
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structural studies and stability of model cysteine containing DNA–protein cross-links†
Kinga
Salus
,
Marcin
Hoffmann
,
Tomasz
Siodła
,
Bożena
Wyrzykiewicz
and
Donata
Pluskota-Karwatka
*
Adam Mickiewicz University in Poznań, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: donatap@amu.edu.pl
First published on 16th February 2017
Abstract
DNA–protein cross-links (DPCs) are bulky, helix-distorting lesions that are formed upon irreversible bonding of proteins to chromosomal DNA in the presence of cross-linking agents. Among a broad range of such agents are α,β-unsaturated carbonyl compounds, which act essentially as bifunctional alkylating agents and form adducts with DNA bases. These adducts can further undergo interactions with other cellular macromolecules leading to the formation of cross-linked products. We synthesized and structurally characterized N-acetylcysteine cross-links formed in the reactions with aldehydic adducts of adenine nucleosides, which possess enol functionality and represent model α,β-unsaturated carbonyl systems. Studies performed by the use of NMR spectroscopy, DFT and ab initio methods established that two of these cross-links exist as rotamers stable at room temperature. Application of Atoms in Molecules Theory enabled hydrogen bonding and other stabilizing interactions within the studied molecules to be estimated. Under physiological conditions the cross-links were found to be relatively stable until Nα-acetyllysine was present in the reaction medium. The presence of this amino acid caused fast transformation of the N-acetylcysteine cross-links into a range of their lysine derivatives. Although instability of the cysteine adduct with acrolein was reported, we showed that the mechanism involved in the gradual decomposition of the N-acetylcysteine cross-links differs from that proposed for acrolein adduct degradation. This demonstrates that in spite of similarities in their structures, numerous α,β-unsaturated carbonyl compounds can interact with nucleophilic biomolecules by different mechanisms leading to structural heterogeneity of the resulting products. Our findings provide an explanation for difficulties in identifying the cysteine containing DPCs in vivo and in vitro, and may be of great importance with respect to detection and isolation of such lesions from biological materials.
Introduction
DNA–protein cross-links (DPCs) are bulky lesions formed when cellular proteins bind covalently to DNA. These lesions distort the DNA helix and can disturb normal DNA–protein interactions leading to impairment of DNA-involving processes such as transcription, replication, repair, recombination and chromatin remodeling.1–3 RNA, due to its structural likeness to DNA, is presumed to exhibit similar reactivity towards protein cross-link formation. RNA–protein cross-links can interfere with processes related to both mRNA and noncoding RNA.4–6 The location of cross-linking on the DNA constituent was found to influence the biological outcomes of DPC formation.7,8 However the biological consequences of DPC formation depend not only on the DNA sequence involved in these lesions, but in a large part, on the protein trapped in it. While early reports suggested that only DNA-binding proteins can undergo crosslinking to DNA, recent studies have implied that any protein can be involved in DPC formation.9,10Proteins related to a number of cellular processes were found to form DPCs. Among them are DNA replication and repair proteins,3,11 architectural, structural and associated proteins, cell cycle proteins and chromatin regulators, proteins responsible for cellular homeostasis and stress response, transcription regulators and RNA splicing components.3,12,13,14–17 In model reactions, lysozyme and serum albumin were also shown to undergo crosslinking to DNA.10,18 Different proteins favor the formation of DPCs with specified DNA sequences.2
DPCs can be induced by exposure to a broad range of environmental agents but also by bifunctional electrophiles of endogenous origin which can react with a nucleophilic site on both DNA and amino acid side chains. Among such electrophiles, α,β-unsaturated carbonyl compounds and 2-oxoaldehydes such as glyoxal and methylglyoxal are known to induce DPCs.9,19 Lysine, arginine, cysteine, histidine, and serine were found to form DPCs.1,2,10 Nevertheless, the majority of reports refer mainly to the N-terminal and lysine side chain amino groups and to the guanidine group as protein structure fragments involved in such lesions. The cysteine residue thiol groups are the most nucleophilic sites in proteins, however the content of these residues in protein surfaces is much lower than those of other amino acids. Moreover cysteine containing DPCs were reported to be less stable than other types of such lesions.10,11 Therefore studies on cysteine DPCs seem to be challenging.
Bis-electrophiles such as 1,2,3,4-diepoxybutane, the key carcinogenic metabolite of 1,3-butadiene and N,N-bis-(2-chloroethyl)-phosphorodiamidic acid as well as N,N-bis-(2-chloroethyl)amine, the metabolites of cyclophosphamide used to treat some kinds of cancer and autoimmune diseases, have been shown to induce the formation of cysteine containing DPCs.14–17,20–22
The lipid peroxidation product, 4-hydoxy-2-nonenal, was demonstrated to modify an active site cysteine residue of protein disulfide isomerase which participates in the maturation of newly synthesized proteins through promoting correct disulfide formation.23
Another α,β-unsaturated aldehyde, acrolein, was found to be responsible for inactivation of protein tyrosine phosphatase 1B by binding covalently to the catalytic cysteine residue at the enzyme active site.24
Cysteine was also, besides lysine, shown to be involved in the DPCs formed by methylglyoxal between dG in the substrate DNA and the exonuclease deficient Klenow fragment of E. coli DNA polymerase I.11 It was found that the yield of the cysteine cross-link was much lower than that of the lysine one, however, the chemical nature of the cross-linked compounds was not determined.11 Glyoxal was also shown to induce the formation of these kinds of cross-links but the aldehyde was found to be a less potent crosslinking agent than methylglyoxal.11 Although DNA–protein crosslinking activity of methylglyoxal was examined in a later study,25 only the structure of the aldehyde cross-link between dG and Nα-acetyllysine was established.25
DPCs can be potentially used in biochemistry and medicine as biomarkers of oxidative/carbonyl stress and of chemical exposure.2 Moreover, controlled formation of DPCs in vitro is a useful tool in proteomics analysis.26,27 Therefore there is a great need for studies aimed at structure determination, formation mechanism elucidation and stability investigation of cysteine containing DPCs.
Recently, we found that aldehydic adducts of adenine nucleosides28,29 (Fig. 1) exhibit potent cross-linking activity.30 We showed that these adducts induce the formation of a range of cross-links while incubating with Nα- and Nε-acetyllysine respectively.30 The enol form predominates for dialdehyde functionality of the adducts in aqueous solution, therefore these chemicals represent model systems suitable for studies on α,β-unsaturated carbonyl compound cross-linking activity. In the present work we used these systems to study their reactivity toward N-acetylcysteine and we identified a range of cross-linked products. Our findings provide insight into the probable structures and stability of similar modifications potentially formed in vivo. They also clearly show that difficulties in identification and structural characterization of cysteine cross-links may result from their instability.
 | ||
| Fig. 1 Structures of the studied adducts: M1Gx-A, M1MGx-Ade, M1MGx-dA and M1MGx-A.29 | ||
Results and discussion
Reactions of M1Gx-A with N-acetylcysteine
M1Gx-A was synthesized as described previously28 (Scheme 1) and subjected to reaction with N-acetylcysteine (NAC) in 0.1 M phosphate buffer (PB) solution at 37 and at 50 °C, respectively, for 10 days. LC-DAD analyses of the reaction mixtures revealed the presence of one major product named M1Gx-A-NAC (Fig. 2), which achieved maximum concentration after 36 h of heating at 50 °C.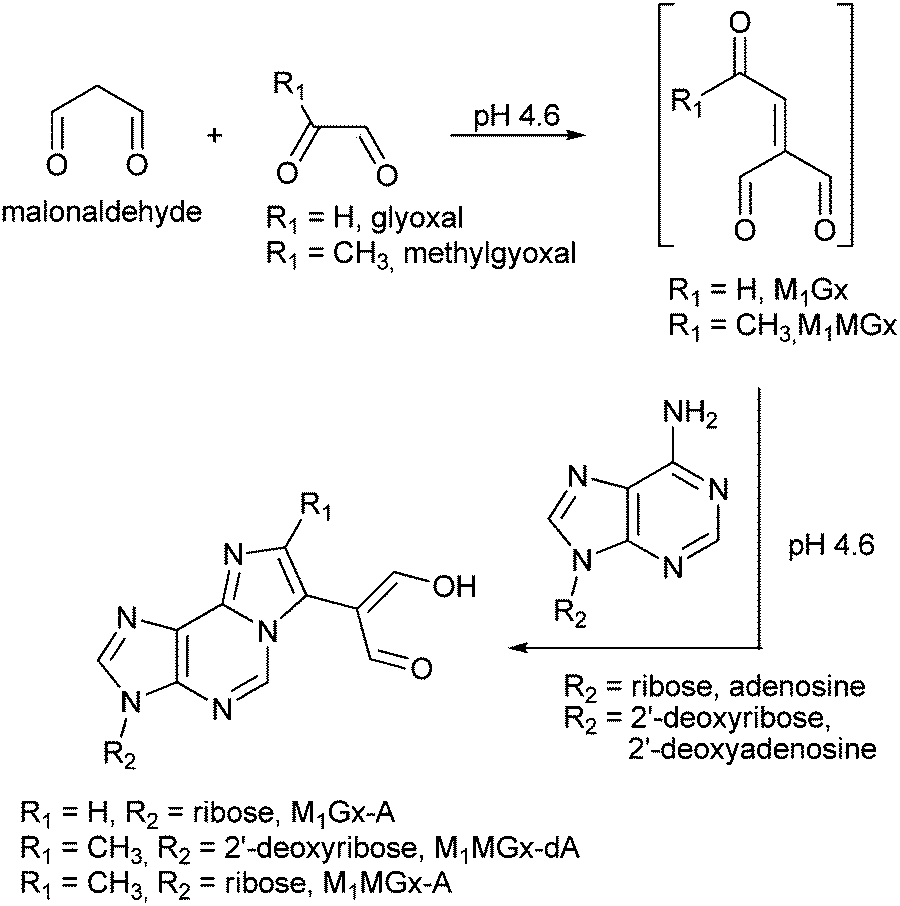 | ||
| Scheme 1 Formation of malonaldehyde-glyoxal and malonaldehyde-methylglyoxal adducts of adenine nucleosides. | ||
 | ||
| Fig. 2 C18 analytical column LC-DAD chromatogram (recorded at λ = 254 nm) of M1Gx-A with N-acetylcysteine reaction mixture held in 0.1 M phosphate buffer solution at 50 °C for 36 h. | ||
Formation of this product was also observed at 37 °C, however, at this temperature the yield of M1Gx-A-NAC was lower. The UV spectrum of this compound exhibited absorption maxima at λ = 234 and λ = 300 nm, and an absorption minimum at λ = 260 nm (Fig. 3).
 | ||
| Fig. 3 UV-Vis spectra of N-acetylcysteine cross-links recorded with a diode array detector as the compounds eluted from the column. (For analysis conditions see the Experimental section.) | ||
Structural data provided by mass spectrometry (m/z 507 [M + H]+, ESI,† Fig. S7) indicated that M1Gx-A-NAC is composed of one N-acetylcysteine molecule and one M1Gx-A molecule. A spectrum resulting from MS/MS analysis, besides the molecular ion signal, showed two fragment ion signals at m/z 375 and 246 (ESI,† Fig. S8). The former signal corresponded to the neutral loss of a ribosyl unit, and the latter resulted from the neutral loss of a ribosyl unit and cleavage of the bond between the sulfur atom and β-carbon atom in the N-acetylcysteine residue (Fig. 4 and ESI,† Fig. S8).
 | ||
| Fig. 4 Representative MS/MS trace for the synthesized cross-link; the positive ion MS/MS spectrum of M1Gx-A-NAC (collision energy 30 eV). | ||
For definitive structural characterization of M1Gx-A-NAC a preparative scale reaction was performed, and the major product was isolated (13 mg, 25% yield) and subjected to spectroscopic studies. The data obtained from the M1Gx-A-NAC NMR spectra (ESI,† Fig. S43–S48) are presented in Table 1 and in part in Fig. 5.
| δ(H) [ppm] | Multiplicity | J H,H [Hz] | δ(C) [ppm] | HMBC | NOESY | |
|---|---|---|---|---|---|---|
| H–C1′ | 6.21 | d | 5.43 | 91.29 | C2, C3a, C2′, C3′, C4′ | H–C2, H–C2′, H–C3′, H–C4′ |
| H–C2′ | 4.86 | m | 76.84 | C1′ | H–C2, H–C1′, H–C3′, Ha–C5′ | |
| H–C3′ | 4.50 | m | 73.25 | C1′, C5′ | H–C2, H–C1′, H–C2′, Ha–C5′, Hb–C5′ | |
| H–C4′ | 4.30 | dd | 4.12; 7.34 | 88.29 | C1′, C2′, C3′, C5′ | H–C1′, Ha–C5′, Hb–C5′ |
| Ha–C5′ | 3.88 | dd | 3.88; 12.72 | 64.25 | C3′, C4′ | H–C2′, H–C3′, H–C4′, Hb–C5′, H–C2 |
| Hb–C5′ | 3.95 | dd | 3.04; 12.73 | C3′, C4′ | H–C3′, H–C4′, Ha–C5′ | |
| H–C2 | 8.49 | s | 143.95 | C3a, C9b, C1′ | H–C1′, H–C2′, H–C3′, Ha–C5′ | |
| C3a | 141.67 | |||||
| H–C5 | 8.63 | s | 139.51 | C3a, C7, C9a, C9b | Ha–Cβ | |
| C7 | 119.23 | |||||
| H–C8 | 7.69 | s | 137.57 | C7, C9a, C1′′, C2′′ | CH3 | |
| C9a | 144.45 | |||||
| C9b | 125.60 | |||||
| C1′′ | 127.63 | |||||
| H–C2′′ | 8.66 | 170.62 | C7, CHO, C1′′, Cβ | CHO, Ha–Cβ, Hb–Cβ, H–Cα | ||
| CHO | 9.52 | s | 194.12 | C7, C1′′, C2′′ | H–C2′′ | |
| H–Cα | 4.57 | dd | 4.33, 7.79 | 57.68 | Cβ, COOH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Ac) O (Ac) |
H–C2′′, Ha–Cβ, Hb–Cβ |
| Ha–Cβ | 3.42 | dd | 7.83, 14.18 | 40.65 | Cα, C2′′, COOH | H–Cα, Hb–Cβ, H–C2′′ |
| Hb–Cβ | 3.61 | dd | 4.33, 14.19 | Cα, C2′′, COOH | H–Cα, Ha–Cβ, H–C2′′ | |
| COOH | 178.30 | |||||
| CO (Ac) |
176.58 | |||||
| CH3 (Ac) | 2.05 | 24.86 | CO (Ac), Cα, Cβ |
H–C8 |
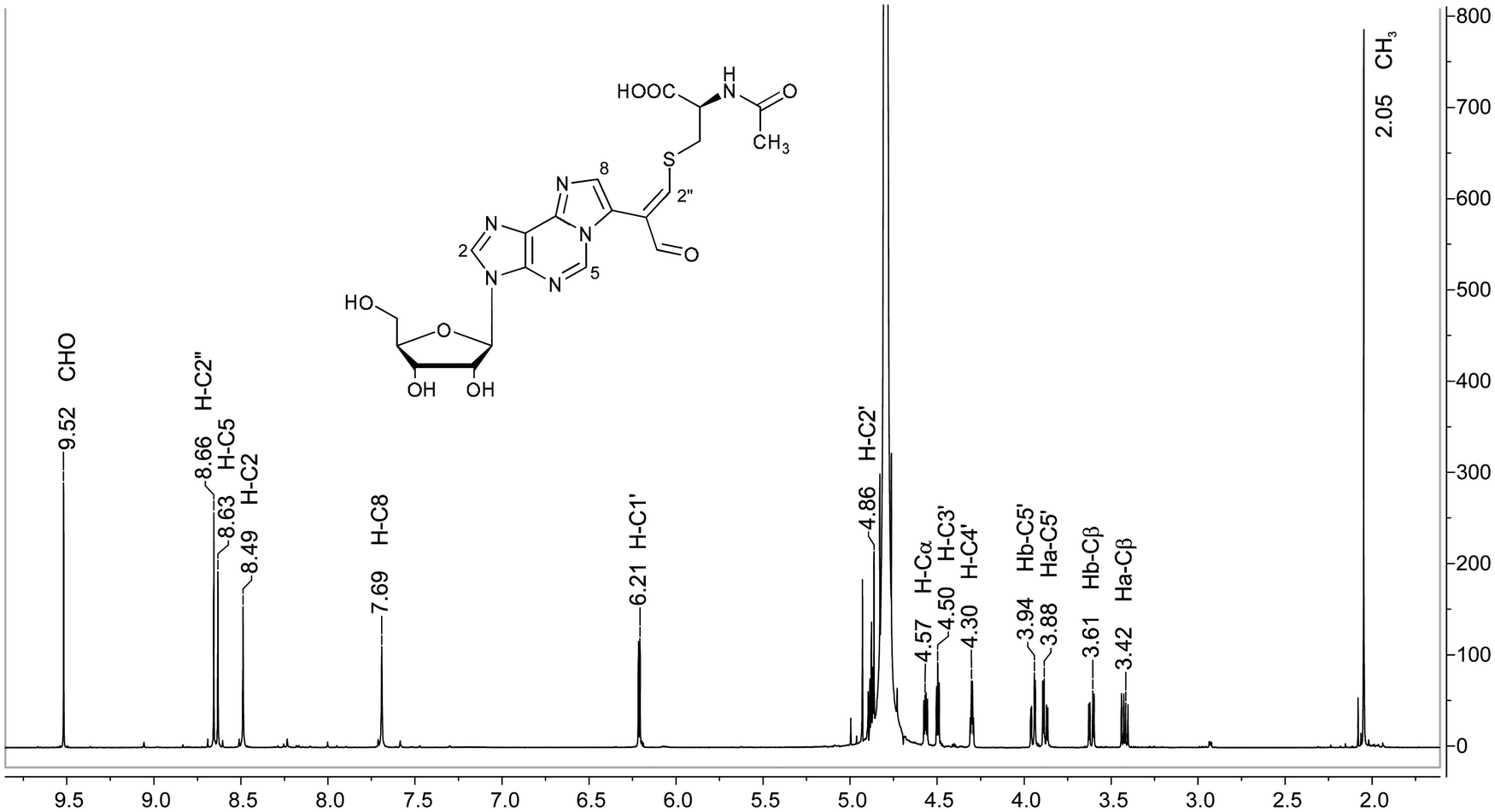 | ||
| Fig. 5 Representative NMR spectrum of the synthesized cross-link; 1H NMR spectrum of M1Gx-A-NAC (D2O). | ||
The singlet one-proton signal observed in the 1H NMR spectrum at 9.52 ppm was assigned to the aldehyde proton based on the chemical shift and 1H-13C correlation with the carbon atom signal at 194.12 ppm. The presence of one aldehyde proton suggested that N-acetylcysteine was attached to the adduct aldehydic functionality. In the 13C NMR spectrum the signal appearing at 119.23 ppm arose from the atom not bonded to the proton and was assigned to the C1′′ carbon atom. The structure proposed for M1Gx-A-NAC (Fig. 6) was confirmed by correlations observed in the HMBC spectrum (Table 1).
 | ||
| Fig. 6 Structures proposed for the cross-links: M1Gx-A-NAC, M1MGx-Ade–NAC and M1MGx-A-NAC. | ||
Reactions of M1MGx-dA with N-acetylcysteine
M1MGx-dA and NAC were held in 0.1 M and 1 M PB solutions at 37 and 50 °C, respectively, for 14 days. The pH of the reaction mixtures was monitored and it was found to be 7.4 for 1 M, and 3 for 0.1 M PB solution. The reaction mixture chromatograms derived from LC-DAD analyses showed the presence of two major products (M1MGx-Ade–NAC and M1MGx-dA–NAC) in 0.1 M PB solution (Fig. 7) and one major product (M1MGx-dA–NAC) in 1 M PB solution (data not shown). | ||
| Fig. 7 C18 analytical column LC-DAD chromatogram (recorded at 254 nm) of M1MGx-dA with NAC reaction mixture held in 0.1 M PB solution at 37 °C for 3 h. | ||
The compounds exhibited very similar UV spectra (M1MGx-Ade–NAC: λmax = 232, 296 nm, λmin = 256 nm; M1MGx-dA–NAC: λmax = 238, 298 nm, λmin = 260 nm) (Fig. 3). The mass spectrum of M1MGx-Ade–NAC displayed a signal at m/z 389 [M + H]+ (ESI,† Fig. S10), while the spectrum of M1MGx-dA–NAC revealed a signal at m/z 505 [M + H]+ (ESI,† Fig. S14). Data derived from the UV and mass spectra (ESI,† Fig. S10–S17) of M1MGx-Ade–NAC and M1MGx-dA–NAC indicated that both compounds were structural analogues which differ only in the presence of a 2′-deoxyribosyl unit. Formation of adenine derivative can be explained by hydrolysis of N-glycosidic bond in the initially formed cross-link and in M1MGx-dA. Moreover, these data suggested that M1MGx-Ade–NAC and M1MGx-dA–NAC consisted of one molecule of N-acetylcysteine and one molecule of the adduct. The isolated M1MGx-Ade–NAC (9 mm, 30% yield) was subjected to spectroscopic studies which confirmed the structure proposed for the compound as shown in Fig. 6. The HMBC spectrum of M1MGx-Ade–NAC exhibited analogous correlations with the HMBC spectrum of M1Gx-A-NAC. (For detailed NMR data see the ESI,† Table S1 and Fig. S49–S53.)
Reaction of M1MGx-A with N-acetylcysteine
Due to the instability of M1MGx-dA under reaction conditions the adenosine analogue of this adduct (M1MGx-A) was used to study its reactivity towards NAC. In the reaction performed in 0.1 M PB solution at 50 °C one major product (M1MGx-A-NAC) was formed (ESI,† Fig. S1). The compound exhibited a very similar UV spectrum (λmax = 238, 298 nm and λmin = 260 nm) to the UV spectra of M1Gx-A-NAC, M1MGx-dA–NAC and M1MGx-Ade–NAC (Fig. 3). Data derived from the mass spectrum (m/z 521 [M + H]+, (ESI,† Fig. S18)) suggested that the product was a structural analogue of the compounds formed in the reaction of M1MGx-dA with NAC. However, contrary to the reaction with M1MGx-dA, no hydrolysis of the N-glycosidic bond in the forming product was observed. The spectrum derived from the M1MGx-A-NAC MS/MS analysis showed fragmentation similar to that observed for M1Gx-A-NAC. Besides the molecular ion signals, two fragment ion peaks at m/z 389 and 260 were present in the spectrum (ESI,† Fig. S19). The former corresponded to the neutral loss of a ribosyl unit, and the latter resulted from the neutral loss of a ribosyl unit and cleavage of the bond between the sulfur atom and β-carbon atom in the N-acetylcysteine residue.The preparative scale reaction of M1MGx-A with NAC was performed and M1MGx-A-NAC was isolated (22 mg, 28% yield) for structural studies (Table 2 and ESI,† Fig. S54–S59). The correlations observed in the HMBC spectrum were crucial for M1MGx-A-NAC structure determination.
| δ(H) [ppm] | Multiplicity | J H,H [Hz] | δ(C) [ppm] | HMBC | NOESY | |
|---|---|---|---|---|---|---|
| H–C1′ | 6.20 | d | 5.44 | 91.79 | C2, C3a, C2′, C3′, C4′ | H–C2, H–C2′, H–C3′, H–C4′ |
| H–C2′ | 4.87 | dd | 5.13; 10.18 | 76.81 | C1′ | H–C2, H–C1′, H–C3′, Ha–C5′, Hb–C5′ |
| H–C3′ | 4.50 | m | 73.31 | C1′, C5′ | H–C2, H–C1′, H–C2′, Ha–C5′, Hb–C5′ | |
| H–C4′ | 4.30 | dd | 3.67; 7.49 | 88.35 | C1′, C2′, C3′, C5′ | H–C1′, Ha–C5′, Hb–C5′ |
| Ha–C5 | 3.90 | dd | 4.26; 12.69 | 64.31 | C3′, C4′ | H–C2′, H–C3′, H–C4′, Hb–C5′, H–C2 |
| Hb–C5′ | 3.94 | dd | 3.06; 12.71 | C3′, C4′ | H–C2′, H–C3′, H–C4′, Ha–C5′ | |
| H–C2 | 8.49 | s (split) | 144.13 | C3a, C9b, C1′ | H–C1′, H–C2′, H–C3′, Ha–C5′ | |
| C3a | 141.98 | |||||
| H–C5 | 8.55 | s (split) | 138.75 | C3a, C7, C9a, C9b | ||
| C7 | 115.30 | |||||
| C8 | 145.10 | |||||
| C9a | 143.75 | |||||
| C9b | 124.83 | |||||
| CH3 | 2.32 | s (split) | 15.63 | C7, C8, C1′′, C2′′, CHO | CHO, C2′′, Hb–Cβ | |
| C1′′ | 127.27 | |||||
| H–C2′′ | 8.74 | s (split) | 171.54 | C7, CHO, Cβ | ||
| CHO | 9.57 | s | 193.25 | C7, C8, C1′′, C2′′ | H–C2′′, CH3 (Ac) | |
| H–Cα | 4.59 | dd (split) | 4.30; 8.37 | 57.68 | Cβ, CO (Ac) |
H–C2′′, Ha–Cβ, Hb–Cβ, CH3 (Ac) |
| Ha–Cβ | 3.41 | dd (split) | 8.34; 14.24 | 40.37 | Cα, C1′′, C2′′, COOH | H–Cα, Hb–Cβ, H–C2′′ |
| Hb–Cβ | 3.66 | dd (split) | 4.29; 13.96 | Cα, C1′′, C2′′, COOH | H–Cα, Ha–Cβ, H–C2′′, CH3 | |
| COOH | 178.29 | |||||
| CO (Ac) |
175.58 | |||||
| CH3 (Ac) | 2.08 | s (split) | 24.90 | CO (Ac) |
H–C2′′, CHO, H–Cα |
Mechanism of the N-acetylcysteine cross-link formation
M1Gx-A-NAC, M1MGx-Ade–NAC and M1MGx-A-NAC are structural analogues of the cross-links of the aldehydic adenine nucleosides adducts with lysine derivatives previously identified in our laboratory.30 Therefore the mechanism proposed for lysine cross-linked product formation is most likely valid also for giving rise to the N-acetylcysteine modification. This mechanism involves Michael addition of the amino acid thiol group to the adducts’ α,β-unsaturated aldehydic functionality, followed by dehydration (Scheme 2) and is in agreement with the well-known rule that for soft nucleophiles such as thiol groups, conjugate addition is a typical reaction with α,β-unsaturated carbonyls.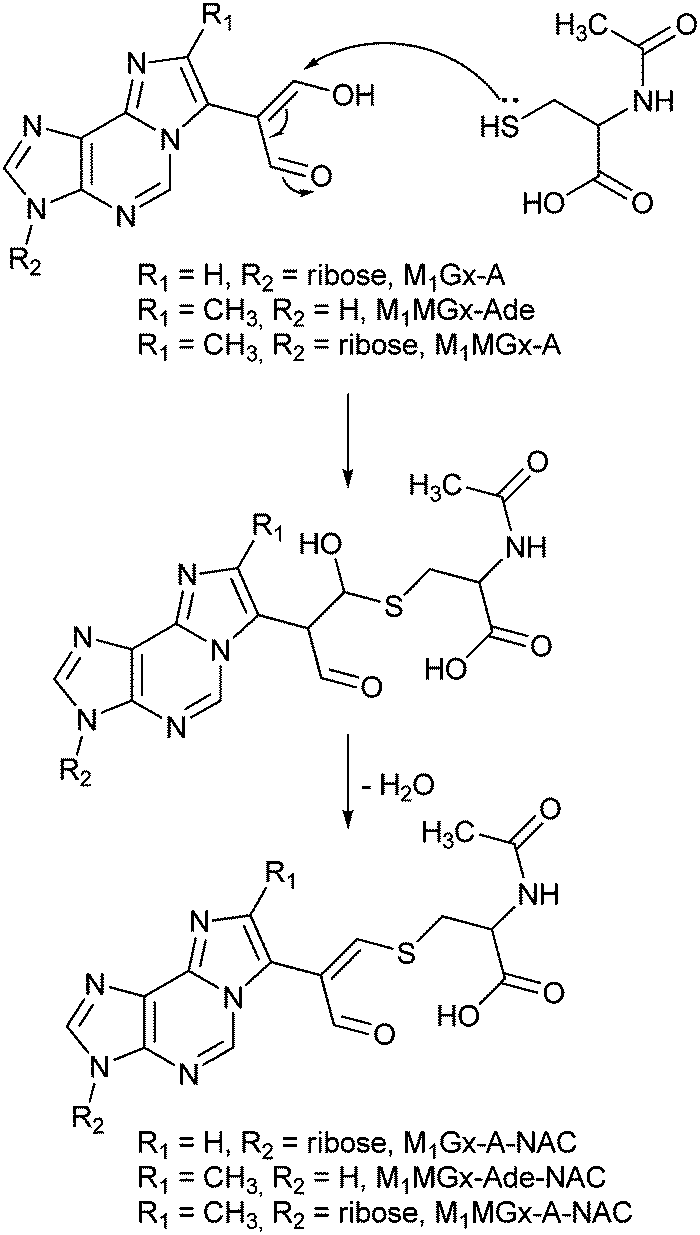 | ||
| Scheme 2 Proposed mechanism of N-acetylcysteine cross-link formation. | ||
N-Acetylcysteine cross-link stability
To determine the stability of the N-acetylcysteine cross-links under physiological conditions pure M1Gx-A-NAC, M1MGx-Ade–NAC and M1MGx-A-NAC were dissolved separately in 0.1 M PB solution (pH 7.4) and stored at 37 °C for 30 days. The compounds’ stability was monitored by using LC and LC-MS systems with diode array detection. M1Gx-A-NAC, M1MGx-Ade–NAC and M1MGx-A-NAC were found to be relatively stable under the applied conditions with half-life times above 20 days. The cross-links underwent slow degradation forming two products (Fig. 8 and ESI,† Fig. S2 and S3). | ||
| Fig. 8 C18 analytical column LC-DAD chromatogram (recorded at 254 nm) of the 0.1 M PB solution of M1Gx-A-NAC held at 37 °C for 17 days. | ||
On the basis of mass and UV spectra, and co-elution with the appropriate adducts used as reference compounds, the products eluted at 8.5 min (Fig. 8), and at 2.7 and 6.2 min (ESI,† Fig. S2 and S3) were identified as the substrate adducts: M1Gx-A, M1MGx-Ade and M1MGx-A, respectively. Compounds with longer retention times (Fig. 8 and ESI,† Fig. S2 and S3) exhibited almost identical UV spectra with λmax at 236 and 272 nm and λmin at 256 nm (Fig. 9). Spectrometric studies were performed and the compounds were found to give rise to protonated molecular ion signals recorded at m/z 361, 243 and 375, respectively (ESI,† Fig. S22, S26 and S30). The corresponding substrate adduct M1Gx-A, M1MGx-Ade and M1MGx-A ion signals were recorded at m/z 362, 244 and 376, respectively (ESI,† Fig. S20, S24 and S28). Such small differences between the appropriate values were very surprising and prompted us to perform ESI-MS/MS analyses using high collision energy for the degradation product derived from M1MGx-Ade–NAC and for M1MGx-Ade. The resulting spectra showed a range of signals arising from similar fragmentation ions (ESI,† Fig. S25 and S27). In the mass spectrum of M1MGx-Ade the signal appearing at m/z 226 corresponded to the neutral loss of water molecules ([M–H2O + H]+, ESI,† Fig. S25). In the mass spectrum of the M1MGx-Ade–NAC degradation product, the signal recorded at m/z 226 was attributed to the neutral loss of an ammonia molecule ([M–NH3 + H]+, ESI,† Fig. S27). Moreover the UV spectra of the degradation products resembled those of cross-links arising from reactions of the aldehydic adducts of adenine nucleosides with Nα-acetyllysine30 (Fig. 9 and 10).
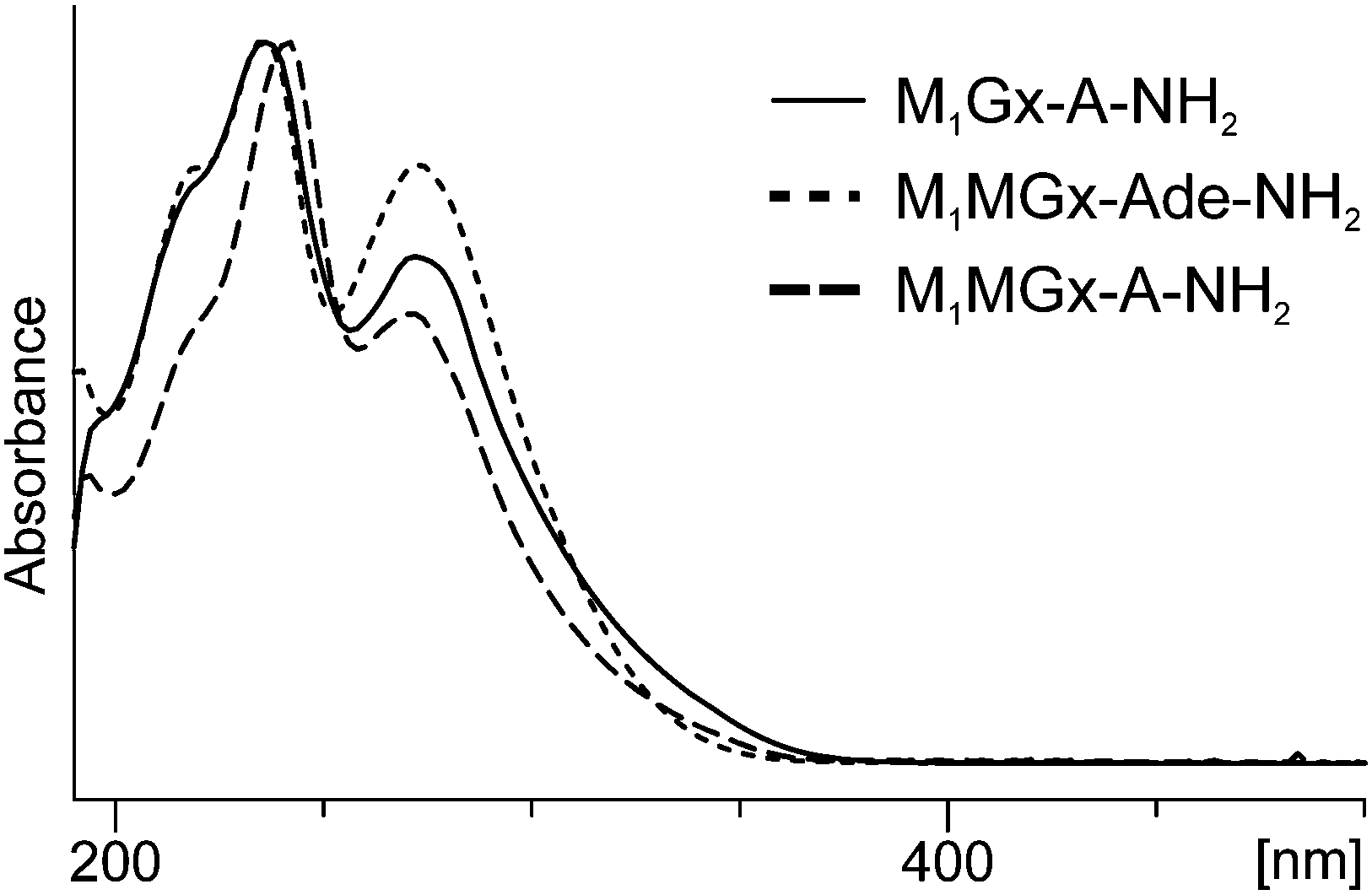 | ||
| Fig. 9 UV-Vis spectra of N-acetylcysteine cross-link degradation products: M1Gx-A-NH2, M1MGx-Ade-NH2 and M1MGx-A-NH2 recorded with a diode array detector as the compounds eluted from the column. (For analysis conditions see the Experimental section.) | ||
 | ||
| Fig. 10 UV-Vis spectra of products formed in the mixture of M1MGx-A-NAC with Nα-acetyllysine. The spectra were recorded with a diode array detector as the compounds eluted from the column. | ||
In these cross-links the free amino group of Nα-acetyllysine is attached to the adducts’ carbonyl carbon atom (Fig. 11B). Based on all these findings we concluded that the degradation products of N-acetylcysteine cross-links represent structures depicted as shown in Fig. 11A.
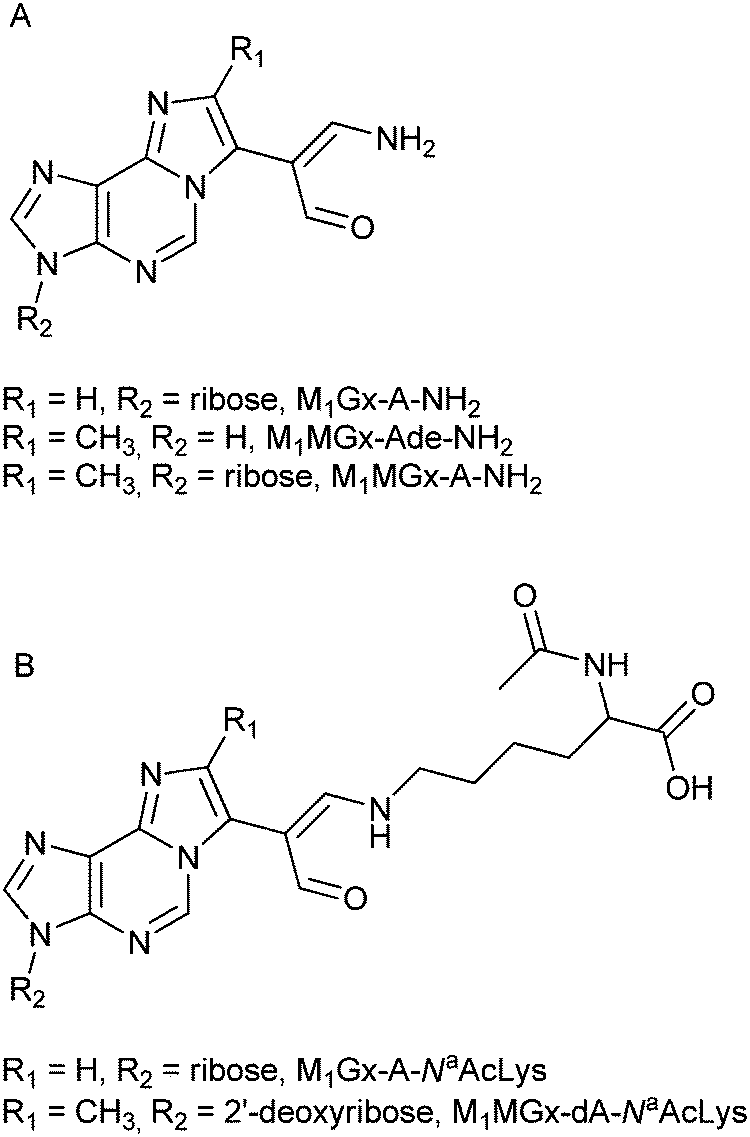 | ||
| Fig. 11 Proposed structures of M1MGx-Ade–NAC, M1MGx-A-NAC and M1MGx-A-NAC degradation products (A), and of known Nα-acetyllysine cross-links with the studied adducts21 (B). | ||
The mechanism responsible for the formation of these products is not clear, however it seems to be likely that they arise from 1,4-addition of ammonia to the cross-link carbon atom C-2′′. Ammonium bicarbonate solution used for isolation and purification of the cross-links is a probable source of ammonia. Although the fractions containing the cross-links devoted to stability studies were evaporated to dryness, the obtained residues might contain ammonia in sufficient amounts for nucleophilic attack. Ammonia could also arise from non-enzymatic oxidation of amino acids31,32 released from the cross-links. The results derived from stability studies of the N-acetylcysteine cross-links performed in the presence of Nα-acetyllysine provide support for a Michael addition of ammonia being involved in the cross-link degradation product formation. These results are also in good agreement with the literature data suggesting that cysteine containing DPCs are less stable than DPCs in which other amino acids are involved.10
N-Acetylcysteine cross-link stability in the presence of Nα-acetyllysine
M1Gx-A-NAC and M1MGx-A-NAC were found to be much more unstable while incubating at 37 °C with Nα-acetyllysine. The half-life time of the cross-links decreased to a few hours. LC-DAD analyses of the reaction mixtures revealed the formation of two products (Fig. 12 and ESI,† Fig. S4). On the basis of mass and UV spectra the compounds were identified as the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:1 cross-linked products of M1Gx-A and M1MGx-A with Nα-acetyllysine previously characterized in our laboratory.30
:2 and 1:1 cross-linked products of M1Gx-A and M1MGx-A with Nα-acetyllysine previously characterized in our laboratory.30
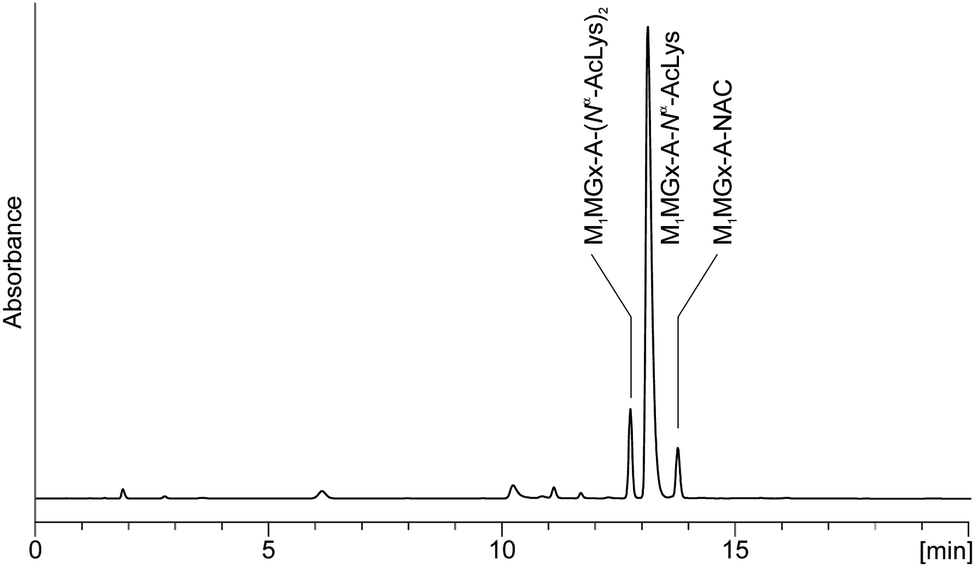 | ||
| Fig. 12 C18 analytical column LC-DAD chromatogram (recorded at 254 nm) of the M1MGx-A-NAC mixture with Nα-acetyllysine held in 0.1 M PB solution at 37 °C for 1 day. | ||
Cai et al.33 examined the reactivity of acrolein towards model peptides containing cysteine, lysine and histidine, and found that in the initial step the reaction pathway involves a Michael addition of the cysteine thiol group. Due to the loss of electron donation from the double CC bond, the aldehyde group in the resulting adduct is more electrophilic than in acrolein and reacts with an amino group forming a Schiff base adduct. This adduct can undergo rearrangement through an intermediate enamine to a more stable Schiff base adduct which contains a CC bond conjugated with the imine group (Scheme 3).33
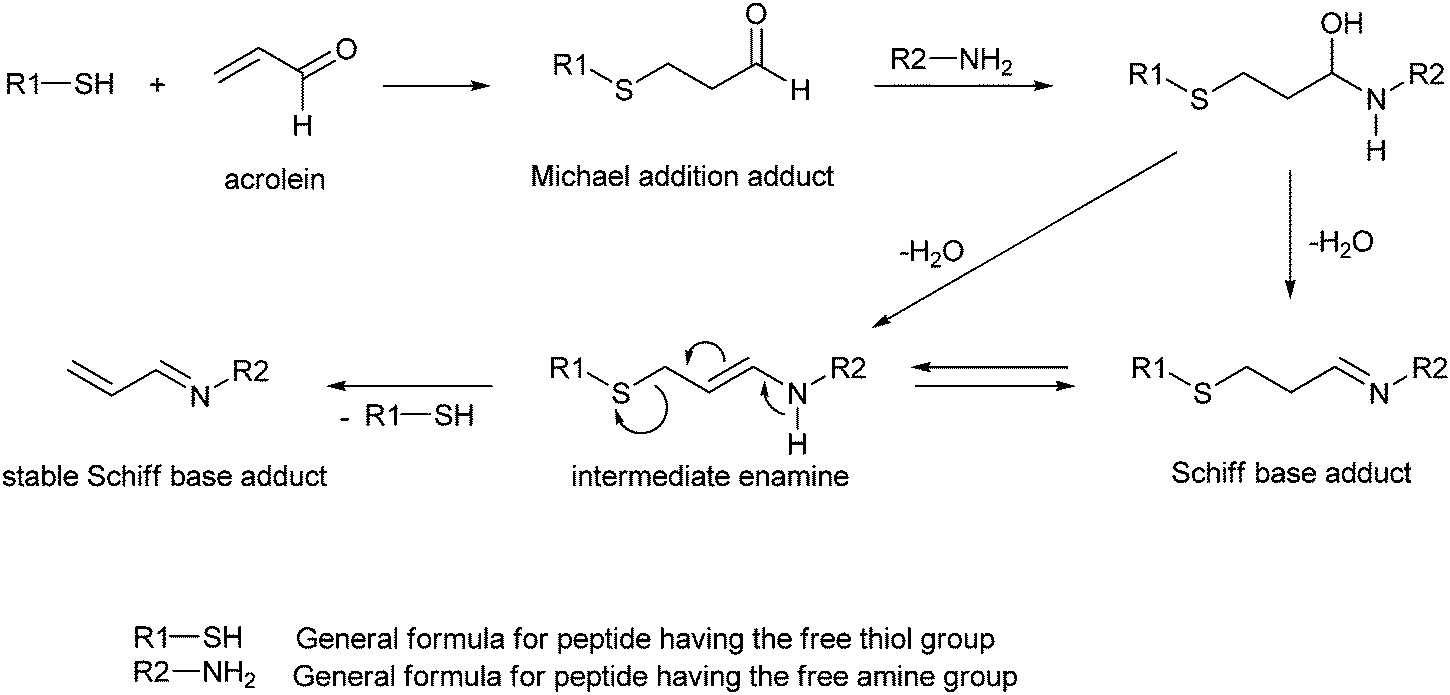 | ||
| Scheme 3 Formation of adducts in the reaction of model peptides with acrolein.33 | ||
This mechanism cannot be involved in the transformation of M1Gx-A-NAC and M1MGx-A-NAC into the Nα-acetyllysine cross-linked products. The N-acetylcysteine derivatives possess substituted α,β-unsaturated aldehydic functionality and therefore are unable to form an enamine intermediate. In the reactions of M1Gx-A and M1MGx-A with Nα-acetyllysine, apart from cross-links resulting from Michael addition also imine-enamine adducts containing two amino acid residues were formed.30 These adducts arose from 1,2-addition of the Nα-acetyllysine amino group to the initially formed Michael addition products, the enamine cross-links, followed by dehydration.30 The imine–enamine adducts are stabilized by a hydrogen bond between the imine nitrogen atom and the proton of the enamine N–H group.30 Such stabilization would not be possible in the products resulting from 1,2-addition of Nα-acetyllysine to M1Gx-A-NAC and M1MGx-A-NAC and consequently formation of such compounds was not observed.
In light of these data we proposed that the mechanistic explanation for the transformation of M1Gx-A-NAC and M1MGx-A-NAC into 1:1 cross-linked products of M1Gx-A and M1MGx-A with Nα-acetyllysine is based on a Michael addition of Nα-acetyllysine to the N-acetylcysteine cross-links in conjunction with N-acetylcysteine elimination (Scheme 4). Formation of 1:2 cross-links occurs as previously described.30
 | ||
| Scheme 4 Mechanism proposed for transformation of the cysteine cross-links into their Nα-acetyllysine derivatives. | ||
To provide additional support for the proposed mechanism, studies on M1Gx-A and M1MGx-A reactivity towards an N-acetylcysteine and Nα-acetyllysine mixture were performed.
M1Gx-A and M1MGx-A reactivity towards a mixture of N-acetylcysteine and Nα-acetyllysine
M1Gx-A and M1MGx-A were separately subjected to simultaneous reactions with N-acetylcysteine and Nα-acetyllysine. LC-DAD and LC-MS analyses revealed that M1Gx-A-NAC and M1MGx-A-NAC were initially formed cross-linked products (ESI,† Fig. S5 and S6). Prolonged reactions led to gradual decline in the amount of N-acetylcysteine cross-links and to the formation of Nα-acetyllysine derivatives which became the main products of the reactions.For the purpose of estimating the difference in energy between the N-acetylcysteine and Nα-acetyllysine cross-links, quantum-chemical investigations using isodesmic model systems were performed. The obtained results showed that the energy of the compound formed by amino group addition is lower than that of the product arising from thiol group addition (Fig. 13). ΔE calculated at the M06/6-311++G(2df,2pd) and M06/aug-cc-pVDZ levels was equal to 4.4 and 5.5 kcal mol−1, respectively, indicating that, in accordance with the experimental data, Nα-acetyllysine cross-links are more stable than their N-acetylcysteine derivatives.
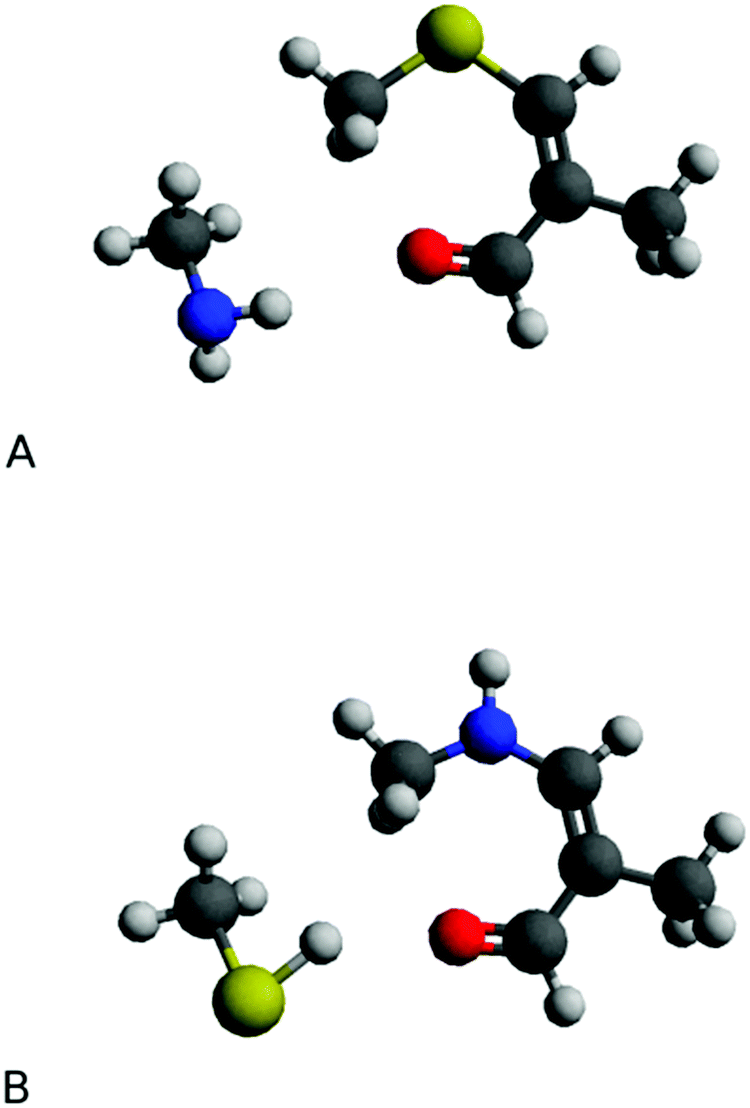 | ||
| Fig. 13 Isodesmic models for cross-links: product resulting from thiol group addition (A), and compound arising from amino group addition (B). (Calculations performed at the M06/6-311++G(2df,2pd) level.) | ||
Variable-temperature 1H NMR studies on M1MGx-Ade–NAC and M1MGx-A-NAC
1H NMR spectra of M1MGx-Ade–NAC and M1MGx-A-NAC recorded at room temperature exhibited doubling of signals (ESI,† Fig. S49 and S54). This phenomenon was also observed in the 13C NMR spectra of both cross-links (ESI,† Fig. S50 and S55). However not all of the carbon and proton signals were duplicated. A very intriguing fact was that no duplication of signals was seen in the spectra of M1Gx-A-NAC (ESI,† Fig. S43 and S44). The three cross-linked products differ only in the substituent at C8; the proton attached to C8 in M1Gx-A-NAC is replaced in M1MGx-Ade–NAC and M1MGx-A-NAC with a methyl group. The presence of this group was supposed to cause hindered rotation over the C7–C1′′ bond resulting in the existence of two rotational conformers (rotamers). To confirm this hypothesis, variable-temperature 1H NMR studies together with computational investigations were performed.The pattern of signal duplication in the 1H NMR spectrum of M1MGx-Ade–NAC (D2O) suggested that at 297 K the cross-link existed as two rotamers (A and B) separated by a rotational energy barrier around the C7–C1′′ bond sufficiently high to prevent fast interconversion of these conformers at room temperature. Both rotamers were detected by the use of signals at 1.96 ppm assigned to methyl protons of the acetyl group as a marker (Fig. 14).
 | ||
| Fig. 14 Fragments of M1MGx-Ade–NAC1H NMR spectra (D2O) recorded at 297.2 K (1), 342.6 K (2), 370.4 K (3), 371.5 K (4) and 372.6 K (5). The split acetyl proton signals at 1.96 coalesced to a singlet at Tc = 371.5 K, and the methyl proton signals at 2.23 ppm coalesced at lower temperatures. | ||
At 297 K the signals appeared as a split singlet due to the existence of the two rotamers in the ratio of 1.44:1.56. The concentration of both conformers was PA = 0.48 and PB = 0.52. The frequency difference, Δν, between the methyl proton signals was 8.42 Hz. This value increased to 8.52 Hz when the sample was cooled to 286 K, but reheating to room temperature resulted in the return to the initial Δν value. As the temperature increased the signals for the two rotamers were still detected, although within particular pairs, the signals broadened and moved closer together. The two signals coalesced to a single one which appeared at 2.63 ppm in the spectrum measured at 371.5 K (Tc) (Fig. 14). The other pairs of signals coalesced at lower temperatures.
The Gibbs free energy of activation for interconversion between the two rotamers was calculated on the basis of NMR studies using Earing's equations modified by Shanan-Atidi and Bar-Eli:34
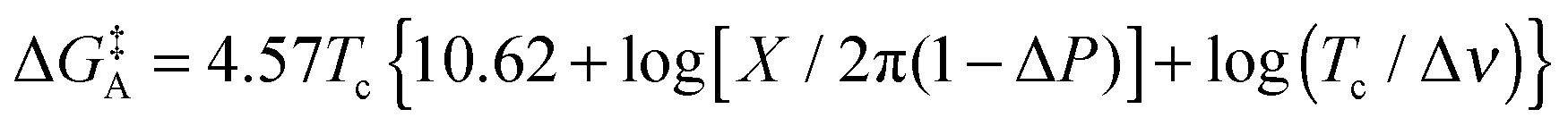
X was obtained by use of the following equation:34

| PA − PB = ΔP = [(X2 − 2)/3]3/2 × 1/X |
The free energy of rotamer A activation was  , and for rotamer B
, and for rotamer B  .
.
The high temperature caused partial decomposition of the studied compound and formation of degradation products being a source of additional signals in the 1H NMR spectra.
Analogous variable-temperature 1H NMR studies were also performed for M1MGx-A-NAC (D2O). The acetyl methyl proton signals which appeared at 1.96 ppm in the spectrum recorded at 279 K were again used as a marker. Two rotational conformers of the cross-link existed nearly in the same ratio as the rotamers of the adenosine analogue (1.43:1.57) (ESI,† Fig. S40). The frequency difference, Δν, between the marker signals was 9.48 Hz. The two signals coalesced to a single peak at 2.67 ppm in the spectrum recorded for the overheated sample at 375 K (Tc). At Tc the coalescence of the split signals was observed for the entire 1H NMR spectrum, although the other pairs of signals coalesced at lower temperatures (ESI,† Fig. S40).
The Gibbs free energy of activation for rotamer A was 19.95 kcal mol−1, and for rotamer B was 20.01 kcal mol−1. The almost identical values were in good agreement with the observation that in the 1H NMR spectrum the paired signals appeared in a nearly 1:1 ratio.
Quantum-chemical studies on M1MGx-Ade–NAC and M1MGx-A-NAC rotamer interconversion
The aim of the quantum chemical investigations was to examine hindered rotation around the C7–C1′′ bond in M1MGx-A-NAC and M1MGx-Ade–NAC. Rotation around this bond is associated with changes in the dihedral angle N6–C7–C1′′–C2′′ values and induces the molecules’ helicity. P helicity corresponds to positive values of this angle, and M helicity to angles of negative values (Fig. 15). TS_0 describes the transition state structure with the dihedral angle N6–C7–C1′′–C2′′ of about 0° (Fig. 16).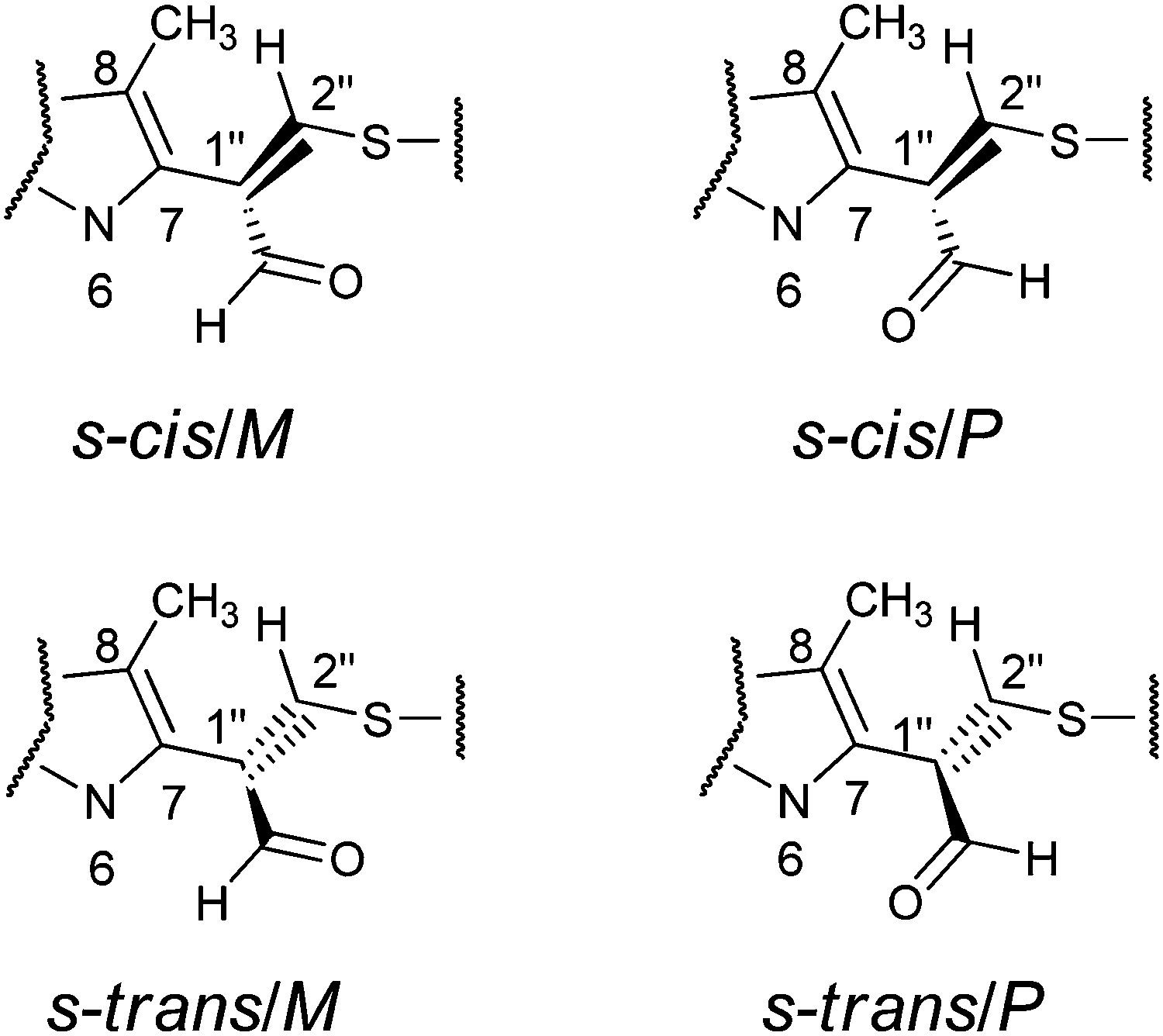 | ||
| Fig. 15 The geometry of conformers in energy minima: s-cis/M, s-cis/P, s-trans/M and s-trans/P. | ||
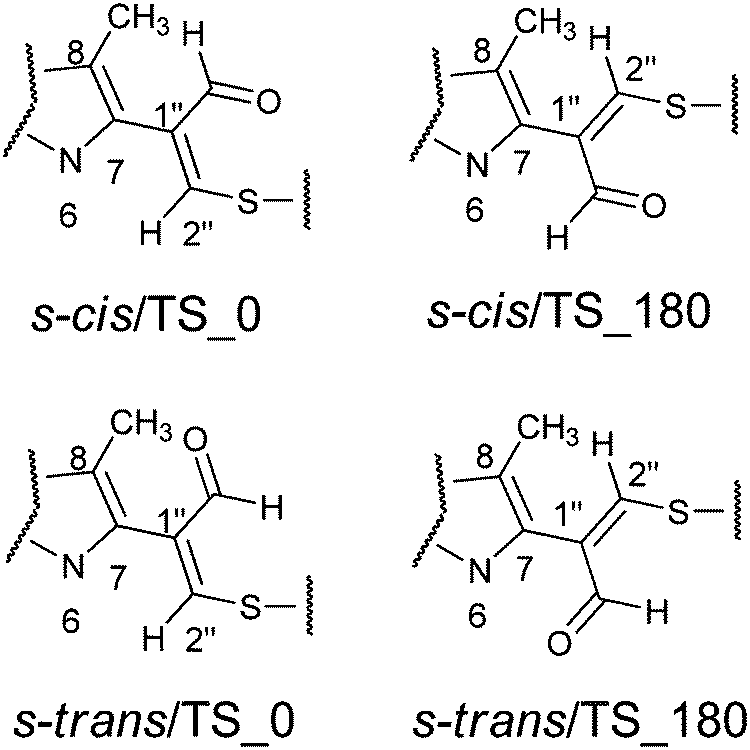 | ||
| Fig. 16 The synperiplanar (TS_0) and antiperiplanar (TS_180) geometry of the transition states with appropriate conformation of the aldehyde group: s-cis/TS_0, s-cis/TS_180, s-trans/TS_0 and s-trans/TS_180. | ||
In this structure atoms within the N6–C7–C1′′–C2′′ fragment adopt synperiplanar geometry. In the transition state structure with the dihedral angle of about 180° (TS_180) atoms within the N6–C7–C1′′–C2′′ fragment adopt antiperiplanar geometry (Fig. 16). The studied molecules differ also in the conformation of the aldehyde group. This group exists in s-cis conformation when the dihedral angle O–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HO–C1′′–C2′′ adopts the value of about 0°, and possess s-trans conformation for this angle of about 180° (Fig. 15 and 16). The rotation of the aldehyde group occurs through transition state structures with dihedral angle O–HO–C1′′–C2′′ values of about 90° (TS_90) and −90° (TS_−90).
HO–C1′′–C2′′ adopts the value of about 0°, and possess s-trans conformation for this angle of about 180° (Fig. 15 and 16). The rotation of the aldehyde group occurs through transition state structures with dihedral angle O–HO–C1′′–C2′′ values of about 90° (TS_90) and −90° (TS_−90).
Quantum-chemical studies in vacuo at the M06/6-31G(d) level of theory showed that the relative energies of the s-trans conformers of M1MGx-Ade–NAC were generally slightly higher (about 1–4 kcal mol−1) than the energies of the appropriate s-cis conformers (Chart 1). Only for N6–C7–C1′′–C2′′ dihedral angle values from −150° to −170° were the s-trans conformers energetically favorable (Chart 1).
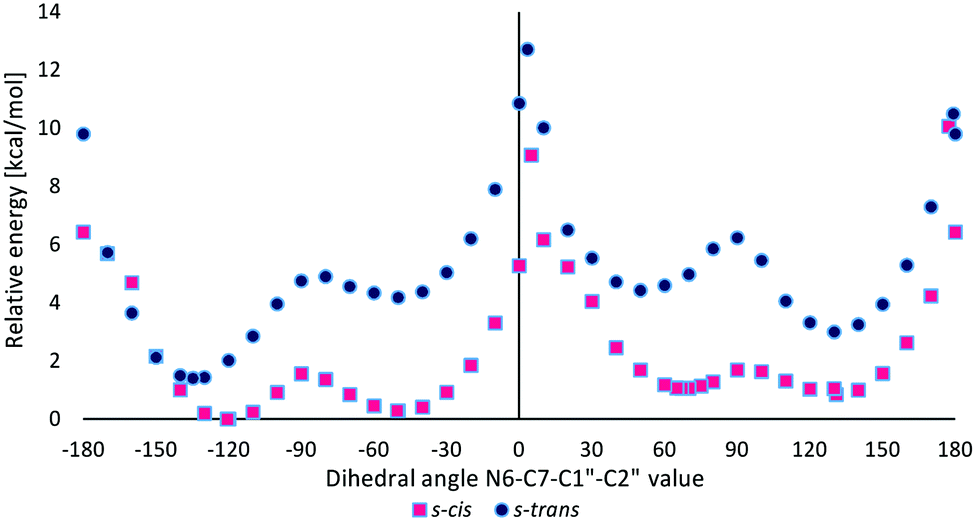 | ||
| Chart 1 The relative energy of s-cis and s-trans conformers of M1MGx-Ade–NAC dependent on the N6–C7–C1′′–C2′′ dihedral angle value calculated at the M06/6-31G(d) level of theory in vacuo. | ||
Two energy minima were found for the s-cis conformers: for a structure with the N6–C7–C1′′–C2′′ dihedral angle value of −120.6° (minimum s-cis/M) and also of 130.8° (minimum s-cis/P) (Chart 1, Table 3 and Fig. 17).
| In vacuo | PCM (water) | |||||
|---|---|---|---|---|---|---|
| Dihedral anglea (°) | ΔE [kcal mol−1] | ΔG [kcal mol−1] | Dihedral anglea (°) | ΔE [kcal mol−1] | ΔG [kcal mol−1] | |
| a N6–C7–C1′′–C2′′ dihedral angle value. | ||||||
| s-cis/M | −120.6 | 0 | 0 | −116.7 | 0 | 0 |
| s-cis/M | 130.8 | 0.85 | 1.08 | 129.0 | 0.66 | 0.28 |
| s-cis/TS_0 | 5.0 | 9.08 | 11.89 | 0.6 | 9.43 | 9.70 |
| s-cis/TS _180 | 177.3 | 10.07 | 12.47 | −176.7 | 10.02 | 11.45 |
| s-trans/M | −134.7 | 1.41 | 2.87 | −130.2 | 0.97 | 1.12 |
| s-trans/P | 130.0 | 3.00 | 2.94 | 128.9 | 1.58 | 0.97 |
| s-trans/TS_0 | 3.4 | 12.72 | 14.45 | −1.1 | 12.78 | 14.04 |
| s-trans/TS _180 | 179.2 | 10.50 | 11.73 | 179.0 | 10.53 | 11.45 |
 | ||
| Fig. 17 Energy minima structures of M1MGx-Ade–NAC and energy barriers of their interconversion (calculated at the M06/6-31G(d) level of theory in vacuo, see the ESI,† Fig. S42 for transition state structures). | ||
The difference in the relative energy between these two minima was very small (0.85 kcal mol−1). This fact was in good agreement with experimental data which indicated that the two rotamers of M1MGx-Ade–NAC existed in almost equal amounts. The energetically preferred transition state structure was found for s-cis conformers with the N6–C7–C1′′–C2′′ dihedral angle value of about 0° (s-cis/TS_0, Table 3). It was characterized by ΔE equal to 9.08 kcal mol−1. The ΔE values of other transition state structures were slightly higher (up to 12.72 kcal mol−1, Table 3). Application of thermal correction led to transition state structures with Gibbs free energies (ΔG) in the range of 11.73–14.45 kcal mol−1 being obtained (Table 3). To take into account the effect of solvent (water), the calculations were performed with the use of the polarizable continuum model (PCM). The obtained results were very similar to those received for the molecules in vacuo (Table 3). ΔE values calculated using the M06/6-31G(d) method did not differ significantly from the values obtained with larger basis sets (M06/6-31++G(d,p) and B3LYP/6-31++G(d,p)) (Table 4). Application of the atom–atom dispersion correction (ωB97X-D) led to slightly higher ΔE values compared to those obtained with the use of standard hybrid functionals (Table 4). The best correlation between the experimentally obtained rotational energy barrier and calculated ΔE values was only achieved using the MP2 perturbation method.
| Dihedral anglea (°) | M06/6-31G(d) | M06/6-31++G(d,p) | B3LYP/6-31++G(d,p) | ωB97X-D/6-31G(d) | MP2/6-31G(d) | |
|---|---|---|---|---|---|---|
| a N6–C7–C1′′–C2′′ dihedral angle value. | ||||||
| s-cis/M | −120.6 | 0.00 | 0.00 | 0.00 | 0.00 | 0.27 |
| s-cis/M | 130.8 | 0.85 | 1.26 | 0.07 | 1.22 | 1.83 |
| s-cis/TS_0 | 5.0 | 9.08 | 9.29 | 9.24 | 10.68 | 15.22 |
| s-cis/TS _180 | 177.3 | 10.07 | 10.73 | 10.32 | 11.87 | 17.73 |
| s-trans/M | −134.7 | 1.41 | 1.52 | 1.34 | 1.06 | 0.00 |
| s-trans/P | 130.0 | 3.00 | 3.23 | 2.73 | 3.05 | 2.08 |
| s-trans/TS_0 | 3.4 | 12.72 | 12.32 | 13.16 | 13.82 | 16.13 |
| s-trans/TS _180 | 179.2 | 10.50 | 11.00 | 10.73 | 11.32 | 14.29 |
Quantum-chemical studies of rotation around the C7–C1′′ bond in M1MGx-A-NAC gave very similar results to those described for M1MGx-Ade–NAC indicating that the presence of the ribosyl unit had no significant impact on the values of energy barriers between the M1MGx-A-NAC conformers, nor on the ΔE values of conformers representing structures of transition states and minima found in the curves of relative energies as a function of the N6–C7–C1′′–C2′′ dihedral angle values (ESI,† Chart S2, Tables S3, S4 and Fig. S41).
Hydrogen bond formation in M1MGx-Ade–NAC and M1MGx-A-NAC based on atoms in molecules theory
Bader's atoms in molecules (AIM) theory35 was applied in order to investigate the intramolecular hydrogen bond formation (or other stabilizing interactions) which may stabilize the rotamers’ structures and inhibit their fast interconversion at room temperature.The topological parameters, such as the electron density at bond critical points (BCPs), ρBCP, its Laplacian, ∇2ρBCP and the density of the total electron energy in BCPs, HBCP, correlate well with the H-bond energy, so they well describe the H-bond strength, but may be applied rather for samples of related compounds.36 For the particular case of O–H⋯O intermolecular hydrogen bonds in water and methanol clusters, a reasonably good linear correlation between the charge density at the BCPs and the strength of the H-bonds was reported.37–39 Thus, analysis of such parameters may be useful for the estimation of the relative strength of hydrogen bonding or other stabilizing interactions.40–43
The analysis of BCP also provides information on the nature of interatomic interaction.44 For shared interactions like covalent and polarized bonds, the Laplacian of electron density is negative because there is concentration of electron density within the atom–atom region. For the interactions between closed-shell systems like van der Waals interactions, ionic ones, and hydrogen bonds, there is the depletion of electron charge within the atom–atom region, and hence the Laplacian is positive. Thus, the sign of the Laplacian may indicate the kind of interaction. There is a very interesting situation in the case of hydrogen bonding where usually the Laplacian is positive; however, for very strong hydrogen bonds like those in H5O2+ or (FHF)−, the Laplacians are negative, respectively, for both H⋯O or H⋯F contacts. This is the strong evidence for the covalent character of hydrogen bonding.45
Rozas et al. have classified hydrogen bonds on the basis of ∇2ρBCP and HBCP values.46 Weak and medium strength hydrogen bonds show both positive ∇2ρBCP and HBCP values. For strong H-bonds, ∇2ρBCP is positive and HBCP is negative. For very strong hydrogen bonds, ∇2ρBCP and consequently the HBCP values are negative. Cremer and Kraka involved the energetic topological parameters into the classification of interactions much earlier.47
To sum up, for samples of related compounds the stronger interactions should exhibit greater value of electron density in BCPs (ρBCP), greater absolute value of the electron density's Laplacian (∇2ρBCP) and lower value of the total electron energy in BCPs (HBCP).
The above mentioned topological parameters for hydrogen bonds and other stabilizing interactions in the investigated structures are collected in Table 5. Only interactions which need to be rearranged in order to initiate the rotation P → M (or reverse) were considered as the ones which may inhibit such interconversion.
| Structure | Rotamer | Complex | ρ BCP | ∇2ρBCP | H BCP |
|---|---|---|---|---|---|
| M1MGx-Ade–NAC | s-cis/M−50 | C5–H⋯OCacetyl |
+0.017 | +0.053 | +0.0002 |
| s-cis/M−120 | C5—H⋯OCacetyl |
+0.008 | +0.032 | +0.0013 | |
| s-cis/P | C8–CH3⋯OCacetyl |
+0.014 | +0.044 | +0.0003 | |
| s-trans/M | C5–H⋯OCaldehyde |
+0.010 | +0.041 | +0.0017 | |
| C5–H⋯OCacetyl |
+0.013 | +0.047 | +0.0010 | ||
| s-trans/P | C5–H⋯OCaldehyde |
+0.012 | +0.048 | +0.0015 | |
| C8–CH3⋯OCacetyl |
+0.014 | +0.042 | +0.0002 | ||
| M1MGx-A-NAC | s-cis/M−50 | C5–H⋯OCacetyl |
+0.013 | +0.047 | +0.0011 |
| C5–H⋯C2′′ | +0.009 | +0.034 | +0.0018 | ||
| N4⋯OCacetyl |
+0.008 | +0.028 | +0.0006 | ||
| C2′–OH⋯OCacetyl |
+0.008 | +0.034 | +0.0011 | ||
| s-cis/M−120 | C5–H⋯OCacetyl |
+0.011 | +0.042 | +0.0012 | |
| s-cis/P | C8–CH3⋯OCacetyl |
+0.014 | +0.045 | +0.0003 | |
| s-trans/M | C5⋯OCaldehyde |
+0.010 | +0.042 | +0.0017 | |
| C5–H⋯O=Cacetyl | +0.014 | +0.050 | +0.0009 | ||
| s-trans/P | C5–H⋯OCaldehyde |
+0.013 | +0.052 | +0.0015 | |
| C8–CH3⋯OCacetyl |
+0.014 | +0.042 | +0.0001 | ||
The considered topological parameters of electron density always have positive values. This indicates that there are no strong interactions which may inhibit rotation and fast interconversion between rotamers P and M at room temperature. However, the most stable conformers s-cis/M can exist as the two stable equivalent forms (having almost the same energy): one with the dihedral angle N6–C7–C1′′–C2′′ of about −50° (s-cis/M−50) and the other one with the angle of about −120° (s-cis/M−120) (both of them are having the M helicity). Within the form having the dihedral angle N6–C7–C1′′–C2′′ of about −50° in compound M1MGx-A-NAC (containing a sugar moiety) there are 4 possible interactions recognized by the AIM analysis (Fig. 18).
 | ||
| Fig. 18 The bond paths (black lines), bond critical points (small green dots), ring critical points (small purple dots) and interatomic surfaces (grey lines) for rotamer s-cis/M−50 of compound M1MGx-Ade–NAC (A) and M1MGx-A-NAC (B). | ||
Probably, all 4 of them simultaneously are capable of inhibiting, to some extent, rotation M → P. What is interesting, is that in the analogous case of compound M1MGx-Ade–NAC, the one without a sugar substituent, such rotamers have only one (C5–H⋯OCacetyl) of the four interactions observed in the compound containing the sugar moiety. This shows the role of sugar substitution in the stabilization of rotamer s-cis/M, where the sugar moiety is involved in interaction (C2′–OH⋯OCacetyl) (Fig. 18). Moreover, the one interaction in rotamer s-cis/M−50 of the compound without the sugar substituent seems to be the strongest one: having the highest value of electron density (+0.017), the highest absolute value of Laplacian (+0.053) and the lowest value of total electron energy (+0.0002).
In the case of rotamers s-cis/P there is only one interaction (C8–CH3⋯OCacetyl) considered as the one which may inhibit such interconversion. Further, conformers having the aldehyde moiety in s-trans configuration (s-trans/M and s-trans/P) are stabilized by two interactions each (Table 5).
The difference in the topological parameter values for the corresponding interactions in compounds M1MGx-Ade–NAC (without sugar) and M1MGx-A-NAC (with sugar) allows us to investigate the impact of the sugar moiety on the strength of the considered interactions. All considered interactions seem to be equal or stronger for the case of compound M1MGx-A-NAC containing a sugar moiety, which indicates that this compound should be less labile in the considered rotation P → M or reverse (assuming the same energy barrier). However, relatively low differences in the discussed numerical values do not allow us to make clear statements.
Conclusions
We studied the reactivity of the aldehydic adenine nucleoside adducts towards N-acetylcysteine and provided structural characterization of cross-links that could be induced by DNA adducts containing carbonyl moieties, in reactions with the cysteine residues of proteins.Bifunctional carbonyl compounds of endogenous origin are considered to be responsible for different types of cross-link formation.48 Such carbonyls are known to form adducts with nucleophilic biomolecules and these adducts can further undergo interactions with other cellular macromolecules giving rise to cross-linked products.49,50 Our investigations show that such mechanisms can be, at least in part, valid also for cysteine containing cross-link formation. It is well known that cysteine residues are involved in the catalytic activity of enzymes. Moreover the cysteine thiol groups take part in disulfide bond formation which plays an important role in protein structure stabilization. Therefore structural modifications of such sites can have broad functional implications. As strong nucleophilic centers, the cysteine thiol groups are very probable targets for electrophilic attack that leads to DPC formation. Despite their ubiquitous presence in living cells, the structural features of DPCs and the mechanisms of their formation still remain to be explored. The literature reports on cysteine containing DPC synthesis and structural determination do not refer to the stability of such lesions. Our results show that stability studies seem to be crucial for establishing the biological importance of cysteine containing DPCs and for clarifying the role that the amino acid plays in their formation. Our results also provide insight into the chemical nature of the cross-links possibly induced by α,β-unsaturated carbonyl compounds between the adenine residues of DNA and cysteine residues of proteins. The highest yield of N-acetylcysteine cross-link formation was observed in reactions performed at a pH of about 3. This fact does not exclude the possibility of such cross-link formation in vivo because they were also found to be formed (in smaller amounts) under physiological conditions. Apart from that different cellular compartments are characterized by individual environments including pH. It is worth noticing that our studies provide the first discussion on the acidity/neutrality of the cysteine containing reaction medium, and pH was shown to be a key factor for the formation of N-acetylcysteine cross-links.
The mechanism proposed for N-acetylcysteine cross-link degradation and transformation gives a clear explanation for difficulties in detecting and isolating such lesions. Fast transformation of the cysteine cross-links occurring in the presence of the lysine was probably the reason for a much lower yield of the methylglyoxal induced cross-links between cysteine residues of DNA Klenow fragment polymerase and guanine residues of the substrate DNA compared to analogous cross-links involving the lysine residues.25 Difficulties in detecting cysteine containing cross-links do not rule out the high probability of such lesion formation in vivo.
Experimental
Materials
1,1,3,3-Tetramethoxypropane (TMP), 2′-deoxyadenosine monohydrate, adenosine, adenine, methylglyoxal solution (40 wt% in H2O), glyoxal solution (40 wt% in H2O), N-acetylcysteine, Nα-acetyllysine, NH4HCO3, ACN (gradient grade for chromatography), and D2O (containing 0.05% TSP-d4) were purchased from Sigma-Aldrich.Chromatographic methods
Progress of the reactions was monitored by LC-DAD and the analyses were performed on an Agilent 1100 series liquid chromatographic system consisting of a binary pump (G1312A), autosampler (G1313A), vacuum degasser (G1379A), diode-array detector (UV; G1315B), 5 μm, 4.6 × 150 mm reversed-phase C18 anal. column (Hypersil BDS, Thermo Scientific) and Agilent ChemStation data handling program (Agilent Technologies). The column was eluted isocratically for 5 min with 0.01 M phosphate buffer, pH 7.1, and then with a gradient from 0% to 20% ACN in 20 min at a flow rate of 1.5 mL min−1.Separation and purification of the synthesized compounds was carried out using an Agilent 1200 Series HPLC system consisting of a binary pump (G1312A), vacuum degasser (G1379B), autosampler (G1329A), thermostated column compartment (G1316A), diode-array detector (UV; G1315B), fraction collector (G1364C), and Agilent ChemStation data handling program (Agilent Technologies). 1.2 mM NH4HCO3 and ACN were used as eluents. The flow was set to 3 mL min−1. Isolation of M1Gx-A, M1MGx-A and M1MGx-dA was performed using a semiprep. 5 μm, 10 × 250 mm (Hypersil BDS, Thermo Scientific) reversed-phase C18 column thermostated at 25 °C. Isolation of M1Gx-A-NAC, M1MGx-A-NAC, M1MGx-dA and M1MGx-Ade–NAC was carried out by the use of a semiprep. 5 μm, 10 × 250 mm (Hypersil GOLD, Thermo Scientific) reversed-phase C18 column thermostated at 20 °C.
ESI-LC/MS and ESI-LC/MS/MS analyses
ESI-LC/MS and ESI-LC/MS/MS analyses were performed with an Agilent 1200 LC system using an Agilent Q-TOF 6540 spectrometer with a DUAL AJS ESI source. Ionization was carried out using nitrogen as the nebulizing/drying gas (8 L min−1, 300 °C, 35 psi) and sheath gas (11 L min−1, 325 °C). The VCap was set to 3500 V, the fragmentor was set to 100 V. Ion polarity mode and collision energy values for MS/MS experiments were chosen individually and given with the appropriate spectra. The analyzed compounds were introduced through the LC system using a reversed phase analytical column (5 μm, 4.6 mm × 150 mm, Thermo Scientific) and eluted isocratically for 3 min with water, and then with a gradient from 0% to 20% of ACN for 20 min with a flow rate of 0.5 min min−1. The thermostat was set to 25 °C.General procedure for M1Gx-A, M1MGx-dA and M1MGx-A synthesis
M1Gx-A, M1MGx-dA, and M1MGx-A were synthesized by using a modified procedure developed earlier in our laboratory.28 Malonaldehyde was prepared in situ by the acidic hydrolysis of 1,1,3,3-tetramethoxypropane (TMP).51 Syntheses of the adducts were accomplished according to the methods published previously.29Studies on M1Gx-A, M1MGx-dA and M1MGx-A reactivity towards N-acetylcysteine (analytical-scale reactions)
M1MGx-dA–NAC HRMS calc. for C21H25N6O7S+ [M + H]+m/z = 505.1500, found m/z = 505.1494 (error 1.2 ppm).
M1MGx-Ade–NAC HRMS calc. for C16H17N6O4S+ [M + H]+m/z = 389.1027, found m/z = 389.1021 (error 1.5 ppm).
Synthesis of cross-links
M1Gx-A-NAC HRMS calc. for C20H23N6O8S+ [M + H]+m/z = 507.1293, found m/z = 507.1283 (error 2.0 ppm).
M1MGx-A-NAC HRMS calc. for C21H25N6O8S+ [M + H]+m/z = 521.1449, found m/z = 521.1448 (error 0.2 ppm).
M1Gx-A HRMS calc. for C15H16N5O6+ [M + H]+m/z = 362.1095, found m/z = 362.1101 (error 1.7 ppm).
M1Gx-A-NH2 HRMS calc. for C15H17N6O5+ [M + H]+m/z = 361.1255, found m/z = 361.1257 (error 0.6 ppm).
M1MGx-Ade HRMS calc. for C11H10N5O2+ [M + H]+m/z = 244.0829, found m/z = 244.0824 (error 2.0 ppm).
M1MGx-Ade-NH2 HRMS calc. for C11H11N6O+ [M + H]+m/z = 243.09889, found m/z = 243.0989 (error 0.04 ppm).
M1MGx-A HRMS calc. for C16H18N5O6+ [M + H]+m/z = 376.1252, found m/z = 376.1247 (error 1.3 ppm).
M1MGx-A-NH2 HRMS calc. for C16H19N6O5+ [M + H]+m/z = 375.1411, found m/z = 375.1408 (error 0.8 ppm).
M1Gx-A-NαAcLys HRMS calc. for C23H30N7O8+ [M + H]+m/z = 532.2150, found m/z = 532.2139 (error 2.1 ppm).
M1Gx-A-(NαAcLys)2 HRMS calc. for C31H44N9O10+ [M + H]+m/z = 702.3206, found m/z = 702.3199 (error 1.0 ppm).
M1MGx-A-NαAcLys HRMS calc. for C24H32N7O8+ [M + H]+m/z = 546.2307. found m/z = 546.2299 (error 1.5 ppm).
M1MGx-A-(NαAcLys)2 HRMS calc. for C32H46N9O10+ [M + H]+m/z = 716.3362, found m/z = 716.3354 (error 1.1 ppm).
NMR spectroscopy
Samples were dissolved in D2O (internal standard: 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt, TSP-d4). The experiments were performed at the temperature of 298 K. All spectra were acquired on a Bruker Avance III DRX system, operating at frequencies of 600.3 MHz (1H) and 150.9 MHz (13C). The spectrometer was equipped with a 5 mm triple-resonance inverse probe head [1H/31P/BB]. High-power 1H and 13C π/2 pulses of 9.00 and 15.00 μs, respectively, were used. 1D and 2D homo- and heteronuclear correlation experiments were carried out by using pulse sequences from the Bruker pulse program library.For the variable-temperature 1H NMR experiments samples of M1MGx-Ade–NAC and M1MGx-A-NAC in D2O were thermostated for 1 h before measurement. 100% ethylene glycol recommended for studies at higher temperatures52 was used for creating a temperature calibrating curve (ESI,† Table S2 and Charts S1, S2).
Calculation methods
The initial structures of M1MGx-A-NAC and M1MGx-Ade–NAC were generated based on average bond lengths and valence angles; dihedral angles for rotatable bonds were sampled randomly. All initial conformers of M1MGx-A-NAC (12) and M1MGx-Ade–NAC (12) were optimized at the M06/6-31G(d) level of theory as isolated molecules. The lowest energy conformers of the s-cis and the s-trans series of both structures were selected for further research. These structures were optimized at the M06/6-31G(d) level in the gas phase with a frozen N6–C7–C1′′–C2′′ dihedral angle in steps of 10°, so full rotation around the C7–C1′′ bond was performed. The lowest energy values found for the conformers of M1MGx-A-NAC and M1MGx-Ade–NAC were taken as a relative energy of 0 kcal mol−1. Transition states TS_0 and TS_180 of the s-cis and s-trans conformers were found and optimized at the same level of theory. The Gibbs free energies were calculated on the basis of energy thermal correction for minima and transition states. To take into account the effect of solvent (water) the minima and transition state structures were reoptimized at the M06/6-31G(d) level using the polarizable continuum model (PCM). Both the relative energies and Gibbs free energies were calculated. The relative energies in single points for isolated molecules were also calculated by using the following methods: M06/6-31++G(d,p), B3LYP/6-31++G(d,p) and MP2/6-31G(d). Geometry optimization and energy calculations were performed by using Gaussian 09.53The energy barriers between rotamers of M1MGx-A-NAC and M1MGx-Ade–NAC calculated by the use of quantum-chemical methods were compared with those obtained on the basis of variable-temperature 1H NMR experiments.
Atoms in molecules theory was used for the investigation of intramolecular interactions in the structures of M1MGx-A-NAC and M1MGx-Ade–NAC in their minima. Parameters: electron density in bond critical points (BCPs), ρBCP, its Laplacian, ∇2ρBCP and density of the total electron energy in BCP, HBCP, were computed using the AIMAll program.54
Acknowledgements
This research was supported in part by PL-Grid Infrastructure (http://www.plgrid.pl/en), and the authors are thankful for help with the figures. The authors thank Perlan Technologies Polska Sp. z o. o. for providing the HPLC/Q-TOF Agilent 1260/6540 instrument.References
- S. Barker, M. Weinfeld and D. Murray, Mutat. Res., 2005, 589, 111–135 CAS.
- N. Y. Tretyakova, A. Groehler and S. Ji, Acc. Chem. Res., 2015, 48, 1631–1644 CrossRef CAS PubMed.
- H. Ide, M. I. Shoulkamy, T. Nakano, M. Miyamoto-Matsubara and A. M. H. Salem, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2011, 711, 113–122 CrossRef CAS PubMed.
- E. J. Wurtmann and S. L. Wolin, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 34–49 CrossRef CAS PubMed.
- K. Möller, J. Rinke, A. Ross, G. Buddle and R. Brimacombe, Eur. J. Biochem., 1977, 76, 175–187 CrossRef.
- P. Casati, Plant Physiol., 2004, 136, 3319–3332 CrossRef CAS PubMed.
- S. Wickramaratne, E. J. Boldry, C. Buehler, Y. C. Wang, M. D. Distefano and N. Y. Tretyakova, J. Biol. Chem., 2015, 290, 775–787 CrossRef CAS PubMed.
- S. Wickramaratne, S. Ji, S. Mukherjee, Y. Su, M. G. Pence, L. Lior-Hoffmann, I. Fu, S. Broyde, F. P. Guengerich, M. Distefano, O. D. Schärer, Y. Y. Sham and N. Tretyakova, J. Biol. Chem., 2016, 291, 23589–23603 CrossRef CAS PubMed.
- C.-Y. Tu, Y.-F. Chen, C.-K. Lii and T.-S. Wang, Toxicol. In Vitro, 2013, 27, 1211–1219 CrossRef CAS PubMed.
- H.-J. C. Chen, W.-L. Chiu, W.-P. Lin and S.-S. Yang, ChemBioChem, 2008, 9, 1074–1081 CrossRef CAS PubMed.
- N. Murata-Kamiya and H. Kamiya, Nucleic Acids Res., 2001, 29, 3433–3438 CrossRef CAS PubMed.
- S. Barker, M. Weinfeld, J. Zheng, L. Li and D. Murray, J. Biol. Chem., 2005, 280, 33826–33838 CrossRef CAS PubMed.
- T. B. Gherezghiher, X. Ming, P. W. Villalta, C. Campbell and N. Y. Tretyakova, J. Proteome Res., 2013, 12, 2151–2164 CrossRef CAS PubMed.
- H. Qiu and Y. Wang, J. Proteome Res., 2009, 8, 1983–1991 CrossRef CAS.
- R. L. Loeber, E. D. Michaelson-Richie, S. G. Codreanu, D. C. Liebler, C. R. Campbell and N. Tretyakova, Chem. Res. Toxicol., 2009, 22, 1151–1162 CrossRef CAS PubMed.
- T. B. Gherezghiher, X. Ming, P. W. Villalta, C. Campbell and N. Y. Tretyakova, J. Proteome Res., 2013, 12, 2151–2164 CrossRef CAS PubMed.
- A. Groehler, P. W. Villalta, C. Campbell and N. Tretyakova, Chem. Res. Toxicol., 2016, 29, 190–202 CrossRef CAS PubMed.
- J. M. Aćimović, B. D. Stanimirović, N. Todorović, V. B. Jovanović and L. M. Mandić, Chem.-Biol. Interact., 2010, 188, 21–30 CrossRef PubMed.
- H. Kasai, N. Iwamoto-Tanaka and S. Fukada, Carcinogenesis, 1998, 19, 1459–1465 CrossRef CAS PubMed.
- R. Loeber, E. Michaelson, Q. Fang, C. Campbell, A. E. Pegg and N. Tretyakova, Chem. Res. Toxicol., 2008, 21, 787–795 CrossRef CAS PubMed.
- R. Loeber, M. Rajesh, Q. Fang, A. E. Pegg and N. Tretyakova, Chem. Res. Toxicol., 2006, 19, 645–654 CrossRef CAS PubMed.
- E. D. Michaelson-Richie, R. L. Loeber, S. G. Codreanu, X. Ming, D. C. Liebler, C. Campbell and N. Y. Tretyakova, J. Proteome Res., 2010, 9, 4353–4367 CrossRef PubMed.
- D. L. Carbone, J. A. Doorn, Z. Kiebler and D. R. Petersen, Chem. Res. Toxicol., 2005, 18, 1324–1331 CrossRef CAS PubMed.
- D. R. Seiner, J. N. LaButti and K. S. Gates, Chem. Res. Toxicol., 2007, 20, 1315–1320 CrossRef CAS PubMed.
- K. V. Petrova, A. D. Millsap, D. F. Stec and C. J. Rizzo, Chem. Res. Toxicol., 2014, 27, 1019–1029 CrossRef CAS PubMed.
- S. Barker, D. Murray, J. Zheng, L. Li and M. Weinfeld, Anal. Biochem., 2005, 344, 204–215 CrossRef CAS PubMed.
- L. L. G. Carrette, T. Morii and A. Madder, Bioconjugate Chem., 2013, 24, 2008–2014 CrossRef CAS PubMed.
- D. Pluskota-Karwatka, A. J. Pawłowicz, M. Bruszyńska, A. Greszkiewicz, R. Latajka and L. Kronberg, Chem. Biodiversity, 2010, 7, 959–974 CAS.
- K. Salus, M. Hoffmann, B. Wyrzykiewicz and D. Pluskota-Karwatka, New J. Chem., 2016, 40, 3875–3884 RSC.
- D. Pluskota-Karwatka, D. Matysiak, M. Makarewicz and L. Kronberg, Eur. J. Org. Chem., 2012, 4797–4804 CrossRef CAS.
- R. Drożdż, J. W. Naskalski and J. Sznajd, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1988, 957, 47–52 CrossRef.
- E. R. Stadtman and B. S. Berlett, J. Biol. Chem., 1991, 266, 17201–17211 CAS.
- J. Cai, A. Bhatnagar and W. M. Pierce, Chem. Res. Toxicol., 2009, 22, 708–716 CrossRef CAS PubMed.
- H. Shanan-Atidi and K. H. Bar-Eli, J. Phys. Chem., 1970, 74, 961–963 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford England: New York, 1st Paperback edn, 1994 Search PubMed.
- S. J. Grabowski, J. Phys. Chem. A, 2001, 105, 10739–10746 CrossRef CAS.
- O. Mó, M. Yáñez and J. Elguero, J. Chem. Phys., 1992, 97, 6628–6638 CrossRef.
- O. Mó, M. Yáñez and J. Elguero, THEOCHEM, 1994, 314, 73–81 CrossRef.
- L. González, O. Mó and M. Yáñez, J. Phys. Chem. A, 1997, 101, 9710–9719 CrossRef.
- E. Espinosa, M. Souhassou, H. Lachekar and C. Lecomte, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 563–572 CrossRef.
- C. Gatti, V. R. Saunders and C. Roetti, J. Chem. Phys., 1994, 101, 10686–10696 CrossRef CAS.
- S. J. Grabowski, J. Mol. Struct., 2001, 562, 137–143 CrossRef CAS.
- J. Murgich, H. J. Franco and G. San-Blas, J. Phys. Chem. A, 2006, 110, 10106–10115 CrossRef CAS PubMed.
- R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- S. J. Grabowski, Chem. Rev., 2011, 111, 2597–2625 CrossRef CAS PubMed.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS.
- D. Cremer and E. Kraka, Croat. Chem. Acta, 1984, 57, 1259–1281 Search PubMed.
- M. P. Stone, Y.-J. Cho, H. Huang, H.-Y. Kim, I. D. Kozekov, A. Kozekova, H. Wang, I. G. Minko, R. S. Lloyd, T. M. Harris and C. J. Rizzo, Acc. Chem. Res., 2008, 41, 793–804 CrossRef CAS PubMed.
- I. D. Kozekov, R. J. Turesky, G. R. Alas, C. M. Harris, T. M. Harris and C. J. Rizzo, Chem. Res. Toxicol., 2010, 23, 1701–1713 CrossRef CAS PubMed.
- M. Sanchez, I. D. Kozekov, T. M. Harris and R. S. Lloyd, Chem. Res. Toxicol., 2005, 18, 1683–1690 CrossRef PubMed.
- K. Stone, A. Uzieblo and L. J. Marnett, Chem. Res. Toxicol., 1990, 3, 467–472 CrossRef CAS PubMed.
- C. Ammann, P. Meier and A. E. Merbach, J. Magn. Reson., 1982, 46, 319–321 CAS.
- Gaussian 09 Citation http://www.gaussian.com/g_tech/g_ur/m_citation.htm, accessed Sep 11, 2015.
- AIMAll (Version 16.01.09), Todd A. Keith, TK Gristmill Software, Overland Park KS, USA, 2016 (aim.tkgristmill.com) http://aim.tkgristmill.com/, accessed May 2, 2016.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj00270j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |