
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystalline salts of metal phthalocyanine radical anions [M(Pc˙3−)]˙− (M = CuII, PbII, VIVO, SnIVCl2) with cryptand(Na+) cations: structure, optical and magnetic properties†
Dmitri V.
Konarev
 *a,
Maxim A.
Faraonov
a,
Alexey V.
Kuzmin
b,
Salavat S.
Khasanov
b,
Yoshiaki
Nakano
c,
Semen I.
Norko
ad,
Mikhail S.
Batov
ad,
Akihiro
Otsuka
c,
Hideki
Yamochi
c,
Gunzi
Saito
e and
Rimma N.
Lyubovskaya
a
*a,
Maxim A.
Faraonov
a,
Alexey V.
Kuzmin
b,
Salavat S.
Khasanov
b,
Yoshiaki
Nakano
c,
Semen I.
Norko
ad,
Mikhail S.
Batov
ad,
Akihiro
Otsuka
c,
Hideki
Yamochi
c,
Gunzi
Saito
e and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, 142432, Russia. E-mail: konarev3@yandex.ru; Fax: +7-496-522-18-52
bInstitute of Solid State Physics RAS, Chernogolovka, Moscow region 142432, Russia
cDivision of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
dMoscow State University, Leninskie Gory, 119991 Moscow, Russia
eToyota Physical and Chemical Research Institute, 41-1, Yokomichi, Nagakute, Aichi 480-1192, Japan
First published on 6th June 2017
Abstract
Radical anion salts of metal phthalocyanines {cryptand[2,2,2](Na+)}[M(Pc˙3−)]˙−·C6H4Cl2 (M = CuII (1), PbII (2), VIVO (3) and SnIVCl2 (4)) and {cryptand[2,2,1](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 (5) have been obtained as single crystals. Phthalocyanine planes form channels to accommodate the cryptand[2,2,2](Na+) cations and solvent C6H4Cl2 molecules in 1–3. Nearly square phthalocyanine layers are formed in salts 4 and 5, which are parallel to the ac plane and have an effective π–π interaction between the Pc˙3− macrocycles. The using of smaller cryptand[2,2,1](Na+) cations in 5, instead of larger cryptand[2,2,2](Na+) cations in 4, allows one to obtain more closely packed Pc layers. Reduction in the salts was centered on the Pc macrocycles, providing alternation of the C–N (imine) bonds in Pc due to partial loss of aromaticity. New bands were also observed in the NIR spectra at 946–1006 nm and the Soret and Q-bands were essentially blue-shifted in the spectra of the salts. The [CuII(Pc˙3−)]˙− and [VIVO(Pc˙3−)]˙− radical anions contain paramagnetic CuII and VIVO centers and the Pc˙3− radical trianions, both having a S = 1/2 spin state. These radical anions were nearly isolated in the salts. Weak antiferromagnetic coupling with a Weiss temperature (θ) of −2 K was observed in 1. The magnetic behavior of 3 was described by a modified singlet–triplet model with a ferromagnetic exchange coupling of J/kB = 7.7 K within [VIVO(Pc˙3−)]˙− and θ = −1.3 K. The stronger antiferromagnetic coupling with a Weiss temperature of θ = −64 K was observed in 4 and was accompanied by the decrease of molar magnetic susceptibility below 35 K due to antiferromagnetic ordering of spins. The magnetic behavior of 4 was described using the Heisenberg model for quasi-square two-dimensional antiferromagnetic coupling between spins with an exchange interaction of J/kB = −18 K.
Introduction
Metal-free and metal-containing phthalocyanines (Pcs) in oxidized or reduced forms can have promising optical, conducting and magnetic properties.1–5 For example, oxidation of axially substituted phthalocyanine [MIIIL2Pc]− anions (M = Co, Fe; L = CN, Cl, Br) allows one to obtain crystalline salts with a π–π stacking columnar arrangement of the macrocycles showing quasi-one-dimensional metallic behavior down to liquid helium temperatures.2–4 Salts with FeIII as a central metal atom show giant magnetoresistance.2–4 Chain compounds with ferrimagnetic ordering between spins were obtained with oxidized manganese phthalocyanine.5Less attention was paid to the preparation of compounds with reduced metal phthalocyanines most likely due to their air and moisture sensitivity. However, some theoretical works predict high conductivity, including even superconductivity, for electron doped metal phthalocyanines.6 At present, powdered anionic Pc compounds can be prepared by doping of metal phthalocyanines by alkali metals in the gas phase.7–9 Several crystalline salts were obtained by metal phthalocyanine reduction with alkali metals or the KC8 reductant in coordinating solvents like tetrahydrofuran or 1,2-dimethoxyethane.10–13 Recently, we have developed a method to prepare crystalline radical anion salts of metal-free and metal-containing phthalocyanines by neutral Pc reduction with sodium fluorenone ketyl in the presence of organic cations. As a result, salts of iron(I) and cobalt(I) phthalocyanine anions whose reduction is centered on metal atoms were obtained.14–16 A series of radical anion (cation+)[M(Pc˙3−)]˙− salts, where M = H2, NiII, CuII, PbII, SnII, TiIVO, VIVO, SnIVCl2, InIIIBr and cation = tetraalkylammonium, were synthesized and structurally characterized.17–20 This allows one to study crystal structures, optical and magnetic properties of radical anion salts with different Pcs as well as to estimate the effect of reduction on their geometry, optical and magnetic properties.
To study how the geometry of metal Pc and size of cations affect the packing of Pc macrocycles in a crystal, we obtained a series of metal phthalocyanine salts with cryptand[2,2,2](Na+) and cryptand[2,2,1](Na+) counter cations, whose compositions are shown in Table 1. Until now, only several metal phthalocyanine salts with cryptand[2,2,2](Na+) cations are known. Those are the salts with iron phthalocyanine [FeI(Pc2−)]− and [Fe0(Pc2−)]2− anions,10,14 {cryptand[2,2,2](Na+)}(H2Pc˙−)·1.5C6H4Cl217 and {cryptand[2,2,2](Na+)}[SnII(Pc˙3−)]˙−·C6H4Cl2.21 Obtained salts are categorized in two types according to the packing manner of Pcs: with channels formed by Pc macrocycles or with closely packed Pc layers. Pcs are nearly isolated in the structures of the first type (1–3) and magnetic interactions are realized only between the spins of the metal center and the Pc˙3− macrocycle. At the same time, salt 4 with a layered structure shows strong coupling between spins with long-range antiferromagnetic ordering below 35 K. We report and discuss the syntheses, crystal structures, optical and magnetic properties of these salts.
| N | Compositiona |
|---|---|
| a Cryptand[2,2,2]: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane; cryptand[2,2,1]; 4,7,13,16,21-pentaoxa-1,10-diazabicyclo[8.8.5]tricosane. | |
| 1 | {Cryptand[2,2,2](Na+)}[CuII(Pc˙3−)]˙−·C6H4Cl2 |
| 2 | {Cryptand[2,2,2](Na+)}[PbII(Pc˙3−)]˙−·C6H4Cl2 |
| 3 | {Cryptand[2,2,2](Na+)}[VIVO(Pc˙3−)]˙−·C6H4Cl2 |
| 4 | {Cryptand[2,2,2](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 |
| 5 | {Cryptand[2,2,1](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 |
Results and discussion
1. Synthesis
Syntheses of 1–3 were carried out in the presence of the organometallic compound [Cp*Mo(CO)2]2, which essentially improved the crystal growth and allows one to obtain high-quality single crystals but was not incorporated in the crystals. Interestingly, similar syntheses with CuIIPc, PbIIPc and VIVOPc without [Cp*Mo(CO)2]2 afforded only polycrystalline products composed of small-sized crystals in low yield.In the case of SnIVCl2Pc, direct reduction with sodium fluoreneone ketyl does not allow the preparation of crystals most probably due to possible substitution of fluorenone ketyl radical anions for chloride anions interfering the formation of good quality crystals. Similar substitution of chloride anions by fluorenone ketyl radical anions is observed, for example, when boron subphthalocyanine chloride is reduced with sodium fluorenone ketyl.22 However, reduction of SnIVCl2Pc with C60˙− provides high-quality single crystals of radical anion salts 4 and 5. Since the (PPN+)[SnIVCl2(Pc˙3−)]˙− salt was previously obtained by this method,19 it is seen that it is suitable for the preparation of crystalline radical anion salts of metal Pcs coordinated with chloride anions at the metal center.
2. Spectra of phthalocyanine salts in the IR and UV-visible-NIR ranges
IR spectral data for the starting compounds and obtained salts are presented in Table S1 and Fig. S1–S4 (ESI†). The IR spectra of 1–4 show a superposition of the absorption bands of parent metal Pcs, cations and solvent molecules (see the ESI†), but some absorption bands of Pcs are essentially shifted or disappeared by the formation of the salts (see Table S1, ESI†).Spectra of starting Pcs and salts 1–4 in KBr pellets in the UV-visible-NIR range are presented in Fig. 1, and Fig. S5 and S6 (ESI†) and are listed in Table 2. New intense bands are observed in the spectra of the salts in the NIR range of 946–1022 nm. These bands are characteristic of metal Pc radical anions, in which an additional electron is delocalized over the Pc macrocycle.17–21 The spectra of the salts based on Fe(I) and Co(I) Pc anions with the reduced metal centers do not show such bands.14–16 Thus, we can conclude that reduction is centered on the Pc macrocycles in 1–4.
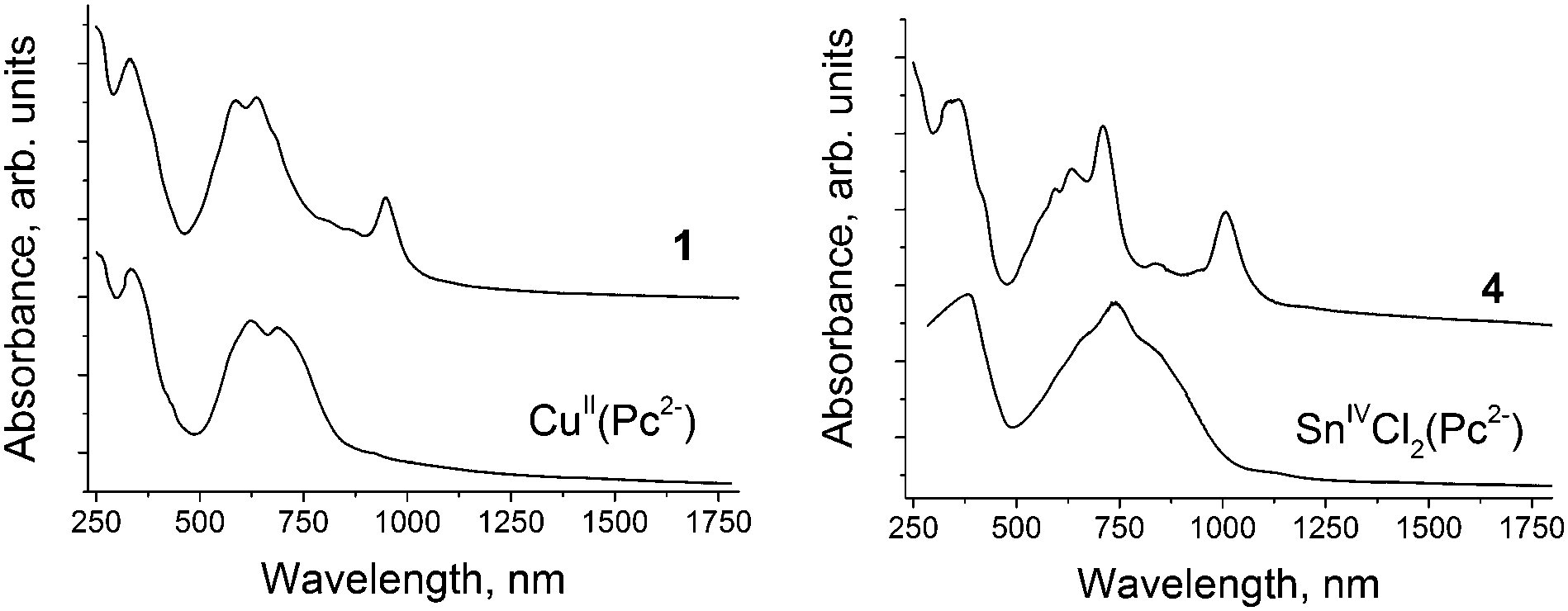 | ||
| Fig. 1 UV-visible-NIR spectra of starting metal phthalocyanines and their radical anion salts 1 and 4 in KBr pellets prepared under anaerobic conditions. | ||
| Compound | Position of the absorption band of phthalocyanines, nm | ||
|---|---|---|---|
| Soret band, nm | Q-band, nm | Radical anion bands, nm | |
| CuII(Pc2−) | 333 | 621, 687 | — |
| {Cryptand[2,2,2](Na+)}[CuII(Pc˙3−)]˙−·C6H4Cl2 (1) | 332 | 585, 636 | 946 |
| PbII(Pc2−) | 345, 432 | 669, 729, 840 | — |
| {Cryptand[2,2,2](Na+)}[PbII(Pc˙3−)]˙−·C6H4Cl2 (2) | 336 | 680, 714 | 1022 |
| VIVO(Pc2−) | 348 | 672, 723 | — |
| {Cryptand[2,2,2](Na+)}[VIVO(Pc˙3−)]˙−·C6H4Cl2 (3) | 334 | 598, 638, 706 | 881, 997 |
| SnIVCl2(Pc2−) | 381 | 670, 740, 848 | — |
| {Cryptand[2,2,2](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 (4) | 348 | 594, 635, 709 | 836, 1006 |
Metal Pcs manifested intense absorption in the UV-visible region, in which the Soret and split Q-bands were located at 333–381 and 620–850 nm, respectively (see Table 2). The formation of 1–4 provided noticeable narrowing of Q-bands and the splitting into two or three bands was still observed in the spectra of the salts (Fig. 1). In all cases, the formation of the salts was accompanied by noticeable blue shifts of Q-bands by 0.04–0.15 eV (15–51 nm) and Soret bands by 0.01–0.31 eV (1–33 nm). Such shifts are generally observed for the metal Pc radical anions, whose Pc macrocycle is reduced. The largest shift of both Soret and Q-bands was observed during the formation of 4 with [SnIVCl2(Pc˙3−)]˙−.
3. Crystal structures of salts 1–5 and molecular structures of [M(Pc˙3−)]˙−
Geometric parameters of metal Pcs in neutral and radical anion states of 1–4 are shown in Table 3.| Compound | Average length of bonds and contacts, Å | Displacement of atoms from the 24-atom Pc plane, Å | ||||
|---|---|---|---|---|---|---|
| M–Npyrrole | C–Npyrrole | C–Nimine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) short/long/difference short/long/difference |
Metal | Pyrrole nitrogen atoms | Outer carbon of phenylene groups | |
| CuII(Pc2−)23 | 1.951(2) | 1.384(2) | 1.354(2) | 0 | 0.032–0.034 | 0.014–0.133 |
| {Cryptand[2,2,2](Na+)}[CuII(Pc˙3−)]˙−·C6H4Cl2 (1), 2 units | 1.950(2) | 1.385(3) | 1.315(4)/1.345(4)/0.030 | 0 | 0.033–0.039 | 0.084–0.230 |
| 1.946(2) | 1.385(3) | 1.310(4)/1.355(4)/0.045 | 0 | 0.031–0.037 | 0.035–0.149 | |
| Octakis((hexylsulfanyl)phthalocyaninato)lead(II)24 | 2.392(3) | 1.368(3) | 1.330(3) | 1.364 | — | — |
| {Cryptand[2,2,2](Na+)}[PbII(Pc˙3−)]˙−·C6H4Cl2 (2), 1 unit | 2.364(2) | 1.379(3) | 1.328(3)/1.346(3)/0.018 | 1.461 | 0.103–0.308 | 0.071–0.945 |
VIVO(Pc2−),25 V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1.595(4) Å) O (1.595(4) Å) |
2.044(4) | 1.387(4) | 1.334(4) | 0.696 | 0.083–0.165 | 0.129–0.549 |
| {Cryptand[2,2,2](Na+)}[VIVO(Pc˙3−)]˙−·C6H4Cl2 (3), 1 unit, VO (1.599(1) Å) |
2.030(1) | 1.389(2) | 1.312(2)/1.348(2)/0.036 | 0.641 | 0.025–0.117 | 0.008–0.559 |
| SnIVCl2(Pc2−)26 | 2.054(2) | 1.375(3) | 1.334(2) | 0 | 0.080–0.010 | 0.002–0.277 |
| {Cryptand[2,2,2](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 (4), 1 unit, Sn–Cl (2.484(5) Å) | 2.047(10) | 1.385(16) | Accuracy is too low to compare bond length | 0.007 | 0.011–0.152 | 0.095–0.442 |
The reduction of metal phthalocyanines was accompanied by slight shortening of equatorial M–N bonds in 1–4 relative to parent Pcs (Table 3). Salts 1 and 3–5 contain Pc macrocycles with nearly planar conformation. In this case only phenylene groups slightly deviate from planarity (Table 3). However, only copper atoms are located exactly in the 24-atom Pc plane in 1. Tin atoms come out insignificantly from this plane (0.007 Å). Lead and vanadium atoms deviate significantly away from the 24-atom Pc plane, and the deviation was equal to 1.461 Å for the lead atoms, which is noticeably enhanced than 1.364 Å for octakis((hexylsulfanyl)phthalocyaninato)lead(II).
The Pc macrocycle has an aromatic 18 π-electron system. Reduction of this macrocycle yields an 19 π-electron system and should partially disrupt the aromaticity of the system. Indeed, Pc has 8 nearly equal C–N(imine) bonds in neutral form (Table 3) whereas alternation of these bonds in 1–3 supports partial loss of aromaticity in the radical trianion Pc˙3− macrocycle. The differences between shorter and longer C–N(imine) bonds are in the range of 0.018–0.045 Å (Table 3). The bonds are positioned in such a way that shorter and longer bonds belong to the oppositely located isoindole units. Such changes in the molecular structure of the reduced Pc macrocycles are characteristic of the [M(Pc˙3−)]˙− salts.18–21
Depending on the geometry of parent metal Pcs, the formation of two types of salts was observed. In the case of the absence of axial ligands or the presence of only one axial ligand in 1–3, structures with channels are formed. The walls of these channels are built with four Pc planes and the channels are occupied by alternating cryptand[2,2,2](Na+) cations and solvent C6H4Cl2 molecules. Similar channel structures were previously found for the radical anion salts of free-base, iron and tin(II) phthalocyanines with the cryptand[2,2,2](Na+) cations.14,17,21 In spite of the channel structure, Pcs form pairs in salts 2 and 3 with one side-by-side van der Waals (vdW) C⋯C contact between Pc˙3− in 3 (Fig. 2b) and without such contacts in 2 (Fig. S7, ESI†). It was found that the [VIVO(Pc˙3−)]˙− radical anions considerably tend to form π–π stacking dimers in the radical anion salts,18,27 but it is not the case for 3. Therefore, this is the first structure of [VIVO(Pc˙3−)]˙− with nearly isolated Pc macrocycles. Chains formed by phthalocyanines can be outlined in 1 along the a axis due to the presence of vdW contacts of 3.39 Å length between them. However, those are only side-by-side vdW C⋯C contacts (Fig. 2a).
 | ||
| Fig. 2 Crystal structures of {cryptand[2,2,2](Na+)}[CuII(Pc˙3−)]˙−·C6H4Cl2 (1) viewed along the b axis (a) and {cryptand[2,2,2](Na+)}[VIVO(Pc˙3−)]˙−·C6H4Cl2 (3) viewed along the a axis (b). Salt 2 is isostructural to 3 (see Fig. S7, ESI†). Short vdW C⋯C contacts between phthalocyanines are shown by green dashed lines. Solvent molecules are not shown for clarity. | ||
In the case of [SnIVCl2(Pc˙3−)]˙− with two axial ligands, closely packed Pc layers parallel to the ac plane are formed in 4 (Fig. 3a). The layers are separated by the cryptand[2,2,2](Na+) cations and solvent C6H4Cl2 molecules (Fig. 3b). Salt 5 is isostructural to 4, but has much more closely packed Pc layers due to the smaller size of the cryptand[2,2,1](Na+) cations (Fig. 4). As a result, the number of vdW contacts between [SnIVCl2(Pc˙3−)]˙− in 5 (30 for each unit, Fig. 4) exceeds the number of such contacts in 4 (14 for each unit, Fig. 3a). These contacts shown by green dashed lines in Fig. 3a and 4 are in the range of 3.28–3.56 Å. That is a first example of such closely packed phthalocyanine layers in the radical anion salts. The distances between tin atoms along the a and c axes are also smaller for the layers in 5 (9.975 and 10.580 Å, respectively) in comparison with those in the layers in 4 (10.217 and 10.702 Å, respectively). Thus, we can affect the packing of Pc layers by changing the size of the cations.
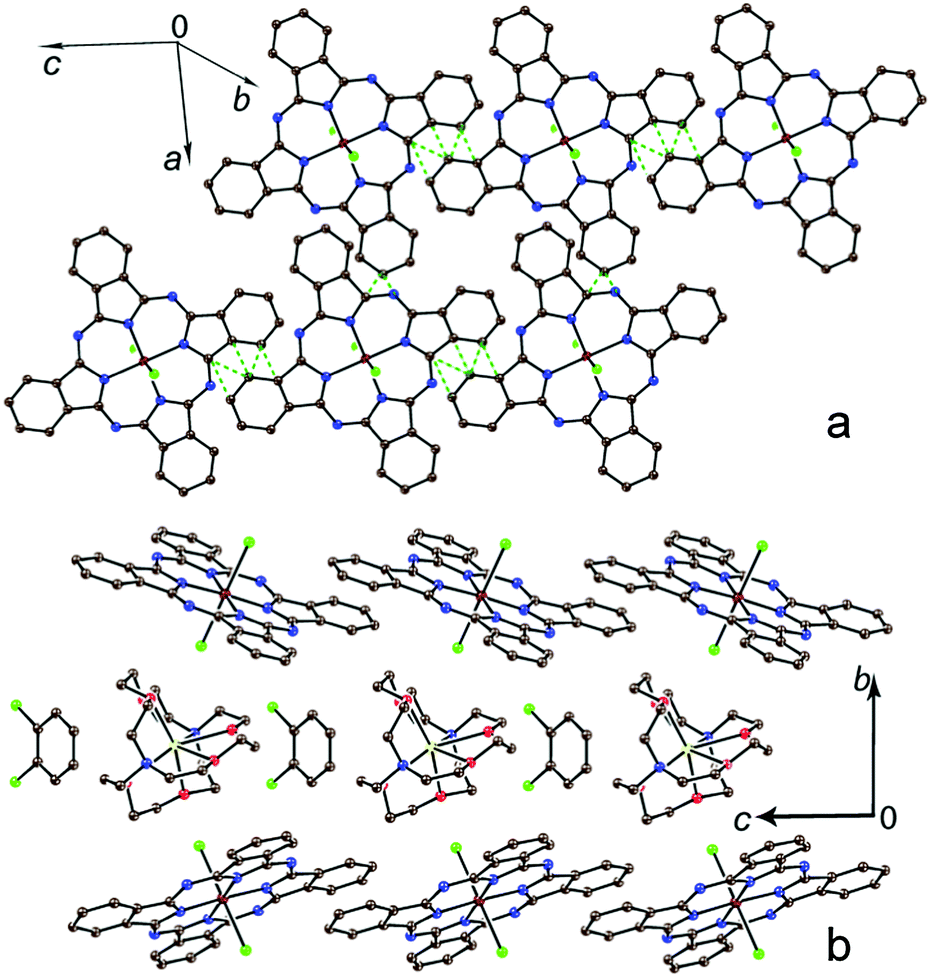 | ||
| Fig. 3 Crystal structure of {cryptand[2,2,2](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 (4). View on the phthalocyanine layer (a) and view along the phthalocyanine layers and the crystallographic a axis (b). vdW C⋯C contacts in the phthalocyanine layers shorter than 3.56 Å are shown by green dashed lines. | ||
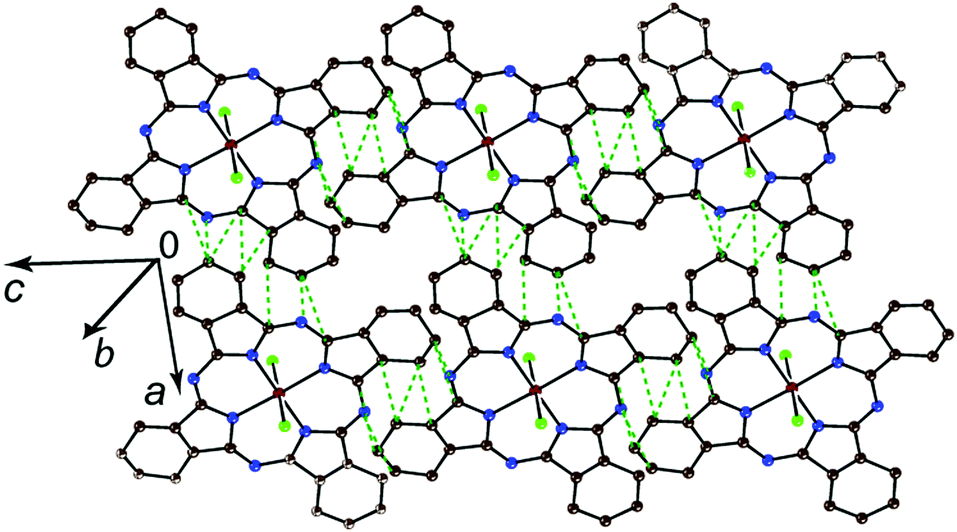 | ||
| Fig. 4 View on the phthalocyanine layer in {cryptand[2,2,1](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 (5). Van der Waals C⋯C contacts in the phthalocyanine layer shorter than 3.56 Å are shown by green dashed lines. | ||
Pc layers in 4 and 5 have almost uniform square arrangement of the [SnIVCl2(Pc˙3−)]˙− radical anions, but according to the number of vdW contacts more closely packed chains can be outlined in 4 along the a axis (Fig. 3a). At the same time, the layers are more uniform in 5 with nearly equal number of vdW contacts in both a and c directions (Fig. 4).
4. Magnetic properties
Magnetic measurements by SQUID and EPR techniques were carried out for polycrystalline samples of 1, 3 and 4 under strictly anaerobic conditions (Table 4).| Compound | SQUID | EPR | |||
|---|---|---|---|---|---|
| μ eff, μB | θ, K | T, K | RT g-factor/ΔH, mT | 4.1 K g-factor/ΔH, mT | |
| 1 | 2.44 | −2 | 10–300 | 2-Component signal | 2-Component signal |
| 2.0522/6.37 | 2.0228/6.38 | ||||
| 2.0794/13.32 | 2.0495/13.49 | ||||
| 3 | 2.19 | +2 | 50–300 | 1.9923/8.91 (99.9%) | 1.9912/4.37 |
| J/kB = 7.7 K | |||||
| θ = −1.3 K | |||||
| 4 | 1.83 | −64 | 70–300 | 1.9933/29.9 | 1.9913/18 |
| J/kB = −18 K | |||||
Magnetic moments of 1 and 3 were 2.44 and 2.19 μB, respectively, at 300 K (Fig. S8a and Fig. 5a), which is in agreement with the contribution of two independent S = 1/2 spins per formula unit (the calculated magnetic moment is 2.45 μB).
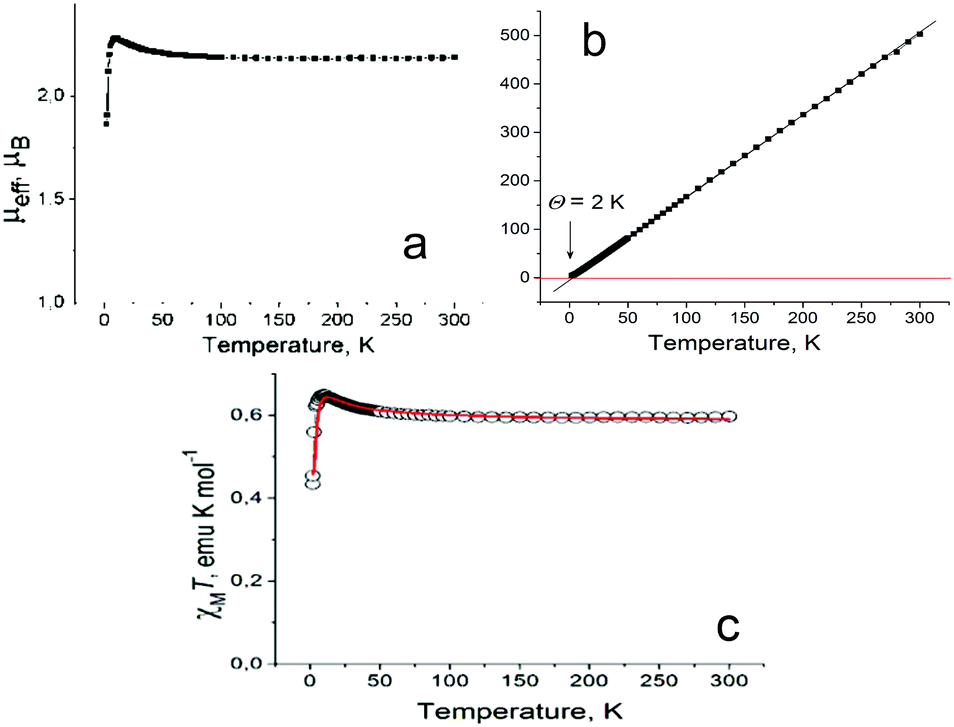 | ||
| Fig. 5 Temperature dependences of effective magnetic moment (a) and reciprocal molar magnetic susceptibility (b) of polycrystalline 3. Fitting of the experimental χMT data (open circles) by the modified S–T model28 with J/kB = 7.7 K, θ = −1.3 K, g = 2 (fix), f = 0.782 (red curve) (c). | ||
Temperature dependences on the χMT values are shown in Fig. S9a and b (ESI†). Since both radical anions contain paramagnetic metal centers, we can suppose that one S = 1/2 spin is localized on the metal atom and the other S = 1/2 spin is delocalized over the Pc˙3− macrocycle. The Weiss temperature for 1 is only −2 K, indicating weak antiferromagnetic coupling between CuII and Pc˙3− spins within one radical anion (Fig. S8b, ESI†). Similarly the salts based on copper(II) phthalocyanine in oxidized Cu(Pc)I or reduced (Bu4N+)2[CuII(Pc˙3−)]˙−(Br−) forms also have close Weiss temperatures of −4.2 and −4 K, respectively, indicating weak antiferromagnetic coupling between spins localized on CuII and delocalized over the Pc macrocycle.4,18
A Weiss temperature of +2 K in 3 determined in the 50–300 K range indicates an apparent weak ferromagnetic coupling between spins (Fig. 5b). The effective magnetic moment of 3 increases below 60 K, and an abrupt decrease is observed below 10 K (Fig. 5a) showing a competition between ferro- and antiferromagnetic interactions in 3. Since the [VIVO(Pc˙3−)]˙− radical anions are nearly isolated in 3, we can suppose that magnetic coupling is realized within the radical anions having two S = 1/2 spins, which can access the triplet and singlet states. Indeed, the magnetic behavior of 3 is described well (Fig. 5c) by the modified singlet–triplet (S–T) model28 with a ferromagnetic exchange interaction between the spins of J/kB = 7.7 K, a Weiss temperature of −1.3 K (antiferromagnetic), a g value of 2 (fixed) and a scaling factor of f = 0.782. Previously, it was shown that the exchange interaction between the VIV and Pc˙3− spins in [VIVO(Pc˙3−)]˙− is also rather weak and has an antiferromagnetic nature. For example, a Weiss temperature of −9.6 K is observed in (Bu4N+)[VIVO(Pc˙3−)]˙−18 also with an isolated arrangement of the macrocycles. The exchange interaction between the VIV and Pc˙3− spins was estimated to be only J/kB = −15.2 K in (Me4P+)[VIVO(Pc˙3−)]˙−(TPC)0.5·C6H4Cl2 (TPC is triptycene) with {[VIVO(Pc˙3−)]˙−}2 π-stacking dimers. In this case the intermolecular coupling between the spins delocalized over the Pc˙3− macrocycles is J/kB = −105 K due to the effective π–π interaction between the macrocycles.27 These data show that despite the two spins being located in one anion at close distances, magnetic coupling between them is rather ineffective and many examples support this supposition. Intermolecular coupling in the presence of π–π interaction between the macrocycles is generally several times greater.
At the same time in both cases (salts 1 and 3) intermolecular coupling is very weak due to the absence of π–π interaction between the macrocycles in the perpendicular arrangement. That is seen if we consider the magnetic behavior of the salt with the radical anions of metal-free phthalocyanine with cryptand[2,2,2](Na+) cations. It has only one S = 1/2 spin delocalized over the Pc˙3− macrocycle and their perpendicular arrangement results in the absence of magnetic coupling (Weiss temperature is in the range of 0 to −1 K).17
Salt 4 has an effective magnetic moment of 1.83 μB at 300 K (Fig. 6a) from one non-interacting S = 1/2 spin per formula unit delocalized on the Pc˙3− macrocycle (the calculated value is 1.73 μB). The temperature dependence of the χMT value is shown in Fig. S9c (ESI†). A Weiss temperature for 4 of −64 K in the 70–300 K range (Fig. 6b) indicates strong antiferromagnetic coupling between spins in the Pc layers (Fig. 3a). The deviation from the Curie–Weiss law to the antiferromagnetic side was observed below 70 K and was accompanied by a decrease of molar magnetic susceptibility below 35 K (Fig. 6c). The temperature behavior of molar magnetic susceptibility of 4 can be described well by the Heisenberg model for quadratic-layer antiferromagnetic coupling between spins29 with an estimated exchange interaction of J/kB = −18 K (Fig. 6c). The use of this model is supported by a nearly square arrangement of [SnIVCl2(Pc˙3−)]˙− in the Pc layers of 4 (Fig. 3a).
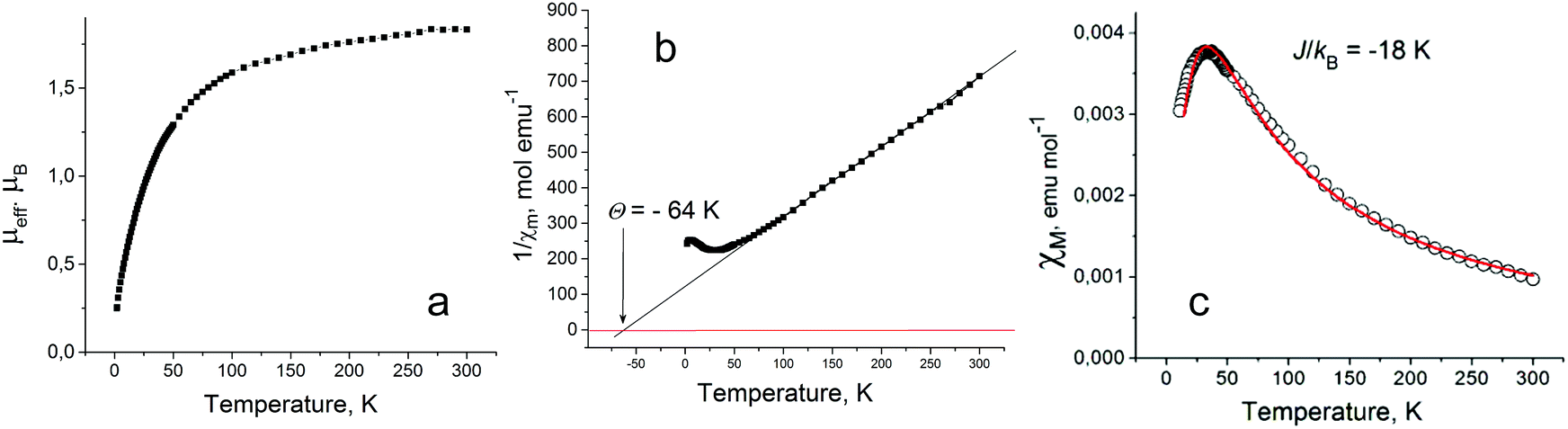 | ||
| Fig. 6 Temperature dependences of effective magnetic moment (a), reciprocal molar magnetic susceptibility (b) and molar magnetic susceptibility (c) of polycrystalline 4. Experimental data were fitted by the Heisenberg model29 for quadratic-layer antiferromagnetic coupling between spins of J/kB = −18 K. | ||
That is a first example of such strong magnetic coupling between spins in the anion-radical phthalocyanine layers which is provided by effective π–π interactions within the layers. Previously even stronger antiferromagnetic coupling was observed only for the isolated π-stacking phthalocyanine {[M(Pc˙3−)]˙−}2 dimers which show an exchange interaction up to J/kB = −183 K.21,27
The EPR spectrum of 1 with two weakly interacting S = 1/2 spins shows two lines with g1 = 2.0522 and ΔH = 6.37 mT, and g2 = 2.0794 and ΔH = 13.32 mT, respectively, at 301 K (Fig. 7a). Previously, two separate EPR signals from Cu2+ (g = 2.194) and Pc˙3− (g = 1.9999–1.9974) were observed for the radical anion (Bu4N+)2[CuII(Pc˙3−)˙−](Br−) salt.18 Since g-factors of the signals in 1 have intermediate values between those characteristic of Cu2+ and Pc˙3−, we can conclude that these signals originate from both paramagnetic Cu2+ and Pc˙3− species having weak exchange interaction. The g-factors in 1 shift to smaller values with almost constant linewidth below 12 K (Fig. S10, ESI†).
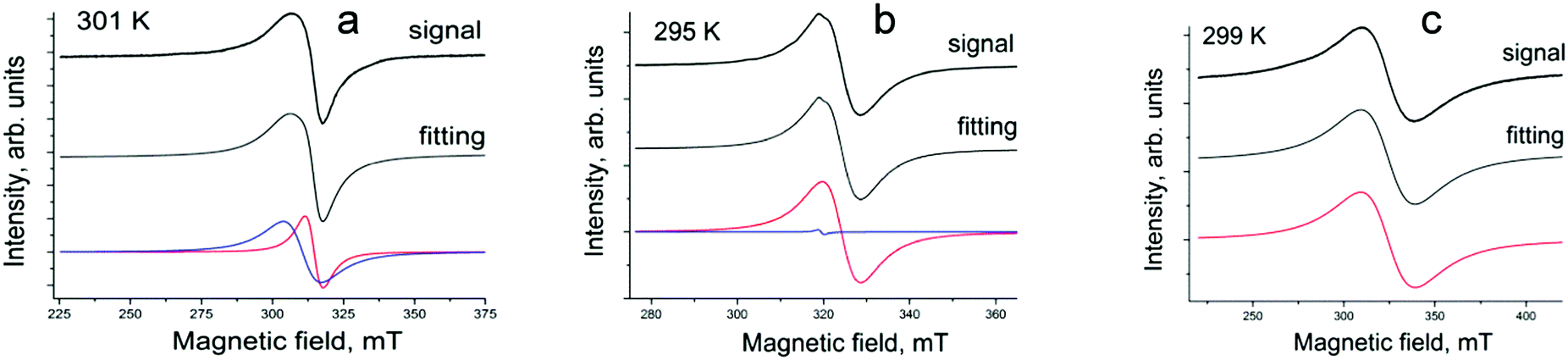 | ||
| Fig. 7 EPR signals observed for polycrystalline {cryptand[2,2,2](Na+)}[CuII(Pc˙3−)]˙−·C6H4Cl2 (1) at 301 K (a); {cryptand[2,2,2](Na+)}[VIVO(Pc˙3−)]˙−·C6H4Cl2 (3) at 295 K (b) and {cryptand[2,2,2](Na+)}[SnIVCl2(Pc˙3−)]˙−·C6H4Cl2 (4) at 299 K (c). Fitting of the signals by one or two Lorentzian lines is shown in the middle and at the bottom (by red and blue curves). | ||
Salt {cryptand[2,2,2](Na+)}[VIVO(Pc˙3−)]˙−·C6H4Cl2 (3) shows two lines at RT with g1 = 1.9923 and ΔH = 8.91 mT, and g2 = 2.0217 and ΔH = 1.44 mT, respectively (Fig. 7b). The integral intensity of the narrower line is only 0.1% from that of the broad line and can be attributed to impurities. The broader main line according to the g-factor value and linewidth can be attributed to paramagnetic VIV and Pc˙3− species having exchange interactions since g-factors of the signals are intermediate between those characteristic of the main signal from VIV in neutral VIVO(Pc2−) (g = 1.9858 and ΔH = 24.9 mT) and the signal from the (H2Pc)˙− salts (g = 2.0033–2.0037 and ΔH = 0.08–0.24 mT).17,18 A similar signals from both VIV and Pc˙3− were observed in (Bu4N+)[VIVO(Pc˙3−)]˙− (g = 1.9918 and ΔH = 7.2 mT at RT)18 and in (Me4P+)[VIVO(Pc˙3−)]˙−(TPC)0.5·C6H4Cl2 (g1 = 1.9954 and ΔH = 25.32 mT, and g2 = 2.0002 and ΔH = 10.49 mT RT).27 The g-factor of the broad signal shifts to smaller values and narrows with the temperature decrease down to 40 K (g = 1.9901 and ΔH = 3.98 mT at 40 K) (Fig. S11, ESI†). Below this temperature, the g-factor shifts slightly to higher values (g = 1.9910 at 4.1 K) without noticeable broadening (ΔH = 4.37 mT at 4.1 K) (Fig. S11, ESI†).
Salt 4 manifests a broad Lorentzian line with g1 = 1.9933 and ΔH = 29.9 mT at 299 K (Fig. 7c) which strongly narrows with the temperature decrease and shifts to smaller g-factors (Fig. S12, ESI†). A similar broad signal is observed in (PPN+)[SnIVCl2(Pc˙3−)]˙−.19 The central metal atom even in the cases of diamagnetic metals, affects the EPR spectra of the radical anions salts. Instead of narrow signals characteristic of (H2Pc)˙− (g-factor of 2.0033–2.0037, linewidth of 0.08–0.24 mT),17,18 broad temperature-dependent signals are observed in the EPR spectra of the radical anion salts based on NiIIPc, PbIIPc, SnIIPc and SnIVCl2Pc.18,19 An increase in the linewidth of the EPR signals is also observed during the transition from the radical anion salt of aluminium(III)phthalocyanines to gallium(III)phthalocyanine and finally to indium(III)phthalocyanine.30 Most probably this effect could be explained by weak transfer of spin density from the Pc˙3− macrocycle to the central metal atom accompanied by the formation of paramagnetic metal centers affecting the EPR spectra of the salts. Similar effects were previously reported for the oxidized NiIIPc species.31,32
Experimental
Materials
CuIIPc (Aldrich, sublimed grade, 99%), PbIIPc (TCI, 95%), VIVOPc (Acros, 85%), and SnIVCl2Pc (TCI) were used as received. Cryptand[2,2,2] and cryptand[2,2,1] were purchased from Aldrich. C60 of 99.9% purity was received from MTR Ltd. The pentamethylcyclopentadienylmolybdenum dicarbonyl dimer ([Cp*Mo(CO)2]2) was purchased from Strem. Sodium fluorenone ketyl was obtained as described.33 Solvents were purified under an argon atmosphere. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure. n-Hexane was distilled over Na/benzophenone. The solvents were degassed and stored in a glove box. The crystals of 1–5 were stored in the glove box. KBr pellets for IR- and UV-visible-NIR measurements were also prepared in the glove box. EPR and SQUID magnetic measurements were performed on polycrystalline samples of 1, 3 and 4 sealed in 2 mm quartz tubes with ambient pressure argon.Synthesis
Crystals of 1–5 were obtained using the diffusion technique. The reaction solutions were filtered into a 50 mL glass tube of 1.8 cm diameter with a ground glass plug, and 30 mL of n-hexane was layered over the solution. Slow mixing of two solvents during 1–2 months provided precipitation of crystals on the walls of the tube. The solvent was decanted from the crystals and they were washed with n-hexane.Salts {cryptand[2,2,2](Na)}[CuIIPc]·C6H4Cl2 (1), {cryptand[2,2,2](Na)}[PbIIPc]·C6H4Cl2 (2), and {cryptand[2,2,2](Na)} [VIVOPc]·C6H4Cl2 (3) were obtained by the reduction of 21.6–30.0 mg of metal Pc (0.042 mmol) with an excess of sodium fluorenone ketyl (16 mg, 0.0789 mmol) in the presence of a stoichiometric amount of cryptand[2,2,2] (16 mg, 0.042 mmol) by stirring for two hours at 100 °C until complete dissolution of metal Pc and the formation of a deep-blue solution. After that, the [Cp*Mo(CO)2]2 dimer (12 mg, 0.0209 mmol) was added to the solutions, and they were additionally stirred at 80 °C for two hours. Then the solutions were cooled down to room temperature, and filtered into the tube for diffusion. Their diffusion into the n-hexane layer produced crystals as black blocks (1, 2) and black prisms (3) in 56–75% yield. All crystals had copper luster characteristic of metal Pc salts. The organometallic compound [Cp*Mo(CO)2]2 was added in the syntheses of the salts to study the possibility of the formation of coordination complexes of metal Pcs with Cp*Mo(CO)2, which were previously prepared starting from the [SnII(Pc˙3−)]˙− and [SnIVCl2(Pc˙3−)]˙− radical anions.21 In the case of CuIIPc, PbIIPc and VIVOPc, coordination complexes were not formed. However, instead of that, high-quality and large single crystals of metal Pc salts were obtained.
{Cryptand[2,2,2](Na)}[SnIVCl2Pc]·C6H4Cl2 (4) and {cryptand[2,2,1](Na)}[SnIVCl2Pc]·C6H4Cl2 (5) were synthesized as previously described for (PPN+)[SnIVCl2(Pc˙3−)]˙−.19 {Cryptand(Na+)}(C60˙−) was generated by the reduction of C60 (30 mg, 0.042 mmol) with a slight excess of sodium fluorenone ketyl (11 mg, 0.054 mmol) in the presence of one equivalent of cryptand[2,2,2] (16 mg, 0.042 mmol) (4) or two drops of cryptand[2,2,1] (5) in 16 mL of o-dichlorobenzene by stirring at 100 °C for 2 hours. As a result, a red-violet solution was formed. The formation of C60˙− was proved by the UV-visible-NIR spectrum. This solution was cooled down to room temperature and filtered into a flask containing 29.2 mg (0.042 mmol) of SnIVCl2(Pc2−). The solution was stirred at 80 °C for 2 hours and overnight at room temperature to produce a deep blue solution, which was filtered into a glass tube for diffusion. The diffusion of the solution into the n-hexane layer produced the crystals as well-shaped black blocks of 4 in 72% yield and 5 in 32% yield. The addition of a stoichiometric amount of [Cp*Mo(CO)2]2 to the {cryptand(Na+)}[SnIVCl2(Pc˙3−)]˙− salt in o-dichlorobenzene solution was accompanied by an immediate color change from deep blue to green, and a previously described coordination compound Cp*Mo(Br)0.83(CO)1.78 [SnII(Pc2−)]·0.5C6H4Cl2 was isolated from this solution, whose structure and properties were described.21
Compositions of 1–5 were determined from X-ray structural analysis on single crystals. The optical microscopic observation of the obtained crystals in a glove box, as well as a sample check of single crystals from each synthesis by X-ray diffraction, showed that only one crystal phase was formed. Elemental analysis cannot be used to determine the composition of the obtained crystals due to their high air sensitivity (i.e., addition of oxygen to the samples during the procedure of elemental analysis).
General
UV-visible-NIR spectra were recorded in KBr pellets on a PerkinElmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra were obtained in KBr pellets with a PerkinElmer Spectrum 400 spectrometer (400–7800 cm−1). EPR spectra were recorded for sealed polycrystalline samples of 1, 3 and 4 from 4 up to 295 K with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat. A quantum design MPMS-XL SQUID magnetometer was used to measure the static magnetic susceptibility of 1, 3 and 4 under a magnetic field of 100 mT in the range of 300–1.9 K upon cooling and heating processes. A sample holder contribution and core temperature independent diamagnetic susceptibility (χd) were subtracted from the experimental values. The χd values for salts 1, 3, and 4 were obtained by the extrapolation of the data (10–300 K, 1), (50–300 K, 3) and (70–300 K, 4) in the high-temperature range by fitting the data with the following expression: χM = C/(T − θ) + χd, where C is the Curie constant and θ is the Weiss temperature. Effective magnetic moments (μeff) were calculated with the formula: μeff = (8·χM·T)1/2.Crystal structure determination
X-ray diffraction data for 1–5 are listed in Table 5. The data were collected on an Oxford diffraction “Gemini-R” CCD diffractometer with graphite monochromated MoKα radiation using an Oxford Instrument Cryojet system. Raw data reduction to F2 was carried out using CrysAlisPro, Oxford Diffraction Ltd. The structures were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX 201634 and Olex2.35 Non-hydrogen atoms were refined in the anisotropic approximation. The positions of hydrogen were calculated geometrically.| Compound | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Emp. formula | C56H56Cl2N10 | C56H56Cl2N10 | C56H56Cl2N10 | C56H56Cl4N10 | C54H52Cl4N10 |
| NaO6Cu | NaO6Pb | NaO7V | NaO6Sn | NaO5Sn | |
| M r [g mol−1] | 1122.53 | 1266.18 | 1125.93 | 1248.58 | 1204.53 |
| Color and shape | Black block | Black block | Black prism | Black block | Black block |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/c | P21 | P21 |
| a, [Å] | 12.5241(8) | 12.73638(18) | 12.7645(1) | 10.2257(4) | 9.97961(16) |
| b, [Å] | 12.6333(10) | 30.5287(4) | 14.9025(2) | 25.3147(11) | 25.3617(6) |
| c, [Å] | 19.1273(10) | 14.2894(2) | 27.6673(3) | 10.6978(4) | 10.5814(2) |
| α, [°] | 81.244(5) | 90 | 90 | 90 | 90 |
| β, [°] | 72.634(5) | 109.2322(16) | 91.874(1) | 96.022(3) | 94.6162(16) |
| γ, [°] | 64.528(7) | 90 | 90 | 90 | 90 |
| V, [Å3] | 2606.6(3) | 5246.00(13) | 5260.14(10) | 2753.95(18) | 2669.46(9) |
| Z | 2 | 4 | 4 | 2 | 2 |
| ρ calc. [g cm−3] | 1.43 | 1.603 | 1.422 | 1.506 | 1.499 |
| μ [mm−1] | 0.592 | 3.389 | 0.363 | 0.727 | 0.745 |
| F(000) | 1168 | 2548 | 2344 | 1278 | 1230 |
| T [K] | 120 | 150 | 120 | 150 | 150 |
| Reflns measured | 21481 |
47083 |
61062 |
15748 |
38755 |
| Max. 2θ, [°] | 58.178 | 52.64 | 56.562 | 52.83 | 52.742 |
| Unique reflns | 11830 |
10687 |
12730 |
8584 | 10865 |
| Parameters | 688 | 1084 | 694 | 409 | 575 |
| Restraints | 0 | 1707 | 0 | 301 | 654 |
| Reflns [Fo > 2(Fo)] | 7575 | 9774 | 10598 |
6627 | 9067 |
| R 1 [Fo > 2σ(Fo)] | 0.057 | 0.0217 | 0.037 | 0.0732 | 0.0806 |
| wR2 (all data) | 0.1048 | 0.0522 | 0.0956 | 0.2327 | 0.2445 |
| G.O.F | 1.021 | 1.031 | 1.022 | 1.084 | 1.083 |
| CCDC | 1528923 | 1528924 | 1528926 | 1528925 | 1529831 |
The crystal structure of 2 contains cryptand[2,2,2](Na+) cations severely disordered between three orientations with the occupancies of 0.541(4), 0.263(4) and 0.196(4). Crystal structures of 4 and 5 were solved with rather low C–C bond length precision (0.012 and 0.018 Å, respectively) which prohibited the discussion on the macrocycle geometries. To keep the anisotropic thermal parameters of the disordered atoms within reasonable limits, the displacement components were restrained using SHELXL instructions of ISOR, SIMU and DELU. That resulted in great numbers of restraints for the refinement of crystal structures of 2, 4 and 5 (Table 5).
Conclusion
A series of radical anion salts with [M(Pc˙3−)]˙−, where M = CuII, PbII, VIVO, and SnIVCl2 with cryptand(Na+) counter cations, was obtained for the first time. The formation of these salts was accompanied by the reduction of Pc macrocycles. That results in the appearance of new NIR bands as well as blue-shifts of the Soret and Q-bands. The aromatic π-system was slightly distorted with the addition of one extra electron to the 18-electron π-system of Pc leading to the alternation of shorter and longer C–N(imine) bonds in the Pc macrocycle. Depending on the geometry of parent metal Pcs (M = CuII, PbII, VIVO), structures with channels accommodating the cryptand[2,2,2] (Na+) cations and solvent C6H4Cl2 molecules were formed. No effective π–π interaction was observed between Pc macrocycles in these crystal structures due to the perpendicular arrangement of the Pc˙3− macrocycles. Salts 1 and 3 demonstrated weak magnetic coupling between spins due to the absence of π–π interactions between Pc˙3−. There is mainly antiferromagnetic coupling between the CuII and Pc˙3− spins in 1 and ferromagnetic coupling between the VIV and Pc˙3− spins in 3. The weakness of this coupling shows ineffective magnetic coupling between two spins localized on the metal centers and delocalized over the Pc macrocycle. There are several examples of such behavior. Tin(IV) dichloride phthalocyanine with two axial ligands formed closely packed Pc layers in 4 and 5. The decrease of the cation size allows one to obtain densely packed Pc layers. The salts with the [SnIVCl2(Pc˙3−)]˙− radical anions containing one S = 1/2 spin per formula unit delocalized over the Pc macrocycle. A layered structure together with effective π–π interactions results in strong antiferromagnetic coupling between spins in 4 with an exchange interaction of J/kB = −18 K. That is a first example of such strong magnetic coupling between spins in the anion-radical phthalocyanine layers. Further work on studying the effect of small cations on the structure and magnetic properties of the salts is in progress.Acknowledgements
This work was supported by Russian Science Foundation grant No. 17-13-01215, and by JSPS KAKENHI Grant Numbers JP15K17901, JP17H05153, JP23225005, and JP26288035.Notes and references
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707 CrossRef CAS.
- D. C. H. Hasegawa, T. Naito, T. Inabe, T. Akutagawa and T. Nakamura, J. Mater. Chem., 1998, 8, 1567 RSC.
- Y. Ethelbhert, M. Matsuda, H. Tajima, A. Kikuchi, T. Taketsugu, N. Hanasaki, T. Naito and T. Inabe, J. Mater. Chem., 2009, 19, 718 RSC.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed.
- D. K. Rittenberg, L. Baars-Hibbe, A. B. Böhm and J. S. Miller, J. Mater. Chem., 2000, 10, 241 RSC.
- E. Tosatti, M. Fabrizio, J. Tóbik and G. E. Santoro, Phys. Rev. Lett., 2004, 93, 117002 CrossRef PubMed.
- S. Margadonna, K. Prassides, Y. Iwasa, Y. Taguchi, M. F. Craciun, S. Rogge and A. F. Morpurgo, Inorg. Chem., 2006, 45, 10472 CrossRef CAS PubMed.
- Y. Taguchi, T. Miyake, S. Margadonna, K. Kato, K. Prassides and Y. Iwasa, J. Am. Chem. Soc., 2006, 128, 3313 CrossRef CAS PubMed.
- P. Gargiani, A. Calabrese, C. Mariani and M. G. Betti, J. Phys. Chem. C, 2010, 114, 12258 CAS.
- M. Tahiri, P. Doppelt, J. Fischer and R. Weiss, Inorg. Chim. Acta, 1987, 127, L1 CrossRef CAS.
- E. W. Y. Wong and D. B. Leznoff, J. Porphyrins Phthalocyanines, 2012, 16, 154 CrossRef CAS.
- J. A. Cissell, T. P. Vaid, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2007, 46, 7713 CrossRef CAS PubMed.
- E. W. Y. Wong, C. J. Walsby, T. Storr and D. B. Leznoff, Inorg. Chem., 2010, 49, 3343 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. V. Simonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2012, 41, 13841 RSC.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2013, 52, 3851 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 9870 RSC.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, A. L. Litvinov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 6810 RSC.
- D. V. Konarev, A. V. Kuzmin, M. A. Faraonov, M. Ishikawa, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2015, 21, 1014 CrossRef CAS PubMed.
- D. V. Konarev, S. I. Troyanov, M. Ishikawa, M. A. Faraonov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Porphyrins Phthalocyanines, 2014, 18, 1157 CrossRef CAS.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2014, 43, 13061 RSC.
- D. V. Konarev, A. V. Kuzmin, Y. Nakano, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2016, 55, 1390 CrossRef CAS PubMed.
- D. V. Konarev, S. I. Troyanov and R. N. Lyubovskaya, CrystEngComm, 2015, 17, 3923 RSC.
- A. Hoshino, Y. Takenaka and H. Miyaji, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 393 Search PubMed.
- P. M. Burnham, M. J. Cook, L. A. Gerrard, M. J. Heeney and D. L. Hughes, Chem. Commun., 2003, 2064 RSC.
- A. J. Ramadan, L. A. Rochford, D. S. Keeble, P. Sullivan, M. P. Ryan, T. S. Jones and S. Heutz, J. Mater. Chem. C, 2015, 3, 461 RSC.
- J. Janczak and R. Kubiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m237 Search PubMed.
- D. V. Konarev, S. S. Khasanov, A. V. Kuzmin, Y. Nakano, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Cryst. Growth Des., 2017, 17, 753 CAS.
- Y. Nakano, T. Yagyu, T. Hirayama, A. Ito and K. Tanaka, Polyhedron, 2005, 24, 2141 CrossRef CAS.
- M. E. Lines, J. Phys. Chem. Solids, 1970, 31, 101 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Asian J., 2017, 12, 910 CrossRef CAS PubMed.
- K. Yakushi, H. Yamakado, A. Yoshitake, N. Kosugi, H. Kuroda, T. Sugano, M. Kinoshita, A. Kawamoto and J. Tanaka, Bull. Chem. Soc. Jpn., 1989, 62, 687 CrossRef CAS.
- T. Ida, H. Yamakado, H. Masuda, K. Yakushi, D. Kanazawa, H. Tajima and H. Kuroda, Mol. Cryst. Liq. Cryst., 1990, 181, 243 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, E. I. Yudanova and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2011, 816 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra of starting compounds and salts 1–4, the crystal structure of 2 and magnetic data for 1, 3 and 4. CCDC 1528923–1528926 and 1529831. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj00530j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |