
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetraalkynylstannanes in the Stille cross coupling reaction: a new effective approach to arylalkynes†
Andrey S.
Levashov
 *a,
Dmitrii S.
Buryi
a,
Olga V.
Goncharova
a,
Valeriy V.
Konshin
a,
Victor V.
Dotsenko
ab and
Alexey A.
Andreev‡
a
*a,
Dmitrii S.
Buryi
a,
Olga V.
Goncharova
a,
Valeriy V.
Konshin
a,
Victor V.
Dotsenko
ab and
Alexey A.
Andreev‡
a
aDepartment of Organic Chemistry & Technologies, Kuban State University, 149 Stavropolskaya str., 350040 Krasnodar, Russian Federation. E-mail: aslevashov@mail.ru
bDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin str., 355009 Stavropol, Russian Federation
First published on 21st February 2017
Abstract
The Stille-type cross coupling reaction with tetraalkynylstannanes was studied in detail for the first time. The reaction provides a simple and effective route towards a variety of arylalkynes. The advantages and limitations of the proposed procedure are discussed.
Introduction
For almost forty years after the pioneering research of John K. Stille, the cross coupling reaction named after him of organic electrophiles with organostannanes has been recognized as a powerful tool for the formation of carbon–carbon bonds (see reviews1–5). The organotin compounds useful in the Stille reaction are mild reagents that tolerate a variety of functional groups and are the reagents of choice for delicate cross coupling syntheses of complex functionalized molecules.5 The Stille reaction has been thoroughly investigated and many advances have been made to expand both the scope and utility of this process.5,6 In contrast to other cross coupling reactions, the Stille reaction has often been found to be effective and relatively undemanding, allowing for harsher conditions, as organostannanes are relatively insensitive to moisture and oxygen.5 On the other hand, the use of organostannanes such as Bu3SnR raises problems with organotin contamination and waste. Both acute and long-term toxicities have been reported for many organotin reagents,7,8 and methods designed to limit or avoid the presence of organotin by-products in reaction products have been developed.9 In general, the toxicity of alkylstannanes decreases as the size of the alkyl groups increases (Me3SnX ∼ Et3SnX ≫ Bu3SnX ≫ Octyl3SnX) and the number of alkyl groups decreases (R3SnX > R2SnX2 > RSnX3);9 R4Sn may reveal enhanced delayed toxicity due to in vivo transformation into R3SnX.10 However, the toxicity strongly depends on the nature of the organic group R.10 Due to the easy hydrolysis of the C(sp)–Sn bond, it is generally accepted that tetraalkynyltin compounds (RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)4Sn are far less toxic than other organotin species having C(sp2)–Sn or C(sp3)–Sn bonds.
C)4Sn are far less toxic than other organotin species having C(sp2)–Sn or C(sp3)–Sn bonds.
Another feature of tetraalkynylstannanes is their high atom economy. A practical disadvantage of the use of tin mono-functional reagents such as R–Sn(alkyl)3 (R is aryl, vinyl or alkynyl) is that a reactant of a high molecular weight is used to introduce a hydrocarbon group of a (relatively) low molecular weight, at the same time producing bulky and highly toxic triorganotin waste. Since each of the four alkynyl fragments in (RCC)4Sn is reactive, tetraalkynyltin compounds may be compared with sodium acetylides with respect to low molecular weight and producing only inorganic Sn(IV) waste of low toxicity. It is noteworthy that, generally, the reactions involving organostannanes (e.g., classical Stille coupling or any other organotin-mediated process) are considered to be of a low atom economy, due to the loss of heavy and toxic tin-containing moieties.11 In other words, the E-factor12 (which is defined as the mass ratio of waste to desired product) of the Stille reaction with R–Sn(alkyl)3 agents is much higher than that expected for coupling reactions with tetraalkynylstannanes, and the latter reactions could be considered as more environmentally benign. The advantages of the use of organotin compounds capable of transferring more than one organyl group are illustrated by the reactions of tetraallylstannane with electrophilic substrates.13 Tetraallylstannane is a gentle nucleophile for allylation reactions and easily reacts with imines,14 aldehydes,15,16 phenacyl bromide,17 other ketones18 or carbon dioxide19 (Scheme 1). In contrast to allyltrialkylstannyl reagents which transfer only one organyl moiety out of four groups on the tin atom, from two to four allyl residues can be utilized in the case of Sn(CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)4.
CH2)4.
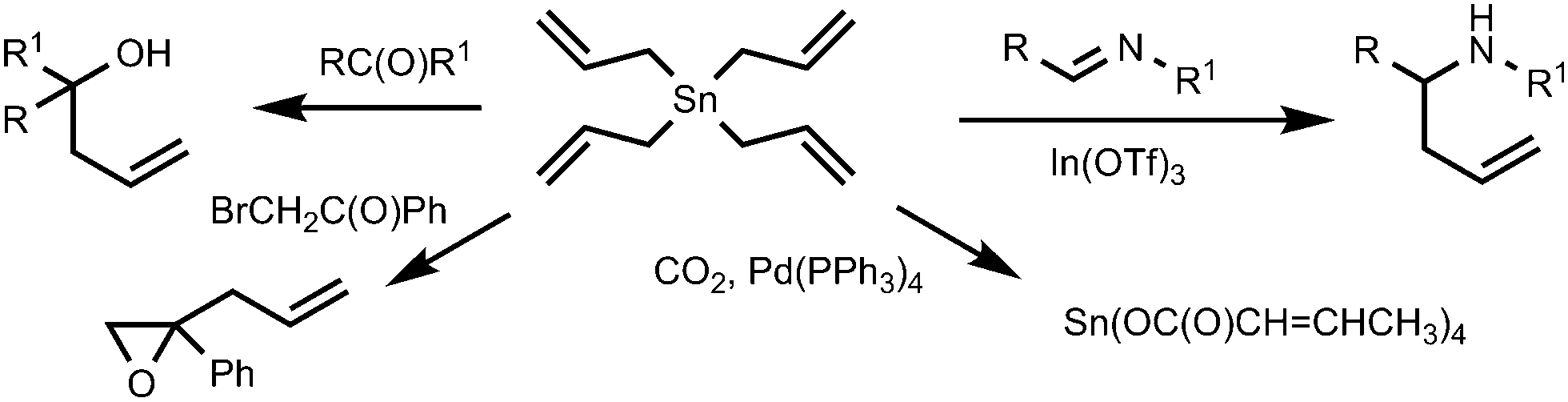 | ||
| Scheme 1 Typical reactions of tetraallylstannane. | ||
The alkynyl fragment can be easily introduced onto a tin atom in different ways. Recently we have developed two convenient and effective methods of synthesis of tetraalkynylstannanes 1 (Scheme 2); the first is based on the direct reaction of terminal alkynes with SnCl4 in the presence of anhydrous ZnCl2 and diethylamine20,21 and the next is based on the reaction of tin tetra(N,N-diethylcarbamate) with phenylacetylene.22 Tetraalkynylstannanes 1 are oily or solid compounds that can be easily purified and isolated in good yields after column chromatography on a silanized silica. Also they are stable enough to be stored in a freezer for months.
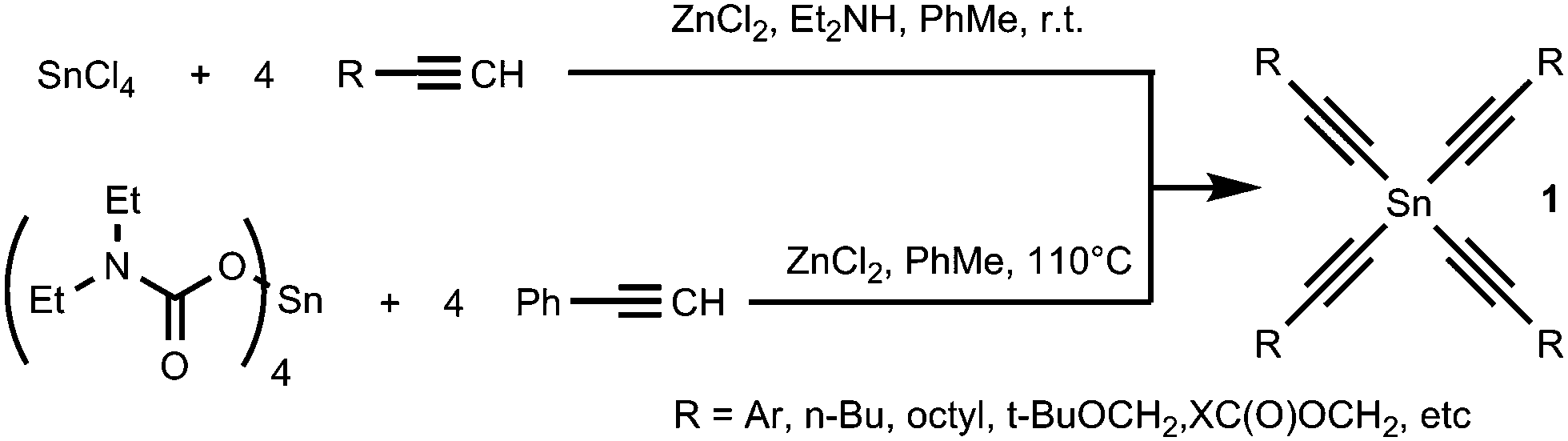 | ||
| Scheme 2 Synthesis of tetraalkynylstannanes 1. | ||
While much is known about the mono-, di- and trialkynyl tin compounds, less is known about the chemistry of tetraalkynyltin compounds. Only a limited number of reactions are reported encompassing (RCC)4Sn as reagents (Scheme 3). Thus, the reactions with Grignard reagents were recognized as a convenient method of smooth transmetallation of tetraalkynylstannanes for the preparation of tri- and dialkynyltins.23,24 The organoboration of (RCC)4Sn with trialkylboranes led to the formation of 1,1′-spirobistannoles 2.25–28 Tetra(phenylethynyl)-tin was reported to be an efficient catalyst of ring-opening polymerization of L-lactide to poly(L-lactide).29 As expected for tetraalkynylstannanes 1, they may also react with acyl chlorides (4 eq.) to afford alkynyl ketones.30
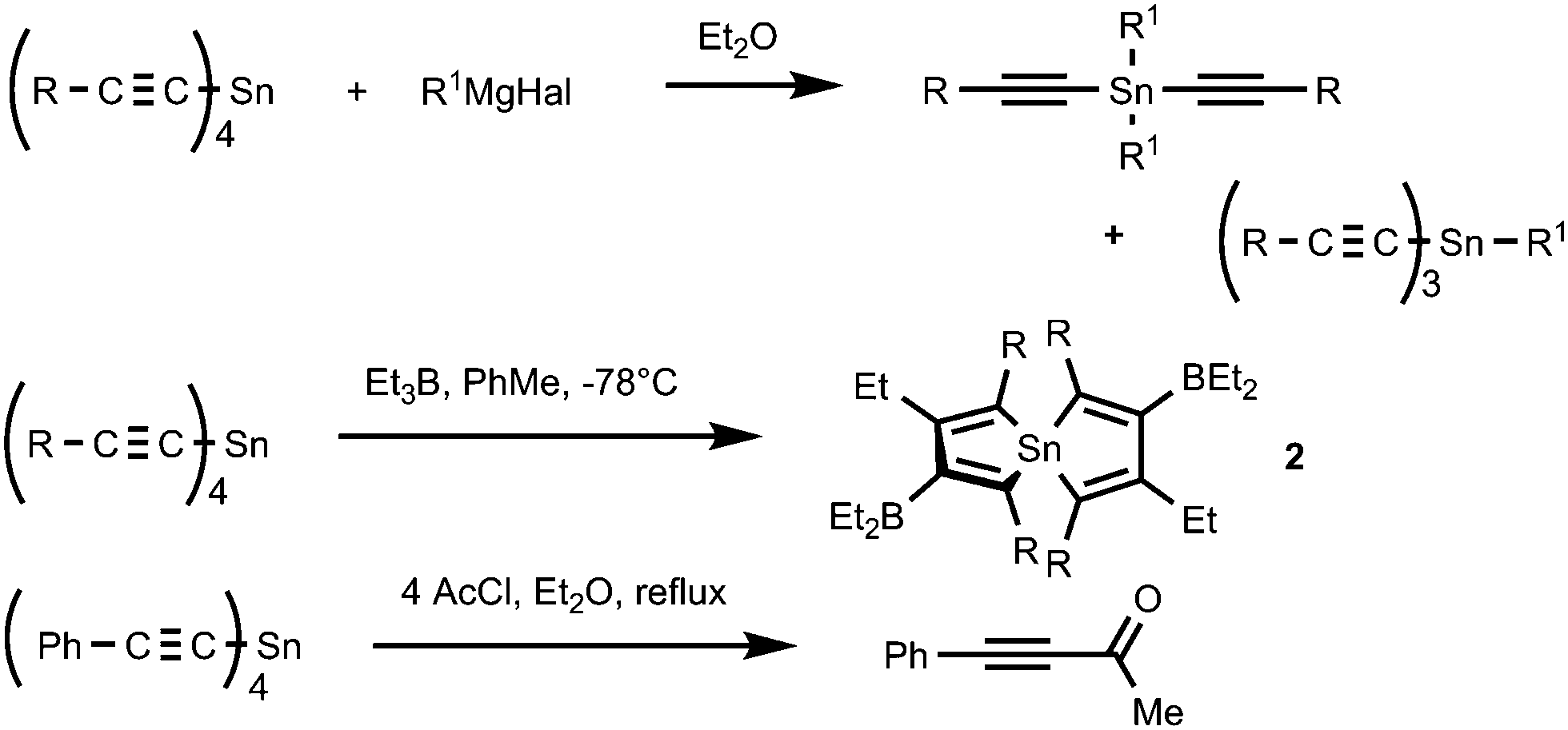 | ||
| Scheme 3 The known reactions of tetraalkynylstannanes 1. | ||
To the best of our knowledge, Stille-type cross coupling reactions with tetraalkynylstannanes have not been described in the literature prior to the present work. Recently, French researchers reported31 the Stille cross coupling reaction of di- or trialkynylstannanes with iodovinylic acids/esters, first introducing a half or a third equivalent of di- or tri-functional organotin compounds. This is the only previous report on Stille cross coupling with multi-functional C(sp)–Sn organotin compounds. In this paper, we wish to report the first example of a Stille-type cross coupling reaction of aryl halides 3 with tetraalkynyltin compounds 1.
Results and discussion
We found that tetraalkynylstannanes 1 easily react with a variety of aryl iodides and bromides under Stille conditions according to the following scheme (Scheme 4):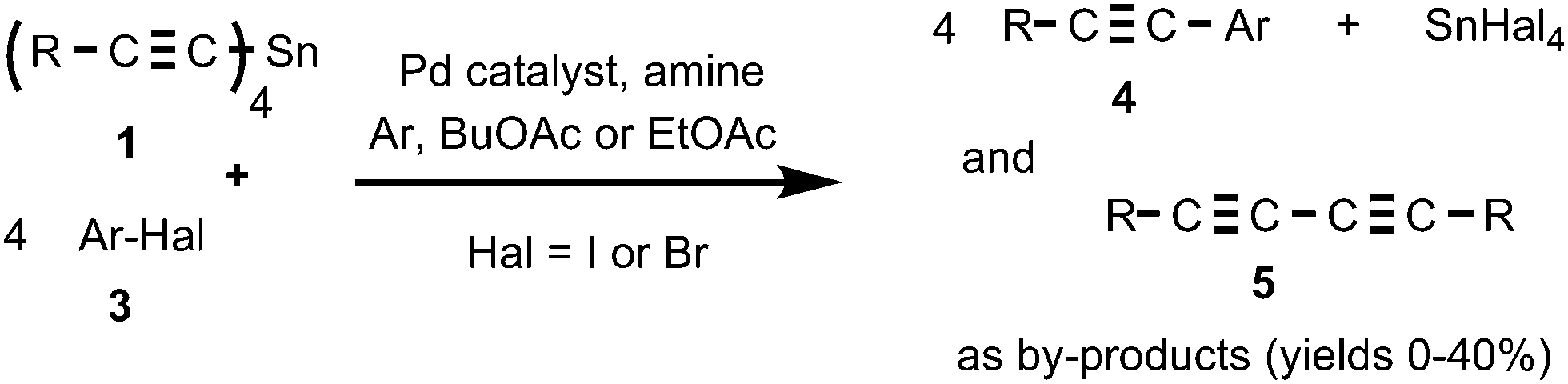 | ||
| Scheme 4 The reaction of tetraalkynylstannanes 1 with aryl halides 3. | ||
The tetraorganylstannanes 1 and aryl halides 3 used in the reaction are shown in Fig. 1. To prevent side reactions such as hydrolysis of stannanes 1 or oxidative couplings, the reactions should be conducted in an inert atmosphere (argon) wherein water and oxygen are excluded. Diaryl diacetylenes 5 are by-products probably derived from the Pd-mediated Glaser-type coupling reaction occurring in the presence of trace oxygen.
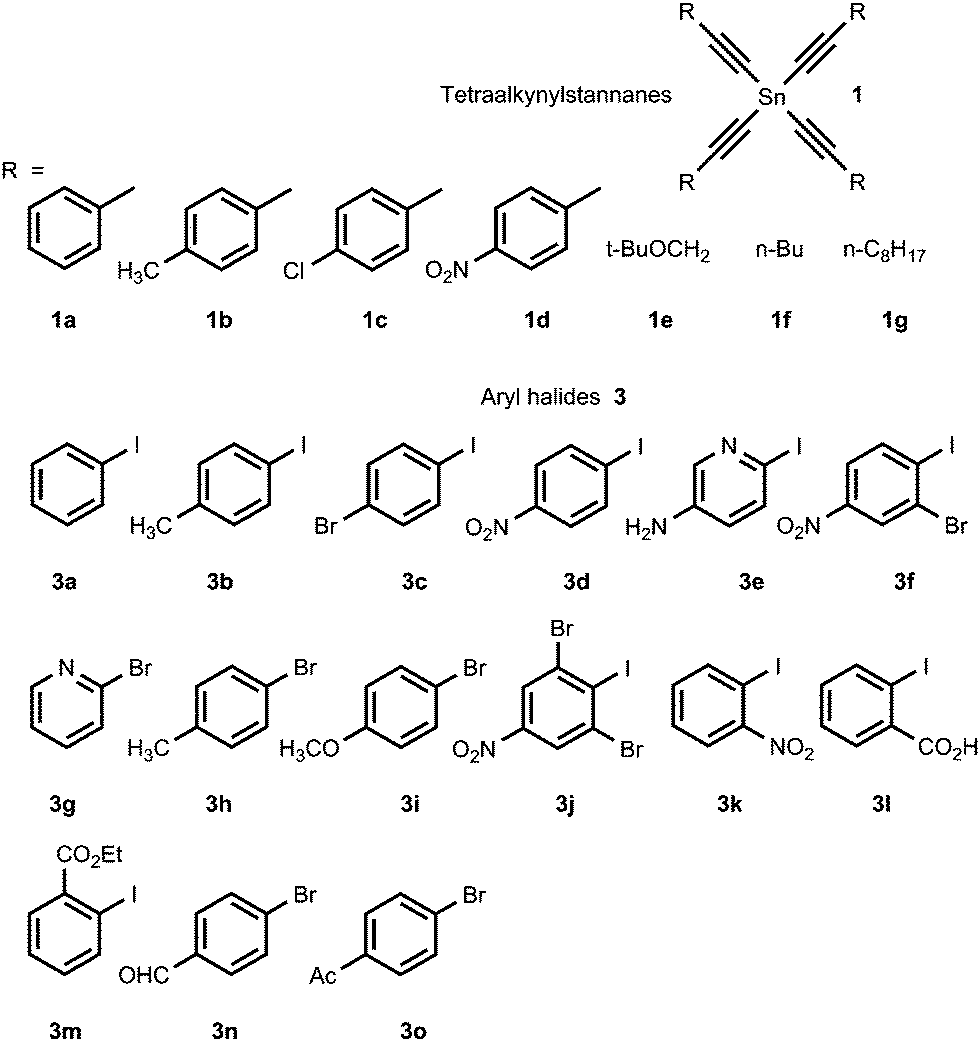 | ||
| Fig. 1 The scope of stannanes 1 and aryl halides 3 used. | ||
A number of attempts have been made to optimize the coupling reaction conditions. We found that a variety of factors may affect the reaction outcome, such as the nature and quantity of amine additive used, the nature of the Pd catalysts and solvents, the temperature and the reaction time. As a model reaction, we examined the coupling of tetra(phenylethynyl)tin 1a with p-nitroiodobenzene 3d under different conditions (Scheme 5). First, we examined the effect of different solvents and amine additives on the yields of the target aryl acetylene 4ad, using Pd(PPh3)2Cl2 as a catalyst. The selected results are summarized in Table 1; the complete set of data is given in ESI,† Table S1.
 | ||
| Scheme 5 The model reaction of tetra(phenylethynyl)tin 1a with p-nitroiodobenzene 3d. | ||
| Solvent | T (°C) | Et2NH | Et3N | DABCO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Yield of 4adb (%) | Yield of 5ab (%) | Timec (h) | Yield of 4adb (%) | Yield of 5ab (%) | Timec (h) | Yield of 4adb (%) | Yield of 5ab (%) | Timec (h) | ||
|
a The reaction conditions were as follows: (PhCC)4Sn 1a (20 mg, 0.038 mmol), 4-NO2C6H4I 3d (0.153 mmol), Pd(PPh3)2Cl2 (5.4 mg, 0.0077 mmol; 5 mol% vs.3d), amine (0.15 mmol), and solvent (2 mL).
b Yields were determined by GC-MS.
c Reaction time when the highest yield was achieved.
|
||||||||||
| Et2O | 35 | 0 | 0 | 5 | 0 | 3 | 5 | 71 | 6 | 5 |
| THF | 80 | — | — | — | 20 | 22 | 5 | 50 | 6 | 1 |
| PhMe | 100 | 55 | 10 | 7 | 27 | 14 | 5 | 87 | 8 | 0.5 |
| MeCN | 85 | 89 | 6 | 5 | 68 | 13 | 5 | 63 | 6.5 | 5 |
| Dioxane | 100 | 84 | 2 | 3 | 44 | 6 | 2 | 78 | 8 | 5 |
| AcOEt | 80 | 91 | 1 | 9 | 87 | 4 | 2 | 87 | 4 | 0.5 |
| AcOBu | 125 | 98 | 2 | 1 | 98 | 2 | 2 | 87 | 5 | 2 |
| AcOBu | 100 | 98 | 2 | 2 | 85 | 2 | 5 | 86 | 5 | 2 |
| Et3N | 80 | — | — | — | 96 | 4 | 2.5 | — | — | — |
| DMF | 100 | — | — | — | 89 | 9 | 2.5 | — | — | — |
As can be seen, the best results were obtained with BuOAc, EtOAc, DMF and pure Et3N as solvents in the temperature range of 80–125 °C, while the use of less polar (dioxane, PhMe) and low-boiling (Et2O) solvents resulted in lower yields of the aryl acetylene 4ad with increased amounts of the diacetylene by-product 5a. Though only a few examples32–34 have been reported for the amine-promoted Stille reaction, we found that the presence of an amine additive is strongly required for the reaction of tetraalkynylstannanes 1 with aryl halides 3. In the absence of an amine no reaction occurs, while even trace amounts give coupling products, albeit in low yields. The nature of the amine additive as well as its amount has a dramatic effect on the reaction course, as shown in Table 2. The best results were obtained with the strong bases such as Et3N, Bu3N, DABCO, and especially with Et2NH and Pr2NH. The application of benzylic amines, piperidine, morpholine and N-methylmorpholine lowered significantly the yield of 4ad, whereas pyridine and ethylene diamine were found to be completely inactive. The full set of data on the amines used is given in ESI,† Table S1. The amine concentration was also important and strongly influenced the reaction rate. Thus, the reaction proceeded more slowly in the presence of 1 eq. Et2NH (the yield of 4ad reached only 66% after 5 h in EtOAc at 80 °C), and quite rapidly when the amount of Et2NH was increased. The use of a large excess of Et2NH under the same conditions produced tolane 4ad in almost quantitative yields after 2 h (Fig. 2).
| Amine | The yield of aryl acetylene 4ada,b (%) | |||||
|---|---|---|---|---|---|---|
| AcOBu | Timec (h) | PhMe | Timec (h) | Dioxane | Timec (h) | |
| a Yields were determined by GC-MS. b The reaction conditions were as follows: 1a (0.038 mmol), 3d (0.153 mmol), Pd(PPh3)2Cl2 (5 mol% vs.3d), amine (0.153 mmol) and solvent (2 mL) at 100 °C. c Reaction time when the highest yield was achieved. | ||||||
| Et2NH | 98 | 2 | 55 | 7 | 84 | 3 |
| Et3N | 85 | 5 | 27 | 5 | 44 | 2 |
| Bu3N | 97 | 3 | 31 | 5 | 87 | 3 |
| DABCO | 68 | 2 | 87 | 0.5 | 78 | 0.5 |
| Isophorone diamine | — | — | 54 | 7 | 86 | 9 |
| Morpholine | — | — | — | — | 54 | 1 |
| N-Methyl-morpholine | 12.5 | 1 | — | — | 73 | 2 |
| Pyridine | — | — | — | — | 0 | 3 |
| Piperidine | 62 | 2 | 49 | 5 | 55 | 2 |
| (CH2)2(NH2)2 | 0 | 3 | — | — | — | — |
| PhCH2NH2 | 41 | 5 | 43 | 5 | 56 | 5 |
| (PhCH2)2NH | — | — | 11 | 3 | 26 | 5 |
| Pr2NH | 99.5 | 1 | — | — | — | — |
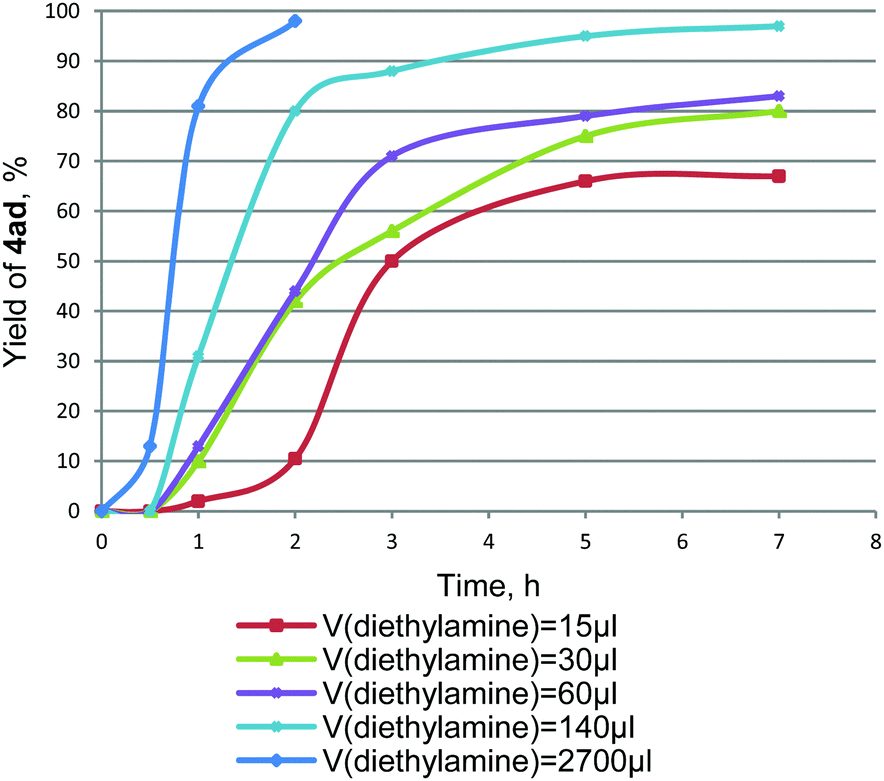 | ||
| Fig. 2 The kinetics of the reaction 1a + 3d → 4ad with different amounts of Et2NH. The reaction conditions were as follows: (PhCC)4Sn 1a (20 mg, 0.038 mmol), 4-NO2C6H4I 3d (0.153 mmol), Pd(PPh3)2Cl2 as the catalyst (5 mol% with respect to 3d), AcOEt (2.0 mL), 80 °C. | ||
To optimize the conditions, we studied the effect of excessive amounts of different amines on the kinetics of the reaction. It was found that other amines were also as effective as Et2NH when they were used in high excess. The results are summarized in Table 3.
| Amine | Volume (μL) | Aryl iodide 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine ratio :amine ratio |
The yieldsb of | Timed (h) | |
|---|---|---|---|---|---|
| 4ad (%) | 5a (%) | ||||
| a Unless otherwise stated, the conditions were as follows: 1a (0.038 mmol), 3d (0.153 mmol), Pd(PPh3)2Cl2 (5 mol% with respect to 3d), BuOAc, 100 °C, total volume solvent + amine = 1000 μL. b Yields were determined by GC-MS. c TMEDA = Me2NCH2CH2NMe2. d Reaction time when the maximum yield was achieved. | |||||
| Et2NH | 900 | 1:57 |
96.4 | 1.5 | 6 |
| Pr2NH | 21 | 1:1 |
99.5 | 0.5 | 1 |
| Pr2NH | 105 | 1:5 |
99.2 | 0.8 | 1 |
| Pr2NH | 210 | 1:10 |
98.8 | 1.2 | 1 |
| Pr2NH | 840 | 1:40 |
98.9 | 1.1 | 1 |
| Pr2NH | 900 | 1:43 |
99.0 | 1.0 | 1 |
| Pr2NH | 1000 | 1:47 |
98.3 | 1.7 | 1 |
| Bu3N | 36 | 1:1 |
75.7 | 2.0 | 4 |
| Bu3N | 182 | 1:5 |
99.5 | 0.5 | 1 |
| Bu3N | 640 | 1:10 |
99.5 | 0.5 | 1 |
| Bu3N | 900 | 1:25 |
83.7 | 9.2 | 4 |
| TMEDAc | 23 | 1:1 |
95.0 | 1.5 | 4 |
| TMEDA | 115 | 1:5 |
97.7 | 2.3 | 1 |
| TMEDA | 229 | 1:10 |
97.2 | 2.8 | 1 |
| TMEDA | 920 | 1:40 |
97.3 | 2.7 | 1 |
| TMEDA | 1000 | 1:43 |
94.0 | 6.0 | 1 |
We have to admit that the mechanistic picture of the Stille reaction is rather complex35 and details cannot be specified with confidence, so the role of an amine and its amount still remains unclear and requires further investigation. We suggest that the reaction proceeds by way of formation of alkynyl-palladium complexes 6 and 7 according to Scheme 6.
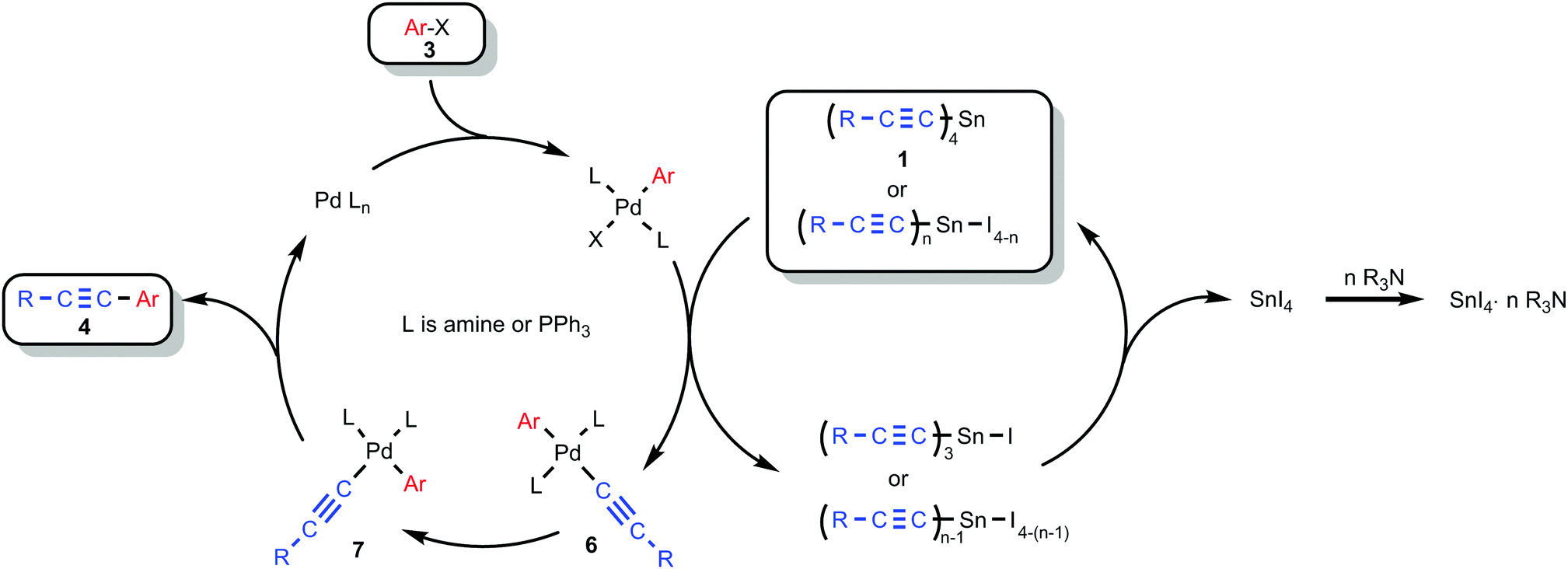 | ||
| Scheme 6 A possible mechanism for the reaction of tetraalkynylstannanes 1 with aryl halides 3. | ||
The effect of different catalysts was studied to determine the best catalytic system. No reaction occurs without a catalyst: thus, when (PhCC)4Sn 1a was added to 4-NO2C6H4I 3d in pure Et3N (80 °C, 2 h), no conversion was observed.
However, when a catalytic amount of CuI was added under the same conditions, a trace amount of the coupling product was detected by GC-MS. It is noteworthy that when (PhCC)4Sn 1a was treated with a 4-fold excess of CuBr2 (THF, 0.5 h, 25 °C), the oxidative Glaser-type coupling product (Ph–CC–)25a was formed in a good yield. The first success came with the use of Pd catalysts, especially Pd(PPh3)2Cl2. To our surprise, the reaction was completely suppressed by the addition of an excess of phosphine ligand. Thus, no reaction between stannane 1a and 1-iodo-4-nitrobenzene 3d occurred in the presence of Pd(PPh3)2Cl2 and PPh3 (5 mol% and 20 mol% with respect to 3d, respectively), while Pd(PPh3)2Cl2 with no PPh3 additive gave the best yields. The results using different Pd catalysts are summarized in Table 4.
| Amine | The yield of 4-NO2C6H4CCPh 4adb (%) |
|||||
|---|---|---|---|---|---|---|
| PdCl2 | Timec (h) | Pd(PhCN)2Cl2 | Timec (h) | Pd(PPh3)2Cl2 | Timec (h) | |
| a The reaction conditions were as follows: 1a (0.038 mmol), 3d (0.153 mmol), Pd catalyst (5 mol% with respect to 3d), amine (0.153 mmol), BuOAc, 100 °C. b Yields were determined by GC-MS. c Reaction time when the maximum yield was achieved. | ||||||
| Et2NH | 13 | 7 | 7 | 5 | 98 | 2 |
| Et3N | 72 | 3 | 71 | 2 | 89 | 7 |
| Bu3N | 60 | 7 | 50 | 5 | 97 | 3 |
| DABCO | 57 | 2 | 56 | 1 | 68 | 2 |
Next, using an optimized protocol (BuOAc or DMF, 100 °C, 5 mol% Pd(PPh3)2Cl2, and an excess (4-fold or more with respect to stannane 1) of an amine additive – Et2NH, Pr2NH, DABCO or Bu3N), we studied the reactivity of different tetraalkynylstannanes 1 and aryl halides 3. As expected, aryl halides bearing electron-withdrawing groups showed the best reactivity and gave the highest yields of acetylenes 4. Selected results are summarized in Table 5; the complete set of data is given in ESI,† Table S1.
C)4Sn 1a under the optimized conditionsa
| Aryl Halides 3 | Solvent | Timeb (h) | Amine | Yield of 4 |
|---|---|---|---|---|
| a Yields were determined by GC-MS. Unless otherwise stated, the reaction conditions were as follows: 1a (0.038 mmol), 3d (0.153 mmol), Pd(PPh3)2Cl2 (5 mol% with respect to 3d), amine (0.153 mmol) and solvent (DMF or BuOAc, 2 mL) at 100 °C. b Reaction time when the maximum yield was achieved. | ||||
| Ph–I 3a | DMF | 2.5 | Et3N | 87 |
| 4-MeC6H4I 3b | DMF | 5 | Et3N | 95 |
| 4-MeC6H4I 3b | BuOAc | 9 | Et2NH | 54 |
| 4-NO2C6H4I 3d | DMF | 2.5 | Et3N | 89 |
| 4-NO2C6H4I 3d | BuOAc | 6 | Et2NH | 98 |
| 4-NO2C6H4I 3d | BuOAc | 1 | Pr2NH | 99.5 |
| 4-NO2C6H4I 3d | BuOAc | 3 | Bu3N | 97 |
| 4-MeC6H4Br 3h | BuOAc | 9 | Et2NH | 11 |
| 4-MeOC6H4Br 3i | BuOAc | 5 | Et2NH | 7 |
| 4-MeOC6H4Br 3i | BuOAc | 3 | DABCO | 15 |
| 2-NO2C6H4I 3k | BuOAc | 10 | Et2NH | 83 |
| 2-IC6H4CO2H 3l | BuOAc | 5 | Et2NH | 0 |
| 2-IC6H4CO2H 3l | Et3N | 4 | Et3N | 63 |
However, when the reaction was carried out using DMF as a solvent instead of BuOAc, the coupling products with aryl halides 3 bearing electron-donating substituents were obtained in good yields. The analogs of (PhCC)4Sn 1a, i.e., tetraalkynylstannanes 1b–g, reacted with 4-NO2C6H4I 3d to form the desired acetylenes 4bd–gd, but the yields were generally lower than those obtained with 1a. The selected results are presented in Table 6. Finally, the Stille coupling products 4 were obtained in 40–93% yields in preparative-scale experiments using conditions similar to those of the kinetic runs and the optimization protocols. The results are given in Table 7.
| Tetraalkynylstannanes 1 | Timeb (h) | Amine | Yields of 4 (%) |
|---|---|---|---|
| a Yields were determined by GC-MS. The reaction conditions were as follows: 1 (0.038 mmol), 3d (0.153 mmol), Pd(PPh3)2Cl2 (5 mol% with respect to 3d), amine (0.153 mmol), BuOAc (2 mL) at 100 °C. b Reaction time when the maximum yield was achieved. | |||
| (PhCC)4Sn 1a |
2 | Et2NH | 98 |
| (t-BuOCH2CC)4Sn 1e |
1 | DABCO | 74 |
| (t-BuOCH2CC)4Sn 1e |
2 | Et2NH | 33 |
| (n-C8H18CH2CC)4Sn 1g |
7 | Et2NH | 50 |
| (n-C8H18CH2CC)4Sn 1g |
5 | DABCO | 52 |
|
|
|||
|---|---|---|---|
| Tetraalkynyl stannane 1 | Aryl halide 3 | Product | Yielda (%) |
|
a Isolated yields are given.
b When 2-Br-4-NO2C6H3I 3f and (PhCC)4Sn 1a were added in a 2:1 ratio, acetylene 4af was obtained in 81% yield.
|
|||
| (PhCC)4Sn 1a |
Ph–I 3a | PhCCPh 4aa |
78 |
| (PhCC)4Sn 1a |
4-MeC6H4I 3b | 4-MeC6H4CCPh 4ab |
81 |
| (PhCC)4Sn 1a |
4-BrC6H4I 3c | 4-BrC6H4CCPh 4ac |
80 |
| (PhCC)4Sn 1a |
4-NO2C6H4I 3d | 4-NO2C6H4CCPh 4ad |
93 |
| (PhCC)4Sn 1a |
2-Br-4-NO2C6H3I 3f | 2-Br-4-NO2C6H3CCPh 4af |
40b |
| (PhCC)4Sn 1a |
2-NO2C6H4I 3k | 2-NO2C6H4CCPh 4ak |
88 |
| (PhCC)4Sn 1a |
2-EtO2CC6H4I 3m | 2-EtO2CC6H4CCPh 4am |
81 |
| (PhCC)4Sn 1a |
4-BrC6H4CHO 3n | PhCCC6H4CHO 4an |
82 |
| (PhCC)4Sn 1a |
4-BrC6H4C(O)Me 3o | PhCCC6H4C(O)Me 4ao |
65 |
| (4-MeC6H4CC)4Sn 1b |
4-NO2C6H4I 3d | 4-MeC6H4CCC6H4NO24bd |
91 |
| (4-ClC6H4CC)4Sn 1c |
4-NO2C6H4I 3d | 4-ClC6H4CCC6H4NO24cd |
82 |
| (t-BuOCH2CC)4Sn 1e |
4-NO2C6H4I 3d |
t-BuOCH2CCC6H4NO24ed |
49 |
| (n-BuCC)4Sn 1f |
4-NO2C6H4I 3d |
n-BuCCC6H4NO24fd |
47 |
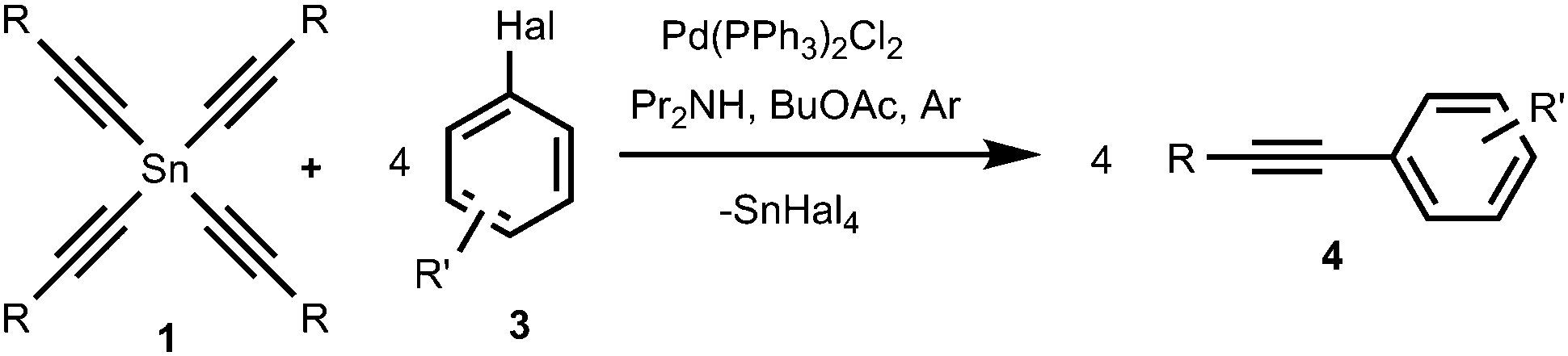
Conclusions
In conclusion, we have developed an effective synthetic protocol based on the Stille cross coupling reaction of easily available tetraalkynylstannanes with aryl halides. The reported method provides atom-economical access to aryl acetylenes and diaryl acetylenes (tolanes) which are valuable reagents for further transformations. The scope and limitations of the reaction were studied and the conditions were optimized.Experimental
Materials and methods
Solvents and starting reagents were thoroughly dried and purified according to common procedures.36 All reactions were carried out and the target compounds were isolated in an argon (99.993%) atmosphere. 1H, 13C, and 119Sn NMR spectra were recorded on a JEOL ECA 400 instrument at operating frequencies of 399.78, 100.52 and 149.08 MHz respectively, in CDCl3 (Aldrich) with reference to TMS or to the residual signals of a solvent (with SnMe4 as a standard for 119Sn NMR). Chemical shifts are given in ppm; coupling constants are given in Hz. IR spectra were recorded on an InfraLUM FT-02 instrument in the range of 400–4200 cm−1 (KBr or HCCl3 solution) and on a Bruker Vertex 70 instrument in ATR (attenuated total reflection) mode. Mass spectra (EI, 70 eV) were obtained on a Shimadzu GCMS-QP 2010 spectrometer. The purity of the compounds was checked by TLC (Sorbfil A plates) with Et2O:hexane (10:1), MeOH:HCCl3 (1:10) or HCCl3:Me2CO (10:1) mixtures as eluents. The spots were visualized with iodine vapors, KMnO4–H2SO4 solution or UV-light. The starting tetraalkynylstannanes 1a–g were obtained according to the reported methods;20,21 the detailed procedures are given in the ESI.†
General procedure for the synthesis of 4-nitrotolane (1-nitro-4-(phenylethynyl)benzene) (4ad) (the model reaction, Scheme 5, Tables 1, 2 and 4)
A 5 mL sealable Wheaton vial was charged with 0.00765 mmol of Pd catalyst (PdCl2, Pd(PPh3)2Cl2, or Pd(PhCN)2Cl2) and 0.153 mmol of the amine additive (Et2NH, Et3N, Bu3N, DABCO, morpholine, etc.). Then the vial was flushed with a stream of dry argon, and subsequently a solution of 0.0382 mmol of (PhCC)4Sn 1a in a dry solvent (1 mL) and a solution of 0.153 mmol of 4-NO2C6H4I in a dry solvent (1 mL) were added through a syringe. The mixture was stirred for the indicated time, and the yield was determined by GC-MS.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C)4Sn (1a).
A dry 25 mL, two-necked, round-bottomed flask equipped with an argon gas inlet tube and a magnetic stirrer was flushed with argon and charged with iodobenzene 3a (212.7 mg, 1.043 mmol), Pd(PPh3)2Cl2 (36.6 mg, 0.052 mmol; 5 mol% vs.3a) and (PhCC)4Sn 1a (150 mg, 0.287 mmol). Then Pr2NH (1.43 mL, 10.43 mmol) and dry BuOAc (6 mL) were added, the solution was degassed by freezing in liquid nitrogen and pumping under vacuum several times, and then it was flushed with argon. The reaction mixture was stirred at 100 °C for 5 h, then allowed to cool and quenched with EtOH (10 mL). The mixture was treated with 0.5 g of silica modified with 3-aminopropyltriethoxysilane, the solvent was removed on a rotary evaporator and the traces of BuOAc were removed in vacuo. The resulting mixture was purified by column chromatography over silica gel (2 g) with pure dry PhMe (50 mL). The eluent was evaporated, the residue was dissolved in n-hexane and purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups; the use of the modified silica gel allowed us to remove easily the by-product SnI4, which appears to be hard to separate when a non-modified silica gel is used as sorbent). Hexane was used as the eluent. Column fractions were analyzed by GC-MS. The eluent was evaporated to give 152.3 mg of tolane 4aa as a colorless crystalline solid (78%, purity by GCMS – 95.3%).
C)4Sn (1a).
A dry 25 mL, two-necked, round-bottomed flask equipped with an argon gas inlet tube and a magnetic stirrer was flushed with argon and charged with iodobenzene 3a (212.7 mg, 1.043 mmol), Pd(PPh3)2Cl2 (36.6 mg, 0.052 mmol; 5 mol% vs.3a) and (PhCC)4Sn 1a (150 mg, 0.287 mmol). Then Pr2NH (1.43 mL, 10.43 mmol) and dry BuOAc (6 mL) were added, the solution was degassed by freezing in liquid nitrogen and pumping under vacuum several times, and then it was flushed with argon. The reaction mixture was stirred at 100 °C for 5 h, then allowed to cool and quenched with EtOH (10 mL). The mixture was treated with 0.5 g of silica modified with 3-aminopropyltriethoxysilane, the solvent was removed on a rotary evaporator and the traces of BuOAc were removed in vacuo. The resulting mixture was purified by column chromatography over silica gel (2 g) with pure dry PhMe (50 mL). The eluent was evaporated, the residue was dissolved in n-hexane and purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups; the use of the modified silica gel allowed us to remove easily the by-product SnI4, which appears to be hard to separate when a non-modified silica gel is used as sorbent). Hexane was used as the eluent. Column fractions were analyzed by GC-MS. The eluent was evaporated to give 152.3 mg of tolane 4aa as a colorless crystalline solid (78%, purity by GCMS – 95.3%).
1H NMR (400 MHz, CDCl3) δ 7.33–7.73 (m, 6H, Ph), 7.52–7.54 (m, 4H, Ph); 13C NMR (100 MHz, CDCl3) δ 89.4, 123.3, 128.2, 128.3, 131.6. IR (KBr, cm−1) νmax 3063, 2928 (C–H, C–C), 1599 (CC). MS (m/z, EI, 70 eV) 178 ([M+], 100), 152 ([M − C2H2]+, 17.6), 77 ([Ph]+, 3.3).
C)4Sn 1a (150 mg, 0.287 mmol). Then Pr2NH (1.43 mL, 10.43 mmol) and dry BuOAc (6 mL) were added, the solution was degassed by freezing in liquid nitrogen and pumping under vacuum several times, and then it was flushed with argon. The reaction mixture was stirred at 100 °C for 5 h, then allowed to cool and quenched with EtOH (10 mL). The mixture was treated with 0.5 g of silica modified with 3-aminopropyltriethoxysilane, the solvent was removed on a rotary evaporator and the traces of BuOAc were removed in vacuo. The resulting mixture was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups). Hexane was used as the eluent. The yield of 4ab was 81% (161.8 mg), white crystalline solid. 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H, Me), 7.114 (d, 3J = 8.0 Hz, 2H, Ar), 7.29–7.35 (m, 3H, Ph), 7.42 (d, 3J = 8.0 Hz, 2H, Ar), 7.50–7.53 (m, 2H, Ph); 13C NMR (100 MHz, CDCl3) δ 21.5, 88.7, 89.6, 120.2, 123.5, 128.1, 128.3, 129.1, 131.6, 132.5, 138.4. IR (KBr, cm−1) νmax 2920.6, 2853.1 (C–H, C–C), 2214.6 (CC), 1595.3 (CC). MS (m/z, EI, 70 eV) 192 ([M+], 100), 115 ([M − Ph]+, 8.3), 77 ([Ph]+, 2.6).
C), 1599.2 (CC). MS (m/z, EI, 70 eV) 258 ([M+ 81Br], 97.2), 256 ([M+ 79Br], 100), 177 ([M − Br]+, 13.6), 77 ([Ph]+, 6.8).
C), 1591.5 (CC), 1346.5 (symm NO2). MS (m/z, EI, 70 eV) 223 ([M+], 100), 177 ([M − NO2]+, 21.1), 77 ([Ph]+, 8.8).
C)4Sn 1a (90 mg, 0.172 mmol). Then Pr2NH (0.43 mL, 3.13 mmol) and dry BuOAc (1.8 mL) were added, the solution was degassed by freezing in liquid nitrogen and pumping under vacuum several times, and then the vial was flushed with argon. The reaction mixture was stirred at 100 °C for 4.5 h, then allowed to cool and quenched with EtOH (2 mL). The mixture was treated with 0.2 g of silica gel modified with 3-aminopropyltriethoxysilane, the solvent was removed on a rotary evaporator and the traces of BuOAc were removed in vacuo. The resulting mixture was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), eluted with hexane, then hexane:PhMe 4:1. Column fractions were analyzed by GCMS. The eluent was evaporated to give 76 mg (81%) of acetylene 4af as a yellow crystalline solid. In addition, the sample could be recrystallized from n-heptane. 1H NMR (400 MHz, CDCl3) δ 7.38–7.42 (m, 3H, Ph), 7.59–7.61 (m, 2H, Ph), 7.68 (d, 3J = 8.2 Hz, 1H, H-6 Ar), 8.20 (dd, 3J = 8.2 Hz, 4J = 2.3 Hz, 1H, H-5 Ar), 8.48 (d, 4J = 2.3 Hz, 1H, H-3 Ar); 13C NMR (100 MHz, CDCl3) δ 86.8, 99.5, 121.8, 122.1, 125.9, 127.6, 128.6, 129.7, 132.0, 132.1, 133.3, 146.9. IR (KBr, cm−1) νmax 3094.2, 3074.9 (C–H, C–C), 2218.4 (CC), 1583.8 (CC), 1340.7 (symm NO2). MS (m/z, EI, 70 eV) 303 ([M+ 81Br], 59.4), 301 ([M+ 79Br], 60.2), 257 ([(M − NO2)+, 81Br], 0.8), 255 ([(M − NO2)+, 79Br], 0.9), 176 (([M − NO2 − Br])+, 100), 77 ([Ph]+, 1.6).
C), 1339.5 (symm NO2). MS (m/z, EI, 70 eV) 223 ([M+], 5.2), 177 ([M − NO2]+, 3.8), 77 ([Ph]+, 89.0).
C)4Sn 1a (150 mg, 0.287 mmol). Then TMEDA (1.56 mL, 10.43 mmol) and dry BuOAc (6 mL) were added, the solution was degassed by freezing in liquid nitrogen and pumping under vacuum several times, and then it was flushed with argon. The reaction mixture was stirred at 100 °C for 5 h, then allowed to cool and quenched with EtOH (10 mL). The mixture was treated with 0.5 g of silica gel modified with 3-aminopropyltriethoxysilane, the solvent was removed on a rotary evaporator and the traces of BuOAc were removed in vacuo. The resulting mixture was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), eluted with hexane, then with hexane:PhMe 4:1 (after diphenyldiacetylene 5a was eluted). Column fractions were analyzed by GCMS. The eluent was evaporated to give 243 mg (purity by GCMS – 87.3%) of benzoate 4am as a yellow solid. The product was further purified with flash chromatography (2 g of silica gel, hexane). Yield 81%. 1H NMR (400 MHz, CDCl3) δ 1.40 (t, 3J = 7.3 Hz, 3H, OCH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 4.42 (q, 3J = 7.3 Hz, 2H, OC2CH3), 7.34–7.39 (m, 4H, Ar), 7.46–7.50 (m, 1H, Ar), 7.56–7.58 (m, 2H, Ar), 7.64 (d, 3J = 8.2 Hz, 1H, Ar), 7.97 (dd, 3J = 7.8 Hz, 4J = 0.9 Hz, 1H, Ar); 13C NMR (100 MHz, CDCl3) δ 14.4, 61.2, 88.3, 94.2, 123.4, 123.6, 127.9, 128.4, 128.5, 130.4, 131.5, 131.7, 132.3, 134.0, 166.4. IR (KBr, cm−1) νmax 3061.4, 2982.3 (C–H, C–C), 2218.4 (CC), 1726.5 (CO). MS (m/z, EI, 70 eV) 250 ([M+], 94.4), 235 ([M − Me]+, 3.0), 222 ([M − CO]+, 100), 221 ([M − Et]+, 29.7), 205 ([M − EtO]+, 36.0), 177 ([M − COOEt]+, 22.1), 77 ([Ph]+, 13.9).
C)4Sn 1a (150 mg, 0.287 mmol), TMEDA (1.56 mL, 10.43 mmol) and dry BuOAc (6 mL). The reaction time was 3 h. The yield of crude aldehyde 4an was 176 mg. For further purification, the sample was recrystallized from n-heptane. Yield 82%, beige crystalline solid. 1H NMR (400 MHz, CDCl3) δ 7.36–7.38 (m, 3H, Ph), 7.54–7.57 (m, 2H, Ph), 7.67 (d, 3J = 8.2 Hz, 2H, Ar), 7.85 (d, 3J = 8.2 Hz, 2H, Ar), 10.00 (s, 1H, CHO); 13C NMR (100 MHz, CDCl3) δ 88.5, 93.5, 122.5, 128.5, 129.0, 129.58, 129.60, 131.8, 132.1, 135.4, 191.4. IR (KBr, cm−1) νmax 3049.3, 2845.8 (C–C, C–H), 2216.1 (CC), 1697.3 (CO), 1599.9 (CC); MS (m/z, EI, 70 eV) 206 ([M+], 100), 205 ([M − H]+, 71.4), 178 ([M − CO]+, 13.3), 77 ([Ph]+, 5.9).
C)4Sn 1a (109.4 mg, 0.209 mmol), TMEDA (0.57 mL, 3.8 mmol) and dry BuOAc (4.5 mL). The product was purified by column chromatography on a silica gel (7 g), eluted with hexane, then hexane:PhMe 4:1 (after diphenyldiacetylene 5a was eluted). The yield of crude ketone 4ao was 129.3 mg (purity by GCMS – 84%). For further purification, the sample was recrystallized from n-heptane. Yield 65%, white crystalline solid. 1H NMR (400 MHz, CDCl3) δ 2.61 (s, 3H, COCH3), 7.36–7.37 (m, 3H, Ph), 7.53–7.56 (m, 2H, Ph), 7.61 (d, 3J = 8.5 Hz, 2H, Ar), 7.93 (d, 3J = 8.5 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 26.6, 88.6, 92.7, 122.7, 128.2, 128.3, 128.5, 128.8, 131.7, 131.8, 136.2, 197.3. IR (KBr, cm−1) νmax 3061.4, 2997.7 (C–H, C–C), 2218.4 (CC), 1680.2 (CO), 1603.0 (CC). MS (m/z, EI, 70 eV) 220 ([M+], 72.5), 205 ([M − CH3]+, 100), 177 ([M − COCH3]+, 27.8), 77 ([Ph]+, 6.4).
C)4Sn 1b (114.1 mg, 0.197 mmol), Pr2NH (0.98 mL, 7.16 mmol) and BuOAc (4.5 mL). The reaction time was 3.5 h. The resulting crude product was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), and subsequently eluted with hexane, hexane:EtOAc 9:1, hexane:EtOAc 4:1 and pure toluene. Recrystallization from toluene yielded 108.6 mg (63.9%) of acetylene 4bd as a white crystalline solid. Another crop of product (49.1 mg, purity by GC-MS – 95%) was obtained from the mother liquor. The total yield of tolane 4bd was 155.2 mg (91%). 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, CH3), 7.18 (d, 3J = 7.8 Hz, 2H, 4-MeC6H4), 7.44 (d, 3J = 7.8 Hz, 2H, 4-MeC6H4), 7.63 (d, 3J = 9.2 Hz, 2H, 4-NO2C6H4), 8.19 (d, 3J = 9.2 Hz, 2H, 4-NO2C6H4); 13C NMR (100 MHz, CDCl3) δ 21.6, 87.1, 95.1, 119.1, 123.6, 129.3, 130.5, 131.8, 132.2, 139.7, 146.9. IR (KBr, cm−1) νmax 2922.5, 2847.3 (C–H, C–C), 2212.6 (CC), 1589.5 (CC), 1342.6 (symm NO2). MS (m/z, EI, 70 eV) 237 ([M+], 100), 191 ([M − NO2]+, 13.6), 176 ([M − Me − NO2]+, 17.1).
C)4Sn 1c (181.7 mg, 0.275 mmol), Pr2NH (1.37 mL, 10.0 mmol) and BuOAc (6 mL). The reaction time was 2.5 h. The resulting crude product was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), and subsequently eluted with hexane, hexane:HCCl3 9:1, hexane:HCCl3 3:2 and hexane:HCCl3 1:1. Recrystallization from toluene yielded 160.9 mg (62%) of acetylene 4cd as a white crystalline solid. Another crop of product (49.8 mg) was isolated from the mother liquor. The total yield was 210.7 mg (82%). 1H NMR (400 MHz, CDCl3) δ 7.36 (d, 3J = 8.2 Hz, 2H, Ar), 7.49 (d, 3J = 8.2 Hz, 2H, Ar), 7.65 (d, 3J = 8.7 Hz, 2H, Ar), 8.22 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 88.4, 93.4, 120.6, 123.7, 129.0, 129.9, 132.3, 133.1, 135.5, 147.2. IR (KBr, cm−1) νmax 3092.3, 2926.4, 2851.1 (C–H, C–C), 2210.7 (CC), 1587.6 (CC), 1346.5 (symm NO2). MS (m/z, EI, 70 eV) 259 ([M+] 37Cl, 32.7), 257 ([M+] 35Cl, 100), 213 ([M − NO2]+ 37Cl, 2.8), 211 ([M − NO2]+ 35Cl, 9.5), 176 ([M − Cl − NO2]+, 82.7).
C)4Sn 1e (180 mg, 0.32 mmol), Pr2NH (1.6 mL, 11.64 mmol) and BuOAc (6 mL). The reaction time was 5.5 h. The resulting crude product was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), and subsequently eluted with hexane and hexane:HCCl3 9:1. The yield of acetylene 4ed was 132 mg (49%), yellow crystalline solid. 1H NMR (400 MHz, CDCl3) δ 1.30 (s, 9H, tBu), 4.34 (s, 2H, OCH2), 7.57 (d, 3J = 9.2 Hz, 2H, Ar), 8.16 (d, 3J = 9.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 27.5, 51.0, 74.8, 83.0, 92.7, 123.5, 130.0, 132.4, 147.1. IR (KBr, cm−1) νmax 3105.8, 2976.5, 2862.7 (C–H, C–C), 2220.3 (CC), 1593.4 (CC), 1342.6 (symm NO2). MS (m/z, EI, 70 eV) 233 ([M+], 0.3), 218 ([M − CH3]+, 10.4), 160 ([M − tBuO]+, 100), 57 ([tBu]+, 60.5).
C)4Sn 1f (121.9 mg, 0.275 mmol), Pr2NH (1.37 mL, 10.0 mmol) and BuOAc (6 mL). The reaction time was 15 h. The resulting product was purified by column chromatography over a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups). Hexane was used as the eluent. The column fractions were concentrated, the unreacted 1-iodo-4-nitrobenzene (3d) was filtered off, and the crude product was again purified under the same column chromatography conditions as previously to give 96.1 mg (47%) of acetylene 4fd as a light yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.96 (t, 3J = 7.3 Hz, 3H, Me), 1.44–1.53 (m, 2H, CH2), 1.58–1.65 (m, 2H, CH2), 2.45 (t, 3J = 6.9 Hz, 2H, CH2C), 7.51 (d, 3J = 8.7 Hz, 2H, Ar), 8.15 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 13.6, 19.3, 22.0, 30.5, 79.3, 96.8, 123.5, 131.3, 132.3, 146.6. IR (KBr, cm−1) νmax 3107.7, 3080.7, 2957.2, 2932.2, 2872.4 (C–H, C–C), 2230.0 (CC), 1592.3 (CC), 1342.6 (symm NO2). MS (m/z, EI, 70 eV) 203 ([M+], 39.9), 188 ([M − CH3]+, 58.3), 174 ([M − CH2CH3]+, 6.6), 157 ([M − NO2]+, 17.9).
3), 4.42 (q, 3J = 7.3 Hz, 2H, OC2CH3), 7.34–7.39 (m, 4H, Ar), 7.46–7.50 (m, 1H, Ar), 7.56–7.58 (m, 2H, Ar), 7.64 (d, 3J = 8.2 Hz, 1H, Ar), 7.97 (dd, 3J = 7.8 Hz, 4J = 0.9 Hz, 1H, Ar); 13C NMR (100 MHz, CDCl3) δ 14.4, 61.2, 88.3, 94.2, 123.4, 123.6, 127.9, 128.4, 128.5, 130.4, 131.5, 131.7, 132.3, 134.0, 166.4. IR (KBr, cm−1) νmax 3061.4, 2982.3 (C–H, C–C), 2218.4 (CC), 1726.5 (CO). MS (m/z, EI, 70 eV) 250 ([M+], 94.4), 235 ([M − Me]+, 3.0), 222 ([M − CO]+, 100), 221 ([M − Et]+, 29.7), 205 ([M − EtO]+, 36.0), 177 ([M − COOEt]+, 22.1), 77 ([Ph]+, 13.9).
C)4Sn 1a (150 mg, 0.287 mmol), TMEDA (1.56 mL, 10.43 mmol) and dry BuOAc (6 mL). The reaction time was 3 h. The yield of crude aldehyde 4an was 176 mg. For further purification, the sample was recrystallized from n-heptane. Yield 82%, beige crystalline solid. 1H NMR (400 MHz, CDCl3) δ 7.36–7.38 (m, 3H, Ph), 7.54–7.57 (m, 2H, Ph), 7.67 (d, 3J = 8.2 Hz, 2H, Ar), 7.85 (d, 3J = 8.2 Hz, 2H, Ar), 10.00 (s, 1H, CHO); 13C NMR (100 MHz, CDCl3) δ 88.5, 93.5, 122.5, 128.5, 129.0, 129.58, 129.60, 131.8, 132.1, 135.4, 191.4. IR (KBr, cm−1) νmax 3049.3, 2845.8 (C–C, C–H), 2216.1 (CC), 1697.3 (CO), 1599.9 (CC); MS (m/z, EI, 70 eV) 206 ([M+], 100), 205 ([M − H]+, 71.4), 178 ([M − CO]+, 13.3), 77 ([Ph]+, 5.9).
C)4Sn 1a (109.4 mg, 0.209 mmol), TMEDA (0.57 mL, 3.8 mmol) and dry BuOAc (4.5 mL). The product was purified by column chromatography on a silica gel (7 g), eluted with hexane, then hexane:PhMe 4:1 (after diphenyldiacetylene 5a was eluted). The yield of crude ketone 4ao was 129.3 mg (purity by GCMS – 84%). For further purification, the sample was recrystallized from n-heptane. Yield 65%, white crystalline solid. 1H NMR (400 MHz, CDCl3) δ 2.61 (s, 3H, COCH3), 7.36–7.37 (m, 3H, Ph), 7.53–7.56 (m, 2H, Ph), 7.61 (d, 3J = 8.5 Hz, 2H, Ar), 7.93 (d, 3J = 8.5 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 26.6, 88.6, 92.7, 122.7, 128.2, 128.3, 128.5, 128.8, 131.7, 131.8, 136.2, 197.3. IR (KBr, cm−1) νmax 3061.4, 2997.7 (C–H, C–C), 2218.4 (CC), 1680.2 (CO), 1603.0 (CC). MS (m/z, EI, 70 eV) 220 ([M+], 72.5), 205 ([M − CH3]+, 100), 177 ([M − COCH3]+, 27.8), 77 ([Ph]+, 6.4).
C)4Sn 1b (114.1 mg, 0.197 mmol), Pr2NH (0.98 mL, 7.16 mmol) and BuOAc (4.5 mL). The reaction time was 3.5 h. The resulting crude product was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), and subsequently eluted with hexane, hexane:EtOAc 9:1, hexane:EtOAc 4:1 and pure toluene. Recrystallization from toluene yielded 108.6 mg (63.9%) of acetylene 4bd as a white crystalline solid. Another crop of product (49.1 mg, purity by GC-MS – 95%) was obtained from the mother liquor. The total yield of tolane 4bd was 155.2 mg (91%). 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, CH3), 7.18 (d, 3J = 7.8 Hz, 2H, 4-MeC6H4), 7.44 (d, 3J = 7.8 Hz, 2H, 4-MeC6H4), 7.63 (d, 3J = 9.2 Hz, 2H, 4-NO2C6H4), 8.19 (d, 3J = 9.2 Hz, 2H, 4-NO2C6H4); 13C NMR (100 MHz, CDCl3) δ 21.6, 87.1, 95.1, 119.1, 123.6, 129.3, 130.5, 131.8, 132.2, 139.7, 146.9. IR (KBr, cm−1) νmax 2922.5, 2847.3 (C–H, C–C), 2212.6 (CC), 1589.5 (CC), 1342.6 (symm NO2). MS (m/z, EI, 70 eV) 237 ([M+], 100), 191 ([M − NO2]+, 13.6), 176 ([M − Me − NO2]+, 17.1).
C)4Sn 1c (181.7 mg, 0.275 mmol), Pr2NH (1.37 mL, 10.0 mmol) and BuOAc (6 mL). The reaction time was 2.5 h. The resulting crude product was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), and subsequently eluted with hexane, hexane:HCCl3 9:1, hexane:HCCl3 3:2 and hexane:HCCl3 1:1. Recrystallization from toluene yielded 160.9 mg (62%) of acetylene 4cd as a white crystalline solid. Another crop of product (49.8 mg) was isolated from the mother liquor. The total yield was 210.7 mg (82%). 1H NMR (400 MHz, CDCl3) δ 7.36 (d, 3J = 8.2 Hz, 2H, Ar), 7.49 (d, 3J = 8.2 Hz, 2H, Ar), 7.65 (d, 3J = 8.7 Hz, 2H, Ar), 8.22 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 88.4, 93.4, 120.6, 123.7, 129.0, 129.9, 132.3, 133.1, 135.5, 147.2. IR (KBr, cm−1) νmax 3092.3, 2926.4, 2851.1 (C–H, C–C), 2210.7 (CC), 1587.6 (CC), 1346.5 (symm NO2). MS (m/z, EI, 70 eV) 259 ([M+] 37Cl, 32.7), 257 ([M+] 35Cl, 100), 213 ([M − NO2]+ 37Cl, 2.8), 211 ([M − NO2]+ 35Cl, 9.5), 176 ([M − Cl − NO2]+, 82.7).
C)4Sn 1e (180 mg, 0.32 mmol), Pr2NH (1.6 mL, 11.64 mmol) and BuOAc (6 mL). The reaction time was 5.5 h. The resulting crude product was purified by column chromatography on a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups), and subsequently eluted with hexane and hexane:HCCl3 9:1. The yield of acetylene 4ed was 132 mg (49%), yellow crystalline solid. 1H NMR (400 MHz, CDCl3) δ 1.30 (s, 9H, tBu), 4.34 (s, 2H, OCH2), 7.57 (d, 3J = 9.2 Hz, 2H, Ar), 8.16 (d, 3J = 9.2 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 27.5, 51.0, 74.8, 83.0, 92.7, 123.5, 130.0, 132.4, 147.1. IR (KBr, cm−1) νmax 3105.8, 2976.5, 2862.7 (C–H, C–C), 2220.3 (CC), 1593.4 (CC), 1342.6 (symm NO2). MS (m/z, EI, 70 eV) 233 ([M+], 0.3), 218 ([M − CH3]+, 10.4), 160 ([M − tBuO]+, 100), 57 ([tBu]+, 60.5).
C)4Sn 1f (121.9 mg, 0.275 mmol), Pr2NH (1.37 mL, 10.0 mmol) and BuOAc (6 mL). The reaction time was 15 h. The resulting product was purified by column chromatography over a mixture of silica gel (6 g) and silica gel modified with 3-aminopropyltriethoxysilane (1 g, 1.14 mmol g−1 of NH2 groups). Hexane was used as the eluent. The column fractions were concentrated, the unreacted 1-iodo-4-nitrobenzene (3d) was filtered off, and the crude product was again purified under the same column chromatography conditions as previously to give 96.1 mg (47%) of acetylene 4fd as a light yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.96 (t, 3J = 7.3 Hz, 3H, Me), 1.44–1.53 (m, 2H, CH2), 1.58–1.65 (m, 2H, CH2), 2.45 (t, 3J = 6.9 Hz, 2H, CH2C), 7.51 (d, 3J = 8.7 Hz, 2H, Ar), 8.15 (d, 3J = 8.7 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3) δ 13.6, 19.3, 22.0, 30.5, 79.3, 96.8, 123.5, 131.3, 132.3, 146.6. IR (KBr, cm−1) νmax 3107.7, 3080.7, 2957.2, 2932.2, 2872.4 (C–H, C–C), 2230.0 (CC), 1592.3 (CC), 1342.6 (symm NO2). MS (m/z, EI, 70 eV) 203 ([M+], 39.9), 188 ([M − CH3]+, 58.3), 174 ([M − CH2CH3]+, 6.6), 157 ([M − NO2]+, 17.9).
Further details on the experimental procedures and spectra are given in the ESI.†
Acknowledgements
VVD is grateful for financial support from the Russian Ministry of Education and Science (State Assignment to Higher Education Institutions, project no. 4.5547.2017/ ).
).
Notes and references
- J. K. Stille, Angew. Chem., Int. Ed., 1986, 25, 508 CrossRef.
- T. N. Mitchell, Synthesis, 1992, 803 CrossRef CAS.
- V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1 CrossRef CAS.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS.
- C. Cordovilla, C. Bartolome, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040 CrossRef CAS.
- M. A. J. Duncton and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1999, 1235 RSC.
- M. Nath, Appl. Organomet. Chem., 2008, 22, 598 CrossRef CAS.
- I. J. Boyer, Toxicology, 1989, 55, 253 CrossRef CAS PubMed.
- E. Le Grognec, J.-M. Chrétien, F. Zammattio and J.-P. Quintard, Chem. Rev., 2015, 115, 10207 CrossRef CAS PubMed.
- M. Pereyre, J.-P. Quintard and A. Rahm, Tin in Organic Synthesis, Butterworths, London, 1986 Search PubMed.
- D. Young, in Tin Chemistry: Fundamentals, Frontiers, and Applications, ed. M. Gielen, A. Davies, K. Pannell and E. Tiekink, John Wiley & Sons, Ltd, 2008, p. 653, ISBN: 978-0-470-51771-0 Search PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- W. J. Scott and A. F. Moretto, Tetraallylstannane, e-EROS Encyclopedia of Reagents for Organic Synthesis, Wiley, 2001 Search PubMed.
- X. N. Wei, L. Y. Liu, B. Wang, W. X. Chang and J. Li, Chin. Chem. Lett., 2009, 20, 40 CrossRef CAS.
- A. Yanagisawa, H. Inoue, M. Morodome and H. Yamamoto, J. Am. Chem. Soc., 1993, 115, 10356 CrossRef CAS.
- X. Zhang, R. Qiu, N. Tan, S. Yin, J. Xia, S. Luo and C.-T. Au, Tetrahedron Lett., 2010, 51, 153 CrossRef CAS.
- I. Pri-Bar, P. S. Pearlman and J. K. Stille, J. Org. Chem., 1983, 48, 4629 CrossRef CAS.
- R. M. Kamble and V. K. Singh, Tetrahedron Lett., 2001, 42, 7525 CrossRef CAS.
- M. Shi, R. Franks and K. M. Nicholas, in Advances in Chemical Conversions for Mitigating Carbon Dioxide. Studies in Surface Science and Catalysis, ed. T. Inui, M. Anpo, K. Izui, S. Yanagida and T. Yamaguchi, Elsevier Science B.V., 1998, vol. 114, p. 165 Search PubMed.
- A. S. Levashov, A. A. Andreev and V. V. Konshin, Tetrahedron Lett., 2015, 56, 1870 CrossRef CAS.
- A. A. Andreev, A. S. Levashov and N. V. Komarov, Method for preparing tri- and tetraorganylalkynyl tin, Pat. RU2317993, 2008 Search PubMed.
- A. S. Levashov, A. A. Andreev, D. S. Buryi and V. V. Konshin, Russ. Chem. Bull., Int. Ed., 2014, 63, 775 CrossRef CAS.
- P. Jaumier, B. Jousseaume and M. Lahcini, Angew. Chem., Int. Ed., 1999, 38, 402 CrossRef CAS.
- P. Jaumier, B. Jousseaume and M. Lahcini, Phosphorus, Sulfur Silicon, 1999, 150–151, 117 CrossRef CAS.
- B. Wrackmeyer and G. Kehr, J. Organomet. Chem., 1995, 501, 87 CrossRef CAS.
- B. Wrackmeyer, G. Kehr, A. Sebald and J. Kümmerlen, Chem. Ber., 1992, 125, 1597 CrossRef CAS.
- B. Wrackmeyer, G. Kehr and R. Boese, Angew. Chem., 1991, 103, 1374 CrossRef CAS.
- B. Wrackmeyer and G. Kehr, Polyhedron, 1991, 10, 1497 CrossRef CAS.
- M. Lahcini, P. M. Castro, M. Kalmi, M. Leskelä and T. Repo, Organometallics, 2004, 23, 4547 CrossRef CAS.
- W. P. Neumann and F. G. Kleiner, Liebigs Ann. Chem., 1968, 716, 29 CrossRef CAS.
- S. Lamandé-Langle, M. Abarbri, J. Thibonnet and A. Duchêne, J. Organomet. Chem., 2009, 694, 2368 CrossRef.
- J.-H. Li, Y. Liang, D.-P. Wang, W.-J. Liu, Y.-X. Xie and D.-L. Yin, J. Org. Chem., 2005, 70, 2832 CrossRef CAS PubMed.
- E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114(16), 6556 CrossRef CAS.
- J. M. Brown, M. Pearson, J. T. B. H. Jastrzebski and G. van Koten, J. Chem. Soc., Chem. Commun., 1992, 1440 RSC.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS.
- W. Armarego and C. Chai, Purification of Laboratory Chemicals, Elsevier Science, Oxford, 2003, p. 608 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data. See DOI: 10.1039/c6nj03905g |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |