
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceβ-Configured clickable [18F]FDGs as novel 18F-fluoroglycosylation tools for PET†
M.
Elgland
a,
P.
Nordeman
b,
T.
Fyrner
a,
G.
Antoni
b,
K. Peter R.
Nilsson
 *a and
P.
Konradsson
*a
*a and
P.
Konradsson
*a
aLinköpings University, IFM – Department of Biology, Chemistry & Physics, SE-581 83 LINKÖPING, Sweden. E-mail: petko@ifm.liu.se; petni@ifm.liu.se
bDepartment of Medicinal Chemistry, Uppsala University, Uppsala SE-75123, Sweden
First published on 18th August 2017
Abstract
In oncology and neurology the 18F-radiolabeled glucose analogue 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) is by far the most commonly employed metabolic imaging agent for positron emission tomography (PET). Herein, we report a novel synthetic route to β-configured mannopyranoside precursors and a chemoselective 18F-fluoroglycosylation method that employ two β-configured [18F]FDG derivatives equipped with either a terminal azide or alkyne aglycon respectively, for use as a CuAAC clickable tool set for PET. The β-configured precursors provided the corresponding [18F]FDGs in a radiochemical yield of 77–88%. Further, the clickability of these [18F]FDGs was investigated by click coupling to the suitably functionalized Fmoc-protected amino acids, Fmoc-N-(propargyl)-glycine and Fmoc-3-azido-L-alanine, which provided the 18F-fluoroglycosylated amino acid conjugates in radiochemical yields of 75–83%. The 18F-fluoroglycosylated amino acids presented herein constitute a new and interesting class of metabolic PET radiotracers.
Introduction
2-Deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) is often referred to as the golden standard in the field of positron emission tomography (PET), where approximately 90% of all PET scans are performed using [18F]FDG.1 As a glucose derivative, [18F]FDG act as a biomarker of glucose metabolism and as such has found widespread use in oncology and neurology, both in clinic and in research.2 When injected into the human body, it preferentially accumulates in tissue that exhibit an increase in glycolysis, e.g., cancer cells. Conversely, diminished glucose consumption due to neuronal cell death in the brain inflicted by neurodegenerative disorders, e.g., Alzheimer's disease, can also indirectly be probed using [18F]FDG. However, despite its prominent role, one major drawback associated with using [18F]FDG is that it suffers from poor tissue selectivity when distinguishing between malignant cells and benign cells that have a high metabolic rate, such as certain cells associated with inflammation or infection.3 Furthermore, since [18F]FDG is a metabolic imaging agent it is not sufficiently capable of detecting tumors found in most prostate cancers, bronchoalveolar-cell carcinoma or renal-cell carcinoma due to the fact that their metabolic rate is relatively low and therefore indistinguishable from normal tissue.4 Consequently, there is a great need for new PET tracers that exhibit both high affinity and selectivity towards a particular biomolecular target. In recent years, the concept of employing [18F]FDG as a prosthetic group for the 18F-radiolabeling of ligands with defined molecular targets in vivo has received considerable attention.5 The rationale is that glycoconjugates (e.g., glycopeptides, glycoproteins) in general exhibit improved pharmacokinetic properties such as increased in vivo kinetics, higher metabolic stability and faster blood clearance compared to their non-glycosylated counterparts.6 Furthermore, the relatively long half-life of 18F (t1/2 = 109.8 min) provides a sufficient amount of time to perform multi-step radiosynthesis followed by a subsequent PET investigation.Several methods have been employed for the ligation of [18F]FDG to a diverse set of ligands, for instance (i) via oxime formation,7 (ii) chemoenzymatic approaches8 as well as using (iii) thiol-selective S-glycosylation.9 More importantly, one of the most established methods is by means of the CuAAC click reaction10,11 which allows for a wide substrate scope using mild conditions.12 Accordingly, in 2009, Maschauer et al.,13 reported the synthesis of a set of 2-O-trifluoromethanesulfonyl-D-mannopyranosides, i.e., precursors of [18F]FDG analogs, that were equipped with either a α-1-O-propargyl (10α), α-1-O-(2-azido)ethyl (6α), α- or β-azido aglycons (16α and 16β respectively, Scheme 1).
 | ||
| Scheme 1 (to the left) Previous work by Maschauer et al. on 18F-radiolabeling of clickable α and β [18F]FDG precursors and (to the right) this work describing the high-yielding 18F-radiolabeling of β-D-mannopyranoside triflates equipped with both azide- and alkyne spacers. | ||
In their study, only minor amounts of the α-configured precursors were radiolabeled (≤14% radiochemical yield (RCY)) which rendered them inapplicable for PET. In contrast, the β-azide precursor (16β) gave a significantly higher RCY (71%) indicating that the β-anomeric configuration is a key criterion for a successful 18F-radiolabelling.
The propensity of the precursors to undergo the displacement by 18F− correlates well with the Richardson–Hough rules from 1967 that predict the SN2 displacement viability in carbohydrate sulfonate derivatives with external nucleophiles.14 The outcome of the reaction is dictated by stereoelectronic effects but also accompanied by steric factors. This theory recently got extended by Hale et al. to encompass seemingly disallowed trifluoromethanesulfonate displacements.15 Briefly, the theory predicts that 2-O-trifluoromethanesulfonyl-β-D-mannopyranosides experience a significantly lower dipolar repulsion at the transition state and will therefore readily undergo the SN2 displacement while the corresponding α-anomer should only give minor substitution or none at all.
The β-azide equipped [18F]FDG (17β, Scheme 1) has successfully been utilized as a 18F-fluoroglycosylation tool to provide [18F]FDG conjugates with a diverse set of ligands, e.g., the RGD peptide targeting the integrin receptor,16,17 folic acid targeting the folate receptor,18 and a diarylpyrazole targeting the NTS-1 neurotensin receptor.19 However, the first generation of clickable [18F]FDGs is rather limited in scope. Ideally, a spacer between the FDG unit and the attached click functional group is desired to prevent any potential steric interference between the FDG unit and the ligand to which it will be attached. In fact, it has been shown that glucose derivatives bearing an anomeric 1,2,3-triazole, generated from the corresponding glucosyl azide, lack two key properties of [18F]FDG, i.e., trapping via O-6 phosphorylation and cell uptake via GLUT-1 transporters.20 This effect is most likely caused by the close proximity of the triazole. Moreover, ideally, in order to be able to radiolabel the full set of available click-enabled ligands, both azide- and alkyne equipped [18F]FDGs should be accessible. However, to date no alkyne equipped [18F]FDG that can be retrieved in a sufficient radiochemical yield is available.
In this work, we report the detailed synthesis of the CuAAC clickable β-configured [18F]FDGs (Scheme 1). In order to evaluate their clickability, the [18F]FDGs were chemically ligated to the suitably functionalized Fmoc-protected amino acids Fmoc-N-(propargyl)-glycine 12 and Fmoc-3-azido-L-alanine 14 by means of the click reaction which successfully provided the expected [18F]FDG conjugates, both rapidly and in good radiochemical yield. Due to the increased cell proliferation of most cancers, protein synthesis is up-regulated and, as a natural consequence, the amino acid uptake is usually significantly increased. The synthesized [18F]FDG amino acid conjugates will therefore serve as the basis for a new class of highly promising metabolic imaging agents. Furthermore, the described 18F-fluoroglycosylation method may well be extended to encompass the radiolabeling of, not only amino acids, but also peptides and proteins.
Results and discussion
Since the fluorination step required to obtain the β-configured [18F]FDG ([18F]7, [18F]11), is accompanied by an SN2 inversion, the triflate precursors (6, 10) are by necessity β-D-mannopyranosides. Unfortunately, β-D-mannopyranosides are well-known to be challenging to synthesize due to the inherently strong preference of conventional mannosyl donors to form α-D-mannopyranosides. This is a result of the simultaneous occurrence of both the α-directing anomeric effect and repulsion between the axial C-2 substituent and the approaching nucleophile.21 Moreover, neighboring group participation of a 2-acyl mannosyl donor also leads to α-mannopyranosides. Fortunately, several ingenious synthetic approaches to circumvent these obstacles have already been devised.A particularly elegant approach was developed by the Danishefsky group during the 1990's, where epoxidation of per-benzylated D-glucal using DMDO, conveniently provided the corresponding 1,2-α-anhydrosugar that subsequently during glycosylation stereoselectively produced the corresponding β-D-glucoside – and simultaneously generated an unprotected hydroxyl group in the 2-position. Subsequent epimerization of this site, following an oxidation–reduction sequence generated the β-D-mannopyranoside.22 In this report we employ a similar synthetic approach which is depicted in Scheme 2. The choosen route requires access to multi-gram quantities of 1,2-anhydrosugar 2α from tri-acetyl-D-glucal 1. While this transformation is commonly achieved using a dilute solution of DMDO in acetone,23 performing this reaction on a large scale poses several practical issues. For instance, the DMDO solution has to be kept rigorously dry, only dilute solutions can be produced (ca. 0.1 M) and furthermore, the scale-up of organic peroxy compounds is potentially hazardous.
 | ||
| Scheme 2 General conditions and reagents: (i) oxone, acetone, TBAHSO4, DCM, satd. aq. NaHCO3, 0 °C to rt. (ii) Cl(CH2)2OH, ZnCl2, DCM, 0 °C to rt. (iii) Propargyl alcohol, ZnCl2, DCM, 0 °C to rt. (iv) NaI, acetone, 70 °C (v) NaN3, DMF, rt. (vi) Tf2O, py, DCM, −15 to 10 °C. (vii) TBANO2, toluene, 50 °C. (viii) DAST, diglyme, 100 °C, 10 min. (ix) TBANO2, toluene, rt. | ||
To circumvent these drawbacks, the Dondoni group, in 2006, developed an epoxidation method for glycals where DMDO is formed in situ from a biphasic buffered solution containing oxone, acetone, satd. NaHCO3 and DCM.24 This protocol was further elaborated on by Lafont et al. in 2011 by the addition of the phase transfer catalyst, tetrabutylammonium hydrogen sulphate (TBAHSO4) that significantly increased the reaction speed as well as the reproducibility.25 By employing the oxone-acetone-TBAHSO4 epoxidation protocol, we conveniently prepared the 1,2-anhydrosugar 2α which was then directly used in the subsequent glycosylation. The glycosylation of 2-chloroethanol or propargyl alcohol, promoted by ZnCl2 in DCM, provided the corresponding β-D-glucosides 3 and 8 in 39% and 29%, respectively over two steps. The predominant by-product formed was the corresponding 1,2-diol hydrolysis product. Converting the 2-chloroethyl glucoside 3 to the corresponding 2-azidoethyl glucoside 4 proved to be a major challenge. Standard conditions for azidation, i.e., NaN3 in DMF at elevated temperatures (80 °C or 110 °C), with or without the additives TBAI or TBAB, consistently resulted in a complex mixture of products due to acetate migration to the unprotected 2-OH, whereas no reaction occurred at ambient temperature. We therefore reasoned that installing a better leaving group might make the reaction proceed more smoothly and thus avoid the accompanying acetate migration. This was realized by converting the 2-chloro compound 3 to the corresponding iodo compound by means of the classical Finkelstein reaction utilizing NaI (2 eq.) in acetone at 70 °C for 48 h. The azidation of the iodoethyl glucoside using NaN3 in DMF now proceeded cleanly at ambient temperature to afford the azidoethyl glucoside 4 in good yield (77%) over two steps. Next, we decided to use the nitrite-mediated Lattrell–Dax inversion for the pending epimerization. The Lattrell–Dax inversion has been extensively studied by the Ramström group26 who concluded that the inversion proceeds reliably – and even stereospecifically – if and only if a vicinal equatorial ester is present. This criterion is suitably met for the glucosides 4 and 8. Standard triflation of glucoside 4 using triflic anhydride followed by inversion using TBANO2 in toluene at ambient temperature over night yielded the β-D-mannopyranosides 5 and 9 cleanly albeit in a moderate yield of 46% and 48% respectively. The β-anomeric configuration was confirmed by evaluating the JC-1,H-1 coupling constant in a coupled 1H–13C NMR experiment and was found to be 157.6 Hz and 159.0 Hz respectively for β-D-mannopyranosides 5 and 9.27 With the desired β-D-mannopyranosides at hand, standard triflation afforded the β-D-mannosyl triflates, i.e., the [18F]FDG precursors 6 and 10 in 48% and 60%, respectively after silica gel chromatography. Subsequently, to afford the FDG analogs as a non-radioactive reference, a typical fluorination protocol using potassium fluoride and the cryptand kryptofix (K[2.2.2]) in MeCN at 85 °C was attempted. Unfortunately, only trace amount of the expected fluorosugar could be observed, most likely as a result of the known poor nucleophilicity of the fluoride ion. Instead, we choose to directly fluorinate the β-D-mannopyranosides bearing a free hydroxyl-group in the second position, 5 and 9, using the common fluorination agent, DAST.28 At first, the reaction was run in DCM at −25 °C to rt over night to give the fluorosugars 7 and 11 in 31% and 19% respectively. The poor yield rendered us to instead perform the reaction in diglyme at 100 °C for 10 min which afforded the fluorosugars 7 and 11 in a significantly increased yield of 66% and 35% respectively. The fluorination proceeded as expected with inversion of configuration, as proven by the characteristic JH-1,H-2 coupling constant of 7.6 Hz for 7 and 7.7 Hz for 11, confirming the gluco-configuration. Furthermore, 1D 19F-NMR revealed a characteristic FDG signal for fluorosugar 7 at −199.7 ppm (ddd, 2JH-2,F-2 = 50.5 Hz, 3JH-3,F-2 = 14.3 Hz, 3JH-1,F-2 = 2.7 Hz), and a similar signal for 11 (ESI,† page 35 and 50 respectively).
To test our hypothesis that a β-configured FDG precursor is radiolabeled to a greater extent than its α-anomeric counterpart, we decided to synthesize the α-FDGs reported by Maschauer et al. to make a direct side-by-side comparison using the same 18F-fluorination set-up. On closer inspection of our devised synthetic route to the β-FDGs we realized that since the epoxidation step is only moderately stereoselective on acetylated glycals (α/β approx., 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1),24 as a consequence, the minor β-1,2-anhydrosugar (2β) would then upon glycosylation stereoselectively be ring-opened to give the sought α-D-mannopyranosides, 4α and 8α, with the second hydroxyl group exposed.
:1),24 as a consequence, the minor β-1,2-anhydrosugar (2β) would then upon glycosylation stereoselectively be ring-opened to give the sought α-D-mannopyranosides, 4α and 8α, with the second hydroxyl group exposed.
As predicted, the glycosylations outlined in Scheme 3 resulted in a diastereomeric mixture of the glycosides 4 and 4α, and 8 and 8α, (dr ∼ 9:1 gluco/manno in both cases) that were separated using preparative HPLC. Spectroscopic data for the α-mannopyranosides was in good agreement with the data reported by Maschauer et al.29 Triflation or fluorination (vide supra) of the isolated α-mannopyranosides then furnished the required precursors (6α and 10α) and the α-configured FDGs (7α and 11α) respectively.
 | ||
| Scheme 3 General conditions and reagents: (i) oxone, acetone, TBAHSO4, DCM, satd. aq. NaHCO3, 0 °C to rt. (ii) Cl(CH2)2OH, ZnCl2, DCM, 0 °C to rt. (iii) Propargyl alcohol, ZnCl2, DCM, 0 °C to rt. (iv) NaI, acetone, 70 °C. (v) NaN3, DMF, rt. (vi) Tf2O, py, DCM, −15 to 10 °C. (vii) DAST, diglyme, 100 °C, 10 min. | ||
18F-Radiolabeling
18F was produced using the 18O(p,n)18F reaction using a scanditronix MC-17 cyclotron. The water was transported to the hot cell and trapped on a QMA which was flushed with MeCN. [18F]Fluoride was eluted using a kryptofix 2.2.2/K2CO3 solution. After azeotropic distillation with MeCN the precursor (6, 6α, 10 or 10α) in dry MeCN was added to the vial which was heated to 85 °C for 30 minutes. After the reaction was finished, an aliquot was taken and analysed by radio-HPLC to determine the non-isolated RCY of the reaction (Table 1). Both β-mannopyranosides 6 and 10 performed well under these conditions and gave RCY of 77 ± 2% and 88 ± 5% for [18F]7 and [18F]11, respectively (n = 2 for both compounds) (Scheme 4). Unfortunately the α-precursors gave no results (0% RCY) using these conditions as only unreacted 18F− could be identified using analytical radio-HPLC.‡ Having established a synthetic protocol to acquire the [18F]FDGs we then turned to investigate their performance in the click reaction. We choose to use the appropriately functionalized Fmoc-protected amino acids, Fmoc-N-(propargyl)-glycine (12) and Fmoc-3-azido-L-alanine (14) as a model system to show the proof of concept. The click reaction was performed in a one pot, two-step process starting from 18F− and 6 or 10. After the reaction, the MeCN was evaporated with a stream of helium after which the protected [18F]7 or [18F]11 was subsequently dissolved in dry DMF, whereto the Cu(I) catalyst, prepared in situ by the addition of aq. CuSO4 (50 μL, 0.20 M) and aq. (+)-L-sodium ascorbate (50 μL, 0.60 M) along with amino acid 12 or 14 (2.5 μmol) were added. The reaction was then heated to 60 °C for 20 min. The RCY of [18F]13 was 75 ± 2% and for [18F]15 83 ± 2% as deducted by analytical-HPLC (n = 2 for each compound). In preparative runs starting with 3 GBq (81 mCi) of fluoride, 820 MBq (22 mCi) of [18F]13 (55% RCY) and 990 MBq (27 mCi) of [18F]15 (66%) was obtained, respectively. Both compounds were pure (RCP > 99%) as deducted by HPLC. The RCY was decay corrected to end of bombardment (EOB) and the total synthesis time was 2 hours. The corresponding non-radioactive FDG amino acid conjugates was synthesized using microwave irradiation at 80 °C for 5 min (ESI,† Scheme S2, page 3). The click reaction between FDG 7 (1.2 eq.) and amino acid 12 (1.0 eq.) and between FDG 11 (1.2 eq.) and amino acid 14 (1.0 eq.) yielded FDG conjugates 13 and 15 in 64% and 86% respectively.|
|
|||
|---|---|---|---|
| Entry | Precursor | Product | RCY (%) |
| 1 | 6 |

|
77 ± 2 |
| 2 | 6α |

|
0 |
| 3 | 10 |

|
88 ± 5 |
| 4 | 10α |
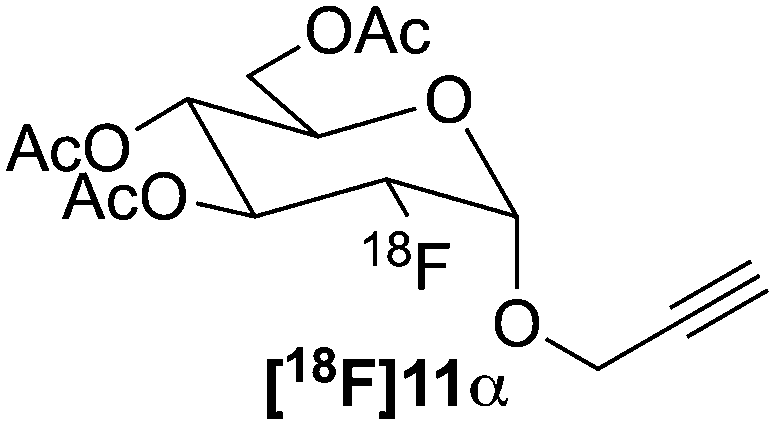
|
0 |

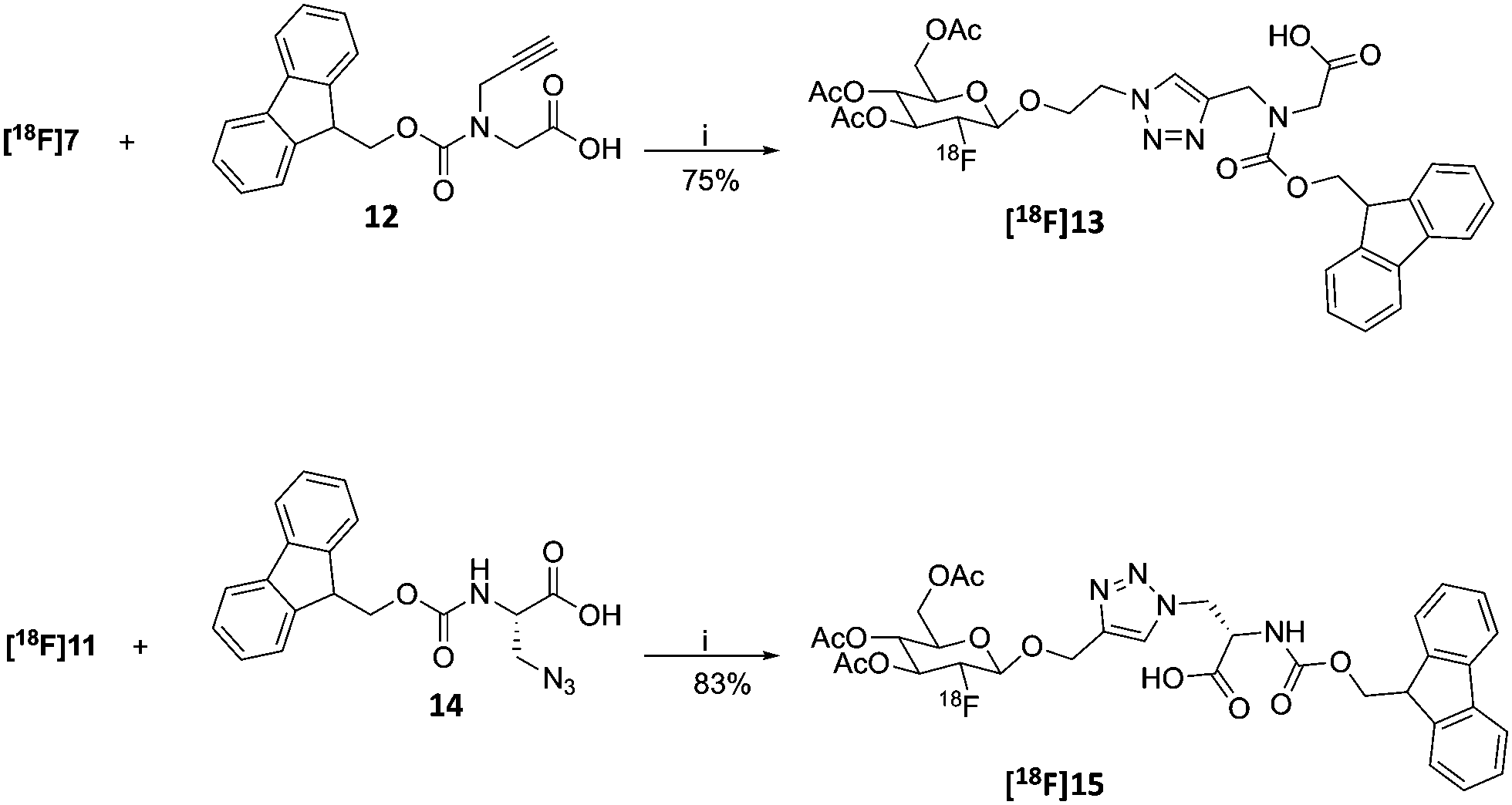 | ||
| Scheme 4 18F-Fluoroglycosylation of amino acids 12 and 14. General conditions and reagents: (i) amino acid (0.25 μmol) aq., CuSO4 (50 μL, 0.20 M), aq., (+)-sodium L-ascorbate (50 μL, 0.60 M), 60 °C, 20 min, DMF. | ||
Conclusions
We have developed a chemoselective 18F-fluoroglycosylation method for PET in vivo imaging that employ the β-configured [18F]FDGs, [18F]7 and [18F]11, as CuAAC clickable prosthetic groups for 18F-labeling. These clickable [18F]FDGs may in principle be readily attached to any click functionalized ligand and thus provide access to a diverse set of 18F-fluoroglycosylated PET radiotracers. We have also demonstrated that the anomeric configuration of the [18F]FDG precursors has a great impact on the radiochemical yield, where the β-configuration is superior to the α-configuration. Furthermore, the clickability of these [18F]FDGs was demonstrated by click coupling to the suitably functionalized Fmoc-protected amino acids, Fmoc-N-(propargyl)-glycine (12) and Fmoc-3-azido-L-alanine (14) in near quantitative RCY, using a simple two-step one-pot protocol. The 18F-fluoroglycosylated amino acids, [18F]13, [18F]15, constitute a new and interesting class of metabolic oncological PET radiotracers. We also recognize that while 18F is excellently suited for PET imaging, the natural isotope, 19F, has almost optimal properties for 19F-NMR, a technique that in recent years has received considerable attention as a means to probe biological mechanisms.30 The developed 18F-fluoroglycosylation method at hand may thus equally well serve as a means to provide novel 19F-NMR probes. Our lab is currently engaged in synthesizing [18F]FDG conjugates targeting cancer and disease associated protein aggregates that will be reported in due course.Acknowledgements
Our work is supported by the Swedish Foundation for Strategic Research and the Swedish Research Council.References
- S. Vallabhajosula, L. Solnes and B. Vallabhajosula, Semin. Nucl. Med., 2011, 41, 246–264 CrossRef PubMed.
- G. Ribeiro Morais, R. A. Falconer and I. Santos, Eur. J. Org. Chem., 2013, 1401–1414 CrossRef CAS.
- L. Jianga, Y. Tub, H. Shia and Z. Cheng, J. Biomed. Res., 2014, 28, 1–12 Search PubMed.
- A. Zhu, D. Lee and H. Shim, Semin. Oncol., 2011, 38, 55–69 CrossRef PubMed.
- S. Maschauer and O. Prante, BioMed Res. Int., 2014, 2014, 1–16 CrossRef PubMed.
- R. Haubner, B. Kuhnast, C. Mang, W. A. Weber, H. Kessler, H.-J. Wester and M. Schwaiger, Bioconjugate Chem., 2004, 15, 61–69 CrossRef CAS PubMed.
- M. Namavari, Z. Cheng, R. Zhang, A. De, J. Levi, J. K. Hoerner, S. S. Yaghoubi, F. A. Syud and S. S. Gambhir, Bioconjugate Chem., 2009, 20, 432–436 CrossRef CAS PubMed.
- O. Prante, K. Hamacher and H. H. Coenen, J. Labelled Compd. Radiopharm., 2007, 50, 55–63 CrossRef CAS.
- O. Prante, J. Einsiedel, R. Haubner, P. Gmeiner, H.-J. Wester, T. Kuwert and S. Maschauer, Bioconjugate Chem., 2007, 18, 254–262 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 114, 2708–2711 CrossRef.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- S. Maschauer and O. Prante, Carbohydr. Res., 2009, 344, 753–761 CrossRef CAS PubMed.
- A. C. Richardson, Carbohydr. Res., 1969, 10, 395–402 CrossRef CAS.
- K. J. Hale, L. Hough, S. Manaviazar and A. Calabrese, Org. Lett., 2014, 16, 4838–4841 CrossRef CAS PubMed.
- S. Maschauer, J. Einsiedel, R. Haubner, C. Hocke, M. Ocker, H. Hübner, T. Kuwert, P. Gmeiner and O. Prante, Angew. Chem., Int. Ed., 2010, 49, 976–979 CrossRef CAS PubMed.
- S. Maschauer, R. Haubner, T. Kuwert and O. Prante, Mol. Pharmaceutics, 2014, 11, 505–515 CrossRef CAS PubMed.
- C. R. Fischer, C. Müller, J. Reber, A. Müller, S. D. Krämer, S. M. Ametamey and R. Schibli, Bioconjugate Chem., 2012, 23, 805–813 CrossRef CAS PubMed.
- C. Lang, S. Maschauer, H. Hübner, P. Gmeiner and O. Prante, J. Med. Chem., 2013, 56, 9361–9365 CrossRef CAS PubMed.
- D. H. Kim, Y. S. Choe, K.-H. Jung, K.-H. Lee, J. Y. Choi, Y. Choi and B.-T. Kim, Arch. Pharmacal Res., 2008, 31, 587–593 CrossRef CAS PubMed.
- H. Dong, Z. Pei, M. Angelin, S. Byström and O. Ramström, J. Org. Chem., 2007, 72, 3694–3701 CrossRef CAS PubMed.
- K. Liu and S. J. Danishefsky, J. Org. Chem., 1994, 59, 1892–1894 CrossRef CAS.
- R. W. Murray and R. Jeyaraman, J. Org. Chem., 1985, 50, 2847–2853 CrossRef CAS.
- P. Cheshev, A. Marra and A. Dondoni, Carbohydr. Res., 2006, 341, 2714–2716 CrossRef CAS PubMed.
- D. Lafont, J. D’Attoma, R. Gomez and P. G. Goekjian, Tetrahedron: Asymmetry, 2011, 22, 1197–1204 CrossRef CAS.
- H. Dong, Z. Pei and O. Ramström, J. Org. Chem., 2006, 71, 3306–3309 CrossRef CAS PubMed.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- P. J. Card, J. Org. Chem., 1983, 48, 393–395 CrossRef CAS.
- S. Maschauer and O. Prante, Carbohydr. Res., 2009, 344, 753–761 CrossRef CAS PubMed.
- E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242–1250 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj00716g |
| ‡ In the original radiolabelling procedure by Maschauer et al. an optimized kryptofix 2.2.2 buffer (K2CO3/KH2PO4) was necessary in order to keep the precursors intact and obtain a RCY. In addition, the precursor load was 15 μM compared to 2 μM used herein. These differences may explain the failed radiolabelling of the α-precursors in our hands. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |