
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative coupling of tetraalkynyltin with aldehydes leading to alkynyl ketones†
Andrey S.
Levashov
 *a,
Nicolai A.
Aksenov
bc,
Inna V.
Aksenova
b and
Valeriy V.
Konshin
a
*a,
Nicolai A.
Aksenov
bc,
Inna V.
Aksenova
b and
Valeriy V.
Konshin
a
aDepartment of Organic Chemistry & Technologies, Kuban State University, 149 Stavropolskaya str., 350040 Krasnodar, Russian Federation. E-mail: aslevashov@mail.ru
bDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin str., 355009 Stavropol, Russian Federation
cPeoples Friendship University Russia, 6 Miklukho Maklaya St, Moscow 117198, Russia
First published on 30th June 2017
Abstract
The reaction of tetraalkynyltin with aldehydes was studied for the first time. The reaction was shown to proceed as a tandem process of nucleophilic addition of tin acetylide to aldehyde followed by Oppenauer-type oxidation of produced tin alcoholates, and may be used as a convenient one-pot approach to acetylenic ketones. The advantages and limitations of the proposed method are discussed.
Introduction
α,β-Acetylenic ketones are valuable reagents for the synthesis of isoxazoles,1,2 chromones,3,4 triazoles,5 quinolones,6 indenones,7–12 thiazoles,13 1,5-benzodiazepines,14 spirocyclic products15etc. (Scheme 1). One of the most straightforward and useful methods to prepare alkynyl ketones is based on a two-step procedure including the reaction of metal acetylides with aldehydes followed by a subsequent oxidation of propargylic alcohols formed.6,16–19 | ||
| Scheme 1 Some applications of alkynyl ketones. | ||
Trialkyl(ethynyl)tin R3SnC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR′ were successfully used in this reaction as reactive acetylene species. The reaction with aldehydes occurs in the presence of InCl3,20 and also in the presence of organoboron and organoaluminum catalysts, to give propargyl alcohols.21,22 Although the results achieved with this method are usually good, all the procedures involving the use of trialkyltin reagents suffer from some drawbacks such as a high E-factor23 (which is defined as the mass ratio of the waste to desired product) and high toxicity of the R3Sn ballast moiety. As far as we know, the literature refers to only one example of the use of other ethynyltin compounds in the reaction with aldehydes; thus, alkynyltin trichloride Cl3SnCCR′, which was generated in situ from 1-alkynes and the SnCl4–Bu3N system, was shown to react with aldehydes.24 It should be noted that this approach is less dangerous and more environmentally friendly since it avoids the use of highly toxic C(sp2)–Sn and C(sp3)–Sn reagents; however, the E-factor still remains too high.
CR′ were successfully used in this reaction as reactive acetylene species. The reaction with aldehydes occurs in the presence of InCl3,20 and also in the presence of organoboron and organoaluminum catalysts, to give propargyl alcohols.21,22 Although the results achieved with this method are usually good, all the procedures involving the use of trialkyltin reagents suffer from some drawbacks such as a high E-factor23 (which is defined as the mass ratio of the waste to desired product) and high toxicity of the R3Sn ballast moiety. As far as we know, the literature refers to only one example of the use of other ethynyltin compounds in the reaction with aldehydes; thus, alkynyltin trichloride Cl3SnCCR′, which was generated in situ from 1-alkynes and the SnCl4–Bu3N system, was shown to react with aldehydes.24 It should be noted that this approach is less dangerous and more environmentally friendly since it avoids the use of highly toxic C(sp2)–Sn and C(sp3)–Sn reagents; however, the E-factor still remains too high.
Recently, we have developed two convenient methods for the preparation of tetraalkynyltin compounds (RCC)4Sn, by a reaction of 1-alkynes either with SnCl4 in the presence of anhydrous ZnCl2 and Et2NH,25,26 or with tin tetra(N,N-diethylcarbamate) in the presence of anhydrous ZnCl2.27 It is noteworthy that although tetraalkynyltin compounds were first prepared as early as the 1950s,28 their synthetic potential was not sufficiently realized. There are only a few reports available on the reactions of tetraalkynyltin reagents with alcohols,29 acids,30 Grignard reagents,31 organoboron compounds32–36 and with aryl halides under Stille-type conditions.37
Tetraalkynyltin compounds, as well as other C(sp)–Sn species, were proved to be superior reagents in terms of toxicity and atom efficiency in comparison with alkyl- and alkenyltin reagents, and could be compared with sodium acetylides with respect to a low molecular weight and a low E-factor. Considering the low toxicity and atom economy, tetraalkynyltins (RCC)4Sn seemed to be good compounds to be used as a source of soft acetylide nucleophiles RCC−. Based on this idea, we suggested that the reaction between tetraalkynyltin compounds and aldehydes would be a good starting point for preparation of propargylic alcohols and α,β-acetylenic ketones. To our knowledge, this approach was not reported by any of the previous workers. It is the aim of the present paper to fill this gap.
Results and discussion
When we studied the reaction of tetraalkynyltin 1 with aliphatic aldehydes 2, we found that, besides the expected products of nucleophilic addition 3, acetylenic ketones 4 were formed (Scheme 2). It was suggested that ketones 4 resulted from the Oppenauer-type oxidation of propargyl alcoholate by a second molecule of an aldehyde 2 (Scheme 3), by analogy with oxidative addition of alkynes to aldehydes in the presence of InBr338 or ZnI2.39 | ||
| Scheme 2 The reaction of tetraalkynyltin with aliphatic aldehydes. | ||
 | ||
| Scheme 3 The suggested mechanism of Oppenauer-type propargyl alcoholate oxidation. | ||
It was found that the reaction does not occur in the absence of Lewis acids as catalysts. The ratio of alcohols 3 and ketones 4 depends mostly on the ratio of the starting reagents and the nature of a solvent used. The effects of the different factors on the ratio of products 3aa/4aa obtained by the reaction of (Ph–CC)4Sn 1a with hexanal 2a are summarized in Table 1. A similar picture was observed when tetraphenylethynyltin 1a was reacted with isobutyraldehyde 2b. The experimental data show that the oxidation rate strongly depends on the solvent polarity. Presumably, a more polar solvent favors the tin atom solvation that resulted in a decrease in complexation of tin atoms with propargyl alcohols and aldehydes 2 leading to the formation of an Oppenauer-type six membered transition state A (Scheme 3). As a result, the oxidation rate is decreased. It should also be noted that when tin tetrachloride was used as a catalyst in 1,2-dichloroethane and an aliphatic aldehyde was used in excess, side reactions occur and aldehyde self-condensation products were detected using GC–MS in the reaction mixture.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4aa in the reaction of (Ph–CC)4Sn 1a with hexanal 2aa
:4aa in the reaction of (Ph–CC)4Sn 1a with hexanal 2aa
| Ratio 1a:2a |
Solvent | Catalyst | Time, h | Ratio 3aa/4aa |
|---|---|---|---|---|
|
a The ratio of products 3aa:4aa was determined using GC-MS. Unless otherwise stated, the reaction conditions were as follows: 1a (0.102 mmol), 2a (0.407 mmol), a catalyst (0.04 mmol) and a solvent (0.5 mL) at 60 °C.
b Here and throughout the paper: DCE = 1,2-dichloroethane.
c An eight-fold excess of hexanal 2a (0.814 mmol) was used.
|
||||
| 1:4 |
DCEb | — | 2 | No reaction |
| 5 | Traces of 3aa | |||
| 1:4 |
DCE | SnCl4 | 2 | 7.5 |
| 5 | 2.7 | |||
| 1:4 |
Dioxane | SnCl4 | 2 | 11.5 |
| 5 | 7.1 | |||
| 1:4 |
THF | ZnCl2 | 2 | 10.7 |
| 5 | 7.1 | |||
| 1:4 |
THF | InCl3 | 2 | 10.1 |
| 5 | 14.5 | |||
| 1:8c |
DCE | SnCl4 | 2 | 3.2 |
| 5 | 1.8 | |||
When aromatic aldehydes were allowed to react with (Ph–CC)4Sn 1a, almost complete oxidation reaction occurs even if the ratio of the starting ethynyltin 1a:aldehyde 2 was 1:4 (no aldehyde excess). Only the addition of an electron donating reagent (Et3N) was required to slow down the oxidation and to give a mixture of alcohol 3 and ketone 4 (Table 2). To explore the effect of Lewis acid on the reaction, all the experiments were conducted with both SnCl4 and ZnCl2.
C–C(O)Ph 4ac in the reaction of tetraphenylethynyltin 1a with PhCHO 2ca
| Ratio 1a:2c |
Solvent | Catalyst | Time, h | Yield of 4ac, % |
|---|---|---|---|---|
|
a Yields were determined by GC-MS. Unless otherwise stated, the reaction conditions were as follows: (Ph–CC)4Sn 1a (0.123 mmol), PhCHO 2c (0.492 mmol), a catalyst (0.05 mmol) and a solvent (0.5 mL), 60 °C.
b An eight-fold excess of benzaldehyde 2c (0.984 mmol) was used.
c Triethylamine (0.25 mmol) was added.
|
||||
| 1:4 |
Dioxane | ZnCl2 | 24 | 4 |
| 1:4 |
Dioxane | SnCl4 | 5 | 2 |
| 1:4 |
DMF | ZnCl2 | 24 | 1 |
| 1:4 |
DMF | SnCl4 | 3 | 0 |
| 1:4 |
THF | ZnCl2 | 3 | 7 |
| 1:4 |
DCE | SnCl4 | 1 | 87 |
| 3 | 99 | |||
| 1:4 |
DCE | ZnCl2 | 1 | 77 |
| 3 | 100 | |||
| 1:4 |
DCE |
ZnCl2
Et3Nc |
1 |
80
(10% 3ac) |
| 1:8b |
DCE | SnCl4 | 1 | 13 |
| 3 | 11 | |||
| 5 | 69 | |||
| 1:4 |
PhMe | SnCl4 | 3 | 98 |
| 1:4 |
PhMe | ZnCl2 | 1 | 100 |
| 1:8b |
PhMe | SnCl4 | 1 | 49 |
| 3 | 86 | |||
They were chosen as Lewis acids due to the successful application in similar reactions with aliphatic aldehydes. It was found that the use of SnCl4 provides a faster reaction, but also causes some resinification and hence is less efficient.
When the amount of SnCl4 was increased to 25 mol%, no significant acceleration of the reaction was observed while the yields of ketone products were a little bit lower (see the ESI† for details). The use of non-polar solvents (PhMe, DCE) resulted in good yields of products 4. Thus, when (Ph–CC)4Sn 1a and PhCHO 2c were taken in a 1:4 ratio, PhCHO was fully converted to acetylenic ketone and PhCH2OH after 1–3 h at 60 °C. Meanwhile, the reaction of (Ph–CC)4Sn 1a with aromatic aldehydes almost does not occur in polar aprotic solvents such as dioxane, THF, or DMF (Table 2), and the starting reagents were detected in the reaction mixture. Next, we run a series of detailed experiments to find an optimal temperature at which the highest preparative yields of the model compound Ph–CC–C(O)Ph 4ac were achieved (Table 3). The reaction between (Ph–CC)4Sn 1a and PhCHO 2c does not occur at room temperature. The temperature in the range of 40–60 °C was determined to be optimal for the reaction. A further increase in the temperature to 80 °C causes a decrease in yields of the target ketones 4 and also results in the formation of side products.
C)4Sn 1a with PhCHO 2ca
| Ratio 1a:2c |
Solvent | Catalyst | T,°C | Time, h | Yield of 4ac, % |
|---|---|---|---|---|---|
|
a Isolated yields are given. Unless otherwise stated, the reaction conditions were as follows: (Ph–CC)4Sn 1a (0.123 mmol), PhCHO 2c (0.492 mmol or 0.984 mmol, depending on the 1a:2c ratio), catalyst (0.05 mmol), and solvent (0.5 mL).
b Acetone (36 μL, 0.492 mmol) was added.
c 2 mL of DCE were used.
d Triethylamine (0.05 mmol) was added.
|
|||||
| 1:4 |
DCE | SnCl4 | 60 | 1.5 | 50 |
| 1:4b |
PhMe | ZnCl2 | 60 | 3 | 45 |
| 1:4b |
DCE | ZnCl2 | 60 | 2 | 50 |
| 1:8 |
DCEc | ZnCl2 | 60 | 5 | 72 |
| 1:8 |
DCE | ZnCl2 | 60 | 5 | 80 |
| 1:8 |
DCEc |
ZnCl2
Et3Nd |
60 | 5 | 56 |
| 1:8 |
DCE |
ZnCl2
Et3Nd |
60 | 5 | 83 |
| 1:8 |
DCE | SnCl4 | 60 | 5 | 71 |
| 1:8 |
PhMe | ZnCl2 | 60 | 5 | 98 |
| 1:8 |
PhMe | ZnCl2 | 40 | 5 | 97 |
| 1:8 |
PhMe | ZnCl2 | 80 | 5 | 88 |
| 1:8 |
PhMe | ZnCl2 | 25 | 5 | 0 |
Then we tried to run the reaction in the presence of acetone as the possible oxidant/hydrogen acceptor for the Oppenauer-type oxidation of the intermediate tin alcoholates. However, the addition of acetone in 4-fold molar excess with regard to tetraphenylethynyltin 1a gave no increase in the yield of ketone 4ac. It is noteworthy that no Favorskii-type acetone alkynylation products were detected.
The yields of target ketones 4 strongly depend on the concentration of the starting compounds (Table 3). This fact is in a good agreement with the previously reported results of oxidative coupling of alkynes with aldehydes in the presence of InBr3 and Et3N.39 Finally, the best yields (up to quantitative) of the model ketone Ph–CC–C(O)Ph 4ac were achieved when the reaction was carried out in toluene at 60 °C using ZnCl2 as a Lewis acid catalyst (Table 3).
All the reactions were conducted at 60 °C in a dry solvent under an argon atmosphere to prevent the hydrolysis of tetraalkynyltin 1. With an improved preparative protocol for the synthesis of ketones 4 in hand, we have prepared and isolated a series of α,β-acetylenic ketones 4. Tetraalkynyltin 1 and aldehydes 2 used in the reaction are shown in Fig. 1. The preparative yields of ketones 4 are shown in Table 4. It is clear from the table that the best results were obtained with aldehydes bearing electron withdrawing groups.
 | ||
| Fig. 1 The scope of tetraalkynyltin 1 and aldehydes 2 used. | ||
| Tetraalkynyltin 1 | Aldehyde 2 | Product | Yield,a % | |
|---|---|---|---|---|
| a Isolated yields are given. b Resinification of the reaction mixture was observed. | ||||
| 1a | 2c | Ph–CC–C(O)Ph |
4ac | 98 |
| 1a | 2d | 4-CF3C6H4C(O)–CC–Ph |
4ad | 97 |
| 1b | 2e | 4-MeC6H4–CC–C(O)C6H4Br-4 |
4be | 85 |
| 1a | 2f | 4-NO2C6H4C(O)–CC–Ph |
4af | 50b |
| 1a | 2g | 4-MeOC6H4C(O)–CC–Ph |
4ag | 55 |
| 1a | 2h | 2,3-(MeO)2C6H3C(O)–CC–Ph |
4ah | 66 |
| 1a | 2i | 4-BzOC6H4C(O)–CC–Ph |
4ai | 83 |
| 1a | 2j | 4-(t-Bu C6H4CO2)C6H4C(O)–CC–Ph |
4aj | 79 |
| 1b | 2k | 4-MeC6H4CC–C(O)C6H4CC–SiMe3 |
4bk | 80 |
| 1b | 2c | 4-MeC6H4–CC–C(O)Ph |
4bc | 57 |
| 1a | 2l | Ph–CC–C(O)C6H4CCPh |
4al | 90 |
| 1a | 2m | Ph–CC–C(O)–thienyl-2 |
4am | 30b |
| 1a | 2n | 5-NO2–thienyl–C(O)–CC–Ph |
4an | 47b |
| 1b | 2o | 5-NO2C4H2OC(O)–CC–C6H4Me-4 |
4bo | 0b |
| 1c | 2c | 4-ClC6H4CC–C(O)Ph |
4cc | 48 |
| 1d | 2c |
n-Bu–CC–C(O)Ph |
4dc | 27 |
We have to note that under the given conditions the reaction of (Ar–CC)4Sn with either 4-NO2C6H4CHO 2f, thiophene-2-carbaldehyde 2m or 5-nitrothiophene-2-carbaldehyde 2n is accompanied by formation of tarry products and resulted in much lower yields of target acetylene ketones 4. The attempts to obtain ketone 4bo starting from 5-nitrofurfural 2o failed because of strong resinification probably due to complexation and side reactions of the aldehyde with ZnCl2. The crystalline structure of the previously unreported alkynyl ketone 4aj was studied using the X-ray diffraction technique (Fig. 2). Crystal data for compound 4aj have been deposited (CCDC 1543712, http://www.ccdc.cam.ac.uk).†
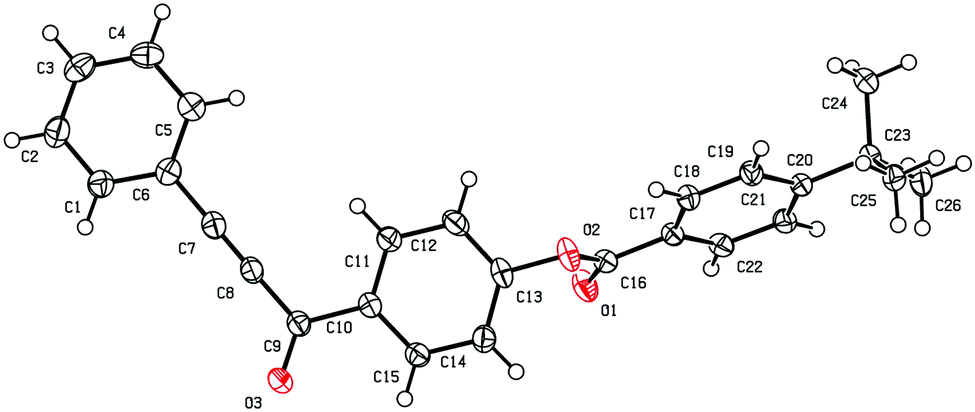 | ||
| Fig. 2 The general view of 4-(3-phenylprop-2-ynoyl)phenyl 4-tert-butylbenzoate 4aj in a crystal. | ||
Conclusions
In summary, we have developed a one-pot, mild and atom-economical method for the preparation of α,β-alkynyl ketones starting from aldehydes and easily available tetraalkynyltin by a sequence involving a nucleophilic addition of a tin acetylide to aldehyde following by the Oppenauer-type oxidation of tin propargyl alcoholates formed. The reaction conditions were optimized, the scope and limitations of the method were explored.Experimental
Materials and methods
Solvents and starting reagents were thoroughly dried and purified according to common procedures.40 All reactions were carried out in an argon (99.993%) atmosphere. 1H, 13C, and 119Sn NMR spectra were recorded on a JEOL ECA 400 instrument at operating frequencies 399.78, 100.52 respectively, in CDCl3 (Aldrich) with reference to TMS or to the residual signals of a solvent. Chemical shifts are given in ppm, coupling constants are given in Hz. IR spectra were recorded on a InfraLUM FT-02 instrument in the range of 400–4200 cm−1 (KBr or HCCl3 solution) and on a Bruker Vertex 70 instrument in ATR (attenuated total reflection) mode. Mass spectra (EI, 70 eV) were obtained on a Shimadzu GCMS-QP 2010 spectrometer. Melting points were measured in open capillaries on a Stuart SMP30 apparatus and are uncorrected. The purity of the compounds was checked by TLC (Sorbfil A plates) with toluene or a hexane:AcOEt (10:1) mixture as an eluent. The spots were visualized with iodine vapors, KMnO4–H2SO4 solution or UV-light. The use of a sorbent consisting of 85% non-modified silica gel and 15% silica gel modified with 3-aminopropyltriethoxysilane (1.14 mmol g−1 of NH2 groups) resulted in a much better separation of the desired acetylenic ketones and interfering by-products on a column.
The starting tetraalkynyltin 1a–d were obtained according to the reported methods.25,27 Aldehydes 2a–h, k, m–o are commercially available (Aldrich). Aldehyde 2l was prepared according to the known procedure.37
4-Formylphenyl benzoate (2i)
Sodium hydrocarbonate (16.0 g, 0.19 mol) was added portionwise to a stirred suspension of 4-HOC6H4CHO (10.0 g, 0.082 mol) in water (100 mL) at room temperature, and the mixture was stirred until the aldehyde is completely dissolved. The solution was cooled on an ice bath and PhC(O)Cl (9.8 mL, 0.085 mol) was added dropwise under vigorous stirring. After all the acyl chloride has been added, the ice bath is removed, and the reaction mixture is then allowed to warm to room temperature. Then the mixture was stirred for 1 h at 25 °C, the precipitated product was filtered off, washed with aqueous NaHCO3 and water. For purification benzoate 2i was reprecipitated from acetone with water. Yield 12.0 g (65%, purity by GCMS 100%). White crystalline solid, m.p. 89.5–90.2 °C. 1H NMR (400 MHz, CDCl3) δ 7.41 (d, 3J = 8.7 Hz, 2H, Ar), 7.51–7.55 (m, 2H, Ar), 7.64–7.68 (m, 1H, Ar), 7.97 (d, 3J = 8.7 Hz, 2H, Ar), 8.19–8.21 (m, 2H, Ar). 13C NMR (100 MHz, CDCl3) δ 122.5, 128.7, 128.9, 130.2, 131.3, 134.0, 155.6, 164.5, 191.0. IR (KBr, cm−1) νmax 3097.5, 3070.5, 3053.2 (C–H, C–C), 2821.7, 2790.9, 2726.3 (C–H, CHO), 1734.9 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COO), 1696.3 (CO, CHO), 1595.1. MS (m/z, EI, 70 eV) 226 ([M+], 0.3), 105 (100), 77 (43), 51 (15).
O, COO), 1696.3 (CO, CHO), 1595.1. MS (m/z, EI, 70 eV) 226 ([M+], 0.3), 105 (100), 77 (43), 51 (15).
4-Formylphenyl 4-tert-butylbenzoate (2j)
Sodium hydrocarbonate (4.0 g, 47.6 mmol) was added portionwise to a stirred suspension of 4-HOC6H4CHO (2.5 g, 20.5 mmol) in water (25 mL) at room temperature, and the mixture was stirred until the aldehyde becomes completely dissolved. The solution was cooled on an ice bath and 4-tert-butylbenzoyl chloride (4.13 g, 21 mmol) was added dropwise under vigorous stirring. Then THF (15 mL) and Bu4NBr (0.1 g) were added, the mixture was stirred for 2 h at room temperature and left overnight. The precipitated solid was filtered off, washed with aqueous NaHCO3 and water. For purification product 2j was reprecipitated from acetone with water. Yield was 5.0 g (87%, purity by GCMS – 98%). The further recrystallization from EtOH gave pure 2j (purity by GCMS – 100%) as white crystalline solid, m.p. 88.3–88.8 °C. 1H NMR (400 MHz, CDCl3) δ 1.37 (s, 9H, Me), 7.40 (d, 3J = 8.7 Hz, 2H, Ar), 7.55 (d, 3J = 8.3 Hz, 2H, Ar), 7.96 (d, 3J = 8.7 Hz, 2H, Ar), 8.13 (d, 3J = 8.7 Hz, 2H, Ar), 10.02 (s, 1H, CHO). 13C NMR (100 MHz, CDCl3) δ 31.1, 35.3, 122.6, 125.7, 126.1, 130.2, 131.2, 134.0, 155.9, 158.0, 164.5, 190.9. IR (KBr, cm−1) νmax 2966.4, 2904.7, 2740.7 (C–H, C–C), 1731.0, 1705.0, 1606.6, 1594.1. MS (m/z, EI, 70 eV) 282 ([M+], 0.1), 267 (0.6), 161 (100), 146 (10.7), 133 (5.2), 118 (13.9), 91 (9.4).General procedure for the reaction of tetra(phenylethynyl)tin 1a with aliphatic aldehydes 2a,b (the model reaction, Scheme 2 and Table 1)
A 2 mL sealable Wheaton vial was charged with 0.04 mmol of Lewis acid (ZnCl2, SnCl4 or InCl3), 0.102 mmol of (PhCC)4Sn 1a and 0.5 mL of a dry solvent. Then the vial was flushed with a stream of dry argon, and 0.407 mmol (or 0.814 mmol if the ratio 1a:2a was set as 1:8) of aliphatics aldehydes 2a or 2b were added subsequently through a syringe. The mixture was stirred for the indicated time (Table 1). The progress of the reaction was monitored using GC-MS (before the analysis, samples taken at regular intervals were quenched with saturated aqueous NH4Cl).
Preparative procedure for the synthesis of 1,3-diphenylprop-2-yn-1-one (4ac)
A 2 mL sealable Wheaton vial was charged with 10.4 mg (0.076 mmol) of anhydrous ZnCl2, PhMe (0.8 mL), 100 mg (0.191 mmol) tetra(phenylethynyl)tin 1a and 155 μL (1.53 mmol) of freshly distilled PhCHO 2c. A reaction mixture was stirred at 60 °C for 5 h, then treated with 1 M aqueous HCl. The product was extracted with HCCl3 and purified using column chromatography (eluent – PhMe). The yield of ketone Ph–CC–C(O)Ph 4ac was 154.6 mg (98%), light yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.39–7.53 (m, 5H, Ar), 7.60–7.69 (m, 3H, Ar), 8.22 (d, J = 7.3 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 86.8, 93.1, 120.1, 128.6, 128.7, 129.5, 130.8, 133.0, 134.1, 136.8, 177.9. IR (liquid film, cm−1) νmax 3099.9, 3082.6, 3059.5, 3034.4, 2199.1 (CC), 1641.6 (CO), 1597.2, 1581.8. MS (m/z, EI, 70 eV) (Irel (%)): 206 (M+, 52), 178 (88), 152 (11), 129 (100), 101 (14), 89 (11), 77 (26), 76 (21), 75 (33), 51 (35).
3-Phenyl-1-[4-(trifluoromethyl)phenyl]prop-2-yn-1-one (4ad)
Acetylenic ketone 4-CF3C6H4C(O)–CC–Ph 4ad was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 64.4 mg (0.123 mmol) of (PhCC)4Sn 1a and 135 μL (0.984 mmol) of 4-(trifluoromethyl)-benzaldehyde 2d. The yield was 130.7 mg (97%), light yellow solid, m.p. 73–74 °C (from EtOH–H2O). 1H NMR (400 MHz, CDCl3) δ 7.42–7.53 (m, 3H, Ar), 7.70 (d, 3J = 8.7 Hz, 2H, Ar), 7.79 (d, 3J = 8.2 Hz, 2H, Ar), 8.32 (d, 3J = 8.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 86.6, 94.5, 119.7, 123.6 (q, 1JC–F = 273.1 Hz), 125.7 (q, 3JC–F = 3.8 Hz), 128.8, 129.8, 131.2, 133.2, 135.2 (q, 2JC–F = 23.6 Hz), 139.5, 176.7. IR (KBr, cm−1) νmax 3059.9, 2202.6 (CC), 1643.3 (CO). MS (m/z, EI, 70 eV) (Irel (%)): 274 (M+, 43), 246 (40), 129 (100), 98 (13), 75 (19).
1-(4-Bromophenyl)-3-(4-methylphenyl)prop-2-yn-1-one (4be)
Acetylenic ketone 4-MeC6H4-CC–C(O)C6H4Br-4 4be was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 71.3 mg (0.123 mmol) of (MeC6H4CC)4Sn 1b and 182 mg (0.984 mmol) of 4-bromobenzaldehyde 2e. The yield was 125.1 mg (85%), light yellow solid, m.p. 110.5–111.5 °C (from EtOH). 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H, CH3), 7.23 (d, 3J = 7.8 Hz, 2H, Ar), 7.58 (d, 3J = 7.8 Hz, 2H, Ar), 7.65 (d, 3J = 8.7 Hz, 2H, Ar) 8.07 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 21.8, 86.5, 94.4, 116.8, 129.4, 129.6, 130.9, 131.9, 133.2, 135.8, 141.8, 176.9. IR (KBr, cm−1) νmax 3085.0, 3030.0, 2914.3, 2855.5, 2194.9 (CC), 1636.5 (CO), 1603.7, 1583.5, 1569.0. MS (m/z, EI, 70 eV) (Irel (%)): 300([M+, 81Br], 27), 398([M+, 79Br], 26), 272 (28, 81Br), 270 (28, 79Br), 190 (10), 189 (19), 143 (100), 115 (16), 95 (18), 89 (16), 75 (15), 63 (11), 50 (9).
1-(4-Nitrophenyl)-3-phenylprop-2-yn-1-one (4af)
Acetylenic ketone 4-NO2C6H4C(O)–CC–Ph 4af was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 64.4 mg (0.123 mmol) of (PhCC)4Sn 1a and 149 mg (0.984 mmol) of 4-nitrobenzaldehyde 2f. The yield was 61.8 mg (50%), light yellow solid, m.p. 160 °C (from toluene). 1H NMR (400 MHz, CDCl3) δ 7.44–7.48 (m, 2H, Ar), 7.52–7.56 (m, 1H, Ar), 7.71 (d, 3J = 8.7 Hz, 2H, Ar), 8.35–4.10 (m, 4H, Ar); 13C NMR (100 MHz, CDCl3) δ 86.6, 95.4, 119.4, 123.9, 128.9, 130.4, 131.5, 133.3, 141.1, 150.9, 175.9. IR (KBr, cm−1) νmax 3113.0, 3048.4, 2193.9 (CC), 1646.1 (CO), 1593.1 (NO2 st as), 1516.0, 1343.4, 1321.2 (NO2 st sy). MS (m/z, EI, 70 eV) (Irel (%)): 251 (M+, 33), 223 (13), 193 (7), 176 (11), 129 (100), 101 (10), 75 (23).
1-(4-Methoxyphenyl)-3-phenylprop-2-yn-1-one (4ag)
Acetylenic ketone 4-MeOC6H4C(O)–CC–Ph 4ag was prepared according to a similar procedure to that for 4ac, using 10.4 mg (0.076 mmol) of anhydrous ZnCl2, PhMe (0.8 mL), 100.0 mg (0.191 mmol) of (PhCC)4Sn 1a and 186.2 μL (1.53 mmol) of 4-methoxybenzaldehyde 2g. The yield was 98.8 mg (55%), light yellow solid, m.p. 98–99 °C (from EtOH). 1H NMR (400 MHz, CDCl3) δ 3.90 (s, 3H, OMe), 6.99 (d, 3J = 8.7 Hz, 2H, Ar), 7.39–7.49 (m, 3H, Ar), 7.67 (d, 3J = 8.7 Hz, 2H, Ar), 8.19 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 55.6, 86.9, 92.3, 113.9, 120.4, 128.7, 130.4, 130.6. 132.0, 133.0, 164.5, 176.7. IR (KBr, cm−1) νmax 3111.7, 2957.7, 2847.8, 2195.9 (CC), 1626.9 (CO), 1599.9, 1594.1, 1570.0. MS (m/z, EI, 70 eV) (Irel (%)): 236 (M+, 100), 208 (99), 193 (73), 165 (53), 129 (75), 104 (12), 101 (17), 92 (15), 75 (29), 63 (19), 51 (15).
1-(2,3-Dimethoxyphenyl)-3-phenylprop-2-yn-1-one (4ah)
Acetylenic ketone 2,3-(MeO)2C6H3C(O)–CC–Ph 4ah was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, PhMe (0.5 mL), 64.4 mg (0.123 mmol) of (PhCC)4Sn 1a and 164 mg (0.984 mmol) of 2,3-dimethoxybenzaldehyde 2h. The yield was 86.4 mg (66%), yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.90 (s, 3H, OMe), 3.99 (s, 3H, OMe), 7.12–7.14 (m, 2H, Ar), 7.36–7.47 (m, 3H, Ar), 7.52–7.57 (m, 1H, Ar), 7.62–7.65 (m, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 56.1, 61.7, 89.5, 91.6, 117.1, 120.6, 122.3, 123.8, 128.6, 130.5, 132.1, 133.0, 149.8, 153.5, 176.9. IR (liquid film, cm−1) νmax 3063.3, 3003.5, 2937.9, 2837.6, 2203.0 (CC), 1645.5, 1624.3, 1593.4, 1577.9. MS (m/z, EI, 70 eV) (Irel (%)): 266 (M+, 26), 255 (14), 207 (25), 165 (25), 152 (36), 151 (29), 150 (22), 135 (14), 129 (100), 126 (24), 122 (79), 115 (23), 107 (18), 101 (23), 75 (50), 51 (48).
4-(3-Phenylprop-2-ynoyl)phenyl benzoate (4ai)
Acetylenic ketone 4-PhC(O)OC6H4C(O)–CC–Ph 4ai was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, PhMe (0.5 mL), 64.4 mg (0.123 mmol) of (PhCC)4Sn 1a and 222.6 mg (0.984 mmol) of 4-formylphenyl benzoate 2i. The yield was 133.3 mg (83%), white solid, m.p. 96.5–97.5 °C (from EtOH). 1H NMR (400 MHz, CDCl3) δ 7.38–7.44 (m, 4H, Ar), 7.47–7.55 (m, 3H, Ar), 7.64–7.71 (m, 3H, Ar), 8.20–8.22 (m, 2H, Ar), 8.29–8.32 (m, 2H, Ar). 13C NMR (100 MHz, CDCl3) δ 86.8, 93.4, 120.1, 122.1, 128.7, 129.0, 130.3, 130.9, 131.2, 133.1, 134.0, 134.6, 155.6, 164.5, 176.8. IR (KBr, cm−1) νmax 3063.8, 2197.8 (CC), 1734.9 (CO, COO), 1635.6 (CO), 1597.9, 1585.4.
4-(3-Phenylprop-2-ynoyl)phenyl 4-tert-butylbenzoate (4aj)
Acetylenic ketone 4-(t-BuC6H4CO2)C6H4C(O)–CC–Ph 4aj was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 64.4 mg (0.123 mmol) of (PhCC)4Sn 1a and 277.8 mg (0.984 mmol) of 4-formylphenyl 4-tert-butylbenzoate 2j. The yield was 148.6 mg (79%), white solid, m.p. 140.8–141.2 °C (from HCCl3–hexane). 1H NMR (400 MHz, CDCl3) δ 1.37 (s, 9H, Me), 7.38 (d, 3J = 8.7 Hz, 2H, Ar), 7.41–7.49 (m, 3H, Ar), 7.54 (d, 3J = 8.7 Hz, 2H, Ar), 7.69 (d, 3J = 8.2 Hz, 2H, Ar), 8.14 (d, 3J = 8.7 Hz, 2H, Ar), 8.30 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 31.1, 35.2, 86.8, 93.3, 120.1, 122.1, 125.7, 126.2, 128.7, 130.2, 130.8, 131.2, 133.1, 134.5, 155.7, 157.9, 164.5, 176.8. IR (KBr, cm−1) νmax 3059.5, 2961.1, 2905.1, 2866.6, 2201.0 (CC), 1734.9 (CO, COO), 1630.1 (CO), 1597.2, 1583.7.
3-(4-Tolyl)-1-{4-[(trimethylsilyl)ethynyl]phenyl}prop-2-yn-1-one (4bk)
Acetylenic ketone 4-MeC6H4CC–C(O)C6H4CC–SiMe34bk was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 71.3 mg (0.123 mmol) of (MeC6H4CC)4Sn 1b and 199 mg (0.984 mmol) of 4-[(trimethylsilyl)ethynyl]benzaldehyde 2k. The yield was 124.3 mg (80%), light yellow solid. M.p. 142.5–144.0 °C (from hexane). 1H NMR (400 MHz, CDCl3) δ 0.28 (s, 9H, Me3Si), 2.41 (s, 3H, CH3), 7.23 (d, 3J = 8.2 Hz, 2H, Ar), 7.57–7.59 (m, 4H, Ar), 8.14 (d, 3J = 8.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 0.21, 21.8, 86.7, 94.3, 98.9, 104.0, 116.9, 128.8, 129.3, 129.5, 132.0, 133.1, 136.3, 141.7, 177.1. IR (KBr, cm−1) νmax 3063.3, 2959.2, 2918.6, 2897.4, 2191.4 (CC), 2160.5 (CC), 1628.1 (CO), 1599.2, 1556.7, 1506.6. MS (m/z, EI, 70 eV) (Irel (%)): 316([M+], 28), 301 (100), 143 (26), 115 (7).
3-(4-Methylphenyl)-1-phenylprop-2-yn-1-one (4bc)
Acetylenic ketone 4-MeC6H4–CC–C(O)Ph 4bc was prepared according to a similar procedure to that for 4ac, using 10.4 mg (0.076 mmol) of anhydrous ZnCl2, toluene (0.8 mL), 110.6 mg (0.191 mmol) of (MeC6H4CC)4Sn 1a and 155.4 μL (1.53 mmol) of benzaldehyde 2c. The yield was 96.0 mg (57%), light yellow solid, m.p. 58.3–59.2 °C (from hexane, with freezing). 1H NMR (400 MHz, CDCl3) δ 2.40 (s, 3H, Me), 7.22 (d, 3J = 7.8 Hz, 2H, Ar), 7.49–7.52 (m, 2H, Ar), 7.57–7.63 (m, 3H, Ar), 8.21 (d, 3J = 8.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 86.8, 93.8, 117.0, 128.6, 129.5, 129.6, 133.1, 134.0, 137.0, 141.6, 178.1. IR (KBr, cm−1) νmax 3059.9, 3031.0, 2187.2 (CC), 1626.9 (CO), 1603.7, 1598.9, 1578.7. MS (m/z, EI, 70 eV) (Irel (%)): 220 (M+, 65), 192 (60), 189 (14), 165 (11), 143 (100), 115 (19), 89 (17), 77 (18), 63 (12), 51 (16).
3-Phenyl-1-[4-(phenylethynyl)phenyl]prop-2-yn-1-one (4al)
Acetylenic ketone Ph–CC–C(O)C6H4CC–Ph 4al was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 64.4 mg (0.123 mmol) of (PhCC)4Sn 1a and 203 mg (0.984 mmol) of 4-(phenylethynyl)benzaldehyde 2l. The yield was 135.2 mg (90%), light yellow solid, m.p. 104.9–105.2 °C (from heptane). 1H NMR (400 MHz, CDCl3) δ 7.35–7.39 (m, 3H, Ar), 7.41–7.51 (m, 3H, Ar), 7.55–7.57 (m, 2H, Ar), 7.65 (d, 3J = 8.7 Hz, 2H, Ar), 7.69 (d, 3J = 8.7 Hz, 2H, Ar), 8.20 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 86.8, 88.7, 93.5, 120.0, 122.5, 128.4, 128.7, 128.9, 129.2, 129.5, 130.9, 131.7, 131.8, 133.1, 136.0. 177.0. IR (KBr, cm−1) νmax 3080.7, 3055.6, 3034.4, 2201.1 (CC), 2164.4 (CC), 1628.1 (CO), 1603.0, 1556.7. MS (m/z, EI, 70 eV) (Irel (%)): 306([M+], 95), 278 (100), 276 (23), 176 (15), 139 (31), 129 (80), 101 (11), 75 (15).
3-Phenyl-1-(2-thienyl)prop-2-yn-1-one (4am)
Acetylenic ketone Ph–CC–C(O)C4H3S-2 4am was prepared according to a similar procedure to that for 4ac, using 10.4 mg (0.076 mmol) of anhydrous ZnCl2, toluene (0.8 mL), 100 mg (0.191 mmol) of (PhCC)4Sn 1a and 143 μL (1.53 mmol) of thiophene-2-carbaldehyde 2m. The product was extracted from a reaction mixture with boiling hexane. The yield was 48.5 mg (30%), light yellow solid, m.p. 57.0–57.6 °C (from hexane, with freezing). 1H NMR (400 MHz, CDCl3) δ 7.10–7.12 (m, 1H, Ar), 7.32–7.43 (m, 3H, Ar), 7.59 (d, 3J = 8.2 Hz, 2H, Ar), 7.65 (d, 3J = 4.6 Hz, 1H, Ar), 7.94 (d, 3J = 3.7 Hz, 1H, Ar); 13C NMR (100 MHz, CDCl3) δ 86.4, 91.7, 119.9, 128.3, 128.7, 130.8, 133.0, 135.0, 135.2, 144.9, 169.7. IR (KBr, cm−1) νmax 3074.9, 2206.8 (CC), 1612.7 (CO), 1595.3, 1579.9, 1516.2. MS (m/z, EI, 70 eV) (Irel (%)): 212 (M+, 71), 184 (100), 152 (23), 139 (23), 129 (77), 111 (11), 101(17), 92 (12), 75 (36), 51 (20).
1-(5-Nitro-2-thienyl)-3-phenylprop-2-yn-1-one (4an)
Acetylenic ketone 5-NO2C4H2SC(O)–CC–Ph 4an was prepared according to a similar procedure to that for 4ac, using 10.4 mg (0.076 mmol) of anhydrous ZnCl2, toluene (0.8 mL), 100 mg (0.191 mmol) of (PhCC)4Sn 1a and 212.8 mg (1.53 mmol) of 5-nitrothiophene-2-carbaldehyde 2n. The product was extracted from a reaction mixture with boiling hexane. The yield was 93.1 mg (47%), yellow solid, m.p. 151.3–152.3 °C (from HCCl3, with freezing). 1H NMR (400 MHz, CDCl3) δ 7.43–7.56 (m, 3H, Ar), 7.68 (d, 3J = 8.2 Hz, 2H, Ar), 7.86 (d, 3J = 4.1 Hz, 1H, Ar), 7.93 (d, 3J = 4,1 Hz, 1H, Ar); 13C NMR (100 MHz, CDCl3) δ 85.6, 95.2, 118.9, 128.2, 128.9, 131.7, 131.9, 133.3, 148.0, 156.8, 169.1. IR (KBr, cm−1) νmax 3107.7, 3092.3, 3065.3, 2193.3 (CC), 1618.5, 1610.8, 1593.4, 1535.5, 1512.4. MS (m/z, EI, 70 eV) (Irel (%)): 257 (M+, 51), 229 (20), 199 (10), 139 (32), 29 (100), 101 (14), 75 (22), 51 (11).
3-(4-Chlorophenyl)-1-phenylprop-2-yn-1-one (4cc)
Acetylenic ketone 4-ClC6H4CC–C(O)Ph 4cc was prepared according to a similar procedure to that for 4ac, using 6.7 mg (0.05 mmol) of anhydrous ZnCl2, toluene (0.5 mL), 81.3 mg (0.123 mmol) of (4-ClC6H4CC)4Sn 1c and 100 μL (0.984 mmol) of PhCHO 2c. The yield was 56.4 mg (48%), light yellow solid, m.p. 105.5–106.5 °C (from hexane). 1H NMR (400 MHz, CDCl3) δ 7.40 (d, 3J = 8.7 Hz, 2H, Ar), 7.49–7.53 (m, 2H, Ar), 7.59–7.65 (m, 3H, Ar), 8.19 (d, 3J = 8.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 87.6, 91.6, 118.6, 128.7, 129.2, 129.6, 134.2, 136.8, 137.2, 177.8. IR (KBr, cm−1) νmax 3082.6, 3053.7, 2922.5, 2203.0 (CC), 2164.4 (CC), 1632.0 (CO), 1616.5, 1599.2, 1589.5, 1579.9. MS (m/z, EI, 70 eV) (Irel (%)): 242([M+] 37Cl, 24), 240 ([M+] 35Cl, 75), 214 (32), 212 (100), 176 (33), 165 (31), 163 (100), 151 (12), 135 (11), 106 (20), 99 (39), 88 (16), 77 (33), 51 (29).
1-Phenylhept-2-yn-1-one (4dc)
Acetylenic ketone CH3CH2CH2CH2–CC–C(O)Ph 4dc was prepared according to a similar procedure to that for 4ac, using 10.4 mg (0.076 mmol) of anhydrous ZnCl2, toluene (0.8 mL), 84.7 mg (0.191 mmol) of (PhCC)4Sn 1a and 155 μL (1.53 mmol) of PhCHO 2c. The yield was 38.9 mg (27%), light yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.97 (t, J = 7.3 Hz, 3H, CH3), 1.46–1.56 (m, 2H, CH2), 1.63–1.71 (m, 2H, CH2), 2.51 (t, J = 7.3 Hz, 2H, CH2), 7.46–7.49 (m, 2H, Ar), 7.58–7.61 (m, 1H, Ar), 8.14 (d, 3J = 8.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 13.5, 18.9, 22.1, 29.9, 79.7, 96.8, 128.5, 129.6, 133.9, 137.0, 178.3. IR (liquid film, cm−1) νmax 3063.3, 2959.1, 2932.1, 2874.3, 2237.7, 2201.0, 1645.5, 1597.2, 1579.9. MS (m/z, EI, 70 eV) (Irel (%)): 186 (M+, 7), 185 (13), 157 (43), 144 (100), 129 (27), 115 (57), 109 (27), 105 (95), 79 (40), 77(73), 66 (32), 53 (26), 51 (39).
X-ray studies of 4-(3-phenylprop-2-ynoyl)phenyl 4-tert-butylbenzoate (4aj)
Single crystals of ketone 4aj were obtained by recrystallization from EtOH. X-ray diffraction studies were performed on an Agilent SuperNova, Dual, Cu at zero, AtlasS2 diffractometer at 100 K. Using Olex2,41 the structure was solved with the ShelXT42 structure solution program using Intrinsic Phasing and refined by the least squares technique using the ShelXL43 refinement package. Crystals of compound 4aj are triclinic, C26H22O3 (M = 382.43), space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No 2), a = 6.7125(3) Å, b = 9.9947(3) Å, c = 15.6535(3) Å, α = 88.139(2)°, β = 87.315(2)°, γ = 80.902(3)°, V = 1035.50(6) Å3, Z = 2, T = 100.00(10) K, μ(CuKα) = 0.630 mm−1, Dcalc = 1.227 g cm−3, 19353 reflections were collected (8.964° ≤ 2θ ≤ 147.682°), of which 4133 were unique (Rint = 0.0373, Rsigma = 0.0216). The final probability factors were: R1 = 0.0374 (I >2σ(I)), wR2 = 0.1038 (all reflections). The full crystallographic data have been placed at the Cambridge Crystallographic Data Center as deposit CCDC 1543712.†
(No 2), a = 6.7125(3) Å, b = 9.9947(3) Å, c = 15.6535(3) Å, α = 88.139(2)°, β = 87.315(2)°, γ = 80.902(3)°, V = 1035.50(6) Å3, Z = 2, T = 100.00(10) K, μ(CuKα) = 0.630 mm−1, Dcalc = 1.227 g cm−3, 19353 reflections were collected (8.964° ≤ 2θ ≤ 147.682°), of which 4133 were unique (Rint = 0.0373, Rsigma = 0.0216). The final probability factors were: R1 = 0.0374 (I >2σ(I)), wR2 = 0.1038 (all reflections). The full crystallographic data have been placed at the Cambridge Crystallographic Data Center as deposit CCDC 1543712.†
Acknowledgements
This publication was financially supported by the Russian Ministry of Education and Science (project no. 4.4892.2017/8.9). N. A. A. and I. V. A. are grateful for financial support from the Russian Ministry of Education and Science (State Assignment to Higher Education Institutions, project no. 4.5547.2017/64 and agreement no. 02.A03.21.0008). This work was performed with the use of equipment of the Research and Educational Centrum “Diagnostics of the structure and properties of nanomaterials”.Notes and references
- D. A. Bulanov, I. A. Novokshonova, V. V. Novokshonov, I. A. Ushakov and I. V. Sterkhova, Synth. Commun., 2017, 47, 335–343 CrossRef CAS.
- R. Harigae, K. Moriyama and H. Togo, J. Org. Chem., 2014, 79, 2049–2058 CrossRef CAS PubMed.
- M. Jonušis, L. Šteinys, R. Bukšnaitienė and I. Čikotienė, Synthesis, 2016, A-1 Search PubMed.
- M. Yoshida, K. Saito, Y. Fujino and T. Doi, Tetrahedron, 2014, 70, 3452–3458 CrossRef CAS.
- T. Ponpandian, S. Muthusubramanian and S. Rajagopal, Eur. J. Inorg. Chem., 2013, 3974–3977 CrossRef CAS.
- J. Shao, X. Huang, X. Hong, B. Liu and B. Xu, Synthesis, 2012, 1798–1808 CAS.
- J. Zhou, G. L. Zhang, J. P. Zou and W. Zhang, Eur. J. Inorg. Chem., 2011, 3412–3415 CrossRef CAS.
- C. Pan, B. Huang, W. Hu, X. Feng and J. T. Yu, J. Org. Chem., 2016, 81, 2087–2093 CrossRef CAS PubMed.
- Y.-K. Song, P.-C. Qian, F. Chen, C.-L. Deng and X.-G. Zhang, Tetrahedron, 2016, 72, 7589–7593 CrossRef CAS.
- A. V Vasilyev, S. Walspurger, M. Haouas, J. Sommer, P. Pale and A. P. Rudenko, Org. Biomol. Chem., 2004, 2, 3483–3489 Search PubMed.
- A. V. Vasil’ev, S. Walspurger, P. Pale, J. Sommer, M. Haouas and A. P. Rudenko, Russ. J. Org. Chem., 2004, 40, 1769–1778 CrossRef.
- A. V. Vasilyev, S. Walspurger, P. Pale and J. Sommer, Tetrahedron Lett., 2004, 45, 3379–3381 CrossRef CAS.
- T. E. Glotova, M. Y. Dvorko, A. I. Albanov, O. N. Kazheva, G. V. Shilov and O. A. D’yachenko, Russ. J. Org. Chem., 2008, 44, 1532–1537 CrossRef CAS.
- L. Yin and L. Wang, Tetrahedron Lett., 2016, 57, 5935–5940 CrossRef CAS.
- A. K. Clarke, M. J. James, P. O. Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 13798–13802 CrossRef CAS PubMed.
- B. Schmidt, M. Riemer and U. Schilde, Eur. J. Inorg. Chem., 2015, 7602–7611 CrossRef CAS.
- S. Ushijima, S. Dohi, K. Moriyama and H. Togo, Tetrahedron, 2012, 68, 1436–1442 CrossRef CAS.
- R. Livingston, L. R. Cox, S. Odermatt and F. Diederich, Helv. Chim. Acta, 2002, 85, 3052–3077 CrossRef CAS.
- T.-T. Jong and S.-J. Leu, J. Chem. Soc., Perkin Trans. 1, 1990, 423 RSC.
- M. Yasuda, T. Miyai, I. Shibata, A. Baba, R. Nomura and H. Matsuda, Tetrahedron Lett., 1995, 36, 9497–9500 CrossRef CAS.
- D. A. Evans, D. P. Halstead and B. D. Allison, Tetrahedron Lett., 1999, 40, 4461–4462 CrossRef CAS.
- E. J. Corey and K. A. Cimprich, J. Am. Chem. Soc., 1994, 116, 3151–3152 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- M. Yamaguchi, A. Hayashi and M. Hirama, Chem. Lett., 1992, 2479–2482 CrossRef CAS.
- A. S. Levashov, A. A. Andreev and V. V. Konshin, Tetrahedron Lett., 2015, 56, 1870–1872 CrossRef CAS.
- A. A. Andreev, A. S. Levashov and N. V. Komarov, RU2317993, 2008.
- A. S. Levashov, A. A. Andreev, D. S. Buryi and V. V. Konshin, Russ. Chem. Bull., 2014, 63, 775–776 CrossRef CAS.
- H. Hartmann and W. Eschenbach, Naturwissenschaften, 1959, 46, 321 Search PubMed.
- M. Lahcini, P. M. Castro, M. Kalmi and M. Leskela, Organometallics, 2004, 20, 4547–4549 CrossRef.
- M. A. Brook, B. Ramacher, C. Dallaire, H. K. Gupta, D. Ulbrich and R. Ruffolo, Inorg. Chim. Acta, 1996, 250, 49–57 CrossRef CAS.
- P. Jaumier, B. Jousseaume and M. Lahcini, Angew. Chem., Int. Ed., 1999, 38, 402–404 CrossRef CAS.
- B. Wrackmeyer and G. Kehr, J. Organomet. Chem., 1995, 501, 87–93 CrossRef CAS.
- B. Wrackmeyer, G. Kehr, A. Sebald and J. Kümmerlen, Chem. Ber., 1992, 125, 1597–1603 CrossRef CAS.
- B. Wrackmeyer, G. Kehr and B. Roland, Chem. Ber., 1992, 125, 643–650 CrossRef CAS.
- B. Wrackmeyer, G. Kehr and R. Boese, Angew. Chem., 1991, 103, 1374–1376 CrossRef CAS.
- B. Wrackmeyer and G. Kehr, Polyhedron, 1991, 10, 1497–1506 CrossRef CAS.
- A. S. Levashov, D. S. Buryi, O. V. Goncharova, V. V. Konshin, V. V. Dotsenko and A. A. Andreev, New J. Chem., 2017, 41, 2910–2918 RSC.
- Y. Ogiwara, M. Kubota, K. Kurogi, T. Konakahara and N. Sakai, Chem. – Eur. J., 2015, 21, 18598–18600 CrossRef CAS PubMed.
- J. Yuan, J. Wang, G. Zhang, C. Liu, X. Qi and Y. Lan, Chem. Commun., 2015, 51, 576–579 RSC.
- W. Armarego and C. Chai, Purification of Laboratory Chemicals, ElsevierScience, Oxford, 2003, p. 608 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, C71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data. CCDC 1543712. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj01376k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |