
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmidoxime platinum(II) complexes: pH-dependent highly selective generation and cytotoxic activity†
Dmitrii S.
Bolotin
 *a,
Marina Ya.
Demakova
a,
Anton A.
Legin
b,
Vitaliy V.
Suslonov
c,
Alexey A.
Nazarov
d,
Michael A.
Jakupec
*b,
Bernhard K.
Keppler
b and
Vadim Yu.
Kukushkin
a
*a,
Marina Ya.
Demakova
a,
Anton A.
Legin
b,
Vitaliy V.
Suslonov
c,
Alexey A.
Nazarov
d,
Michael A.
Jakupec
*b,
Bernhard K.
Keppler
b and
Vadim Yu.
Kukushkin
a
aInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab., 7/9, Saint Petersburg, Russian Federation. E-mail: d.s.bolotin@spbu.ru
bInstitute of Inorganic Chemistry and Research Platform “Translational Cancer Therapy Research”, University of Vienna, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: michael.jakupec@univie.ac.at
cCenter for X-ray Diffraction Studies, Saint Petersburg State University, Universitetskii Pr., 26, Saint Petersburg, Russian Federation
dDepartment of Chemistry, Moscow State University, Leninskie Gory, 1,3, Moscow, Russian Federation
First published on 1st June 2017
Abstract
The reaction of cis-[PtCl2(Me2![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) O)2] with 1 equiv. of each of the amidoximes RC(NH2)
O)2] with 1 equiv. of each of the amidoximes RC(NH2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH in neutral media in MeOH results in the formation of complexes cis-[PtCl2{RC(NH2)
NOH in neutral media in MeOH results in the formation of complexes cis-[PtCl2{RC(NH2)![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) OH}(Me2O)] (5 examples; 83–98% isolated yields). In the presence of 2 equiv. of NaOH in MeOH solution, the reaction of cis-[PtCl2(Me2O)2] with 1 equiv. of each of the amidoximes RC(NH2)NOH leads to [Pt{RC(H)N
OH}(Me2O)] (5 examples; 83–98% isolated yields). In the presence of 2 equiv. of NaOH in MeOH solution, the reaction of cis-[PtCl2(Me2O)2] with 1 equiv. of each of the amidoximes RC(NH2)NOH leads to [Pt{RC(H)N![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) }(Me2O)2] (7 examples; 74–95% isolated yields). All new complexes were characterized by C, H, and N elemental analyses, HRESI+-MS, IR, 1H, 13C{1H}, and CP-MAS TOSS 13C{1H} NMR spectroscopies, and additionally by single-crystal XRD (for seven species). The cytotoxic potency of six compounds was determined in the human cancer cell lines CH1/PA-1, A549, SK-BR-3, and SW480. Generally, the second class of complexes containing chelating amidoximato ligands shows much higher cytotoxicity than the non-chelate amidoxime analogs, despite the lack of easily exchangeable chlorido ligands. Especially, the complex [Pt(p-CF3C6H4C(H)N)(Me2O)2] displays a remarkable activity in the inherently cisplatin resistant SW480 cell line (0.51 μM vs. 3.3 μM).
}(Me2O)2] (7 examples; 74–95% isolated yields). All new complexes were characterized by C, H, and N elemental analyses, HRESI+-MS, IR, 1H, 13C{1H}, and CP-MAS TOSS 13C{1H} NMR spectroscopies, and additionally by single-crystal XRD (for seven species). The cytotoxic potency of six compounds was determined in the human cancer cell lines CH1/PA-1, A549, SK-BR-3, and SW480. Generally, the second class of complexes containing chelating amidoximato ligands shows much higher cytotoxicity than the non-chelate amidoxime analogs, despite the lack of easily exchangeable chlorido ligands. Especially, the complex [Pt(p-CF3C6H4C(H)N)(Me2O)2] displays a remarkable activity in the inherently cisplatin resistant SW480 cell line (0.51 μM vs. 3.3 μM).
Introduction
Platinum-based anticancer drugs are among the most widely used chemotherapeutics and applied in the treatment of a wide range of malignant tumors.1 Specifically in the treatment of testicular cancer, the introduction of cisplatin increased the survival rate to more than 90% of the diagnosed cases. The platinum chemotherapeutics cisplatin and the second and third generation compounds carboplatin and oxaliplatin, respectively, are now the first line of treatment for bladder, testicular, ovarian, head/neck and colorectal cancers.2 However, severe toxic effects such as nausea, nephro- and neurotoxicity, and resistance of common cancer types to Pt compounds as well as resistance acquired during treatment limit their application to a great extent.3The classic structure–activity relationships for platinum compounds are based on platinum being present as PtII or PtIV centers with two cis-configured leaving groups and two stable am(m)ine ligands coordinated to the metal.4 While the complexes are supposed to remain in their original composition during administration and circulation in the bloodstream, the low chloride concentration in the cell allows a chlorido/aqua exchange to occur. Sequential replacement of the leaving ligands leads to positively charged complexes that interact initially electrostatically with the negatively charged phosphate backbone of DNA before a second step of ligand exchange reactions leading to the platination of DNA. Platinum is found most commonly attached to guanine residues in the major groove and to a lesser extent to adenine moieties. In particular, the presence of adjacent guanines residues has been found to be advantageous to support the binding event between the platinum center and DNA, leading to DNA lesions that cannot be repaired easily and trigger apoptosis.5
The nature of the leaving ligand(s) has been shown to be crucial for reducing the side effects, and this has especially been demonstrated for the second and third generation compounds, carboplatin and oxaliplatin, which feature the negatively charged cyclobutanedicarboxylato and oxalato ligands, respectively.5c,6 However, there are also neutral ligands that can easily be substituted under physiological conditions. Ligands such as DMF and (CH3)2SO have been widely used in the preparation of complexes and their metal species are often identified as intermediates.6c,7 Furthermore, (CH3)2SO is found in the ruthenium-based anticancer agent NAMI-A, which was studied for its antimetastatic effects in two clinical trials before being ruled out recently.8
More recently, interest has grown in the so-called rule-breaker compounds that do not follow the classic structure–activity relationships recognized for the established anticancer agents and their close analogs. Such rule-breakers are for example trans-configured PtII complexes, but also multinuclear platinum compounds have been investigated and the latter even reached clinical trials due to their potential to overcome cisplatin resistance at least in cell culture. Such non-classic compounds have been suggested to interact with DNA in a different manner from the platinum cis-complexes and thereby would have a different mode of action that may help to overcome the drawbacks of platinum-based chemotherapy.9
These considerations led us to the design and preparation of amidoxime and amidoximato PtII complexes. Oxime complexes have been shown in the past by us and others to exhibit anticancer activity in an in vitro setting.10 Moreover, organic compounds featuring the amidoxime functional group have also been shown to be anticancer active.11 The synthesis of the platinum complexes was performed starting from the platinum precursor cis-[PtCl2(Me2O)2] and the amidoximes in methanol and in the presence of NaOH. Depending on the conditions, either the [PtCl2(amidoxime-κ1-Noxime)(Me2O)] or the [Pt(amidoximato-κ2-O,Namide)(Me2O)2] complexes were obtained. The cytotoxicity of three species of each class was screened in the human cancer cell lines CH1/PA-1, A549, SK-BR-3, and SW480, where the MTT assay revealed only marginal activity for the three amidoxime complexes, moderate activity for two of three amidoximato complexes, but a remarkably high cytotoxic potency for the complex bearing a p-trifluoromethylbenzamidoximate ligand, especially in the SW480 cell line. All our studies are consistently disclosed in sections that follow.
Results and discussion
Platinum(II) ketoxime complexes have been extensively studied for the last two decades and data related to these species indicated that ketoximes easily coordinate to the platinum(II) center by the N atom.10d,12 Such platinum(II) complexes featuring N-bound oximes react with excess platinum(II) to obtain two- or trinuclear species featuring μ2-κ1-N-κ1-O oxime ligands.10b In spite of that, amidoxime platinum(II) complexes are still remarkably less explored, whereas amidoxime complexes of other metals are studied as well as conventional oxime complexes.13 Thus, only four platinum(II) complexes featuring amidoxime ligands are presented in the literature (Fig. 1).14 Complexes A (ca. 100% yield) and B (50% yield) were obtained via the action of the corresponding amidoximes upon the binuclear complexes [Pt(μ2-Cl)(SnCl3)(PEt3)]2 or [Pt(μ2-Cl)(Ac)(C(Me)OH)]2, respectively. Complexes C (the cycle is phenyl or ferrocenyl; 25 and 18% yields) were obtained by the reaction of RC(NH2)NOH (R = Ph, Fc) with cis-[PtCl2(Me2O)2] in MeOH upon reflux for 12–16 h.
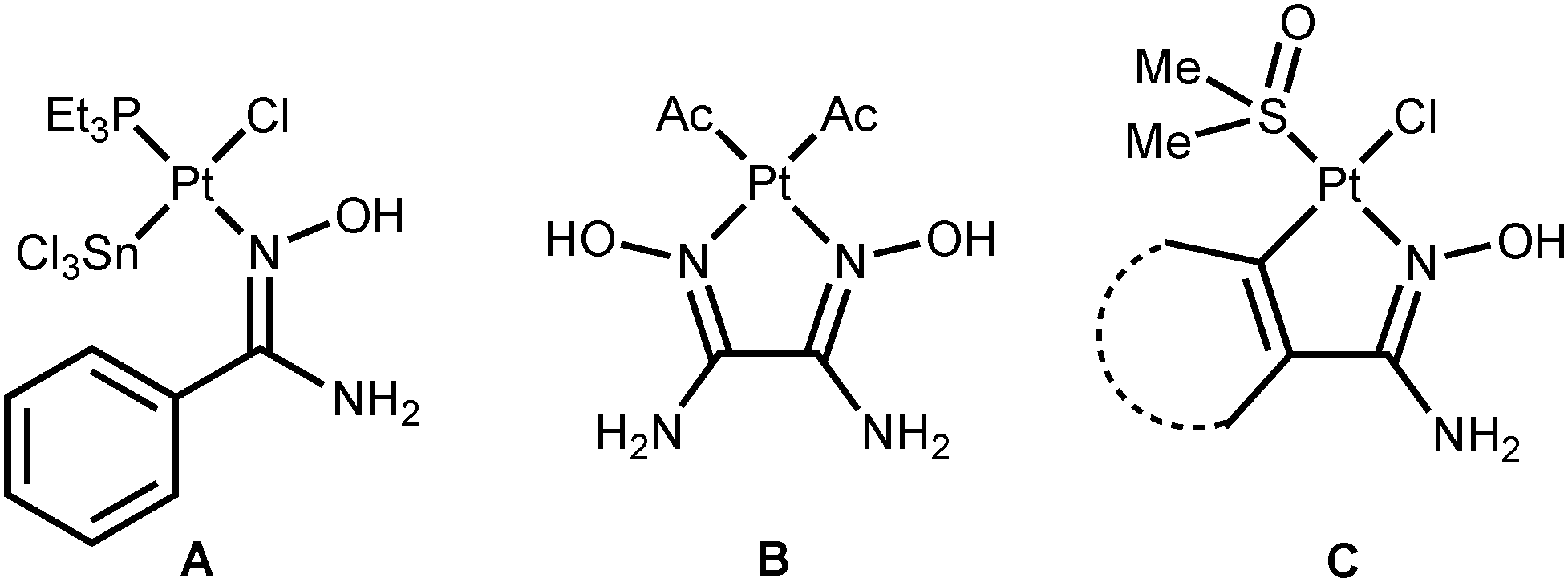 | ||
| Fig. 1 Previously reported platinum(II) amidoxime complexes. | ||
The four known amidoxime complexes feature exclusively Noxime-ligated amidoximes and the NH2 moiety of amidoxime ligands is unligated to the platinum(II) stipulating the amidoximes play a role of conventional oxime ligand. No examples of platinum(II) amidoxime complexes featuring the Namide-bound ligands were previously reported. Moreover, as analysis of literature data indicated,13 amidoxime species have at least four types of ligation in mononuclear complexes (Fig. 2) and the selective preparation of amidoxime complexes with an exact coordination mode is still an open problem.
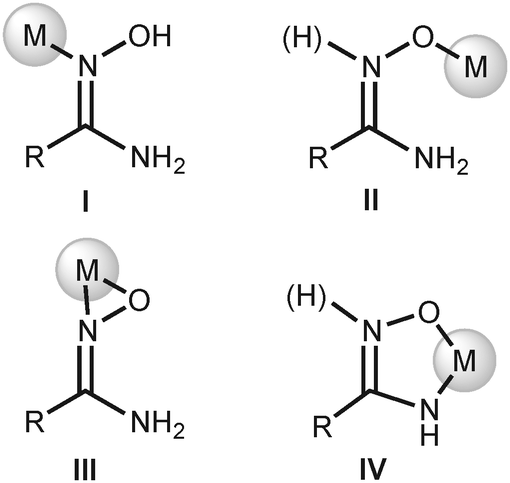 | ||
| Fig. 2 Coordination modes of amidoximes in their mononuclear complexes.13 | ||
The generation of type II complexes proceeds either with oxophilic metal centers or in the presence of a base due to the deprotonation of the O atom resulting in an increase of its nucleophilicity. The coordination of amidoximes by the O atom in basic media commonly is accompanied by the deprotonation of the NH2 moiety with consequent chelation of the metal center giving type IV complexes. Type III complexes are mostly realized at electron-deficient metal centers, viz., early transition metals or f-elements.
In this work, we decided to comprehensively investigate the preparation of amidoxime platinum(II) complexes with a particular purpose of finding selective routes for the generation of complexes with a well-defined coordination mode of amidoxime ligands. We planned the selective preparation of the complexes of types I, II, and IV by variation of the acidity of the reaction mixtures, whereas complexes of type III are hardly accessible at a rather electron-rich platinum(II) center. Previously, it was reported that the amidoxime OH group can be effectively deprotonated by NaOH in alcohol media15 and, concurrently, amidoximes can be protonated at the oxime N atom by strong acids.13 Because of these reasons, we decided to use NaOH and TfOH to vary the acidity of the reaction mixtures and to employ MeOH as a solvent.
Synthesis and characterization of amidoxime complexes 3a–b and 3d–f
As the starting materials for this study, we addressed, on the one hand, cis-[PtCl2(Me2O)2]16 (1) and, on the other hand, the aliphatic, aromatic, and heteroaromatic amidoximes RC(NH2)NOH (R = Me 2a, Et 2b, t-Bu 2c, Ph 2d, p-CF3C6H42e, p-O2NC6H42f, p-NC5H42g). The reaction of 1 equiv. of any one of the amidoximes 2a–b or 2d–f with complex 1 proceeds in MeOH at 65 °C for 5 min and results in the formation of amidoxime platinum(II) complexes 3a–b or 3d–f that were isolated in good to excellent yields (83–98%; Scheme 1a). The rate of this reaction is decreased by additions of TfOH (1 equiv.) and 3a–b or 3d–f forms for 1 h in 15–20% lower yields, which could be explained by simultaneous Tiemann rearrangement (for recent works, see ref. 17) of the amidoximes in the reaction mixtures (traces of corresponding ureas RNHC(O)NH2 were detected by 1H NMR). Complexes 4a–g (Scheme 1b) were not detected in the reaction mixtures by HRESI-MS and 1H NMR.
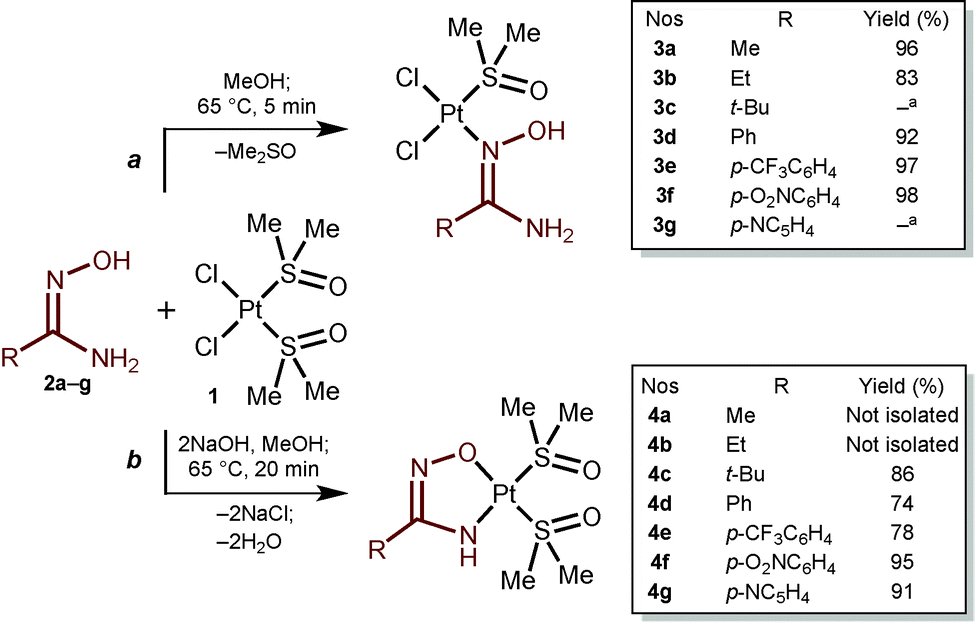 | ||
| Scheme 1 Generation of amidoxime and amidoximate platinum(II) complexes. a Analytical data are not sufficient for full characterization. | ||
Complexes 3a–b and 3d–f give satisfactory C, H, and N elemental analyses for the proposed formulas. These species were also characterized by HRESI+-MS, IR, 1H, 13C{1H}, and CP-MAS TOSS 13C{1H} NMR spectroscopies, and additionally by X-ray diffraction (XRD). Compounds 3a–b and 3d–f are stable in the solid state at RT and upon heating decompose in the range of 142–185 °C.
A characteristic feature of the positive-mode high-resolution ESI mass spectra of 3a–b and 3d–f is the availability of sets of peaks related to fragmentation and quasi-ions corresponding to [M − 2Cl + H]+, [M − Cl]+, [M − Cl − H + Na]+, [M + Na]+, [M + K]+, [2M − 2Cl − 2H + Na]+, [2M − Cl]+, [2M + Na]+, [2M + K]+, and [3M − 2Cl − 2H + Na]+.
In the IR spectra of 3a–b and 3d–f, we observed two or three medium-strong to very strong bands in the region of 3480–3215 cm−1, which can be attributed to the O–H and N–H stretches, and a set of weak to medium bands at 3187–2785 cm−1 assignable to the C–H stretches.18 All spectra also display one very strong band in the 1663–1655 cm−1 region specific for the ν(CN) of the amidoxime moiety13 and one medium to very strong band at 1138–1105 cm−1 characteristic of the ν(SO) stretches of sulfur-bound sulfoxides.19 In addition, the spectrum of 3f displays two very strong bands at 1520 and 1346 cm−1 specific for asymmetric and symmetric ν(NO) bands of the NO2 moiety.18
Complexes 3a–b and 3d–f are almost insoluble in the common deuterated organic solvents (CDCl3, CD2Cl2, (CD3)2CO, and D2O) apart from (CD3)2SO and CD3OD. In (CD3)2SO, the exchange of ligated (CH3)2SO with the solvent molecules occurs for 5 min, and therefore the spectra were recorded in CD3OD. A characteristic feature of the 1H NMR spectra is the non-equivalence of the methyl groups of the Me2SO ligand. Thus, the spectra of 3a–b exhibit one unresolved broad singlet at 3.45 ppm (3a) or two singlets at 3.46 and 3.44 ppm (3b), whereas the spectra of 3d–f, derived from aromatic amidoximes 2d–f, display two singlets at 3.42–3.37 and 2.83–2.59 ppm. In 3d–f relatively to 3a–b, the high-field shift (0.59–0.78 ppm) of one of the methyl groups of the Me2SO ligand should be associated with the shielding effect of the aromatic rings (see X-ray structure determinations).
The 13C{1H} NMR spectra in CD3OD (3a–b and 3d–e) exhibit a low-field signal at 162.29–157.89 ppm, which is attributed to the CNOH carbon of the amidoxime moiety,13 whereas in the high-field the spectra display two singlets attributed to the methyl groups of the Me2SO ligand at 43.31–42.95 and 42.98–42.55 ppm, which agree well with the two singlets observed in the 1H NMR spectra. Compound 3f exhibits poor solubility in CD3OD leading to a poor-quality 13C{1H} NMR spectrum even with long acquisition time and it was characterized by solid-state CP-MAS TOSS 13C{1H} NMR. The spectrum displays one singlet at 155.19 ppm from the C atom of the carbamidoxime moiety and four singlets of the methyl groups of Me2SO ligand in the region of 46.59–40.18 ppm. Duplication of signals related to the Me2SO ligand is probably due to two different conformations of 3f in the solid sample.
Synthesis and characterization of amidoximate complexes 4a–g
The reaction of 1 equiv. of any one of the amidoximes 2a–g with complex 1 in the presence of 2 equiv. of NaOH proceeds in MeOH at 65 °C for 20 min and leads to amidoximate platinum(II) chelated complexes 4a–g (83–98%; Scheme 1b). Compounds 4a–b were not separated from some by-products due to their similar solubilities in organic solvents and in H2O and also because of decomposition of the mixture on silica gel. However, the formation of 4a and 4b was confirmed by HRESI+-MS (4a: 424.0313 [M + H]+, calcd 424.0323; 4b: 438.0470 [M + H]+, calcd 438.0480). Complexes 4a–g form in a wide range of amidoxime and NaOH ratios. These complexes were isolated (for 4c–g) from the reaction mixtures or detected in the mixtures (for 4a–b see Fig. S26–S29, ESI†) when 1–10 equiv. of 2a–g and 1–4 equiv. of NaOH were used. It was concluded that, on the one hand, the yields of 4c–g do not significantly depend on the quantity of 2a–g when it was taken in more than 1.1-fold excess. On the other hand, the yields gradually increase with increasing relative quantity of NaOH up to 2.5-fold excess. Upon further addition of the base, the yields decreased for 10–20% in the case to 4-fold excess of NaOH. In these experiments, the formation of the corresponding open-chain species 3 was not detected by HRESI-MS and 1H NMR. Only when 0.1–2 equiv. of NaOH are used, the reaction of 1 with 2 leads to mixtures of the corresponding complexes 3 and 4.Complexes 4c–g give satisfactory C, H, and N elemental analyses for the proposed formulas. These species were also characterized by HRESI+-MS, IR, 1H, and 13C{1H} NMR spectroscopies, and additionally by XRD (for 4c and 4f). Compounds 4c–g are stable in the solid state at RT and upon heating decompose in the range of 189–218 °C.
A characteristic feature of the positive-mode high-resolution ESI mass spectra of 4c–g is the availability of sets of peaks related to fragmentation [M − Me2SO + H]+ (4e) and the quasi-ions [M + H]+ and [M + Na]+. The availability of a high-intensity set of peaks from [M + H]+, which was not observed for 3a–b and 3d–f, is probably due to protonation of the basic N atom of the oxime group, which is occupied by the platinum center in 3a–b and 3d–f.
In the IR spectra of 3c–g, we observed from one to three weak-medium to medium bands in the region of 3420–3295 cm−1, which can be attributed to the N–H stretches, and a set of weak to medium bands at 3013–2870 cm−1 assignable to the C–H stretches.18 All spectra also display one weak to medium band in the 1638–1597 cm−1 region assigned to the ν(CN) of the amidoximate moiety13 and one strong to very strong band at 1130–1126 cm−1 characteristic of the ν(SO) of sulfur-bound sulfoxides.19 In addition, the spectrum of 4f displays two strong bands at 1516 and 1341 cm−1 specific for asymmetric and symmetric NO stretches of the NO2 moiety.18
Complexes 3c–g are soluble in the most common deuterated organic solvents, but in (CD3)2SO, the exchange of ligated (CH3)2SO with the solvent molecules was observed for 15 min and therefore their 1H NMR spectra were recorded in CDCl3. A characteristic feature of the 1H NMR spectra is the availability of a singlet flanked with satellites (JPtH2 = 88–112 Hz) in the region 5.52–4.85 ppm assignable to the amide H. Another characteristic feature is the presence of two singlets with satellites (JPtH3 = 12–24 Hz) related to two Me2SO ligands.
The 13C{1H} NMR spectra in CDCl3 (4c–e and 4g) exhibit a low-field signal at 168.94–159.19 ppm, which is attributed to the HN–CNO carbon of the amidoximate moiety,13 whereas in the high-field, the spectra display two singlets flanked with satellites (4c–e: JPtC2 = 32–39 Hz; 4g: not observable due to low solubility) attributed to the methyl groups of the Me2SO ligand at 47.02–46.96 and 46.39–46.23 ppm. Compound 4f exhibits poor solubility in CDCl3 and in other common deuterated solvents leading to a poor-quality 13C{1H} NMR spectrum for 12 h acquisition and it was characterized by solid-state CP-MAS TOSS 13C{1H} NMR. The spectrum of 4f displays three characteristic singlets at 161.75, 45.47, and 41.01 ppm assignable to the HN–CNO carbon and two OS(CH3)2 carbons.
X-ray structure determinations
The molecular structures of 3a–b, 3d·MeOH, 3e–f, 4c, and 4f indicate that all coordination polyhedra exhibit typical square-planar geometries (Fig. 3). In the structures of 3a–b, 3d·MeOH, and 3e–f, all bond angles around the PtII centers are close to 90°, whereas the structure of 4c and 4f display the O(1)–Pt(1)–N(2) angles that fall into the interval 79.95–80.58°. Distortion of these angles is probably due to the chelation of the amidoximate ligands. The Pt–Cl [2.2969(9)–2.3301(17) Å] and Pt–S [2.2025(16)–2.2426(13) Å] distances exhibit values characteristic of usual PtII–Cl and PtII–S bonds.20 The Pt(1)–N(1) bonds are in the range of 2.008(5)–2.020(3) Å, which is typical for Pt–Namidoxime species.21 The S–O bond lengths [1.469(4)–1.487(5) Å] are characteristic of the sulfinyl groups of S-bound sulfoxides.22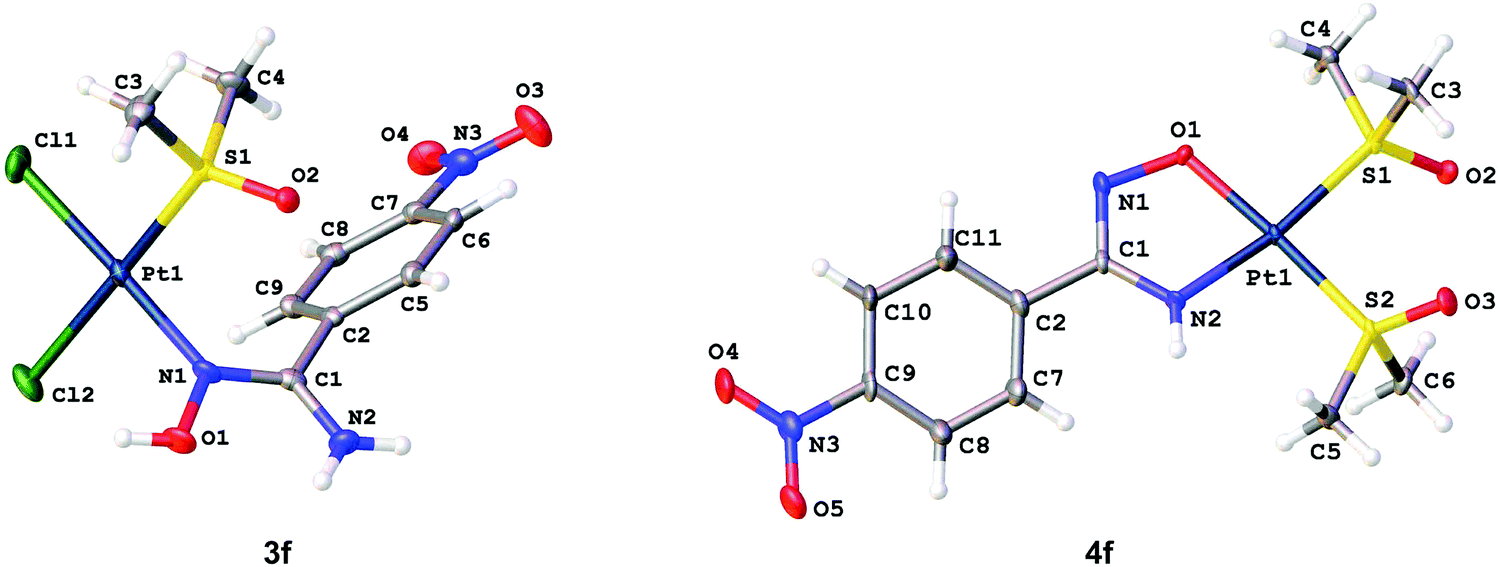 | ||
| Fig. 3 Molecular structures of 3f (left) and 4f (right) showing the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
In amidoxime and amidoximate ligands of 3a–b, 3d·MeOH, 3e–f, 4c, and 4f, the O(1)–N(1), N(1)–C(1), and N(2)–C(1) distances are equal to 1.38(2)–1.437(6), 1.289(8)–1.309(4), and 1.326(5)–1.365(8) Å, respectively, and these values are specific for amidoxime and amidoximate complexes.13 The inspection of bond length values indicate that O(1)–N(1) is a single bond,18 whereas the N(1)–C(1) and N(2)–C(1) bonds have transitive orders between single and double bonds, but the former is rather a double bond and the latter has more single bond character.18
Owing to steric reasons, in 3d·MeOH and 3e–f, the torsion angles between the aromatic rings and the carbamidoxime moiety are in the range of 49.76–54.92°, which indicates partial delocalization between these two groups. In 4f, the torsion angles between the groups in two crystallographically independent types of molecules are 15.60 and 22.04° favoring π-conjugation. In 3d·MeOH and 3e–f, one of the methyl groups of the ligated Me2SO is located above a plane of the aromatic rings with the C6⋯C(4) distances equal 3.642(4)–3.716(5) Å, which lead to a high-field shift of the CH3 signals in the 1H NMR spectra.
The crystal structures of 3a–b and 3e–f display intermolecular H-bonds between the oxygen atom of the Me2SO ligand and one of the amide hydrogen atoms [O(2)⋯N(2) 2.914–2.961 Å; O(2)⋯H–N(2) 139.71–152.21°]. In addition, in 3a an intermolecular H-bond was observed between the Cl(1) ligand and the HO moiety of the amidoxime ligand [Cl(1)⋯O(1) 3.117 Å; Cl(1)⋯H–O(1) 174.01°], whereas an intermolecular H-bond between the O atom of the Me2SO ligand and the HO moiety of the amidoxime ligand was found in 3b [O(1)⋯O(2) 2.749 Å; O(1)–H⋯O(2) 171.62°]. In 3d·MeOH, three intermolecular H-bonds were observed, viz. O(1)–H⋯O(3) [O(1)⋯O(3) 2.664 Å; O(1)–H⋯O(3) 176.31°], N(2)–H⋯O(3) [N(2)⋯O(3) 2.975 Å; N(2)–H⋯O(3) 162.83°], and O(3)–H⋯O(2) [O(3)⋯O(2) 2.765 Å; O(3)–H⋯O(2) 161.44°].
Scope and limitations of the reactions
1H NMR monitoring of the reaction between 2c and 1 in CD3OD at RT or 65 °C indicates that the reaction initially (the first 5 min) leads to the formation of at least four unidentified compounds, which, in particular, degrade for 24 h giving a broad mixture of products. Slow evaporation of the reaction mixture gave several crystals of {tBuC(NH2)N(H)OH}[PtCl3(Me2SO)], whereas the main part is an oily residue. The structure of the mono-sulfoxide complex was verified by single-crystal X-ray diffraction (see Table S2 in the ESI†). A plausible explanation of these observations is steric hindrances provided by the tert-butyl substituent at the oxime N atom preventing the coordination of 2c to the platinum(II) center.
1H NMR monitoring of the reaction between 2g and 1 in CD3OD at RT or 65 °C indicates the formation of a broad spectrum of unidentified products. In the HRESI+-MS spectra, sets of peaks associated with 3g (503.9614 [M + Na]+, calcd 503.9626; 926.9742 [2M − Cl]+, calcd 926.9782; 984.9367 [2M + Na]+, calcd 984.9363) were found, but all these peaks could also be due to the isomeric [PtCl2(Me2O){C5H4C(NH2)NOH}]; the coordination mode of the amidoxime ligand is typical for 4-pyridyl carbamidoxime.13 Owing to the similar solubilities of the compounds in the mixture and their decomposition on silica gel, no pure components were isolated. We associate the formation of the mixture of products with the availability of an additional nucleophilic center, i.e. the pyridyl N atom, which competes with the amidoxime N atom in coordination to the metal center and provides some side-reactions.
No formation of the bis-amidoxime complexes [PtCl2{RC(NH2)OH}2] or [PtCl{RC(NH2)OH}2(Me2O)]Cl was detected even in the presence of 10-fold excess of any one of the amidoximes 2a–g relative to 1 in MeOH at RT or at 65 °C for 10 d. Under these conditions, only the formation of 3a–b and 3d–f was detected and these complexes were isolated in 70–85% yields and no sets of peaks related to the bis-amidoxime complexes were observed in HRESI+-MS spectra. In addition, all attempts of the generation of [PtCl{RC(NH2)OH}2(Me2SO)](OTf) by treatment of 3a–b or 3d–f with 1 equiv. of AgOTf in MeOH from −20 to 65 °C were unsuccessful due to the formation of broad mixtures of products that we failed to separate.
The amidoximes featuring strong donor substituents R (R = 4-morpholyl, p-Me2NC6H4, and p-MeOC6H4) react with 1 at various temperatures in the range from 0 to 65 °C in MeOH, Me2CO, and CHCl3 giving a broad spectrum of unidentified products and they could not be utilized in the reactions described above.
Comparative studies of cytotoxic activity
The antiproliferative activity of six complexes was studied in vitro in four human cancer cell lines by means of the colorimetric MTT assay. Species featuring the chelating amidoximate ligand (4c–e) exhibit higher cytotoxicity in all investigated cell lines compared to their open-chain amidoxime analogues (3b, 3d–e) (Table 1).| No. | IC50 (μM) | |||
|---|---|---|---|---|
| CH1/PA-1 | SW480 | A549 | SK-BR-3 | |
| a Data taken from ref. 10b. | ||||
| 3b | 130 ± 44 | >320 | >320 | 202 ± 103 |
| 3d | 150 ± 37 | 272 ± 72 | >320 | 240 ± 78 |
| 3e | 129 ± 26 | 157 ± 30 | >320 | 227 ± 21 |
| 4c | 19 ± 5 | 19 ± 4 | 99 ± 7 | 18 ± 2 |
| 4d | 11 ± 1 | 7.9 ± 0.4 | 78 ± 6 | 13 ± 1 |
| 4e | 0.96 ± 0.25 | 0.51 ± 0.13 | 30 ± 4 | 2.3 ± 0.3 |
| Cisplatina | 0.14 ± 0.3 | 3.3 ± 0.4 | 1.3 ± 0.4 | — |
Complexes 3b and 3d–e display very low activity in these cell lines with IC50 values consistently exceeding 100 μM and are hence about two orders of magnitude less active than the prototypic oxime complex cis-[PtCl2(Me2COH)2] containing two acetoxime ligands in CH1/PA-1 and SW480 cells.23 In contrast, most IC50 values of chelate congeners 4c–e (except for A549 cells) are in the low micromolar to submicromolar range and hence at least one order of magnitude lower than those of the former complexes. In particular, complex 4e bearing a p-trifluoromethylbenzamidoximate ligand shows by far the highest cytotoxic potency and displays a remarkable activity in the inherently cisplatin resistant SW480 cell line (0.51 μM vs. 3.3 μM, Table 1). This indicates that the trifluoromethyl substituent in the para position, which differentiates 4e from 4d (bearing an unsubstituted phenyl group), is highly favorable for cytotoxicity, enhancing it by another order of magnitude in CH1/PA-1 and SW480 cells, whereas no such effect could be observed for 3e in comparison to 3d.
The results for both classes of complexes are surprising, insofar as the presence of two readily exchangeable chlorido ligands in the cis position is not associated with high cytotoxicity here, whereas the lack of easily exchangeable ligands is. An opposite relationship between the activity of the chelate and open-chain forms was in fact previously shown for the sister class of platinum(II) compounds featuring 1,3-dihydroxyacetone oxime ligands.10c In contrast, one of the open-chain and not a chelate species had been shown to overcome the resistance of SW480 cells to cisplatin. It has to be borne in mind, however, that the two classes studied here are no such close analogs that can be converted into each other by (de)protonation and chloride subtraction/addition, but additionally differ by the presence of a second dimethyl sulfoxide instead of a chlorido ligand in the amidoximato complexes. In line with current knowledge on platinum drugs, it is reasonable to assume that opening of the chelates is a prerequisite for high biological activity of the amidoximato complexes and that this opening may be favored by trans effects exerted by the sulfur donors destabilizing the opposite bonds.
These results are intriguing in the light of the known impact of dimethyl sulfoxide on the biological activity of platinum compounds. When used as a solvent for cisplatin or carboplatin (both having two chlorido ligands), (CH3)2SO causes a profound deactivation of the drug due to ligand exchange reactions, whereas the activity of oxaliplatin (having a chelating oxalate as a leaving group) remains virtually unaffected or is slightly enhanced for unknown reasons.19 A report on the antiproliferative activity of platinum(II) complexes of the type [PtL2(Me2O)2] (where L2 is a chelating ligand or L is a monodentate ligand) against a set of human cancer cell lines clearly indicates low activity of these complexes relative to non-dimethyl sulfoxide analogs.24 On the other hand, (CH3)2SO ligands are supposed to favor the cellular uptake of metal complexes, and the increase in activity of 4c–e might therefore also arise from the additional (CH3)2SO ligand in this group, which may enhance the permeability of the drugs through lipid membranes.25
Final remarks
The possibility of pH-dependent highly selective generation of the open-chain amidoxime and chelated amidoximate platinum(II) complexes, cis-[PtCl2{RC(NH2)OH}(Me2O)] and [Pt{RC(H)N}(Me2O)2], respectively, has been demonstrated. Although the coordination chemistry of amidoximes has been extensively studied,13 only one example of coordination isomerism of amidoximes in mononuclear complexes has previously been known, viz. the UO22+ center, amidoximes exhibited κ1-O26 and κ2-O,Noxime27 coordination modes in neutral and basic media, respectively.
The cytotoxic properties of the novel complexes of both types were examined in four human cancer cell lines (CH1/PA-1, SW480, A549, and SK-BR-3). It was found that chelate amidoximate complexes are significantly more cytotoxic than the open-chain species. Notably, complex 4e displays appreciably enhanced cytotoxicity in the intrinsically cisplatin-resistant SW480 cell line as compared to cisplatin, indicating a promising potential to overcome at least some forms of cisplatin resistance.
Further development of selective preparation of amidoxime complexes of other metals and studies of the cytotoxic activity of complexes featured ligands that structurally similar to amidoximes (amidrazones and hydroxamic acids) are underway in our group.
Experimental section
Materials and instrumentation
Solvents were obtained from commercial sources and used as received. Amidoximes 2a–g were synthesized accordingly to the literature methods.28 Melting points were measured on a Stuart SMP30 apparatus in capillaries and not corrected. Microanalyses (C, H, N) were carried out on a Euro EA3028-HT instrument. Electrospray ionization mass-spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization (ESI) source. The instrument was operated both in negative and positive ion modes over a m/z range 50–3000. The nebulizer gas flow was 0.4 bar and the drying gas flow was 4.0 L min−1. For HRESI, complexes were dissolved in MeOH. In the isotopic pattern, the most intensive peak is reported. Infrared spectra (4000–400 cm−1) were recorded on a Shimadzu IR Prestige-21 instrument in KBr pellets. 1H and 13C{1H} NMR spectra were measured on Bruker Avance 400 and Bruker Avance III 500 spectrometers at 294–298 K; residual solvent signals were used as the internal standard. The solid-state CP-MAS TOSS 13C{1H} NMR spectra were measured on a Bruker Avance III WB 400 with magic angle spinning at 6 kHz frequency.X-Ray structure determinations
XRD experiments were carried out using Agilent Technologies “Xcalibur” (for 3a–b, 3d·MeOH, 3e–f, and 4c) and “Supernova” (for 4f) diffractometers with monochromated MoKα or CuKα radiation, respectively. Crystals were fixed on a micro mount, placed on diffractometer and measured at a temperature of 100 K. The unit cell parameters (Tables S1 and S2, ESI†) were refined by least square techniques in the 2θ range of 5.6–55.0 for MoKα and 9.0–150.0 for CuKα. The structures have been solved using the Superflip29 structure solution program using charge flipping and refined with the ShElXL30 refinement incorporated in the OLEX2 program package.31 The carbon-bound H atoms were placed in calculated positions and included in the refinement in the ‘riding’ model approximation, with Uiso(H) set to 1.2Ueq(C) and C–H 0.97 Å for the CH2 groups, Uiso(H) set to 1.5Ueq(C) and C–H 0.96 Å for the CH3 groups, Uiso(H) set to 1.2Ueq(C) and C–H 0.93 Å for the CH groups, Uiso(H) set to 1.2Ueq(N) and N–H 0.88 Å for the NH and NH2 groups and Uiso(H) set to 1.5Ueq(O) and O–H 0.82 Å for the OH groups. Empirical absorption correction was applied in the CrysAlisPro32 program complex using spherical harmonics (for 3a–b, 3d·MeOH, 3e–f, and 4c), implemented in the SCALE3 ABSPACK scaling algorithm. For 4f, numerical absorption correction based on Gaussian integration over a multifaceted crystal model was applied (Tables S1 and S2, ESI†).Cell lines and culture conditions
The cytotoxicity tests were performed in four human cancer cell lines. CH1/PA-1 cells (identified via STR profiling as PA-1 ovarian teratocarcinoma cells by Multiplexion, Heidelberg, Germany; compare ref. 33) were kindly provided by Lloyd R. Kelland (CRC Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, UK). SW480 (human adenocarcinoma of the colon) and A549 (human non-small cell lung cancer) cells were obtained from Brigitte Marian (Institute of Cancer Research, Department of Medicine I, Medical University of Vienna, Austria) and SK-BR-3 (human adenocarcinoma of the mammary gland) cells from Evelyn Dittrich (General Hospital, Medical University of Vienna, Austria). Cell monolayer adherent cultures were grown in 75 cm2 culture flasks (Starlab, Germany) in complete medium [i.e., minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies, Germany), 1% sodium pyruvate, 1% non-essential amino acids from 100× ready-to-use stock and 2% L-glutamine (all purchased from Sigma-Aldrich, Austria)]. Cell cultures were incubated at 37 °C in a moist atmosphere containing 5% CO2 in air.Cytotoxicity tests in cancer cell lines
The cytotoxic activity in vitro was determined by means of the colorimetric microculture MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide). For this purpose, cells were harvested from culture flasks by trypsinization and seeded into 96-well plates (Starlab, Germany) at densities of 1.25 × 103 cells per well (CH1/PA-1), 2 × 103 cells per well (SW480), 3 × 103 cells per well (A549) and 5 × 103 cells per well (SK-BR-3) in volumes of 100 μL per well. Cells were allowed to settle and proliferate for 24 h before exposure to the drugs. Stock solutions of each complex were prepared in (CH3)2SO, appropriately diluted in complete medium (not to exceed a concentration of 0.5% (CH3)2SO in cells) and instantly added into the plates (100 μL per well). After continuous exposure for 96 h, drug solutions were replaced with 100 μL per well of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 MTT/RPMI 1640 solution (MTT solution, 5 mg mL−1 of MTT reagent in phosphate-buffered saline; RPMI 1640 medium, supplemented with 10% heat-inactivated fetal bovine serum and 2% L-glutamine). After incubation for 4 h, the medium/MTT mixtures were removed, and the formazan crystals formed by viable cells were dissolved in (CH3)2SO (150 μL per well). Optical densities were measured at 550 nm using a microplate reader (ELx808 Absorbance Microplate Reader, Bio-Tek, USA), using a reference wavelength of 690 nm to correct for unspecific absorption. The quantity of viable cells was expressed in relation to the untreated control and 50% inhibitory concentrations (IC50) were calculated from concentration–effect curves by interpolation. Evaluation is based on means from at least three independent experiments, each comprising triplicates per concentration level.
:6 MTT/RPMI 1640 solution (MTT solution, 5 mg mL−1 of MTT reagent in phosphate-buffered saline; RPMI 1640 medium, supplemented with 10% heat-inactivated fetal bovine serum and 2% L-glutamine). After incubation for 4 h, the medium/MTT mixtures were removed, and the formazan crystals formed by viable cells were dissolved in (CH3)2SO (150 μL per well). Optical densities were measured at 550 nm using a microplate reader (ELx808 Absorbance Microplate Reader, Bio-Tek, USA), using a reference wavelength of 690 nm to correct for unspecific absorption. The quantity of viable cells was expressed in relation to the untreated control and 50% inhibitory concentrations (IC50) were calculated from concentration–effect curves by interpolation. Evaluation is based on means from at least three independent experiments, each comprising triplicates per concentration level.
Synthetic work
Preparation of amidoxime complexes 3a–b and 3d–f
Powder of cis-[PtCl2(Me2O)2] (1) (84.5 mg; 200 μmol) was added into a stirred solution of any one of the amidoximes 2a–b and 2d–f (200 μmol) in MeOH (3 mL). The suspension was left to stand at 65 °C under stirring for 5 min, whereupon the homogeneous solution formed was cooled to RT and the solvent was evaporated at RT in vacuo. The oily residue was crystallized under CHCl3 (1 mL) and separated by centrifugation. The precipitate was washed with two 1.5 mL portions of Et2O and dried in air at 65 °C. For the characterization of 3a–b and 3d–e, see the ESI.†
3f. Yield: 98% (102.9 mg). Mp: 183 °C (dec). Anal. calcd for C9H13N3Cl2O4PtS: C, 21.65; H, 3.52; N, 6.39. Found: C, 21.73; H, 3.41; N, 6.48. HRESI+-MS (MeOH, m/z): 453.0172 ([M − 2Cl − H]+, calcd 453.0191), 489.9923 ([M − Cl]+, calcd 489.9946), 547.9517 ([M + Na]+, calcd 547.9524), 1014.9577 ([2M − Cl]+, calcd 1014.9580), 1072.9157 ([2M + Na]+, calcd 1072.9161). IR (KBr, selected bonds, cm−1): 3439(s), 3335(s) ν(O–H) and ν(N–H); 3185(m), 3107(w), 3079(w), 3001(w-m), 2918(w-m) ν(C–H); 1661(vs) ν(CN); 1520(vs) ν(NO)as; 1346(vs) ν(NO)s; 1136(s) ν(SO). 1H NMR (CD3OD, δ): 8.43 (d, 2H, CH), 8.19 (d, 2H, CH), 3.42 (s, 3H, CH3), 2.83 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 155.19 (C(NH2)NOH), 149.60, 148.08, 146.40, 138.11, 133.02, 129.73, 123.36, 122.12 (Ar), 46.59, 44.17, 42.66, 40.18 (s, CH3). Crystals of 3f suitable for X-ray diffraction were obtained by slow evaporation of MeNO2 solution at RT in air.
Preparation of amidoximate complexes 4c–g
Powder of cis-[PtCl2(Me2O)2] (1) (84.5 mg; 200 μmol) was added into a stirred solution of any of the amidoximes 2a–g (200 μmol) and NaOH (20.0 mg; 500 μmol) in MeOH (3 mL). The suspension was left to stand at 65 °C under stirring for 20 min, whereupon the suspension formed was cooled to RT and the solvent was evaporated at RT in vacuo. The solid obtained was washed by one 0.5 mL portion of cold (−20 °C) MeOH and two 1.5 mL portions of Et2O, and dried in air at 65 °C. For the characterization of 4c–e and 4g, see the ESI.†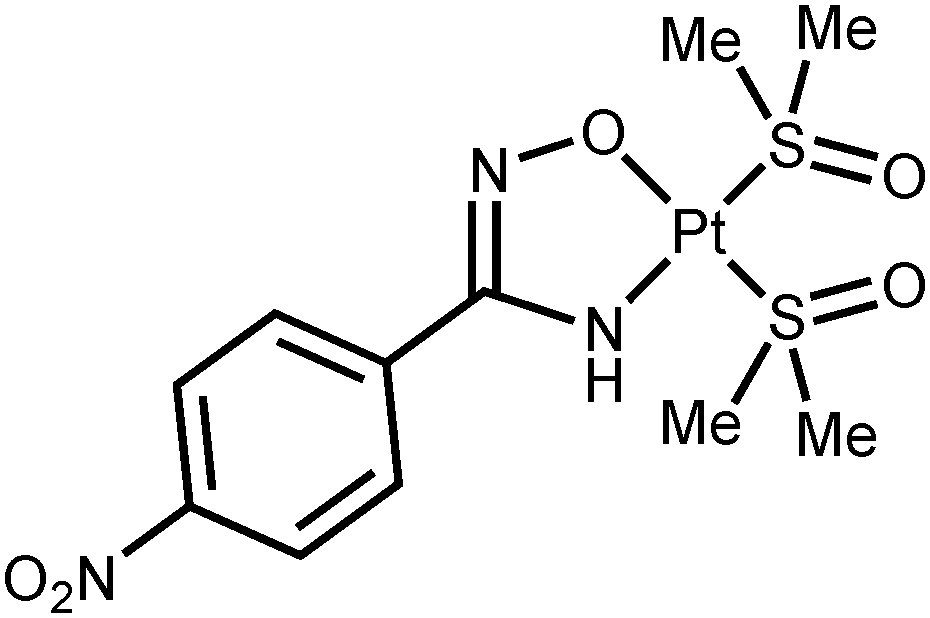
4f·H2O. Yield: 95% (104.1 mg). Mp: 210 °C (dec). Anal. calcd for C11H17N3O5PtS2·H2O: C, 24.09; H, 3.49; N, 7.66. Found: C, 23.99; H, 3.54; N, 7.65. HRESI+-MS (m/z): 531.0306 ([M + H]+, calcd 531.0331), 553.0104 ([M + Na]+, calcd 553.0150). IR (KBr, selected bonds, cm−1): 3414(m), 3335(m) ν(N–H); 2990(m), 2913(m) ν(C–H); 1597(m) ν(CN); 1516(m-s) ν(NO)as; 1341(s) ν(NO)s; 1130(vs) ν(SO). 1H NMR (CDCl3, δ): 8.20 (d, 2H, CH), 7.92 (d, 2H, CH), 5.27 (s + d, JPtH2 = 108 Hz, br, 1H, NH), 3.58 (s + d, JPtH3 = 20 Hz, 6H, S(CH3)2), 3.55 (s + d, JPtH3 = 20 Hz, 6H, S(CH3)2). CP-MAS TOSS 13C{1H} NMR (CDCl3, δ): 161.75 (C(NH)NO), 148.61, 145.56, 135.64, 123.33 (Ar), 45.47 (S(CH3)2), 41.01 (S(CH3)2). Crystals of 4f suitable for X-ray diffraction were obtained by slow evaporation of MeOH solution at RT in air.
Acknowledgements
The authors are grateful to the Russian Foundation for Basic Research (project 16-03-00573), the Scientific Council of the President of the Russian Federation (project MK-473.2017.3), and RAS (program 1.14P) for financial support. Physicochemical studies were performed at the Magnetic Resonance Research Center, Center for X-ray Diffraction Studies, Center for Chemical Analysis and Materials Research, and Chemistry Educational Center of Saint Petersburg State University.References
- (a) A. V. Klein and T. W. Hambley, Ligand Design in Medicinal Inorganic Chemistry, John Wiley & Sons, Ltd., 2014, pp. 9–45 Search PubMed; (b) S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed; (c) N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- (a) T. Boulikas, A. Pantos, E. Bellis and P. Christofis, Anticancer Therapeutics, John Wiley & Sons, Ltd., 2008, pp. 55–78 Search PubMed; (b) J. Zhang, Y. Ke and J. Shen, Shanghai Yiyao, 2013, 34, 52–59 Search PubMed; (c) M. G. Apps, E. H. Y. Choi and N. J. Wheate, Endocr.-Relat. Cancer, 2015, 22, R219–R233 CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- (a) V. Brabec and J. Kasparkova, Metal Compounds in Cancer Chemotherapy, Research Signpost, 2005, pp. 187–218 Search PubMed; (b) D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed; (c) S. Ahmad, A. A. Isab and S. Ali, Transition Met. Chem., 2006, 31, 1003–1016 CrossRef CAS; (d) S. J. Berners-Price, L. Ronconi and P. J. Sadler, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 65–98 CrossRef CAS; (e) M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2261 CrossRef CAS; (f) B. Wu, G. E. Davey, A. A. Nazarov, P. J. Dyson and C. A. Davey, Nucleic Acids Res., 2011, 39, 8200–8212 CrossRef CAS PubMed; (g) J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470–4495 CrossRef CAS PubMed.
- (a) F. Arnesano, A. Pannunzio, M. Coluccia and G. Natile, Coord. Chem. Rev., 2015, 284, 286–297 CrossRef CAS; (b) X. Wang, X. Wang and Z. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed; (c) J. Pracharova, T. Radosova Muchova, E. Dvorak Tomastikova, F. P. Intini, C. Pacifico, G. Natile, J. Kasparkova and V. Brabec, Dalton Trans., 2016, 45, 13179–13186 RSC.
- (a) A. D. Ryabov, Ross. Khim. Zh., 1996, 40, 33–42 CAS; (b) A. S. de Pascali, A. Muscella, S. Marsigliante, M. G. Bottone, G. Bernocchi and F. P. Fanizzi, Pure Appl. Chem., 2013, 85, 355–364 Search PubMed; (c) F.-U. Rahman, A. Ali, R. Guo, Y.-C. Zhang, H. Wang, Z.-T. Li and D.-W. Zhang, Dalton Trans., 2015, 44, 2166–2175 RSC; (d) M. Serratrice, L. Maiore, A. Zucca, S. Stoccoro, I. Landini, E. Mini, L. Massai, G. Ferraro, A. Merlino, L. Messori and M. A. Cinellu, Dalton Trans., 2016, 45, 579–590 RSC; (e) C. M. A. Müller, M. V. Babak, M. Kubanik, M. Hanif, S. M. F. Jamieson, C. G. Hartinger and L. J. Wright, Inorg. Chim. Acta, 2016, 450, 124–130 CrossRef.
- (a) E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 CrossRef CAS PubMed; (b) C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed; (c) A. Bergamo and G. Sava, Dalton Trans., 2007, 1267–1272 RSC; (d) I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692–697 CrossRef CAS; (e) A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed; (f) S. Pillozzi, L. Gasparoli, M. Stefanini, M. Ristori, M. D'Amico, E. Alessio, F. Scaletti, A. Becchetti, A. Arcangeli and L. Messori, Dalton Trans., 2014, 43, 12150–12155 RSC; (g) S. Komeda and A. Casini, Curr. Top. Med. Chem., 2012, 12, 219–235 CrossRef CAS PubMed; (h) S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201–214 CrossRef CAS PubMed.
- (a) N. Farrell, Cancer Invest., 1993, 11, 578–589 CrossRef CAS PubMed; (b) N. P. Farrell, Chem. Soc. Rev., 2015, 44, 8773–8785 RSC; (c) T. C. Johnstone, S. J. Lippard and K. Suntharalingam, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- (a) Y. Y. Scaffidi-Domianello, K. Meelich, M. A. Jakupec, V. B. Arion, V. Y. Kukushkin, M. Galanski and B. K. Keppler, Inorg. Chem., 2010, 49, 5669–5678 CrossRef CAS PubMed; (b) Y. Y. Scaffidi-Domianello, A. A. Legin, M. A. Jakupec, A. Roller, V. Y. Kukushkin, M. Galanski and B. K. Keppler, Inorg. Chem., 2012, 51, 7153–7163 CrossRef CAS PubMed; (c) Y. Y. Scaffidi-Domianello, A. A. Legin, M. A. Jakupec, V. B. Arion, V. Y. Kukushkin, M. Galanski and B. K. Keppler, Inorg. Chem., 2011, 50, 10673–10681 CrossRef CAS PubMed; (d) D. A. Erdogan and S. Ozalp-Yaman, J. Mol. Struct., 2014, 1064, 50–57 CrossRef CAS; (e) K. Ossipov, Y. Y. Scaffidi-Domianello, I. F. Seregina, M. Galanski, B. K. Keppler, A. R. Timerbaev and M. A. Bolshov, J. Inorg. Biochem., 2014, 137, 40–45 CrossRef CAS PubMed; (f) Y. Benabdelouahab, L. Munoz-Moreno, M. Frik, I. de la Cueva-Alique, M. A. El Amrani, M. Contel, A. M. Bajo, T. Cuenca and E. Royo, Eur. J. Inorg. Chem., 2015, 2295–2307 CrossRef CAS PubMed; (g) H. Meyer, M. Brenner, S.-P. Hoefert, T.-O. Knedel, P. C. Kunz, A. M. Schmidt, A. Hamacher, M. U. Kassack and C. Janiak, Dalton Trans., 2016, 45, 7605–7615 RSC; (h) M. Kubanik, W. Kandioller, K. Kim, R. F. Anderson, E. Klapproth, M. A. Jakupec, A. Roller, T. Sohnel, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2016, 45, 13091–13103 RSC.
- (a) I. Racz, K. Tory, F. J. Gallyas, Z. Berente, E. Osz, L. Jaszlits, S. Bernath, B. Sumegi, G. Rabloczky and P. Literati-Nagy, Biochem. Pharmacol., 2002, 63, 1099–1111 CrossRef CAS PubMed; (b) T. Kardon, G. Nagy, M. Csala, A. Kiss, Z. Schaff, P. L. Nagy, L. Wunderlich, G. Banhegyi and J. Mandl, Anticancer Res., 2006, 26, 1023–1028 CAS; (c) K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas, E. A. Varella and D. N. Nicolaides, Curr. Pharm. Des., 2008, 14, 1001–1047 CrossRef CAS PubMed.
- (a) D. S. Bolotin, A. S. Novikov, I. E. Kolesnikov, V. V. Suslonov, Y. Novozhilov, O. Ronzhina, M. Dorogov, M. Krasavin and V. Y. Kukushkin, ChemistrySelect, 2016, 1, 456–461 CrossRef CAS; (b) Y. Kaya, C. Icsel, V. T. Yilmaz and O. Buyukgungor, J. Mol. Struct., 2015, 1095, 51–60 CrossRef CAS; (c) E. Y. Bulatov, T. G. Chulkova, I. A. Boyarskaya, V. V. Kondratiev, M. Haukka and V. Y. Kukushkin, J. Mol. Struct., 2014, 1068, 176–181 CrossRef CAS.
- D. S. Bolotin, N. A. Bokach and V. Y. Kukushkin, Coord. Chem. Rev., 2016, 313, 62–93 CrossRef CAS.
- (a) A. B. Goel, S. Goel and D. Vanderveer, Inorg. Chim. Acta, 1981, 54, L5–L6 CrossRef CAS; (b) A. D. Ryabov, G. M. Kazankov, I. M. Panyashkina, O. V. Grozovsky, O. G. Dyachenko, V. A. Polyakov and L. G. Kuz'mina, Dalton Trans., 1997, 4385–4391 RSC; (c) T. Kluge, M. Bette, T. Rüffer, C. Bruhn, C. Wagner, D. Ströhl, J. Schmidt and D. Steinborn, Organometallics, 2013, 32, 7090–7106 CrossRef CAS.
- N. Dürüst, M. A. Akay, Y. Dürüst and E. Kiliç, Anal. Sci., 2000, 16, 825–827 CrossRef.
- (a) V. Y. Kukushkin, A. J. L. Pombeiro, C. M. P. Ferreira, L. I. Elding and R. J. Puddephatt, Inorg. Synth., 2002, 33, 189–196 CAS; (b) Y. N. Kukushkin, Y. E. Vyaz'menskii, L. I. Zorina and Y. L. Pazukhina, Zh. Neorg. Khim., 1968, 13, 1595–1599 CAS.
- (a) T.-C. Chien, C.-H. Wang, T.-H. Hsieh, C.-C. Lin, W.-H. Yeh and C.-A. Lin, Synlett, 2015, 1823–1826 CrossRef; (b) D. S. Bolotin, K. I. Kulish, N. A. Bokach, G. L. Starova, V. V. Gurzhiy and V. Y. Kukushkin, Inorg. Chem., 2014, 53, 10312–10324 CrossRef CAS PubMed.
- L. J. Bellamy, The infra-red spectra of complex molecules, Methuen and co. Ltd, John Wiley and sons, Inc, London, New York, 1958 Search PubMed.
- E. D. Risberg, J. Mink, A. Abbasi, M. Y. Skripkin, L. Hajba, P. Lindqvist-Reis, É. Bencze and M. Sandström, Dalton Trans., 2009, 1328–1338, 10.1039/b814252a.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, 12, S1–S83 RSC.
- (a) D. S. Bolotin, M. Y. Demakova, A. S. Novikov, M. S. Avdontceva, M. L. Kuznetsov, N. A. Bokach and V. Y. Kukushkin, Inorg. Chem., 2015, 54, 4039–4046 CrossRef CAS PubMed; (b) D. S. Bolotin, N. A. Bokach, M. Haukka and V. Y. Kukushkin, ChemPlusChem, 2012, 77, 31–40 CrossRef CAS; (c) D. S. Bolotin, N. A. Bokach, M. Haukka and V. Y. Kukushkin, Inorg. Chem., 2012, 51, 5950–5964 CrossRef CAS PubMed.
- Y. Y. Scaffidi-Domianello, A. A. Nazarov, M. Haukka, M. Galanski, B. K. Keppler, J. Schneider, P. Du, R. Eisenberg and V. Y. Kukushkin, Inorg. Chem., 2007, 46, 4469–4482 CrossRef CAS PubMed.
- S. Zorbas-Seifried, M. A. Jakupec, N. V. Kukushkin, M. Groessl, C. G. Hartinger, O. Semenova, H. Zorbas, V. Y. Kukushkin and B. K. Keppler, Mol. Pharmacol., 2007, 71, 357–765 CrossRef CAS PubMed.
- (a) M. D. Hall, K. A. Telma, K. E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed; (b) P. Bergamini, L. Marvelli, G. Spirandelli and E. Gallerani, Inorg. Chim. Acta, 2016, 439, 35–42 CrossRef CAS; (c) T. A. K. Al-Allaf, L. J. Rashan, A. S. Abu-Surrah, R. Fawzi and M. Steimann, Transition Met. Chem., 1998, 23, 403–406 CrossRef CAS.
- R. Notman, M. Noro, B. O'Malley and J. Anwar, J. Am. Chem. Soc., 2006, 128, 13982–13983 CrossRef CAS PubMed.
- E. G. Witte and K. S. Schwochau, Inorg. Chim. Acta, 1984, 94, 323–331 CrossRef CAS.
- S. Vukovic, L. A. Watson, S. O. Kang, R. Custelcean and B. P. Hay, Inorg. Chem., 2012, 51, 3855–3859 CrossRef CAS PubMed.
- (a) J. K. Augustine, V. Akabote, S. G. Hegde and P. Alagarsamy, J. Org. Chem., 2009, 74, 5640–5643 CrossRef CAS PubMed; (b) C. C. Lin, T. H. Hsieh, P. Y. Liao, Z. Y. Liao, C. W. Chang, Y. C. Shih, W. H. Yeh and T. C. Chien, Org. Lett., 2014, 16, 892–895 CrossRef CAS PubMed.
- (a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; (b) L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- CrysAlisPro, 1.171.36.20; Agilent Technologies: Santa Clara, CA, 2012.
- C. Korch, M. A. Spillman, T. A. Jackson, B. M. Jacobsen, S. K. Murphy, B. A. Lessey, V. C. Jordan and A. P. Bradford, Gynecol. Oncol., 2012, 127, 241–248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and spectra of 3a–b, 3d–f, and 4c–g, X-ray structure determinations and crystal data of 3a–b, 3d–f, 4c, 4f and [2cH][PtCl3(Me2O)], and concentration–effect curves of 3b, 3d–e and 4c–e. CCDC 1475377, 1475378, 1475381, 1475389, 1475399, 1475400, 1475446 and 1475683. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj00982h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |