
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of the Zn(II)–cyclen complex with aminomethylphosphonic acid: original simultaneous potentiometric and 31P NMR data treatment†
Ingrida
Rostášová
a,
Mária
Vilková
b,
Zuzana
Vargová
 *a,
Róbert
Gyepes
cd,
Miroslava
Litecká
a,
Vojtěch
Kubíček
c,
Ján
Imrich
b and
Ivan
Lukeš
c
*a,
Róbert
Gyepes
cd,
Miroslava
Litecká
a,
Vojtěch
Kubíček
c,
Ján
Imrich
b and
Ivan
Lukeš
c
aDepartment of Inorganic Chemistry, Faculty of Science, P. J. Šafárik University, Moyzesova 11, SK-041 54 Košice, Slovak Republic. E-mail: zuzana.vargova@upjs.sk
bNMR Laboratory, Faculty of Science, P. J. Šafárik University, Moyzesova 11, SK-041 54 Košice, Slovak Republic
cDepartment of Chemistry, Faculty of Education, J. Selye University, Bratislavská cesta 3322, SK-945 01 Komárno, Slovak Republic
dDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, CZ-128 43 Prague 2, Czech Republic
First published on 13th June 2017
Abstract
The interaction of aminomethylphosphonic acid (H2amp) with the Zn(II)–cyclen complex and the formation of ternary complexes was studied by potentiometry and 31P NMR titrations. Data evaluation of each of the methods separately was found to be insufficient. Thus, data obtained from the both methods were simultaneously treated with the computer program OPIUM. The determined stability constants indicated only weak (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K ∼ 3) coordination of aminomethylphosphonic acid through the phosphonate group. The chelating coordination mode with the participation of the amine group was not confirmed. In excess of Zn(II)–cyclen, the formation of dinuclear complexes, in which two Zn(II)–cyclen units are bridged by a phosphonate group, was observed and their presence was confirmed by mass spectrometry. Such a coordination motif is typical for phosphonates in the solid state and also for phosphatases.
K ∼ 3) coordination of aminomethylphosphonic acid through the phosphonate group. The chelating coordination mode with the participation of the amine group was not confirmed. In excess of Zn(II)–cyclen, the formation of dinuclear complexes, in which two Zn(II)–cyclen units are bridged by a phosphonate group, was observed and their presence was confirmed by mass spectrometry. Such a coordination motif is typical for phosphonates in the solid state and also for phosphatases.
Introduction
More than 300 enzymes containing Zn(II) in their centers have been described. To understand the role of Zn(II) as well as other transition metal ions in mechanistic pathways of the metalloenzymes, model complexes have been recently designed, synthesized and studied. The coordination site of the Zn(II) ion in the enzymes is formed by side chains of amino acids such as histidine or aspartate and its geometry results from the secondary or tertiary structure of the protein. It is very difficult to prepare such a coordination site artificially and, thus, in the models the coordination vicinity of the donor atoms is simplified.1–3 A number of ligands have been utilized and tested. Most of them are based on an aza-macrocyclic core4–6 and among them, cyclen is commonly used.Investigation of the Zn(II)–cyclen system has been mostly focused on the recognition of small organic molecules occurring in organisms,5–8 on nucleic acids, which have been summarized previously,9 and also on phosphates.10,11 In our group, we have studied the interaction of the Zn(II)–cyclen system with amino acids12 and dipeptides.13 The high affinity of amino acids for the Zn(II)–cyclen system can be explained by significant non-bonding contributions (hydrophobic and/or π–π stacking).12 The study of dipeptide interaction indicated carbonyl–carboxylate chelate binding in the acidic range and carbonyl–amine chelate or only terminal amino group coordination in the slightly alkaline range.13
This paper continues with our investigation of the Zn(II)–cyclen interaction with bioactive molecules. Here, we deal with interaction with a phosphorus analogue of glycine, i.e. aminomethylphosphonic acid (H2amp).
Natural aminophosphonates have been found in many different organisms, from prokaryotes to eubacteria and fungi, mollusks, insects and others.14–17 They play an important role in the interaction with many metalloenzymes. Many natural as well as synthetic aminophosphonates bind strongly in the active site of enzymes and act as inhibitors of their function.18 The aminomethylphosphonate binding mode toward metal ions has been extensively studied and summarized.19–21 Generally, differences in basicity, charge, the electron-releasing effect and the size of relevant donor groups between aminophosphonates and aminocarboxylates result in different complexing properties. Aminophosphonates offer the possibility of monodentate coordination of the ligand via the phosphonate group, forming stable protonated complexes. In the case of chelate formation, the thermodynamic stability of complexes of aminophosphonates is usually higher than that of aminocarboxylates, which is caused, at least in the first approximation, by the enhanced basicity of the ligands.
Investigation of metal–phosphonate systems is often hampered by the formation of insoluble complexes and, thus, also the aminomethylphosphonate complexing ability towards Zn(II) has not attracted much attention.22 Protonated [Zn(Hamp)]+, neutral [Zn(amp)] and anionic [Zn(OH)(amp)]− species were identified and their coordination modes were proposed.
Here, we report the results of the study of ternary complexes of the Zn(II)–cyclen system with aminomethylphosphonic acid. The system was studied by a combination of potentiometry, (1H, 13C, 31P) NMR titration and mass spectrometry.
Experimental
Reagents and solutions
All solutions were prepared using deionized water. Aminomethylphosphonic acid (H2amp) was synthetized according to the previously described procedure.23 Cyclen was obtained from CheMatech (France). Zn(NO3)2·4H2O was purchased from Lachema. The others chemicals were of analytical purity.Potentiometric titration
Potentiometric measurements were accomplished using a PHM 220 pH-meter, an ABU 901 autoburette and a GK 2401 B combination electrode (Radiometer) in a glass vessel (10 cm3) thermostatted at 25 ± 0.1 °C at an ionic strength of I = 0.1 M (KNO3). An inert atmosphere was ensured by constant flow of nitrogen gas. Precision calibration was accomplished using 0.028 M HNO3 and 0.192 M KOH in the pH range 1.8–12.0, with the pH-meter yielding E values. The relation between E and −log[H+] can be described as:| E = E0 − S(−log[H+]) + ja[H+] + jb(pKw/[H+]), |
The protonation constants of the ligands (H2amp or cyclen) were determined at cL = 0.004 M. For the determination of the stability constants in the binary systems, the metal:ligand molar ratios were 1:1, 1:2, 1:4 for the Zn(II)–amp system and 1:1 for the Zn(II)–cyclen system (cZn = 0.004 M). In the ternary system, the Zn:cyclen:amp ratio was 1:1:1 (cZn = 0.004 M). Due to slow complexation kinetics (4 to 5 hours were required to reach equilibrium), the binary system Zn(II)–cyclen and the ternary system were studied by the out-of-cell method with an equilibration time a 3 days.24 Three titrations were performed for each system and each ratio. Typical titration curves are shown in Fig. S4 (ESI†).
1H, 13C and 31P NMR characterization
1H, 13C and 31P NMR spectra were recorded on a Varian VNMRS 600 MHz spectrometer (resonance frequency for 1H = 599.870 MHz, 13C = 150.836 MHz, and 31P = 242.836 MHz). Samples were prepared in H2O and measured with a D2O insert containing 0.5% t-BuOH (1.25 ppm) as a reference. 1H spectra were measured using a PRESAT pulse sequence. NMR measurements were accomplished for free H2amp, for the binary system Zn(II)–amp and for the ternary system Zn(II)–cyclen–amp. The initial concentration of individual species was 0.05 M for both binary and ternary systems. Cyclen was used in 10% excess. For the ternary system, Zn(II) and cyclen solution were mixed and left for 4 h at RT before H2amp solution addition. The pH was adjusted with 0.1 M aq. NaOH and 0.1 M aq. HNO3.31P NMR titration
The stock solution of the Zn(II)–cyclen complex was prepared by dissolving cyclen in Zn(NO3)2 solution. To assure full complexation, the ligand was used in 10% excess and the solution was heated at 80 °C overnight. The aliquot part of the stock solution was mixed with an appropriate amount of H2amp solution to reach H2amp–Zn(II) ratios of 2:1 to 1:12. The concentration of H2amp in each sample was camp = 20 mM. For each ratio, several samples were prepared in the pH range 4–10 (adjusted with 0.1 M aq. NaOH or 0.1 M HNO3). Finally, 31P NMR spectra were recorded.
Data treatment
The protonation constants and the stability constants of the complexes in the binary systems (Zn(II)–cyclen and Zn(II)–amp) were determined from potentiometry data. The stability constants of the ternary complexes were determined by simultaneous treatment of potentiometry data and data from 31P NMR titration. All data were processed using the program OPIUM.25 The protonation constants of ligands β110, β210 and β310 are concentration constants and are defined as β110 = [HL]/[H][L], β210 = [H2L]/[H]2[L], β310 = [H3L]/[H]3[L], (pK3 = logβ110, pK2 = logβ210–logβ110, pK1 = logβ310–logβ210). The stability constants in binary systems are defined as βpqr = [HpLqMr]/[H]p[L]q[M]r. In the ternary system, the stability constants are defined as βpqrs = [HpL1qL2rMs]/[H]p[L1]q[L2]r[M]s (L1 = cyclen and L2 = amp).
Mass spectrometry
Stock solutions of ZnCl2, H2amp and cyclen were mixed to reach cZn = ccyclen = 1 mM and camp = 1–10 mM or camp = 1 mM and cZn = ccyclen = 1–10 mM. A solution of NMe4OH was added to reach pH ∼ 7 and ESI-MS spectra were measured on a Bruker Esquire 3000 apparatus in the positive mode.Synthesis of [Zn(Hamp)2]·4H2O
Zinc perchlorate hexahydrate (134 mg, 0.360 mmol) in water (10 mL) was slowly added into an aqueous solution (10 mL) of cyclen (62 mg, 0.360 mmol). After standing for 24 hours, an equimolar amount of H2amp (40 mg, 0.360 mmol) in water (10 mL) was added and the solution was kept at room temperature (pH = 4.5). After five days, very fragile colourless crystals, suitable for X-ray analysis, were obtained.X-ray crystallography
Single-crystal X-ray measurements of the complex [Zn(Hamp)2]·4H2O was performed on a Nonius Kappa four-circle CCD diffractometer equipped with a Bruker APEX II detector and using graphite monochromated MoKα radiation (λ = 0.71073 Å). Diffraction data were processed using the diffractometer software.26 The structure model was refined by full-matrix least-squares on F2 using SHELXL 97.27 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included in their ideal positions and refined isotropically. The structural figures were drawn using the DIAMOND 3.0 software.28 Crystallographic data and processing parameters are given in Table 1.| [Zn(H2O)4(Hamp)2] | |
|---|---|
| Empirical formula | C2H17N2O10P2Zn |
| M r | 356.49 |
| Crystal colour | White |
| Crystal system | Orthorhombic |
| Space group | Pca/21 |
| Unit cell dimensions | |
| a (Å) | 9.9779(4) |
| b (Å) | 12.0890(5) |
| c (Å) | 10.2249(5) |
| α (°) | 90.00 |
| β (°) | 90.00 |
| γ (°) | 90.00 |
| V c (Å3) | 1233.36 |
| Molecules per cell, Z | 4 |
| D calc (g cm−3) | 1.920 |
| μ (mm−1) | 2.295 |
| 2θmax | 55° |
| Measured reflections | 10197 |
| Independent reflections | 2824 |
| R 1[I > 2σ(I)] | 0.0237 |
| wR2 | 0.0489 |
Results and discussion
Speciation and stability constants
To describe the interaction modes of H2amp with the Zn(II)–cyclen species, NMR spectra were measured in solutions equilibrated at different pH values. In the 1H NMR spectra of free H2amp and those obtained for Zn(II)–amp system, one doublet signal of CH2 group (coupling to 31P with J = 13.2 Hz) was observed (Fig. S1 in ESI†). Similarly, the 13C NMR spectra show a doublet with J = 141.8 Hz (Fig. S2 in ESI†). In the 31P NMR spectra, one signal of the phosphorus nucleus (coupling from 1H) was noted (Fig. S3 in ESI†).In the ternary system, the changes in the 1H NMR spectra of cyclen (Fig. 1) are similar to those previously reported.12,24 The signal of free cyclen disappears at pH 3.5–4 due to Zn(II) complexation. The two multiplets of the Zn(II)–cyclen complex remain unchanged over the whole pH range, which indicates the same coordination mode of cyclen in all species. In the ternary system, H2amp shows a characteristic pH dependence of δH and δC, however, the differences from free H2amp and from the binary Zn(II)–amp system are not significant and, so, they do not provide clear hints of the coordination mode (Fig. 2A and B). Both 1H and 13C NMR spectra show similar trends as observed in systems with glycine (Gly),12 in which analogous species [Zn(cyclen)(HGly)]2+ and [Zn(cyclen)(Gly)]+ were determined. More information could be obtained from 31P NMR data (Fig. 2C). The changes observed for free H2amp are consistent with previous results.29 The decrease of the δP at pH 5–6 indicates intramolecular interaction of the deprotonated phosphonate group with the protonated amino group. The largest changes in the 31P NMR shift are caused by deprotonation of the amino group at pH > 9. The binary system Zn(II)–amp could not be studied at pH above 7 due to the precipitation of the polymeric hydroxido species. However, a significant decrease of the δP at pH 5–6 could be observed. Comparison with distribution diagrams (Fig. S6 in ESI†) shows that the change is associated with deprotonation of the amino group as result of N,O-chelate coordination of the ligand. So, the decrease of the δP results from the formation of a cyclic structure.
 | ||
| Fig. 1 pH changes of the 1H NMR spectra of the ternary Zn(II)–cyclen–amp system. camp = cZn = 0.05 M, ccyclen = 0.06 M, 25 °C. | ||
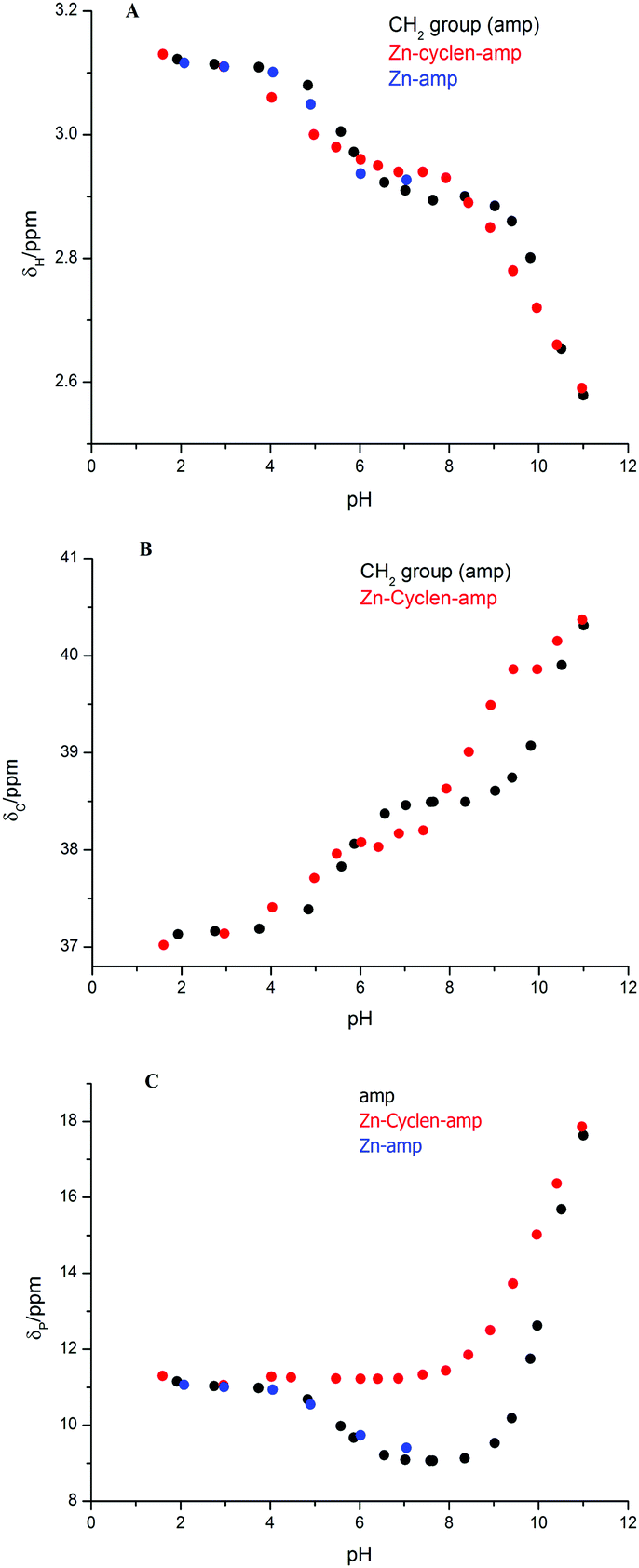 | ||
| Fig. 2 pH dependence of the 1H (A), 13C (B) and 31P (C) NMR shifts of H2amp. Free H2amp (black), binary Zn(II)–amp system (blue) and ternary Zn(II)–cyclen–amp (red) system. camp = cZn = ccyclen = 0.05 M, 25 °C. | ||
In the ternary system, the pH dependence of δP is different; δP remains constant up to pH 8. The increase at higher pH results from deprotonation of the amino group. The absence of the δP drop at pH 5–6 indicates that H2amp should be coordinated only through the phosphonate group in the ternary system. The phosphonate group might be coordinated in a bidentate mode as suggested by Kimura30 or a monodentate mode with additional interactions with cyclen amine groups through hydrogen bonds.
The determination of the stability constants of the ternary complexes requires detailed and precise characterization of all subsystems under identical conditions. So, the protonation constants of the ligands (cyclen and H2amp) as well as the stability constants in the binary systems (Zn(II)–amp and Zn(II)–cyclen) had to be redetermined (Tables S1–S4 in ESI†).
The protonation constants of H2amp as well as the stability constants of the complexes with Zn(II) (Tables S1 and S2 in ESI†) were found to be close to those previously reported.22,31 The complexes in the Zn(II)–amp system are formed in the pH range 4–10. The distribution diagrams are shown in Fig. S5 and S6 (ESI†). The dominant species at pH 7–8 is the [Zn(amp)] complex. The protonated complex [Zn(Hamp)]+ is present in low abundance (about 10%). The derived value of induced deprotonation logβ111 − logβ011 = 6.03 is significantly lower than the pK value of free amp due to the high stability of the complex. It indicates the formation of the N,O-chelate. Above pH ∼ 8, the formation of the hydroxido-species [Zn(amp)(OH)]− and less abundant [Zn(amp)2]2− is observed.
Because of the slow formation kinetics of macrocyclic complexes, the “out-of-cell” method was used for potentiometric titration of the binary system Zn(II)–cyclen. The protonation constants and stability constants of the complexes (Tables S3 and S4 in ESI†) are in very good agreement with those previously reported.24 The distribution diagrams are shown in Fig. S7 and S8 in the ESI.† The dominant species in the pH range 5–7 is non-protonated [Zn(cyclen)]2+. The protonated complex [Zn(Hcyclen)]3+ was identified in low abundance at pH ∼ 4. In the alkaline range, the coordinated water molecule is deprotonated forming hydroxido complexes [Zn(cyclen)(OH)]+ and [Zn(cyclen)(OH)2].
The ternary system Zn(II)–cyclen–amp was studied by a combination of potentiometry and 31P NMR. Each of the methods separately was found to be insufficient. The data obtained from potentiometry have shown the low stability of the ternary complexes; and mostly, the formation of the complexes is not associated with a change in the protonation state of the ligands (e.g. coordination of the deprotonated phosphonate). 31P NMR titration is more suitable as it allows utilizing some components in a large excess. It leads to a higher abundance of the ternary complexes and better determination of their stabilities. However, the studied system contains many species differing in their protonation states and NMR titration could hardly distinguish between these species. So, only the combination of both techniques gave reliable speciation as well as stability constants of the ternary system. Data were treated with the program OPIUM25 that allows simultaneous treatment of data obtained by various analytical techniques.
Potentiometry was performed using the “out-of-cell” method with equimolar concentrations of all components. 31P NMR titration was performed at various pH values 4–10 and at amp:Zn ratios from 2:1 to 1:12. In each sample, cyclen was used in 10% molar excess over Zn(II) to assure full complexation. The results show significant differences between the 31P NMR spectra of free H2amp and those measured in the presence of the Zn(II)–cyclen complex (Fig. 3). The stability constants presented in Table 2 and Table S5 in the ESI† represent the best model obtained from simultaneous fitting of the potentiometry and NMR data.
 | ||
| Fig. 3 Ternary system Zn(II)–cyclen–amp. Dependence of the 31P NMR chemical shift of H2amp on pH for various amp:Zn ratios (the numbers in the plot give the ratio for each series; camp = 20 mM, 25 °C). In each sample, cyclen was used in 20% molar excess over Zn(II). The curves were constructed using the 31P NMR shifts of individual species calculated simultaneously with the determination of stability constants. | ||
| Equilibrium | logK |
|---|---|
| [Zn(cyclen)]2+ + Hamp− → [Zn(cyclen)(Hamp)]+ | 3.05 |
| [Zn(cyclen)]2+ + amp2− → [Zn(cyclen)(amp)] | 3.3 |
| [Zn(cyclen)(amp)] + H+ → [Zn(cyclen)(Hamp)]+ | 9.8 |
| [Zn(cyclen)(amp)] + [Zn(cyclen)]2+ → [{Zn(cyclen)}2(amp)]2+ | 3.06 |
| [{Zn(cyclen)}2(amp)(OH)]+ + H+ → [{Zn(cyclen)}2(amp)]2+ | 9.4 |
The distribution diagrams (Fig. 4) show that the only ternary species [Zn(cyclen)(Hamp)]+ is formed in significant abundance. This species is present in the pH range 4–9. Its abundance ranges from 60% at equimolar concentrations of all components to >90% at a 5-fold excess of Zn(II) and cyclen. At higher pH, the ternary complex undergoes deprotonation yielding [Zn(cyclen)(amp)]. However, its abundance does not exceed 20% even at a 5-fold excess of Zn(II) and cyclen over H2amp. The low abundance of the [Zn(cyclen)(amp)] complex is given by the competition of the hydroxide anion. Water molecules coordinated to the metal ion in the [Zn(cyclen)]2+ complex are deprotonated at pH ∼ 8. At higher pH, the coordination of the hydroxide anion dominates over the formation of ternary complexes which leads to an increasing abundance of free monoprotonated Hamp (Fig. 4A) and low abundance of the [Zn(cyclen)(amp)] complex.
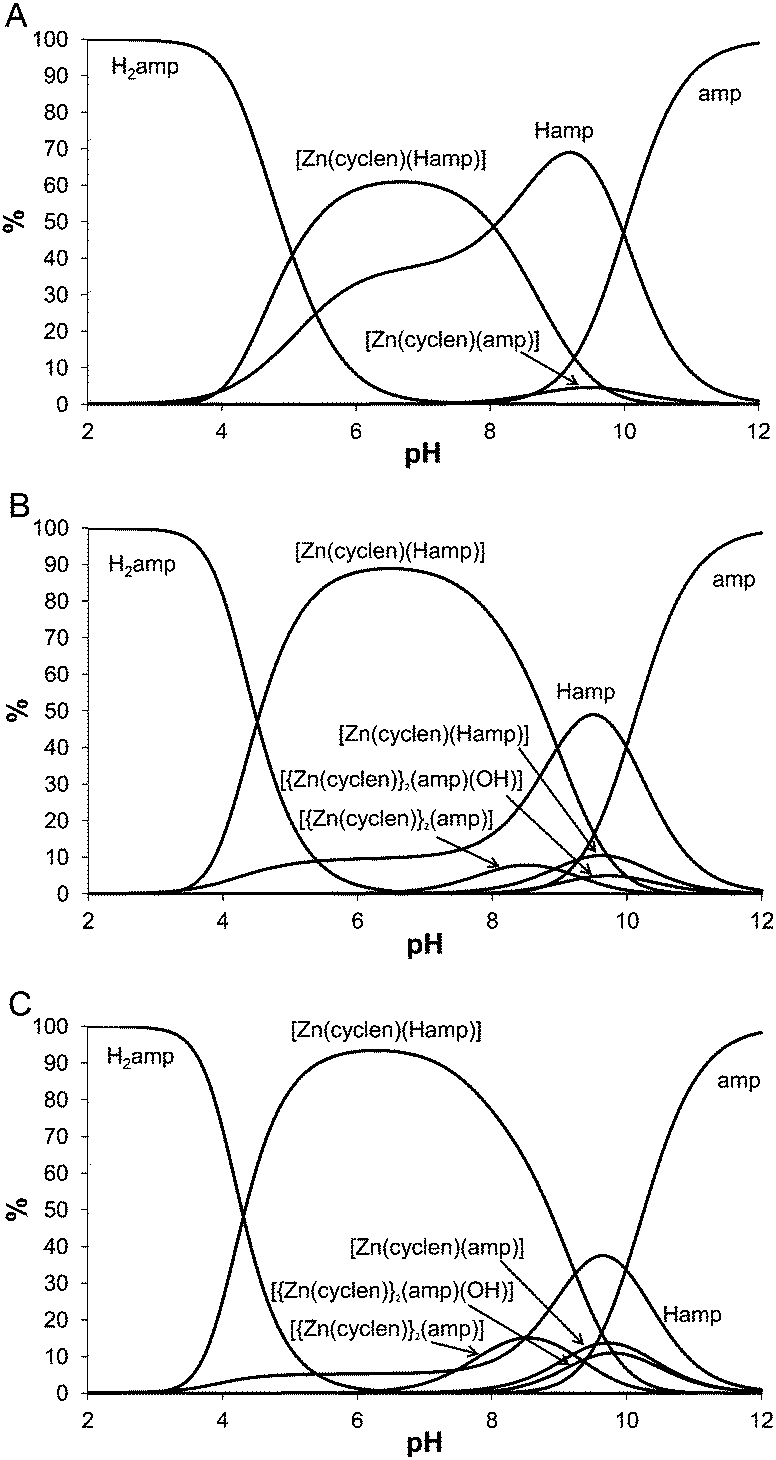 | ||
| Fig. 4 Distribution of aminomethylphosphonate in the ternary system Zn(II)–cyclen–amp (A: camp = ccyclen = cZn = 4 mM; B: camp = 4 mM, ccyclen = cZn = 12 mM; C: camp = 4 mM, ccyclen = cZn = 20 mM; I = 0.1 M, 25 °C). | ||
At an excess of [Zn(cyclen)]2+ over H2amp, dinuclear complexes with stoichiometries [{Zn(cyclen)}2(amp)]2+ and [{Zn(cyclen)}2(amp)(OH)]+ are formed. However, their abundance is low (<20%) even at a 5-fold excess of the Zn(II)–cyclen complex over H2amp (Fig. 4C).
The formation of the ternary complexes is also evidenced by the ESI-MS spectra of the Zn(II)–cyclen–amp system (Fig. S9, ESI†). Under equimolar conditions, the dominant detected ion is [Zn(cyclen)Cl]+ (m/z = 271) and a minor signal of [Zn(cyclen)(Hamp)]+ (m/z = 346) was observed. At a 10-fold excess of H2amp, the latter signal becomes dominant indicating an increased abundance of the ternary complex in the system. At a 10-fold excess of the Zn(II)–cyclen complex, the detected signal centered at m/z = 581 confirms the formation of the dinuclear species [{Zn(cyclen)}2(amp)].
The speciation is similar to that previously reported in ternary systems of Zn(II)–cyclen with glycine and alanine except for the dinuclear species.12 On the other hand, such species in which an amino acid connects two [Zn(cyclen)]2+ units was observed in the solid-state structure of [{Zn(cyclen)}2(picolinic acid)]+.24 The value of the protonation constant of the ternary complex (logK = 9.8) is close to the first protonation constant of H2amp (logK = 9.99). So, [Zn(cyclen)(Hamp)]+ is protonated on the amino group. The constants describing the coordination of non-protonated amp2− and monoprotonated Hamp− to [Zn(cyclen)]2+ are 3.3 and 3.05, respectively. Similar values indicate negligible role of the amino group in the coordination. As suggested above, monodentate or bidentate coordination only through the phosphonate group could be expected in both species. It would point to the bidentate coordination of deprotonated phosphonate in both [Zn(cyclen)(Hamp)]+ and [Zn(cyclen)(amp)] species as expected by Kimura30 or to the oxygen interaction of the O–P group with the NH group of cyclen.
The positive charge localized on the protonated amino group in [Zn(cyclen)(Hamp)]+ does not allow the approach of the additional [Zn(cyclen)]2+ unit. So, the dinuclear species are formed only after amine deprotonation. The constants describing the coordination of the first and the second [Zn(cyclen)]2+ unit are also similar (3.3 and 3.06, respectively). It is in agreement with the high ability of the phosphonate groups for the bridging of the metal ions. Deprotonation of the coordinated water molecule leads to the dinuclear hydroxido species. The corresponding protonation constant (logK = 9.4) is lower than that of the amino group in [Zn(cyclen)(Hamp)]+. It shows a preferential coordination of the hydroxide anion that does not allow the N,O-bidentate coordination of H2amp. It might also indicate that the hydroxide anion also bridges the two Zn(II) ions. The suggested structures of the ternary species are shown in Scheme 1, and the structures of amp and the Zn(II)–cyclen species identified in the studied systems are shown in Scheme S1 (ESI†).
 | ||
| Scheme 1 Suggested structures of species identified in the ternary system Zn(II)–cyclen–amp. | ||
Similar systems with amino acids Zn(II)–cyclen–Gly or Ala show stronger coordination (logK = 4.1 and 4.0, respectively).12 It does not correspond to the basicity of the amino groups of the ligands and so, it also excludes the participation of the amino group in metal binding. If we compare the sterical requirements of phosphonate and carboxylate groups, phosphonate is bulkier and both, P–C and P–O, bonds are longer. Thus, phosphonate occupies more space than carboxylate, which could lead to a preferential square-pyramidal coordination sphere of Zn(II). On the other hand, the O–P–O angle is smaller than in carboxylates and the bidentate coordination of phosphonate and the formation of an octahedral sphere is also possible.
There are many aminophosphonates and aminophosphinates that are used and studied as enzyme inhibitors in medicine or agriculture. The results presented here show that the interaction of aminophosphonates with the Zn(II) centre of the model complex is rather weak and even weaker interaction could be expected for aminophosphinates. The described coordination interaction is insufficient for effective binding of the inhibitor in the enzyme active site. It indicates that the binding of aminophosphonates and aminophosphinates in the enzyme active centre must be dominantly realized through hydrogen bonds and/or through non-bonding interactions with amino acid side chains. As the geometry of each enzyme active site is unique, it implicates a high specificity of the enzyme–inhibitor interaction for aminophosphonate and aminophosphinate inhibitors. It also explains, e.g. the low animal toxicity and the high selectivity of the most utilized herbicide glyphosate (phosphonomethylglycine).
Zn(II)–amp coordination modes in the solid state
Despite many attempts to prepare crystals of the studied complexes, only single crystals of [Zn(Hamp)2]·4H2O were isolated as the substance with the lowest solubility product. The solid-state structure is shown in Fig. S10 in the ESI† and the principal bond lengths and angles and the list of possible hydrogen bonds are summarized in Tables S6 and S7 in the ESI.† The complex crystallizes in the orthorhombic space group Pca/21 with four formula units per cell. The central Zn(II) atom coordination sphere is formed by four oxygen atoms of the phosphonate part of the aminomethylphosphonate ligand. The coordination polyhedron around the metal centre can be described as a tetrahedron with bond lengths of Zn–O1 1.915(2), Zn–O3 1.940(2), Zn–O4 1.955(2), Zn–O5 1.933(2) Å and bond angles around 104.9° (Table S6 in ESI†). The typical feature of the phosphonate coordination way, bridging two metal ions with two oxygen atoms of the phosphonate group and the formation of a polymeric network, was also found in this structure. As this study shows, the formation of the bridged structure is not only the result of the crystal packing, but also originates from the Zn(II)–phosphonate interaction in solution. However, this crystal structure was determined previously32 and all parameters and bond lengths of both determinations were found to be very similar.The resulting structure of the Zn(II)–amp complex is sensitive to reaction conditions as found for the published structures. The analogous reaction between ZnCl2 and H2amp upon gentle heating led to a polymeric product in which the coordination sphere was formed with three oxygen atoms from three phosphonates and one chloride anion.33 The formation of the chelate was observed in the case of derivatives with a substituted amino group. The hydrothermal reaction of (1-phenylethyl) aminomethylphosphonic acid with ZnSO4·7H2O and NaCl in a 1:1:0.5 molar ratio, adjusted to pH = 5.7 at 140 °C, afforded crystals in which the unit contains two independent Zn(II) ions, two phosphonate ligands, and one chloride anion. Zn1 possess the O3N1 coordination sphere in which one ligand molecule is coordinated in the N,O-chelating mode and two ligand molecules are coordinated through one phosphonate oxygen atom. The coordination sphere around Zn2 is formed by three phosphonate oxygen atoms and one chloride anion.34
Conclusion
The interaction of aminomethylphosphonic acid (H2amp) with the Zn(II)–cyclen complex was studied as a model for enzyme interactions with phosphonates. The stabilities of the complexes were evaluated by a combination of potentiometry and multinuclear NMR. Aminomethylphosphonic acid binds weakly to the Zn(II)–cyclen complex in both monoprotonated and deprotonated forms. Taking into account both the trends based on the solid-state structures and thermodynamic constants determined in this study, coordination only through the phosphonate group is suggested for both mononuclear species [Zn(cyclen)(Hamp)]+ and [Zn(cyclen)(amp)]. In dinuclear species [{Zn(cyclen)}2(amp)]2+, the phosphonate group bridges two [Zn(cyclen)]2+ units. This coordination way is probably more favorable than the formation of [Zn(cyclen)(amp)]. The coordination of the amine group is not expected. However, amine as well as phosphonate could further stabilize the structure through hydrogen-bonds with cyclen nitrogen atoms. A similar interaction was previously reported in ternary systems with amino acids.35 The dinuclear species were not described in the solutions of ternary complexes with amino acids12 and dipeptides,13 which documents the high bridging ability of the phosphonate group. The dinuclear species were reported only for ternary complexes with picolinic and nicotinic acids in the solid state.24 In the alkaline range, the dinuclear hydroxide complex [{Zn(cyclen)}2(amp)(OH)]+ is formed.Acknowledgements
Financial support was provided by a grant of the Ministry of the Education of the Slovak Republic (grant number KEGA 002UPJŠ-4/2015).References
- T. Heinisch and T. R. Ward, Eur. J. Inorg. Chem., 2015, 3406 CrossRef CAS.
- K. E. Dalle and F. Meyer, Eur. J. Inorg. Chem., 2015, 3391 CrossRef CAS.
- D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897 CrossRef CAS.
- P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2012, 256, 953 CrossRef CAS.
- E. Kimura, Acc. Chem. Res., 2001, 34, 171 CrossRef CAS PubMed.
- S. Aoki and E. Kimura, Comprehensive Coordination Chemistry, Elsevier, Amsterdam, 2004, vol. 8, p. 601 Search PubMed.
- T. Koike, E. Kimura, I. Nakamura, Y. Hashimoto and M. Shiro, J. Am. Chem. Soc., 1992, 114, 7338 CrossRef CAS.
- S. Aoki and E. Kimura, J. Am. Chem. Soc., 2000, 122, 4542 CrossRef CAS.
- S. Aoki and E. Kimura, Chem. Rev., 2004, 104, 769 CrossRef CAS PubMed.
- S. Aoki, K. Iwaida, N. Hanamoto, M. Shiro and E. Kimura, J. Am. Chem. Soc., 2002, 124, 5256 CrossRef CAS PubMed.
- S. Aoki, D. Kagata, M. Shiro, K. Takeda and E. Kimura, J. Am. Chem. Soc., 2004, 126, 13377 CrossRef CAS PubMed.
- Z. Vargová, E. Balentová, M. Walko, L. Arabuli, P. Hermann and I. Lukeš, J. Mol. Recognit., 2011, 24, 295 CrossRef PubMed.
- I. Rostášová, M. Vilková, Z. Vargová, M. Walko, M. Almáši, J. Imrich, P. Hermann and I. Lukeš, J. Mol. Recognit., 2015, 28, 211 CrossRef PubMed.
- B. Nowack, Water Res., 2003, 37, 2533 CrossRef CAS PubMed.
- S. V. Kononova and M. A. Nesmeyanova, Biochemistry, 2002, 67, 184 CAS.
- G. DellaCioppa, S. C. Bauer, B. K. Klein, D. M. Shah, R. T. Fraley and G. Kishore, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 6873 CrossRef CAS.
- Q. Li, M. J. Lambrechts, Q. Zhang, S. Liu, D. Ge, R. Yin, M. Xi and Z. You, Drug Des., Dev. Ther., 2013, 7, 635 CAS.
- V. P. Kukhar and H. R. Hudson, Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity, John Wiley and Sons, New York, 2000 Search PubMed.
- T. Kiss, I. Lázár and P. Kafarski, Met.-Based Drugs, 1994, 1, 247 CrossRef CAS PubMed.
- T. Kiss, I. Lázár, V. P. Kukhar and H. R. Hudson, Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity, Wiley, New York, 2000, p. 285 Search PubMed.
- J. Rohovec, P. Vojtíšek, I. Císařová, P. Hermann and I. Lukeš, J. Chem. Soc., Dalton Trans., 1996, 2685 RSC.
- M. Wozniak and G. Nowogrocki, Talanta, 1979, 26, 1135 CrossRef CAS PubMed.
- R. Tyka and G. A. Hagele, Synthesis, 1984, 218 CrossRef CAS.
- Z. Vargová, J. Kotek, J. Rudovský, J. Plutnár, R. Gyepes, P. Hermann, K. Györyová and I. Lukeš, Eur. J. Inorg. Chem., 2007, 3974 CrossRef.
- M. Kývala and I. Lukeš, 1995. International Conference, Chemometrics 95, Pardubice, Česká republika, p. 63, University of Pardubice, Pardubice, 3rd–7th July 1995, the full version of “OPIUM” is available (free of charge) on: http://web.natur.cuni.cz/˜kyvala/opium.html.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- K. Brandenburg, DIAMOND, Version 2.1e, Crystal Impact GbR, Bonn, 2000 Search PubMed.
- T. G. Appleton, J. R. Hall, A. D. Harris, H. A. Kimlin and I. J. McMahon, Aust. J. Chem., 1984, 37, 1833 CrossRef CAS.
- T. Koike and E. Kimura, J. Am. Chem. Soc., 1991, 113, 8935 CrossRef CAS.
- P. Hermann and I. Lukeš, J. Chem. Soc., Dalton Trans., 1995, 2605 RSC.
- P. Fenot, J. Darriet, C. Garrigou-Lagrange and A. Cassaigne, J. Mol. Struct., 1978, 43, 49 CrossRef CAS.
- C. R. Samanamu, E. N. Zamora, J. L. Montchamp and A. F. Richards, J. Solid State Chem., 2008, 181, 1462 CrossRef CAS.
- X. G. Liu, S. S. Bao, Y. Z. Li and L. M. Zheng, Inorg. Chem., 2008, 47, 5525 CrossRef CAS PubMed.
- Z. Vargová, R. Gyepes, L. Arabuli, K. Györyová, P. Hermann and I. Lukeš, Inorg. Chim. Acta, 2009, 362, 3860 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1528770. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj00254h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |