigni, L. S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385–391 RSC.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Berlin, 2007 Search PubMed.
- A. F. Henwood, A. K. Bansal, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuelb and E. Zysman-Colman, J. Mater. Chem. C, 2016, 4, 3726–3737 RSC.
- X. Yang, X. Xu, J. S. Dang, G. Zhou, C. L. Ho and W. Y. Wong, Inorg. Chem., 2016, 55, 1720–1727 CrossRef CAS PubMed.
- E. Baranoff, J. H. Yum, M. Graetzel and M. K. Nazeeruddin, J. Organomet. Chem., 2009, 694, 2661–2670 CrossRef CAS.
- Z. Ning, Q. Zhang, W. Wu and H. Tian, J. Organomet. Chem., 2009, 694, 2705–2711 CrossRef CAS.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- Q. Zhao, M. Yu, L. Shi, S. Liu, M. Shi, Z. Zhou, C. Huang and F. Li, Organometallics, 2010, 29, 1085–1091 CrossRef CAS.
- C. H. Lin, Y. Y. Chang, J. Y. Hung, C. Y. Lin, Y. Chi, M. W. Chung, C. L. Lin, P. T. Chou, G. H. Lee, C. H. Chang and W. C. Lin, Angew. Chem., Int. Ed., 2011, 50, 3182–3186 CrossRef CAS PubMed.
- S. Lee, S. O. Kim, H. Shin, H. J. Yun, K. Yang, S. K. Kwon, J. J. Kim and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14321–14328 CrossRef CAS PubMed.
- S. Mondal, S. K. Seth, P. Gupta and P. Purkayastha, J. Phys. Chem. C, 2015, 119, 25122–25128 CAS.
- S. Serroni, S. Campagna, G. Denti, A. Juris, M. Venturi and V. Balzani, J. Am. Chem. Soc., 1994, 116, 9086–9091 CrossRef CAS.
- L. Hammarström, F. Barigelletti, L. Flamigni, M. T. Indelli, N. Armaroli, G. Calogero, M. Guardigli, A. Sour, J. P. Collin and J. P. Sauvage, J. Phys. Chem. A, 1997, 101, 9061–9069 CrossRef.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- L. F. Gildea and J. A. G. Williams, Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, U.K., 2013 Search PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- H. Chao, R.-H. Li, C.-W. Jiang, L.-N. Ji and X.-Y. Li, J. Chem. Soc., Dalton Trans., 2001, 1920–1926 RSC.
- P. Srinivasan, R. H. Mason, J. R. G. MacNeil and B. J. MacLean, Inorg. Chim. Acta, 2011, 366, 116–121 CrossRef CAS.
- F. Gao, H. Chao, F. Zhou, B. Peng and L.-N. Ji, Inorg. Chem. Commun., 2007, 10, 170–173 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Photochemistry and Photophysics of Coordination Compounds: Iridium, in Topics in Current Chemistry, ed. V. Balzani, S. Campagna, Springer, 2007, 281, 143–203 Search PubMed.
- B. Gholamkhass, K. Koike, N. Negishi, H. Hori, T. Sano and K. Takeuchi, Inorg. Chem., 2003, 42, 2919–2932 CrossRef CAS PubMed.
- G. F. Manbeck, E. Fujita and J. J. Concepcion, J. Am. Chem. Soc., 2016, 138, 11536–11549 CrossRef CAS PubMed.
- F. Gao, X. Chen, F. Zhou, L.-P. Weng, L.-T. Guo, M. Chen, H. Chao and L.-N. Ji, Inorg. Chim. Acta, 2009, 362, 4960–4966 CrossRef CAS.
- S. K. Seth, S. Mandal and P. Purkayastha, Polyhedron, 2015, 95, 14–23 CrossRef CAS.
- E. E. Méndez, C. Crespo-Hernández, R. Figueroa, R. Arce and E. Quiñones, J. Photochem. Photobiol., A, 2001, 142, 19–24 CrossRef.
- V. Martínez-Junza, A. Rizzi, K. A. Jolliffe, N. J. Head, M. N. Paddon-Row and S. E. Braslavsky, Phys. Chem. Chem. Phys., 2005, 7, 4114–4125 RSC.
- S. Y. Takizawa, K. Shimada, Y. Sato and S. Murata, Inorg. Chem., 2014, 53, 2983–2995 CrossRef CAS PubMed.
- G. F. Manbeck, E. Fujita and J. J. Concepcion, J. Am. Chem. Soc., 2016, 138, 11536–11549 CrossRef CAS PubMed.
- S. Takizawa, K. Shimada, Y. Sato and S. Murata, Inorg. Chem., 2014, 53, 2983–2995 CrossRef CAS PubMed.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647–6653 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |