
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimeric molecular clips based on glycoluril†
Jan
Sokolov
,
Tomas
Lizal
and
Vladimir
Sindelar
 *
*
Department of Chemistry and RECETOX, Masaryk University, Kamenice 5, 62500 Brno, Czech Republic. E-mail: sindelar@chemi.muni.cz
First published on 13th June 2017
Abstract
Glycoluril dimers 4a and 4b were prepared and successfully transformed to glycoluril dimeric clips 7a and 7b. 1H NMR spectroscopy was used to reveal that the dimeric clip 7b with methyl substituents in its structure exists as a mixture of two distinct conformers in D2O at ambient temperature, while 7a possesses only one stable conformer under the same conditions due to its bulky phenyl substituents. The coalescence temperatures for 7b and its precursor 5b were determined. Both dimeric clips 7a and 7b formed complexes with methylviologen in D2O in which each of two clip-binding sites was occupied by one molecule of the guest. Thus, this study uncovers possible use of glycoluril dimers 4a and 4b as general building blocks for glycoluril-based supramolecular host molecules with two binding sites in their structure.
Introduction
Cucurbiturils are an important class of supramolecular host molecules that are prized for their ability to strongly and selectively bind organic molecules in water.1–6 These macrocycles consist of glycoluril units connected by methylene bridges. Recent progress in the chemistry of cucurbiturils enables the preparation of their functionalized derivatives,7–13 which can be covalently attached to various substrates and thus can be used for real applications. Along with the synthesis of cucurbituril derivatives, other supramolecular hosts based on glycoluril have been prepared, including anionic receptors – bambusurils14–16 – and acyclic glycoluril oligomers.17–19 The latter group of compounds features high affinities toward anaesthetics, nitrosamines and various drugs.20–22 Despite recent progress in cucurbituril synthesis, there have been only two reported approaches dealing with the preparation of cucurbituril dimers. The first is based on the condensation of acyclic glycoluril hexamers with tetraaldehydes;23,24 the second relies on the conjugation of two monofunctionalized cucurbiturils.25 These cucurbituril dimers have been used as building blocks for the construction of supramolecular polymers and oligomers.We decided to investigate a different approach for the preparation of dimeric cucurbiturils and their acyclic analogs. Our work was inspired by compound 4a (Scheme 1) previously reported in the literature.26,27 This compound contains two glycoluril units connected through phenylene links. Therefore, we wanted to investigate whether this structural motif could be suitable for the construction of cucurbituril-based compounds. As a first approximation of a cucurbituril derivative, we selected to prepare dimeric clip 7a. The successful preparation of this compound should uncover the possibility of attaching methylene bridges to nitrogen atoms of sterically hindered glycoluril units of 4a.
 | ||
| Scheme 1 Synthesis of molecular clips 7a and 7b. Conditions: (a) urea, PhMe/TFA, reflux, 12 h, 96%; (b) CH2O, 7 M HCl, reflux, 12 h, 91%; (c) hydroquinone, TFA, reflux, 6 h, 69%; (d) 9, NaOH, water/dioxane, 25 °C, 12 h, 68%; (e) (1) n-BuLi, THF, −78 °C, 1 h, (2) MeI, 0–25 °C, 12 h, 95%; (f) RuCl3, NaIO4, CCl4/MeCN/water, 25 °C, 2 h, 55%; (g) urea, 0.3 M HCl, 25 °C, 48 h, 77%; (h) CH2O, 7 M HCl, 25 °C, 36 h, 47%; (i) 10, TFA/Ac2O, 95 °C, 3 h, 59%. | ||
Results and discussion
The starting building block 4a for this dimeric clip has already been reported.26,27 According to the literature, compound 4a can be prepared by condensation of urea or urea derivative 8 (Fig. 1) with compound 3a in refluxing formic or acetic acid with a catalytic amount of sulfuric acid. However, when the synthesis was repeated in our laboratory under these conditions, only compound 9 with one glycoluril moiety was formed.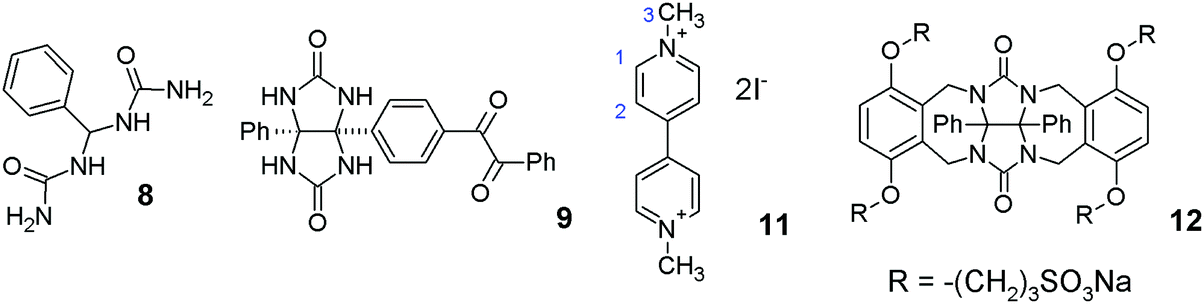 | ||
| Fig. 1 Structures of selected compounds discussed in this work. | ||
Fortunately, the reaction of urea with 3a in toluene in the presence of TFA provided dimer 4a in 95% yield. Compound 4a was then transformed to cyclic ether 5a by its reaction with paraformaldehyde in aqueous HCl (Scheme 1). Compound 5a was further reacted with hydroquinone to provide dimeric molecular clip 6 bearing OH groups on its terminal aromatic walls in 69% yield. Clip 6 was alkylated using 1,3-propanesultone to give water-soluble clip 7a in 68% yield.
The phenyl substituent present in glycoluril dimer 4a can be a source of low reactivity and low solubility of this compound, as previously reported on the corresponding glycolurils for cucurbituril syntheses.8 Thus, we decided to prepare dimer 4b in which two phenyls are replaced by methyl substituents. The synthesis of 4b required precursor 3b, which was not commercially available (unlike 3a) and had to be prepared from 1,4-diethynylbenzene (1) (Scheme 1). Alkyne 1 was deprotonated with n-butyllithium and subsequently treated with methyl iodide to give 2. Compound 2 was oxidized to 3b using an RuCl3/NaIO4 mixture. Having compound 3b in our hands, we performed its reaction with urea in an aqueous HCl/CH2Cl2 emulsion to obtain glycoluril dimer 4b, which was transformed into cyclic ether 5b by its reaction with paraformaldehyde. Each of the glycoluril units of 5b bears methyl groups in the methine position, which are significantly smaller compared to phenyl groups in 5a. Cyclic ether 5b can react directly with compound 10 yielding molecular clip 7b in 59% yield. This approach was significantly less efficient in the case of 5a as bulky phenyl groups lower the reactivity of the ether groups towards the aromatic ring. This observation is in agreement with previously reported synthesis.16
Conformational properties
When recording the 1H NMR spectra of compounds 5b and 7b at 30 °C in DMSO, we observed two signals instead of the expected one signal for each of their protons. These two sets of signals can be attributed to two distinct conformers, which were likely enabled by the rotation of two single C–C bonds connecting the central phenylene group to the glycoluril moieties. The rotation was slow on the NMR timescale at 30 °C; thus, the conformers were distinguished by their unique NMR spectra. Steric hindrance between methyl groups (attached to the methine carbon atoms of the glycoluril units) and the side walls of glycoluril units were likely the reason why the rotation was restricted. We decided to label the first conformer syn (the methyl groups located on the same side of the central phenylene group) and second anti (the methyl groups on opposite sides) (Fig. 2). Unfortunately, we could not distinguish which set of signals in the NMR spectra belonged to which conformer. Conformers of 5b, as well as of 7b, are present in a ratio of 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :42, which is based on the integration of the corresponding signals in the NMR spectra. The ratio corresponds to a 0.2 kcal mol−1 energy difference (ΔE) between the two conformers. The situation is different in the case of glycoluril dimer 4b, which shows only one set of signals under the same conditions. This indicates that the rotation is fast on the NMR timescale as it is not restricted, due to the absence of the methylene side-groups featuring in 5b and 7b. Glycoluril dimer 4a and its derivatives 5a and 7a also showed only one set of signals in their NMR spectra. However, these compounds distinguish themselves from 4b, 5b, and 7b by the presence of phenyl instead of methyl groups in the two methine positions of the glycoluril dimer core. We assume that two phenyl groups are too bulky to be fitted in the syn conformation. This is why 4a and its derivatives adopt only the sterically favorable anti conformation.
:42, which is based on the integration of the corresponding signals in the NMR spectra. The ratio corresponds to a 0.2 kcal mol−1 energy difference (ΔE) between the two conformers. The situation is different in the case of glycoluril dimer 4b, which shows only one set of signals under the same conditions. This indicates that the rotation is fast on the NMR timescale as it is not restricted, due to the absence of the methylene side-groups featuring in 5b and 7b. Glycoluril dimer 4a and its derivatives 5a and 7a also showed only one set of signals in their NMR spectra. However, these compounds distinguish themselves from 4b, 5b, and 7b by the presence of phenyl instead of methyl groups in the two methine positions of the glycoluril dimer core. We assume that two phenyl groups are too bulky to be fitted in the syn conformation. This is why 4a and its derivatives adopt only the sterically favorable anti conformation.
 | ||
| Fig. 2 Optimized structures of anti (A) and syn (B) conformers of compound 5b obtained at the B3LYP/6-311G level of theory. | ||
In order to gain information on the energy characteristics of the conformational changes in 5b, the 1H NMR spectra in DMSO-d6 with temperature rising to 130 °C were recorded (Fig. 3). It was discovered that some signals in the NMR spectra show coalescence at a different temperature from the others. The coalescence temperature of signals 2b and 4 was 90 °C, while 1, 2a and 3b had already coalesced at 60 °C. The reason why there are two coalescence temperatures observed instead of one may be that compound 5b possesses two C–C bonds around which the rotation occurs. Due to this, there is a possibility of two different kinds of rotational movements: (i) rotation of phenylene with respect to the glycoluril units and (ii) rotation of one glycoluril unit with respect to the other. The signals of protons 1 and 4 were used for the calculation of Gibbs activation energies of both rotations (ΔG‡) using the Eyring equation. The resulting values of ΔG‡ are shown in Table 1. The whole calculation is described in detail in the ESI.†
 | ||
| Fig. 3 1H NMR spectra (500 MHz, DMSO-d6) of compound 5b recorded at 30 °C (A), 60 °C (B), 90 °C (C), and 130 °C (D). * denotes the signals of major conformers. # denotes the signals of minor conformers. | ||
| Compound | |||||
|---|---|---|---|---|---|
| 5b | 7b | ||||
| Signals | 1 | 4 | 1 | 4 | |
| ΔG‡ (kcal mol−1) | Major → minor | 18.42 | 18.95 | 21.05 | 21.14 |
| Minor → major | 18.20 | 18.71 | 20.81 | 20.89 | |
In contrast to 5b, dimeric molecular clip 7b showed only a single coalescence temperature, 110 °C. The values of ΔG‡ of 7b calculated using singlets 1 and 4 were greater than those of 5b (Table 1). This means that the xylylene side walls of the clip 7b are a source of more significant steric hindrance than the cyclic ether groups of 5b. In the case of compound 4b, the ΔG‡ might be determined by measurement of the NMR spectra at low temperature. However, compound 4b was insoluble in common organic solvents including DMF. The only suitable NMR solvent for this compound was DCOOD. The melting point of formic acid is 8.4 °C and therefore we could not measure the low-temperature spectra and determine the ΔG‡ of compound 4b.
Supramolecular properties
The ability of dimeric clips 7a and 7b to act as supramolecular hosts was investigated by 1H NMR spectroscopy. Inclusion complexes between viologens with cucurbiturils as well as their acyclic versions have been previously described. However, interactions of molecular clips resembling those present in dimeric clips 7a and 7b with viologen have not been reported. Thus, methylviologen 11 was selected as a suitable guest for supramolecular studies with dimeric clips 7a and 7b and their interactions in D2O (Fig. 4). | ||
| Fig. 4 1H NMR (300 MHz, D2O) spectra of 11 in the absence (A) and in the presence of 0.13 equiv. (B), 0.25 equiv. (C), 0.38 equiv. (D), 0.50 equiv. (E), 0.63 equiv. (F), 0.75 equiv. (G), 1.00 equiv. (H), 1.50 equiv. (I), 2.00 equiv. (J), and 3.00 equiv. (K) of 7b. * denotes the signals of 7b. | ||
The aromatic signals of 11 underwent an upfield shift after addition of clips 7a and 7b to the guest solution. This indicates the formation of an inclusion complex between the clips and methylviologen. Since compounds 7a and 7b have two sites that could encapsulate guest 11, it was expected that these compounds would form 1:2 complexes, in which each clip arm holds one guest molecule 11. To prove the expected binding mode, the stoichiometry of the complex was determined by the continuous variation method (Job's plot). The maximum of Job's curve appeared approximately at x-coordinate = 0.66 which proves that 1:2 complexation is predominant at equilibrium (Fig. S19 and S20, ESI†). This binding mode was further visualized by a model obtained by quantum chemical calculations (Fig. 5).
 | ||
| Fig. 5 Optimized structures of the 7a·112 complex obtained using the PM6 method. | ||
A plot of the chemical shifts of signals 1 and 2 of the guest was fitted to the binding isotherm of the 1:2 binding model to obtain the corresponding association constants (Table 2). Unlike clip 7a, the situation in the case of clip 7b was complicated by its ability to adopt two different conformers. In aqueous solution, the conformers were always in a ratio of 58:42; the equilibrium was not affected by the presence of guest 11. We were not able to determine the association constants for separate conformers due to the presence of four binding sites (each conformer has two indistinguishable binding sites) during titration with 11. However, if the concentration of 11 was kept constant during the titration and the concentration of 7b was varied, it was possible to use the signals of guest 11 to establish the association constants describing the affinity of 11 towards an equilibrium mixture of both conformers of 7b. The association constants determined for complexes 7a·112 and 7b·112 are summarized in Table 2.
| Host | β (M−2) | K 1 (M−1) | K 2 (M−1) | 4 K2/K1 |
|---|---|---|---|---|
| 7a | (2.11 ± 0.41) × 107 | (1.28 ± 0.25) × 104 | (1.66 ± 0.34) × 103 | 0.52 |
| 7b | (1.31 ± 0.42) × 107 | (2.15 ± 0.19) × 104 | (6.3 ± 1.4) × 102 | 0.12 |
Hosts 7a and 7b bind methylviologen 11 with a very similar overall affinity and in both cases the binding is associated with a negative cooperativity. The observed negative cooperativity can be rationalized by the inclusion of the guest molecule in the first binding side of the dimeric clip which causes conformational changes at the second binding site of the host. However, in our case the binding sites are separated by a rigid phenylene moiety. Therefore, we do not expect that the negative cooperativity is a result of conformational changes. We presume that binding of the first cation 11 affects the distribution of electron density in the host molecule, which makes it less attractive for another guest 11. Thus, electronic effects, rather than conformational changes, are the reason for the negative cooperativity. Finally, we determined the binding between 11 and model compound 12 to be (8.82 ± 0.77) × 103 M−1 which is in good agreement with the values obtained for binding sites in dimeric clips.
Conclusion
We prepared compounds 7a and 7b as the first examples of glycoluril-based dimeric molecular clips. Clip 7b, as well as other compounds derived from glycoluril dimer 4b, can adopt two different conformations, which can be distinguished by means of NMR. On the other hand, free rotation is forbidden in compounds based on 4a due to the presence of the bulky phenyl substituent. Both dimeric clips 7a and 7b form complexes with methylviologen in a 1:2 ratio and with submillimolar stability in water. Our work demonstrated that structural motifs represented by compounds 4a and 4b can be converted to supramolecular hosts with two binding sites. Thus, a similar strategy should be applicable to the synthesis of dimers of acyclic glycoluril oligomers or cucurbituril dimers. The syntheses of these compounds are under development in our laboratory.
Experimental section
General methods
Starting materials were purchased from commercial suppliers and used without further purification. NMR spectra were recorded using a Bruker Avance 500 spectrometer operating at frequencies of 500.13 MHz (1H) and 125.77 MHz (13C), and a Bruker Avance 300 spectrometer operating at frequencies of 300.13 MHz (1H) and 75.48 MHz (13C). Both spectrometers were equipped with a BBFO probe. High-resolution mass spectra were recorded on an Agilent 6224 Accurate-Mass TOF LC-MS spectrometer using a multimode ESI/APCI ion source and a KDS Model 100 Series manual pump for sampling.Synthetic procedures and characterization
:42 ratio. 1H NMR (500 MHz, DMSO-d6) major conformer: δ 7.63 (s, 4H), 5.40 (d, J = 11.3 Hz, 4H), 5.32 (d, J = 11.2 Hz. 4H) 5.01 (d, J = 11.2 Hz, 4H), 4.62 (d, J = 11.3 Hz, 4H), 1.18 (s, 6H). Minor conformer: δ 7.62 (s, 4H), 5.39 (d, J = 11.3 Hz, 4H), 5.32 (d, J = 11.2 Hz, 4H), 5.03 (d, J = 11.2 Hz, 4H), 4.57 (d, J = 11.3 Hz, 4H), 1.27 (s, 6H). 13C NMR (126 MHz, DMSO-d6) major conformer: δ 157.56, 134.84, 128.73, 77.87, 73.96, 71.30, 70.57, 18.77. Minor conformer: δ 157.56, 134.59, 128.73, 77.89, 73.89, 71.30, 70.57, 19.14. HR-MS (APCI−): m/z [C24H26N8O7 + Cl]− observed: 589.1570, m/z [C24H26N8O7 + Cl]− calculated: 589.1568.
Competing financial interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Czech Science Foundation (13-15576S) and the MEYS CR (projects LM2011028 and LO1214). Jan Sokolov acknowledges the Brno PhD Talent Scholarship program funded by the Brno City Municipality. We thank Dr Robert Vícha (Tomas Bata University in Zlín) for helpful discussion.References
- W. A. Freeman, W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1981, 103(24), 7367–7368 CrossRef CAS.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122(3), 540–541 CrossRef CAS.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36(8), 621–630 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44(31), 4844–4870 CrossRef CAS PubMed.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2(4), 1213 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115(22), 12320–12406 CrossRef CAS PubMed.
- D. Lucas, T. Minami, G. Iannuzzi, L. Cao, J. B. Wittenberg, P. Anzenbacher and L. Isaacs, J. Am. Chem. Soc., 2011, 133(44), 17966–17976 CrossRef CAS PubMed.
- B. Vinciguerra, L. Cao, J. R. Cannon, P. Y. Zavalij, C. Fenselau and L. Isaacs, J. Am. Chem. Soc., 2012, 134(31), 13133–13140 CrossRef CAS PubMed.
- B. Vinciguerra, P. Y. Zavalij and L. Isaacs, Org. Lett., 2015, 17(20), 5068–5071 CrossRef CAS PubMed.
- L. Gilberg, M. S. A. Khan, M. Enderesova and V. Sindelar, Org. Lett., 2014, 16(9), 2446–2449 CrossRef CAS PubMed.
- M. M. Ayhan, H. Karoui, M. Hardy, A. Rockenbauer, L. Charles, R. Rosas, K. Udachin, P. Tordo, D. Bardelang and O. Ouari, J. Am. Chem. Soc., 2015, 137(32), 10238–10245 CrossRef CAS PubMed.
- S. Y. Jon, N. Selvapalam, D. H. Oh, J.-K. Kang, S.-Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125(34), 10186–10187 CrossRef CAS PubMed.
- N. Zhao, G. O. Lloyd and O. A. Scherman, Chem. Commun., 2012, 48(25), 3070 RSC.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49(13), 2378–2381 CrossRef CAS PubMed.
- V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13(15), 4000–4003 CrossRef CAS PubMed.
- M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54(1), 276–279 CrossRef CAS PubMed.
- M. Stancl, M. Hodan and V. Sindelar, Org. Lett., 2009, 11(18), 4184–4187 CrossRef CAS PubMed.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, Org. Lett., 2009, 11(17), 3918–3921 CrossRef CAS PubMed.
- D. Ma, B. Zhang, U. Hoffmann, M. G. Sundrup, M. Eikermann and L. Isaacs, Angew. Chem., 2012, 124(45), 11520–11524 CrossRef.
- B. Zhang and L. Isaacs, J. Med. Chem., 2014, 57(22), 9554–9563 CrossRef CAS PubMed.
- D. Ma, G. Hettiarachchi, D. Nguyen, B. Zhang, J. B. Wittenberg, P. Y. Zavalij, V. Briken and L. Isaacs, Nat. Chem., 2012, 4(6), 503–510 CrossRef CAS PubMed.
- T. Minami, N. A. Esipenko, B. Zhang, M. E. Kozelkova, L. Isaacs, R. Nishiyabu, Y. Kubo and P. Anzenbacher, J. Am. Chem. Soc., 2012, 134(49), 20021–20024 CrossRef CAS PubMed.
- J. B. Wittenberg, P. Y. Zavalij and L. Isaacs, Angew. Chem., Int. Ed., 2013, 52(13), 3690–3694 CrossRef CAS PubMed.
- J. B. Wittenberg and L. Isaacs, Supramol. Chem., 2014, 26(3–4), 157–167 CrossRef CAS.
- M. Zhang, L. Cao and L. Isaacs, Chem. Commun., 2014, 50(94), 14756–14759 RSC.
- A. A. Bakibaev, T. I. Savchenko, V. D. Filimonov, A. Yu. Yagovkin and A. N. Novikov, Zh. Org. Khim., 1988, 24(12), 2581–2584 CAS.
- A. A. Bakibaev and A. Yu. Yagovkin, Zh. Org. Khim., 1994, 30(1), 133–135 CAS.
- Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52(49), 12975–12979 CrossRef CAS PubMed.
- V. K. Singh, H. Sharma, S. K. Singh and L. Gangwar, Chem. Biol. Drug Des., 2013, 82(1), 119–124 CAS.
- R. P. Sijbesma, A. P. M. Kentgens and R. J. M. Nolte, J. Org. Chem., 1991, 56(10), 3199–3201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of all compounds, NMR titration data, and DFT calculation details (PDF). See DOI: 10.1039/c7nj00969k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |