
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid[4]arenes with anthracene units and tuneable cavities†
Tomasz
Boinski
and
Agnieszka
Szumna
 *
*
Institute of Organic Chemistry, Polish Academy of Science, Kasprzaka 44/52, 01-244 Warsaw, Poland. E-mail: agnieszka.szumna@icho.edu.pl
First published on 5th April 2017
Abstract
We present the synthesis of hybrid[4]arenes via a one-pot reaction of 1,4,5,8-tetramethoxyanthracene and 1,3-dimethoxybenzene with formaldehyde catalyzed by TFA. Two new C2h-symmetric macrocycles having different substitution patterns at anthracene units were obtained. Both X-ray structure analysis and molecular modelling reveal a parallel arrangement of the anthracene units with an interplane distance of 4.8 Å. A comparison of UV and fluorescence spectra of hybrid[4]arene and a monomeric anthracene unit indicates that the energies and intensities of the absorption bands (scaled to the same number of chromophores) remain similar. However, for the macrocycle the quantum yield of emission is increased, which may originate from its rigid structure. Hybrid[4]arene was post-macrocyclization modified by the Diels–Alder reaction with benzyne and a macrocycle with an expanded cavity was obtained. The expanded cavity macrocycle is able to selectively complex and solubilize pyridinium iodide in chloroform.
Introduction
Macrocyclic skeletons composed of anthracene fragments expose large, hydrophobic, electron-rich panels into macrocyclic cavities. This feature greatly enhances complexation properties and additionally allows for easy detection of complexation events due to the inherent fluorescence of anthracene units.1 In comparison with other polyaromatic building blocks, anthracenes present a reasonable compromise between availability, chemical modifiability, structural rigidity and solubility. Therefore, over the years they have been widely used as building blocks for cyclophanes – hydrocarbons composed of aromatic units bridged by n-alkyl (n ≥ 2) chains.2–6 More recently anthracene units were also used as parts of cage-shaped anthraphanes,7 hybrid imidazolium–anthracene macrocycles8,9 and shape-persistent tubes.10–15 These polyaromatic macrocycles effectively complex various fluorescent dyes, aromatic carbonyl derivatives14 and fullerenes.16 An oxidized form of anthracene – anthraquinone – has likewise been used for the construction of macrocyclic compounds using imine forming reactions.17,18 Quite intriguingly, anthracene-based building blocks have not been incorporated as parts of calixarenes – macrocycles with aromatic panels linked by short methylene bridges. In this paper we present our approach towards synthesis of calixarene-type macrocycles using anthracene building blocks and towards post-synthetic modification of their cavity.Among the methods of synthesis of calixarene type hybrid macrocycles19–21 only two are reported to be reversible. The first one is based on the Friedel–Crafts reaction of dialkoxybenzyl bromides under dry conditions.22 The second one utilizes a TFA-catalyzed reaction between electron-rich aromatics and formaldehyde in chlorinated solvents at high temperatures.23–25 The reversibility of the macrocyclization reaction opens new synthetic possibilities as it allows for amplification of the thermodynamically most stable structures and allows for a template-type synthesis. Indeed, we recently demonstrated a high yielding, solvent-templated synthesis of pillar[5]arenes23 and synthesis of new calix[4]naphthalenes25 using this approach. Additionally, a dynamic covalent approach facilitated the synthesis of hybrid macrocycles consisting of different building blocks within a macrocyclic ring via a one-pot reaction.24 Recently, we further increased the dimensions of the building blocks aiming at the synthesis of anthracene-based macrocycles using this methodology. Herein, we report the structures and complexation properties of macrocycles obtained using such a method.
Results and discussion
Initially, we tested the possibility of obtaining homo-anthracene macrocycles, i.e. macrocycles consisting of identical anthracene building blocks. We used 1,4,5,8-tetramethoxyanthracene 2 and paraformaldehyde in TFA/DCE (5% vol/vol) at 80 °C. We have previously reported that under such conditions the macrocyclization reaction of benzene analogs is reversible.24 In the case of 2 only undefined polymeric products were obtained, suggesting that homo-macrocycles are not thermodynamically preferred over polymeric products under current conditions. In the next step anthracene monomer 2 was combined with 1 (a building block based on a benzene ring) in a one pot reaction with formaldehyde in DCE catalysed by TFA. Two macrocyclic products 3 and 4 were isolated from the reaction mixture. Both products are hybrid[4]arenes that contain two anthracene-based units and two 1,3-dimethoxybenzene units, as was established based on NMR and mass spectra (calculated mass 1095.4295 for [C68H64O12Na]+, found 1095.4291). Optimization of the synthetic procedure (Table 1) resulted in the formation of 3 in 10% yield. The yield of 3 is only slightly dependent on the initial concentration of substrates; however, it requires at least 5% of the catalyst and sufficient time for equilibration. These results are in agreement with the thermodynamic control of the reaction. In contrast, macrocyclic product 4 was obtained only in trace amounts, independent of the conditions, which may result from its lower thermodynamic stability.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 (M)
C2 (M)1H and 13C NMR spectra of 3 reveal the presence of only ¼ of the maximum number of signals of the anthracene units and ½ of the maximum number of signals of the benzene units (ESI†). Methylene bridges are present as two doublets with a geminal coupling constant of 16.1 Hz. 1H–13C HSQC and 1H–13C HMBC spectra indicate that methylene bridges link the macrocycle at the 2,6 positions of the anthracene unit and the 4,6 positions of the benzene unit. These characteristics suggest a C2h-symmetric structure for 3 with a two-fold axis passing through the centres of the anthracene units and a perpendicular mirror plane crossing the benzene units. In agreement with this symmetry, there are three signals from CH3 groups present in the spectrum. The signal at 4.00 ppm was assigned to resorcinol methyl groups, more shielded signals at 3.74 and 3.54 were assigned to methyl groups from the anthracene building block positioned respectively meta and ortho in relation to the bridge. The modelled structure of 3 calculated by molecular mechanics is presented in Fig. 1b and c. The anthracene faces are positioned perpendicularly to the main macrocyclic plane (defined by four bridging methylene groups) and parallel to each other (distance between the planes is ca. 4.8 Å). According to this model, free rotation of the anthracene units along the connection points is hindered and consequently prohibits interconversion of the macrocyclic ring. In contrast, the benzene units can flip, but only by 180°. This model is in agreement with non-equivalency of protons at the methylene bridges (exhibiting geminal coupling due to hindered rotation of the anthracene units) and with considerable upfield shift of the signals of the methyl groups positioned at the anthracene units (due to close face-to-face proximity to these units).
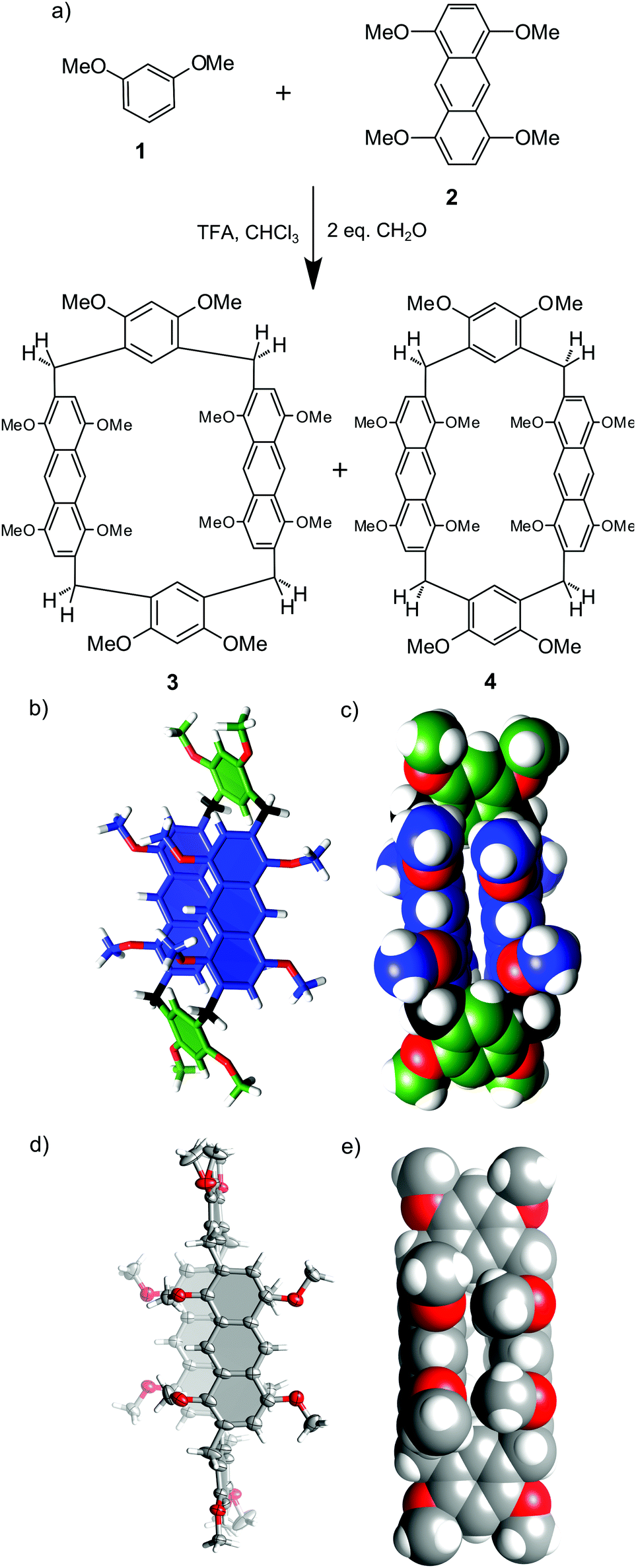 | ||
| Fig. 1 (a) Synthesis of hybrid[4]arenes 3 and 4; (b) calculated structure of 3 (3D structure optimization module in ChemSketch); (c) van der Waals representation of the calculated structure of 3; (d) X-ray structure of 4 – ORTEP view (e) X-ray structure of 4 – van der Waals representation. | ||
Hybrid[4]arene 4 was obtained in trace amounts and, due to purification problems, was not characterized in solution. However, we were able to obtain single crystals suitable for X-ray analysis from an enriched sample (Fig. 1d and e). In hybrid[4]arene 4 methylene bridges link the anthracene units at the 2,7 positions and the benzene units at the 4,6 positions. The molecule has a Ci crystallographic symmetry, but in the solution the symmetry of this molecule is expected to be higher (C2h with a two-fold axis passing through the centres of the benzene units and a perpendicular mirror plane crossing the anthracene units). The shape of the macrocyclic cavity is similar to that in the modelled structure of 3: two anthracene faces are parallel to each other and perpendicular to the main macrocyclic plane, while benzene rings are parallel to the main macrocyclic plane.
A comparison of UV and fluorescence spectra of hybrid[4]arene 3 and monomeric unit 2 indicates that the energies and intensities of the absorption bands (scaled to the same number of chromophores) do not change considerably (Fig. 2). However, for hybrid[4]arene 3, the quantum yield of emission increased, which may have originated from its rigid structure.
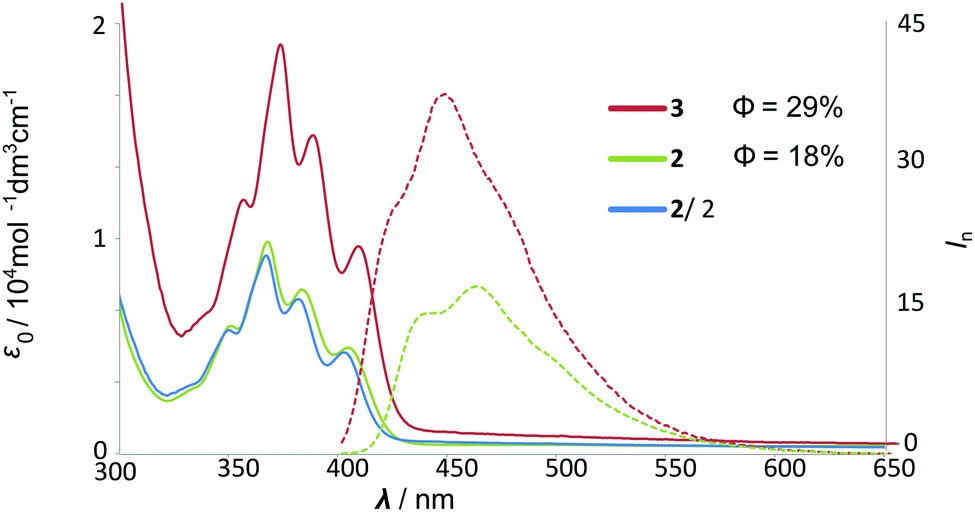 | ||
| Fig. 2 UV-Vis and fluorescence spectra of monomer 2 and macrocycle 3 in chloroform (for the full-range spectrum see the ESI†). | ||
Due to parallel positions of anthracene faces, the cavity of macrocycle 3 is narrow. In order to tune the cavity's shape 3 was modified by the Diels–Alder reaction with benzyne (Fig. 3). Benzyne was in situ generated from benzene-diazonium-2-carboxylate and reacted with 3 to yield 5 in 60% yield. The connection pattern and C2h symmetry of 5 were confirmed by NMR. Molecular modeling indicates that the molecular cavity in 5 is expanded as compared with 3 (Fig. 3b and c).
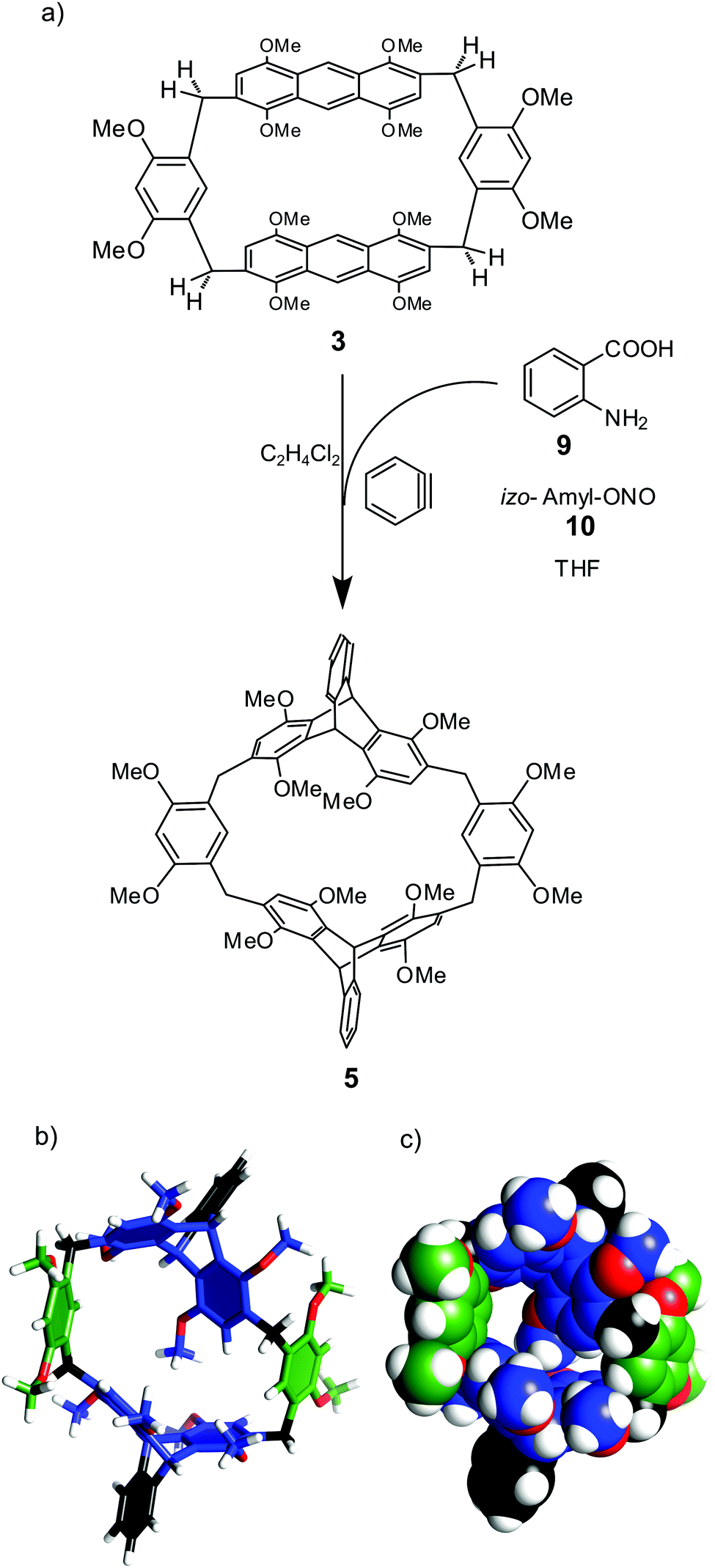 | ||
| Fig. 3 (a) Modification of hybrid[4]arene 3 by the Diels–Alder reaction; (b) calculated structure of 5 (3D structure optimization module in ChemSketch); (c) van der Waals representation of the calculated structure of 5. | ||
Macrocycles 3 and 5 were tested for complexation of rac-6, 7 and 8 (Fig. 4–6 and Table 2). The results indicate that both macrocycles interact with rac-6 and 7. For rac-6 the most affected proton during complexation is Hc (downfield shift, Fig. 4), while for 7 the most affected proton is Ha (upfield shift, Fig. 5). These differences suggest different modes of interactions for these two guests that may involve hydrogen bond-type interactions in the case of imidazolium based guest rac-6 (e.g. between the most acidic proton Hc and ethereal oxygen atoms) and π-stacking interactions in the case of 7 (electron-poor pyridinium ring and electron-rich anthracene rings). 1H NMR titrations indicate that the experimental points can be reasonably fitted only assuming simultaneous formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 (H:G). These qualitative and quantitative observations are consistent with the out-of-cavity complexation of guests rac-6 and 7 by macrocycles 3 and 5. Complexation experiments using fluorescence monitoring were performed for 3; however, no effective changes were detected. A different complexation behavior was observed towards guest 8. Guest 8 is poorly soluble in chloroform and it remains virtually undissolved in the presence of macrocycle 3. However, in the presence of macrocycle 5 (0.4 mM) up to 2 equivalents of 8 can be dissolved in chloroform (Fig. 6a). All protons of guest 8 experience pronounced upfield shifts upon complexation. Molecular modelling indicates that the expanded cavity of 5 may accommodate a whole molecule of guest 8 (Fig. 6b).
:1 and 1:2 (H:G). These qualitative and quantitative observations are consistent with the out-of-cavity complexation of guests rac-6 and 7 by macrocycles 3 and 5. Complexation experiments using fluorescence monitoring were performed for 3; however, no effective changes were detected. A different complexation behavior was observed towards guest 8. Guest 8 is poorly soluble in chloroform and it remains virtually undissolved in the presence of macrocycle 3. However, in the presence of macrocycle 5 (0.4 mM) up to 2 equivalents of 8 can be dissolved in chloroform (Fig. 6a). All protons of guest 8 experience pronounced upfield shifts upon complexation. Molecular modelling indicates that the expanded cavity of 5 may accommodate a whole molecule of guest 8 (Fig. 6b).
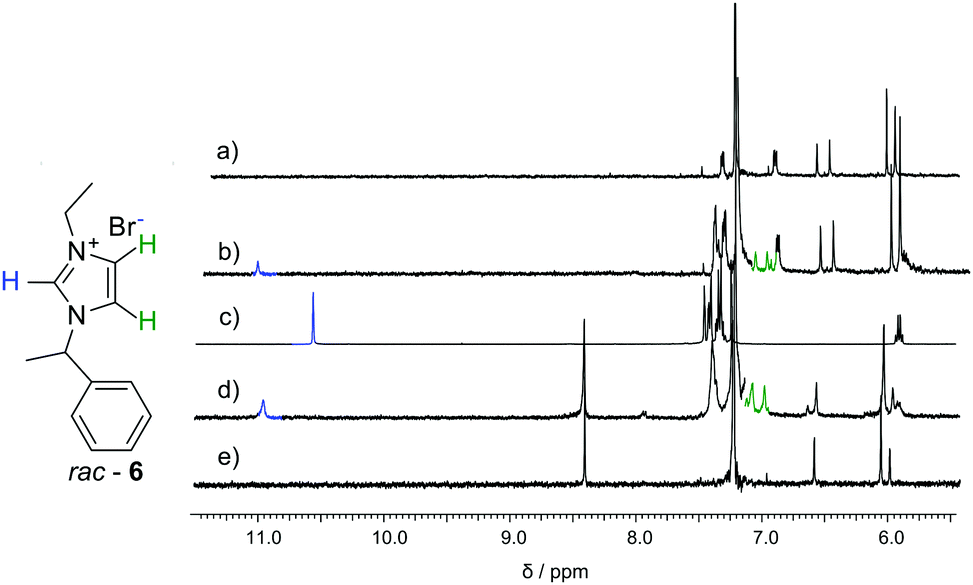 | ||
| Fig. 4 NMR spectra of (a) 3 (0.83 mM); (b) 3 (0.83 mM) + rac-6 (0.95 mM); (c) rac-6 (5.64 mM); (d) 5 (0.59 mM) + rac-6 (0.57 mM); (e) 5 (0.59 mM), all in CDCl3 at 298 K. | ||
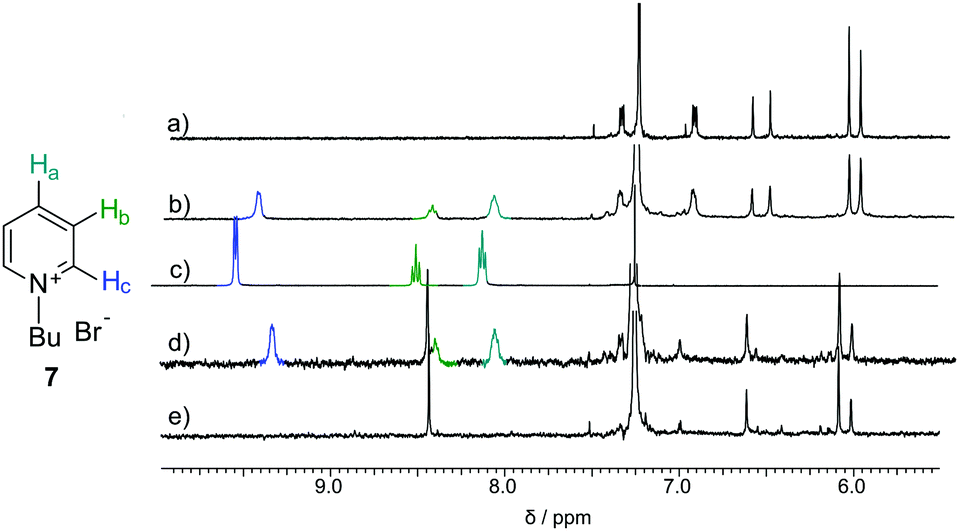 | ||
| Fig. 5 NMR spectra of (a) 3 (0.57 mM); (b) 3 (0.57 mM) + 7 (0.51 mM); (c) 7 (19.57 mM); (d) 5 (0.59 mM) + 7 (0.86 mM); (e) 5 (0.59 mM) all in CDCl3. | ||
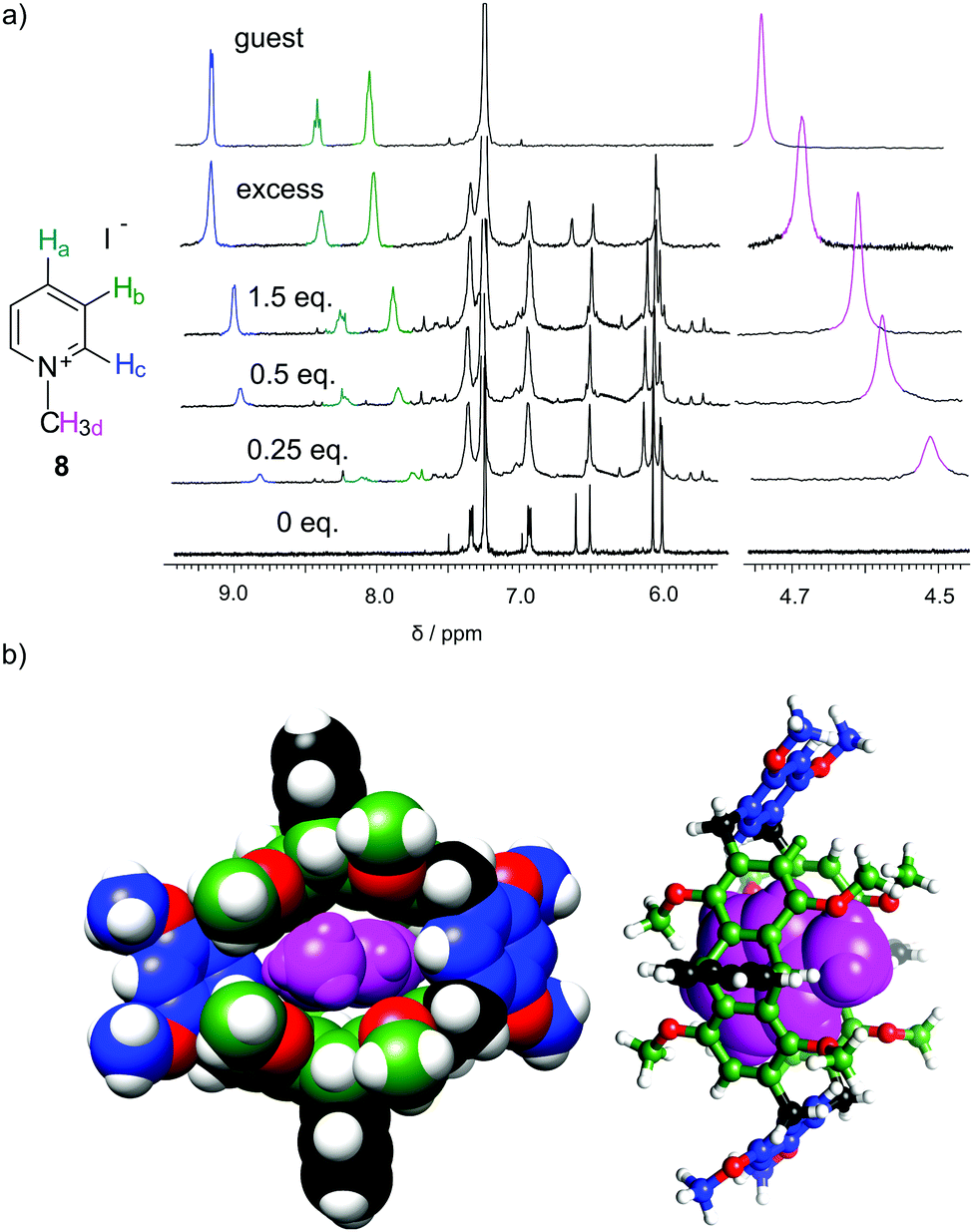 | ||
| Fig. 6 (a) 1H NMR spectra for complexation of methyl pyridinium iodate 8 by macrocyclic host 5 (CDCl3, 298 K); (b) modelled structure of the complex (MM+ force field). | ||
| Host | Guest rac-6 | Guest 7 | ||
|---|---|---|---|---|
| log(β1) | log(β2) | log(β1) | log(β2) | |
| 3 | 2.1 | 6.0 | 4.9 | 8.9 |
| 5 | 2.9 | 6.3 | 3.3 | 8.4 |
In conclusion we have shown that an acid catalyzed condensation between 1,4,5,8-tetramethoxyanthracenes and formaldehyde leads to macrocyclic products only in the presence of additional benzene-based building blocks. This procedure results in the formation of anthracene-containing hybrid[4]arenes with a parallel arrangement of the anthracene units. The shape of the cavity in anthracene-based hybrid[4]arenes is tunable by the Diels–Alder reaction. The macrocycle with an expanded cavity is able to selectively complex pyridinium iodide in chloroform.
Experimental
3: To a solution of 2 (2.5 mmol, 0.745 g) and 1 (2.5 mmol, 0.345 g) in DCE (50 ml) paraformaldehyde (5 mmol, 0.150 g) and TFA (2.5 ml) were added. The reaction was heated at reflux for 4 h under an argon atmosphere. During the reaction the color changed from yellow to dark green. The reaction was cooled to rt and an excess of Na2CO3 was added to neutralize TFA. The mixture was filtered off and evaporated to dryness. The product was isolated by chromatography (silica gel, 30 g, CHCl3/ethyl acetate from 100/0 to 100/7). Yield 10% (115 mg). 1H NMR (CDCl3, 400 MHz, 303 K): 8.44 (2H, s), 6.62 (1H, s), 6.09 (2H, s), 6.02 (2H, s), 4.20 (2H, d, J2 = 16.1 Hz), 4.00 (6H, s), 3.83 (2H, d, J2 = 16.1 Hz), 3.74 (6H, s), 3.54 (6H, s). 13C NMR (CDCl3, 125 MHz, 303 K): 156.3, 151.7, 147.9, 130.2, 125.4, 124.8, 121.1, 114.8, 104.5, 61.9, 55.8, 55.2, 28.3. HR MS (ESI) calcd for C56H56O12Na ([M + Na]+) 943.3669; found: 943.3665.Benzenediazonium-2-carboxylate
Anthranilic acid 9 (1 mmol, 137 mg) and trichloroacetic acid (15 μl) were stirred in dry THF (15 ml) under argon in a 25 ml flask. To this solution iso-amyl nitrite 10 (0.22 ml, 1.7 mmol) was added over a period of 1–2 minutes. A mildly exothermic reaction occurred and the reaction mixture was stirred for a further 1.5 hours maintained at 18–25 °C. A transient orange to brick-red precipitate appeared which was slowly converted to the tan product. Then the mixture was cooled to 10 °C and the product was collected by filtration on a plastic funnel and washed on the funnel with cold dry THF until the washings were colourless (Caution! The filter cake should not be allowed to become dry). Benzene-diazonium-2-carboxylate was then washed with two portions of 1,2-dichloroethane (2.5 ml each) to displace THF and the solvent-wet material was used in the next step.5: A solution of 3 (0.1 mmol, 92 mg) in DCE (25 ml) was prepared in a two necked 50 ml flask equipped with a condenser and a septum under an argon atmosphere. The solution was heated to reflux. The solvent-wet benzenediazonium-2-carboxylate was suspended in DCE (10 ml) and the freshly prepared slurry was slowly added to the reaction mixture over 45 min. The reaction was kept under reflux overnight. Then the solvent was removed by evaporation and the resulting solid was subjected to chromatography (silica gel, 5 g, CHCl3/ethyl acetate from 100/0 to 100/3). Yield 60% (64 mg). 1H NMR (CDCl3, 400 MHz, 303 K): 7.33 (dd, 4H, J3 = 5.4 Hz, J4 = 3.2 Hz), 6.95 (dd, 4 H, J3 = 5.4 Hz, J4 = 3.2 Hz), 6.62 (s, 2 H), 6.52 (s, 2 H), 6.07 (s, 4 H), 6.00 (s, 4 H), 3.93 (d, 4 H, J2 = 16.6 Hz), 3.81 (s, 12 H) 3.66 (d, 4 H, J2 = 16.6 Hz) 3.63 (s, 12 H), 3.55 (s, 12 H). 13C NMR (CDCl3, 100 MHz, 303 K): 156.7, 150.5, 147.5, 146.0, 139.5, 132.5, 131.7, 131.2, 124.9, 123.7, 120.0, 110.0, 95.4, 61.7, 55.8, 55.7, 41.6, 29.3. HR MS (ESI) calcd for C64H64O12Na ([M + Na]+) 1095.4295; found: 1095.4291.
Crystal data for 4
C57H57Cl3O12 (4 × CHCl3), M = 1040.37, space group C2/c, V = 5099.6(10) Å3, Z = 4, Dc = 1.355 g cm−3, F000 = 2184, CuKα radiation, λ = 1.54184 Å, T = 173(2) K, 2θmax = 143.8°, 8910 reflections collected, 4904 unique (Rint = 0.0208). Final GooF = 1.345, R1 = 0.1234, wR2 = 0.3838, R indices based on 2501 reflections with I > 2σ(I) (refinement on F2), 360 parameters, 18 restraints. Lp and absorption corrections applied, μ = 2.159 mm−1. CCDC 1525253 contains the supplementary crystallographic data for this paper.Acknowledgements
This work was supported by the National Science Center (grant 2013/09/N/ST5/00907).Notes and references
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885 RSC.
- B. Slepetz and M. Kertesz, J. Am. Chem. Soc., 2013, 135, 13720 CrossRef CAS PubMed.
- S. R. Jezowski, L. Zhu, Y. Wang, A. P. Rice, G. W. Scott, C. J. Bardeen and E. L. Chronister, J. Am. Chem. Soc., 2012, 134, 7459 CrossRef CAS PubMed.
- R. Kutsumizu, H. Shinmori and T. Takeuchi, Tetrahedron Lett., 2007, 48, 3225 CrossRef CAS.
- Y. Tobe, N. Utsumi, S. Saiki and K. Naemura, J. Org. Chem., 1994, 59, 5516 CrossRef CAS.
- A. Iwama, T. Toyoda, M. Yoshida, T. Otsubo, Y. Sakata and S. Misumi, Bull. Chem. Soc. Jpn., 1978, 51, 2988 CrossRef CAS.
- M. Servalli, N. Trapp, M. Worle and F. G. Klarner, J. Org. Chem., 2016, 81, 2572 CrossRef CAS PubMed.
- N. Ahmed, B. Shirinfar, I. Geronimo and K. S. Kim, Org. Lett., 2011, 13, 5476 CrossRef CAS PubMed.
- N. Ahmed, B. Shirinfar, I. S. Youn, A. Bist, V. Suresh and K. S. Kim, Chem. Commun., 2012, 48, 2662 RSC.
- K. Hagiwara, M. Otsuki, M. Akita and M. Yoshizawa, Chem. Commun., 2015, 51, 10451 RSC.
- K. Hagiwara, M. Akita and M. Yoshizawa, Chem. Sci., 2015, 6, 259 RSC.
- Z. Li, Y. Sei, M. Akita and M. Yoshizawa, Chem. – Asian J., 2014, 9, 1016 CrossRef CAS PubMed.
- K. Yazaki, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 5179 CrossRef CAS PubMed.
- K. Yazaki, N. Kishi, M. Akita and M. Yoshizawa, Chem. Commun., 2013, 49, 1630 RSC.
- K. Hagiwara, Y. Sei, M. Akita and M. Yoshizawa, Chem. Commun., 2012, 48, 7678 RSC.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H. F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976 CrossRef CAS PubMed.
- Y. Okazawa, K. Kondo, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2005, 127, 16404 CrossRef PubMed.
- T. Ide, D. Takeuchi, K. Osakada, T. Sato and M. Higuchi, J. Org. Chem., 2011, 76, 9504 CrossRef CAS PubMed.
- B. Gao, L. L. Tan, N. Song, K. Li and Y. W. Yang, Chem. Commun., 2016, 52, 5804 RSC.
- B. Hua, L. Shao, J. Zhou and G. Yu, New J. Chem., 2016, 40, 4756 RSC.
- J. Zhou, J. Yang, B. Hua, L. Shao, Z. Zhang and G. Yu, Chem. Commun., 2016, 52, 1622 RSC.
- M. Holler, N. Allenbach, J. Sonet and J. F. Nierengarten, Chem. Commun., 2012, 48, 2576 RSC.
- T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419 CrossRef CAS.
- T. Boinski, A. Cieszkowski, B. Rosa and A. Szumna, J. Org. Chem., 2015, 80, 3488 CrossRef CAS PubMed.
- T. Boinski, A. Cieszkowski, B. Rosa, B. Leśniewska and A. Szumna, New J. Chem., 2016, 40, 8892 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and ESI MS spectra and titration experiments. CCDC 1525253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj00039a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |