
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanism of electropolymerization of nickel(II) salen type complexes†
Danuta
Tomczyk
 *a,
Wiktor
Bukowski
b,
Karol
Bester
b,
Paweł
Urbaniak
a,
Piotr
Seliger
a,
Grzegorz
Andrijewski
a and
Sławomira
Skrzypek
a
*a,
Wiktor
Bukowski
b,
Karol
Bester
b,
Paweł
Urbaniak
a,
Piotr
Seliger
a,
Grzegorz
Andrijewski
a and
Sławomira
Skrzypek
a
aDepartment of Inorganic and Analytical Chemistry, University of Łódź, 91-403 Łódź, ul.Tamka 12, Poland. E-mail: tomczyk@chemia.uni.lodz.pl
bFaculty of Chemistry, Rzeszów University of Technology, 35-959 Rzeszów, Al. Powstańców W-wy 6, Poland
First published on 1st February 2017
Abstract
Ni(II) complexes with (±)-trans-N,N′-bis(salicylidene)-1,2-cyclohexanediamine ([Ni(salcn)]), and its methyl ([Ni(salcn(Me))]) and tert-butyl ([Ni(salcn(Bu))]) derivatives have been synthesized. Anodic electropolymerization has been used to form electrodes modified by polymer films. FTIR and FTIR ATR methods showed that the obtained films consist of polymers of phenyl–phenyl type. The structure of poly[Ni(salcn)] is built with 1,2,4- and 1,2,3,5-substituted chains. A three-step process of the oxidation of the complexes and their films has been ascertained on the basis of cyclic voltammetry measurements. Furthermore we have also detected the oxidized species which probably serve as intermediates for polymer formation. The influence of the substituent in the phenolate moiety on the type of reaction after which the dimerization and polymerization reaction occurs has been evidenced. Furthermore the substituent dependence on the stability of the phenoxyl radical complex and the Ni(II)-phenoxonium cation has been noticed. For the [Ni(salcn(Bu))] complex, an additional electropolymerization step – the adsorption of the reagent on the electrode surface – has been observed.
Introduction
Complexes of the transition metal ions with Schiff bases and other ligands consisting of oxygen and/or nitrogen donor atoms, especially Ni(II) ions with salen type ligands,1–4 have been under investigation for years, due to their optical properties and application in analysis, catalysis and electrocatalysis. The reduced5 as well as oxidised6 forms of Ni(II)-salen type complexes are used. The oxidation process of Ni(II)-salen type complexes depends on the solvent used. In strong donor solvents such complexes are oxidised to Ni(III)-salen type complexes.7,8 In weak donor solvents the oxidation process depends also on the presence or absence of a substituent in the p-position of the phenolate moiety. The complexes with a blocked p-position undergo reaction to phenoxyl radicals and bis-phenoxyl radicals,9–13 while those with a free p-position go through an electropolymerization process and finally form electroactive polymeric films.14–18The mechanism of anodic electropolymerization is a subject of discussion among scientists. Audebert et al.16,17 and Goldsby et al.14 explained that this process is based on the ligand. After the first oxidation step the phenoxyl radicals undergo a coupling reaction resulting in C–C bond polymers. Freire, Hillman et al.19,20 have also shown that the anodic polymerization process is focused on the ligand and leads to a polymer consisting of o- and p-bound phenyl rings. Metal ions do not take direct part in the charge transfer through the polymer film but they are forming the bridge stabilising the process of charge transfer between biphenylene moieties. On the other hand Popeko and Timonov et al.21,22 and Dahm et al.17 suggested that the Ni(II)-salen type complex oxidation mechanism is based on the metal ion. They found that at the first step, the oxidation of the complexes leads to a Ni(III)–phenolate complex. According to their studies the oxidation process is possible due to the donor–acceptor interaction between the monomeric ligand and the oxidised metal centre of the other monomer. Such types of interactions result in stack polymer formation. Dahm et al.17 have also investigated the next step of anodic electropolymerization of Ni(II)-salen type complexes with free p-positions. A two-electron irreversible process was ascribed to the formation of phenyl–phenyl bonding between two non-covalent bonded monomers. This process causes the more compact structure of the polymer films. The whole investigated compounds14–22 consisted of complexes of salen and its derivatives which differ in substituents within the ethylene imine bridge as well as in the phenolate moieties.
Our investigations concern the Ni(II) complexes with salen derivatives consisting of a cyclohexane moiety in the imine bridge and of different substituents within the aromatic rings.
The aim of the study was characterisation of the electropolymerization mechanism of electrodes modified with polymer films of a (±)-trans-N,N′-bis(salicylidene)-1,2-cyclohexanediaminenickel(II) ([Ni(salcn)]) complex and with polymer films of its two derivatives substituted in the o-positions of the phenolate moieties with tert-butyl ([Ni(salcn(Bu))]) or methyl ([Ni(salcn(Me))]) substituents (Scheme 1).
 | ||
| Scheme 1 Complexes presented in this study. | ||
In our previous work,23 performed under similar conditions, for the di-tert-butyl derivative of the investigated complexes ([Ni(salcn(Bu))] and [Ni(salcn)]) the non-polymerized oxidation mechanism has been characterised. We evidenced that in CH2Cl2 solution the ((±)-trans-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminenickel(II) complex, [Ni(salcn(2Bu))]) is oxidized in a three-step process successively to Ni(II)-mono-phenoxyl radicals, Ni(II)-bis-phenoxyl radicals and Ni(II)-bis-phenoxonium cations. For Ni(II)-mono-phenoxyl radicals we proved the delocalization phenomenon.
In this paper we also investigate the mechanism of oxidation of the ligand substituted in the o-position with methyl substituents, H2(salcn(Me)).
The study was carried out on the basis of cyclic voltammetry, UV-VIS, NMR and FTIR measurements of the ligands and their complexes as well as the FTIR ATR measurements of the polymer films deposited on the electrode surface.
Experimental
Materials
(±)-trans-1,2-Diaminocyclohexane and salicylaldehyde, 99% were purchased from Aldrich; nickel(II) acetate tetrahydrate Ni(AcO)2·4H2O (reagent grade), potassium hexacyanoferrate(III) (analytical grade) and ethanol (EtOH) 95% were obtained from POCh. Methylene chloride (CH2Cl2) was of HPLC grade, from Baker. Tetrabutylammonium hexafluorophosphate (TBAH) and tetrabutylammonium perchlorate (TBAP) were of analytical grade, from Fluka. All of the compounds were used without purification. The solutions of the complexes were freshly prepared before the measurements.Ligands and complexes
The method of the synthesis of the salen ligands (±)-trans-N,N′-bis(salicylidene)-1,2-cyclohexanediamine (H2salcn), (±)-trans-N,N′-bis(3-tert-butylsalicylidene)-1,2-cyclohexanediamine (H2salcn(Bu)), (±)-trans-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine (H2salcn(2Bu)) and their complexes as well as their elemental analyses, NMR data, melting points and their synthetic yields were previously described.23The data for a ligand with methyl substituents and its Ni(II) complex are presented below:
(±)-trans-N,N′-bis(3-methylsalicylidene)-1,2-cyclohexanediamine (H2salcn(Me))
Anal. calc. for C22H26N2O2: C, 75.40%; H, 7.48%; N, 7.99%. Found: C, 75.39%; H, 7.50%; N, 7.99%. 1H-NMR (CD2Cl2): δ = 13.47 (s, 2H), 8.28 (s, 2H), 7.13 (dd, J = 7.3 Hz, J = 1.0 Hz, 2H), 7.02 (dd, J = 7.5 Hz, J = 1.0 Hz, 2H), 6.72–6.69 (m, 2H), 3.37–3.30 (m, 2H), 2.18 (s, 6H), 1.96–1.90 (m, 2H), 1.88–1.82 (m, 2H), 1.77–1.67 (m, 2H), 1.52–1.45 (m, 2H). Yield: 1.459 g, 78%.
(±)-trans-N,N′-Bis(3-methylsalicylidene)-1,2-cyclohexanediaminenickel(II) [Ni(salcn(Me))]
Anal. calc. for C22H24N2NiO2: C, 64.90%; H, 5.94%; N, 6.88%. Found: C, 64.87%; H, 5.92%; N, 6.87%. 1HNMR (CD2Cl2): δ = 7.42 (s, 2H), 7.12–7.09 (m, 2H), 7.01 (dd, J = 7.8 Hz, J = 1.3 Hz, 2H), 6.48–6.42 (m, 2H), 3.16–3.09 (m, 2H), 2.47–2.37 (m, 2H), 2.16 (s, 6H), 1.95–1.85 (m, 2H), 1.38–1.28 (m, 4H). The complex decomposed into CDCl3. Yield: 0.57 g, 74%.
Instruments and procedures
The elemental CHN analysis was performed using a Vario EL III analyzer.Magnetic studies were performed on the magnetic susceptibility balance of Sherwood Scientific Ltd at 298 K.
NMR spectra were recorded using a Bruker AM 500 MHz spectrometer.
FTIR and FTIR-ATR spectra were recorded on a Nicolet 8700 Thermo Scientific.
FTIR spectra were recorded using a standard KBr pellet technique.
FTIR-ATR spectra were recorded for electrodes modified with the polymers obtained after the electropolymerization process conducted in CH2Cl2/TBAH (0.1 mol dm−3) and CH2Cl2/TBAP (0.1 mol dm−3) solutions. Such modified electrodes were washed and conditioned in CH2Cl2 for 10 min and then they were dried at room temperature in air atmosphere for 5 h in order to remove as much of the solvent as possible from the film structures.
UV-Vis spectra were performed on a Jasco V630 spectrophotometer in standard quartz (0.995 cm) cells, using solutions of H2(salcn(Me)) and [Ni(salcn(Me))] (10−5–2 × 10−4 mol dm−3) in CH2Cl2.
Cyclic voltammetry (CV) was performed using an AUTOLAB PGSTAT 10 Eco Chemie in a three-electrode system. A Pt disk electrode (MINERAL) with an area of 0.1865 cm2 and a Pt disk electrode modified with polymer nickel complexes were used as working electrodes. The electrochemical surface area of the Pt disk electrode was determined by voltammetry using potassium hexacyanoferrate(III) solution as a standard.24
Ag/AgCl (1 mol dm−3 NaCl) was used as a reference electrode connected to the bulk of the solution by a Luggin capillary and a Pt plate as a counter electrode. Measurements were recorded in CH2Cl2/TBAH (0.1 mol dm−3) and solutions containing 10−3 mol dm−3 of the ligand or 10−3 mol dm−3 of the complex. Voltammograms were recorded in the potential range of 0 V to 2.1 V, at sweep rates between 0.01 and 2 V s−1.
All electrochemical and spectroscopic studies were carried out in an argon atmosphere at a temperature of 295 K. The Pt electrodes were cleaned in concentrated HNO3 and polished with an aqueous suspension of 0.05 μm alumina before each measurement.
The electroactivity of the modified electrodes was investigated in the supporting electrolyte, the same as was used in the electropolymerization process.
The modified electrode, Ptpoly, was rinsed with CH2Cl2 and conditioned (10 minutes) in CH2Cl2/TBAH solution (0.1 mol dm−3) – which was different from that used during the electropolymerization process. Such conditioning was performed to remove from the film structure the complex particles that were not removed by washing.
Thus, 1st and 2nd voltammetric cycles recorded on the Ptpoly electrode in supporting electrolyte solution showed no difference in contrast to such curves obtained in the process carried out on a non-preconditioned Ptpoly electrode.
All electroactive polymer films are yellow and are insoluble in the investigated solvent.
Results and discussion
General characteristics of the complexes
Complexes were obtained in the form of crystalline deposits. [Ni(salcn)] and [Ni(salcn(Me))] are pale orange-brown, and [Ni(salcn(Bu))] is brown. The complexes are stable both in solid form and as a solution in CH2Cl2. In solvents with higher coordination properties, such as acetonitrile, only [Ni(salcn)] can be dissolved. None of the investigated complexes are soluble in protic solvents.Spectroscopic and magnetic studies of the ligand and its complex
Shown in Table 1 are the most characteristic absorption bands ascribed to O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, C–N and C–O stretching vibrations in the ligand with methyl substituents and its complex. The shift of the O–H stretching vibration in the ligand toward the lower frequencies (2900 cm−1), relative to characteristic bands for O–H groups not intramolecularly interacting with other atoms, shows the intermolecular interactions of O–H groups with N atoms.25–27 The lack of this band on the spectrum of the complex is connected with the deprotonation process during complex formation.25 The shift of the characteristic imine band (1624 cm−1) in the complex by 22 cm−1 toward the lower frequencies relative to the ligand is related to the participation of the azomethine nitrogen in nickel ion coordination.26,28,29 Also, offset bands at 1400 cm−1 and 1281 cm−1 corresponding to C–N and C–O stretching frequencies, respectively, towards higher frequencies (shifted by 11 cm−1 for N–O and 35 cm−1 for C–O) are the consequence of both azomethine nitrogen atom and phenolate oxygen atom participation in the complexation process.26,28
C, CN, C–N and C–O stretching vibrations in the ligand with methyl substituents and its complex. The shift of the O–H stretching vibration in the ligand toward the lower frequencies (2900 cm−1), relative to characteristic bands for O–H groups not intramolecularly interacting with other atoms, shows the intermolecular interactions of O–H groups with N atoms.25–27 The lack of this band on the spectrum of the complex is connected with the deprotonation process during complex formation.25 The shift of the characteristic imine band (1624 cm−1) in the complex by 22 cm−1 toward the lower frequencies relative to the ligand is related to the participation of the azomethine nitrogen in nickel ion coordination.26,28,29 Also, offset bands at 1400 cm−1 and 1281 cm−1 corresponding to C–N and C–O stretching frequencies, respectively, towards higher frequencies (shifted by 11 cm−1 for N–O and 35 cm−1 for C–O) are the consequence of both azomethine nitrogen atom and phenolate oxygen atom participation in the complexation process.26,28
| cm−1 | |
|---|---|
| λ (nm), ε′ = 10−4ε (dm3 mol−1 cm−1), sh = shoulder. | |
| H2salcn(Me) | 2900(s, br), 1624(s), 1400(m), 1281(m) |
| [Ni(salcn(Me))] | 1602(s), 1411(m), 1316(s) |
| λ(ε′) | |
|---|---|
| H2salcn(Me) | 256(2.07), 318(0.76), 417sh(0.02) |
| [Ni(salcn(Me))] | 263(7.86), 320(0.76), 344(0.71), 387(0.40), 411(0.63), 442(0.36), 547(0.01) |
The ligand complexing characteristic features30–33 are also evidenced on the 1H-NMR and UV-VIS-NIR spectra of the complex. The shift of the corresponding signals versus the uncomplexed ligand signals (see Section Ligands and complexes and Table 1) as well as the presence of the CT bands at 344 and 387 nm, and bands corresponding to d–d transfer in nickel ions (411, 442 and 547 nm) (Table 1) clearly support the presented hypothesis. Furthermore the bands within the VIS region indicate the low-spin electron configuration of d8 ions in square planar ligand fields.
Measurements of magnetic susceptibility of the complex, at a temperature of 273 K, showed the diamagnetic nature of the compound, that confirms low-spin, square planar symmetry around the Ni(II) ion.
FTIR ATR of films
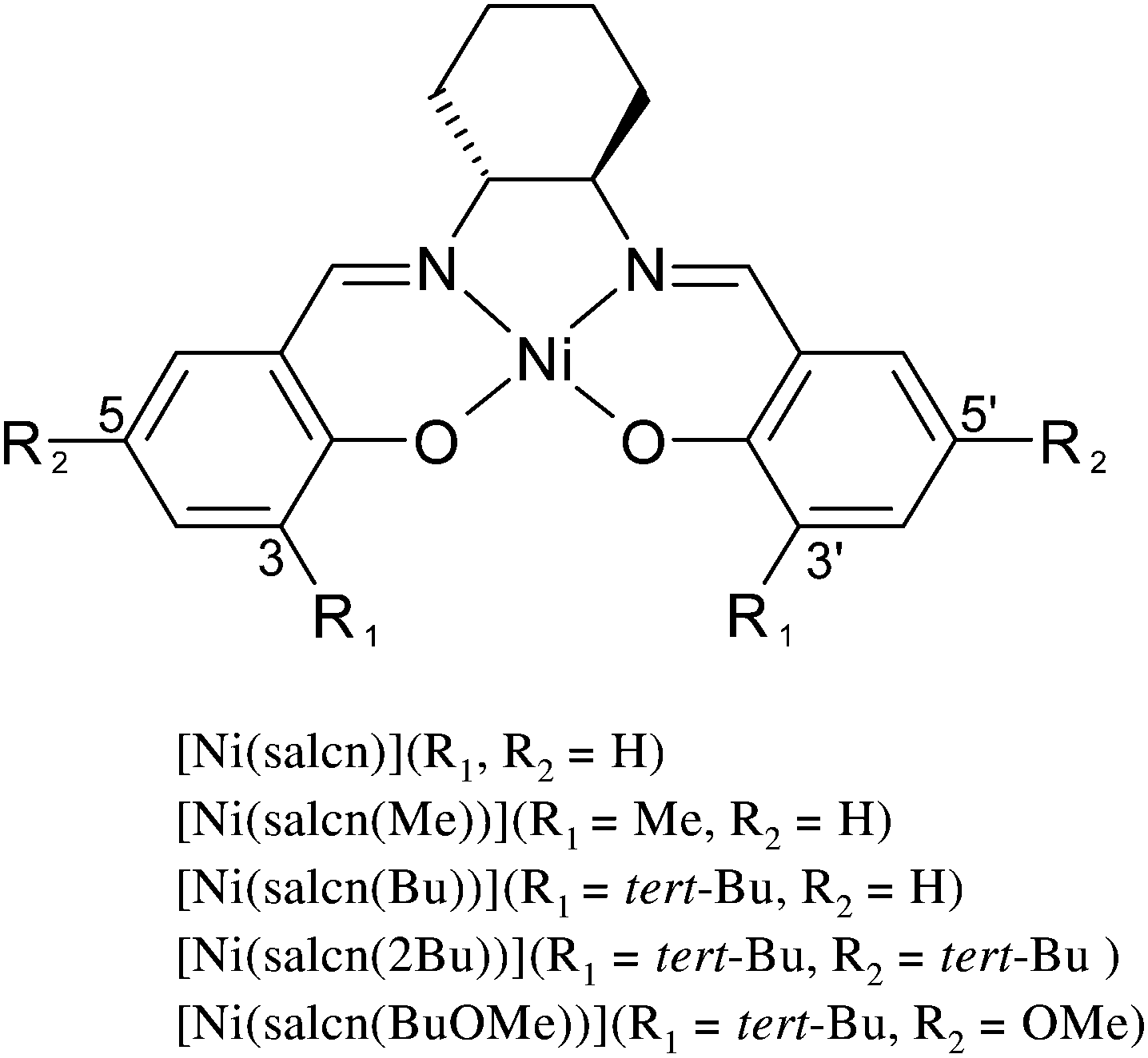
The analysis of the film spectra was done on the basis of comparison of the FTIR ATR spectrum taken for the selected film with the FTIR spectrum of the corresponding complex compound. It is shown in the [Ni(salcn)] and poly[Ni(salcn)] examples. The observed film bands (Fig. 2) are lower than the corresponding bands for complex species (Fig. 1). Such behaviour may be connected with a difference in refractive index in the complex and in films which affects the penetration of the evanescent wave and hence absorbance. Furthermore, some bands of the films are slightly shifted (max. 10 cm−1). In Table S1 (ESI†) the frequencies of all the bands for the investigated complexes and films are presented.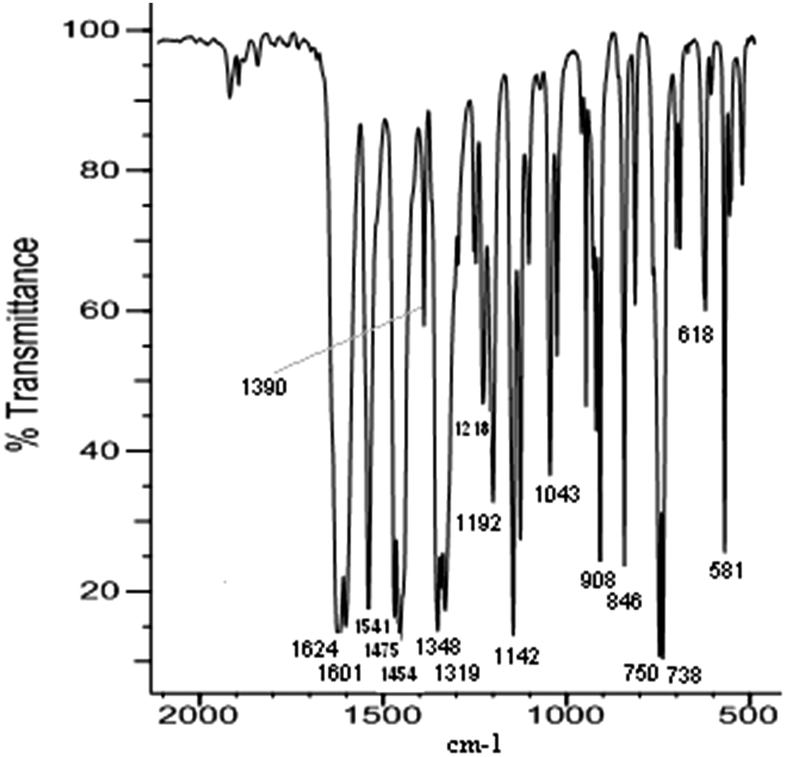 | ||
| Fig. 1 FTIR spectra of [Ni(salcn)]. | ||
On the film spectra there are also more bands observed than in the case of the complex. Part of them corresponds to the presence of a supporting electrolyte and solvent molecules within the structure of the film. A relatively strong broad band at 844 cm−1 and a medium band at 568 cm−1 (Fig. 2A) may be attributed to the PF6− anion,34 while the ClO4− anion35 is characterized by a strong and broad band ranging from 1220 to 1000 cm−1, with the maximum at 1092 cm−1. Furthermore the broad band of medium intensity within 780–600 cm−1 with the maximum at 702 cm−1 (Fig. 2B) may also be attributed to the perchlorate anion. On the spectrum of the film electrosynthesized in ClO4− solution the broad band at 1092 cm−1 masks the region which is visible on the film synthesized in PF6− solution and vice versa (band at 844 cm−1 originated from PF6− masks the range 900–800 cm−1). Such choice of the supporting electrolytes applied during the electrosyntheses allows in both cases to observe all of the bands observed in the case of a complex spectrum on the spectra taken for film species (Fig. 2A and B), excluding the ones at 1335, 1227 and 912 cm−1 (Fig. 1). The latter bands are superimposed on the bands at 1319(m), 1218(m, sh) and 908(w) cm−1 (Fig. 2), respectively, present also on the spectrum of the complex. The other bands observed for the film are within the range of C–C aromatic in plane stretching vibrations36 (Fig. 2A, band at 1562(m) cm−1), within the range of aromatic C–H in plane bending vibrations36,37 (Fig. 2, bands at 1276(m, sh), 1247(m), 1177(m, sh), and 978(w) cm−1), within the range of aromatic C–H out of plane bending36,37 (Fig. 2B, bands at 892(m), 874(w, sh), 818(w), and 804(w) cm−1) and within the range of C–C aromatic out of plane bending36 (Fig. 2A, band at 703(w, sh) cm−1). Among the mentioned bands, the ones at 1276(m, sh), 892(m) (Fig. 2B) and 703(w, sh) cm−1 (Fig. 2A) appear at frequencies characteristic also for CH2Cl2 bands.38,39
 | ||
| Fig. 2 FTIR ATR spectra of poly[Ni(salcn)] electrosynthesized in: (A) Bu4NPF6 and (B) Bu4NClO4. | ||
The band at 703(w, sh) cm−1 (Fig. 2A) is broad and clearly complicated indicating that its origin can be ascribed both to solvent and to C–C out of plane bending vibrations. The fourth band characteristic for this type of solvent occurs at 750 cm−1 and is strong.38,39
On the spectrum of the complex (Fig. 1) the band present at 750(s) cm−1 corresponds to o-substituted aromatic rings, thus the band at that frequency on the film spectrum (Fig. 2B, 750(w) cm−1) can not be ascribed solely to the presence of CH2Cl2. The very low intensity of the two characteristic CH2Cl2 bands, 1276(m, sh) cm−1 and 750(w) cm−1 (Fig. 2B), indicates that there is only a tiny amount of solvent within the film structure. This fact is reasonable taking into account the low boiling point of CH2Cl2 (39.6 °C) as well as the electrode preparation procedure for FTIR ATR measurements. Thus, the much higher intensity band at 892(m) cm−1 (Fig. 2B) for CH2Cl2 indicates that its origin is connected with both the solvent and the aromatic C–H out of plane bending36,39 vibrations.
The presence of additional bands of the films within the range of aromatic compound vibrations (Fig. 2A and B, 1562(m), 1276(m, sh), 1247(m), 1177(m, sh), 978(w), 892(m), 874(w), 818(w), 804(w), 703(w, sh) cm−1), with the exception of the band at 1276(m, sh) cm−1, corresponds to the vibration of phenolate moieties and is a consequence of changes in their surroundings, resulting from the formation of new covalent bonds between aromatic rings. An important evidence of changes in the external aromatic ring vicinity is the extra bandwidth observed within the range of phenyl ring in plane vibration (Fig. 2A, 1562(m) cm−1), indicating the conjugated aromatic ring system36,40,41 and hence the phenyl–phenyl type of polymerization.
The comparison of the intensities of bands observed for the complex form with the ones corresponding to the film additionally confirms the presented electropolymerization mechanism. The most common bands have been compared (Table S1, ESI†), with the exception of the fingerprint region, in which the overlay of bands occurs and hence their interpretation may be erroneous. In the film, the bands corresponding to the phenyl ring in plane vibration (Fig. 2A, 1541(m, sh) and 1475(m, sh) cm−1) decreased significantly (more than twice). Such behaviour is probably connected with the biggest stiffening of the structure in the area of the coupling. Moreover, large differences in band intensities (1601(m, sh), 1541(m, sh) and 1475(m, sh) cm−1, Fig. 2A) support the hypothesis that the mechanism of electropolymerization is of phenyl–phenyl type (all corresponding bands for the complex are equally strong).
The bands ascribed to the CH2 bending vibration in cyclohexane42 (Fig. 2A, 1454(m) cm−1) and the band corresponding to C–N43 stretching vibrations (Fig. 2A, 1390(m) cm−1) have been slightly reduced (approximately 1.5 times). This confirms, what is obvious, the lack of involvement of these groups in the coupling.
The fact that the band intensity (Fig. 2A, 1624(s) cm−1) corresponding to the CN stretching vibrations44 (the group that could be involved in the delocalization of the charge) has not changed at all excludes the participation of these bonds in the coupling.
Another band observed on the spectrum of the film, whose intensity was reduced to the greatest extent, is the band corresponding to C–O vibrations (Fig. 2A, 1319(m) cm−1).43 Taking into account the unchanged intensity of the band associated with the CN stretching vibrations (Fig. 2A, 1624(s) cm−1) one can suppose that the delocalization of the charge occurs through C–O bonding, omitting the CN bond. Such a conduction pathway was previously shown for the salen complex with a substituted ethylene imine bridge.19
On the basis of the above analysis one can attempt to assign bands appearing at 518(w) and 581(m) cm−1 (Fig. 1 and 2), that are absent on the spectrum of the uncomplexed ligand and thus might be attributable to Ni–O and Ni–N bonding, respectively. The band at 581(m) cm−1 for the film decreased significantly indicating the participation of the band corresponding to this bandwidth in the coupling process. Taking into account the considered direction of delocalization, this band may correspond to Ni–O bonding.
The changes of bands in the area of the C–H out of plane bending vibrations in the aromatic ring (900–581 cm−1) as well as the CC out of plane bending vibrations in the aromatic ring (675![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 730 cm−1) indicate the change in the substitution of the aromatic ring.36 For the complex, in this region, there are two strong bands at 750(s) and 738(s) cm−1 (Fig. 1) confirming the o-substituted structure of the coordination compound. The third band of medium intensity at 846(m) cm−1 originates from the ring stretching in the cyclohexane moiety.42 On the spectrum of the film (Fig. 2A) (this is also the area of one of the bands observed for the solvent), both bands are also visible, but they are very weak, indicating the presence of a small amount of the complex in the structure of the film. Additionally, within the region corresponding to the substitution pattern of the aromatic ring there are five more bands (892(m), 874(m), 818(w), 804(w) (Fig. 2B) and 703(w, sh) (Fig. 2A)), absent in the spectrum of the sole complex. Bands at 874(w, sh) and 818(w) cm−1 (Fig. 2B) may correspond to the 1,2,4-substituted form.36 This assignment may be justified because the peak at 874(w, sh) cm−1 is superimposed on the 892(m) cm−1 band, hence the actual intensity is likely to be small. This means that the ratio of the intensity of pair of bands corresponding to such substitution (874(w, sh) and 818(w) cm−1) suits the ratio presented in the literature.36 Another pair of bands at 804(w) (Fig. 2B) and 703(w, sh) cm−1 (Fig. 2A) could confirm the 1,2,3 substitution36, but the fact that they were noticed on different spectra impedes formulating such hypotheses. Whereas the most intense, in the group, band at 892(m) cm−1 (Fig. 2B) associated with the presence of trace amounts of solvent is far too intense for the sole origin of CH2Cl2 and may be attributed to the 1,2,3,5 substituted form of the compound.36
730 cm−1) indicate the change in the substitution of the aromatic ring.36 For the complex, in this region, there are two strong bands at 750(s) and 738(s) cm−1 (Fig. 1) confirming the o-substituted structure of the coordination compound. The third band of medium intensity at 846(m) cm−1 originates from the ring stretching in the cyclohexane moiety.42 On the spectrum of the film (Fig. 2A) (this is also the area of one of the bands observed for the solvent), both bands are also visible, but they are very weak, indicating the presence of a small amount of the complex in the structure of the film. Additionally, within the region corresponding to the substitution pattern of the aromatic ring there are five more bands (892(m), 874(m), 818(w), 804(w) (Fig. 2B) and 703(w, sh) (Fig. 2A)), absent in the spectrum of the sole complex. Bands at 874(w, sh) and 818(w) cm−1 (Fig. 2B) may correspond to the 1,2,4-substituted form.36 This assignment may be justified because the peak at 874(w, sh) cm−1 is superimposed on the 892(m) cm−1 band, hence the actual intensity is likely to be small. This means that the ratio of the intensity of pair of bands corresponding to such substitution (874(w, sh) and 818(w) cm−1) suits the ratio presented in the literature.36 Another pair of bands at 804(w) (Fig. 2B) and 703(w, sh) cm−1 (Fig. 2A) could confirm the 1,2,3 substitution36, but the fact that they were noticed on different spectra impedes formulating such hypotheses. Whereas the most intense, in the group, band at 892(m) cm−1 (Fig. 2B) associated with the presence of trace amounts of solvent is far too intense for the sole origin of CH2Cl2 and may be attributed to the 1,2,3,5 substituted form of the compound.36
However, due to the fact that bands attributed to C–H bending out of plane in the aromatic ring (900–581 cm−1) and CC bending out of plane in the aromatic ring are observed in the fingerprint region, we cannot draw conclusions regarding the phenolate moieties. So the main criterion of determining the way of substituting the phenolate moieties are bands within the phenyl ring in plane vibration regions. A much stronger band at 1601(m, sh) cm−1 (Fig. 2A) (in comparison with the other ones at 1562(m), 1541(m, sh) and 1475(m, sh) cm−1) indicates non-linear polyphenyl molecules,36,45 which gives the possibility of forming a crosslinked polymer. The nature of the observed overtones (1670–2000 cm−1)46 may also prove the existence of this form of the polymer. A signal in this area, closer to the lower range of overtones (Fig. 2A, 1730(vw) cm−1), indicates a tetra-substituted aromatic ring.46 For the complex the higher intensity of the overtones occurs in the centre part of the range and is present in the form of three bands (Fig. 1) confirming the o-position substitution of the phenolate moiety. Considering all the features of the presented spectra of films one can argue that the resulting poly[Ni(salcn)] consists of 1,2,4- and 1,2,3,5-chains and may also contain 1,2,3 tri-substituted aromatic rings as well as molecules of the complex, the supporting electrolyte and the solvent. The possibility of obtaining a cross-linked polymer is very crucial because the polymer obtained from a complex with a smaller ligand such as salen, which has been investigated by Audebert et al.,16,17 polymerized only in the o-position. The reason for the possibility of creating a crosslinked polymer by [Ni(salcn)] is probably the presence of the cyclohexane moiety in the imino bridge, affecting the increase in the distance between the polymer chains.
Voltammetric study of electropolymerization of complexes
step)) very close to the oxidation potential of [Ni(salcn(2Bu))] (0.85 V in TBAP/CH2Cl2),23 so one can conclude that this peak corresponds to the analogical process which takes place during the oxidation of [Ni(salcn(2Bu))].
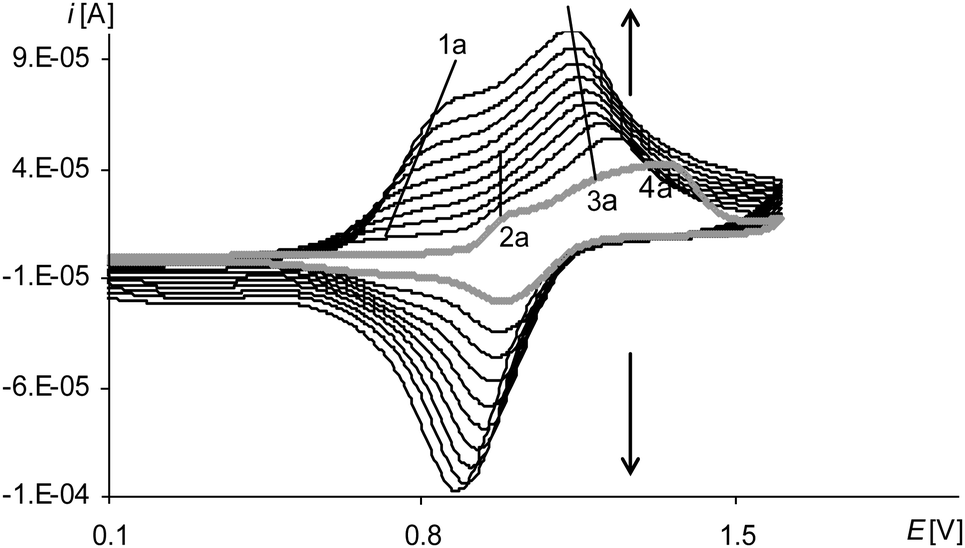 | ||
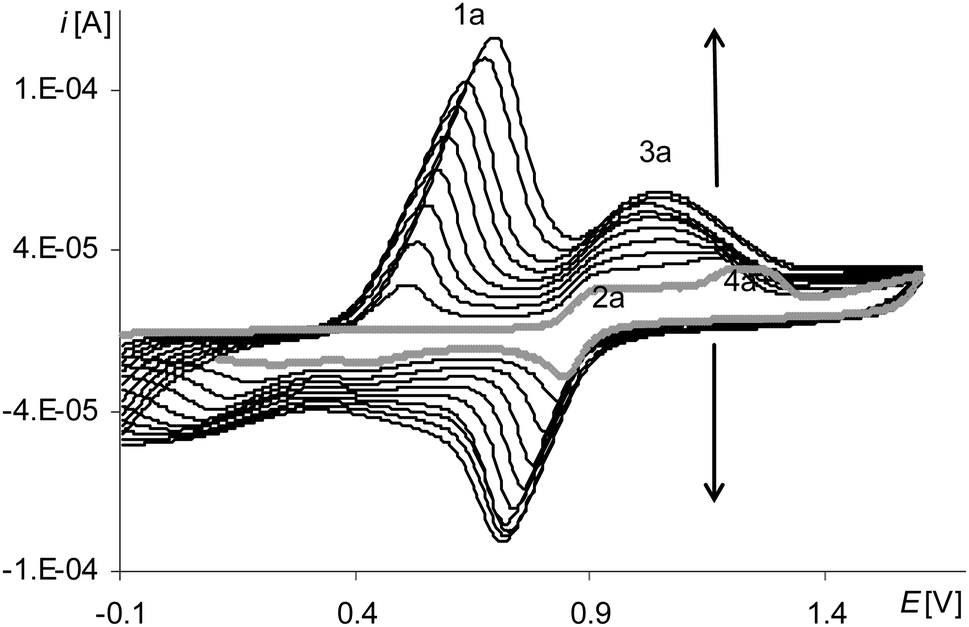
| Fig. 3 Cyclic voltammograms of [Ni(salcn)] in TBAH/CH2Cl2, 0–1.6 V, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; gray line – 1st scan, black lines – 2nd–10th scan. | ||
The other complexes during the first step undergo the oxidation process at higher potential values (Table 2, Epa(Istep), Fig. S2, ESI†). Taking into account that among previously investigated unpolymerized complexes,23 various substituents have not influenced the mechanism and only caused the shifts of the peak potentials for the subsequent steps (in the case of [Ni(salcn(2Bu))] and [Ni(salcn(BuOMe))] the difference of Epa(Istep) was 0.15 V23) hence the first oxidation step of [Ni(salcn)] and [Ni(salcn(Me))] (Table 2, Epa(Istep)) may be also connected with the oxidation of the phenolate moiety to the Ni(II)-phenoxyl radical complex. Another oxidation mechanism, based on the central ion oxidation, as was indicated by Dahm18 and Timonov21,22 for the complexes with a shorter imine bridge, has been noticed in the DMSO solutions.23 The reason is it is probably connected with the coordination properties of the solvent, which stabilizes the thermodynamically unstable Ni(III) ion.
For [Ni(salcn)] and [Ni(salcn(Me))] complexes after the first stage of the process a subsequent reaction takes place. This fact was determined on the basis of electropolymerization processes, carried out within the potential regions limited to the potential of the first anodic peak. The results of these electropolymerization processes are yellow films on electrode surfaces, the same as the ones obtained when the process was conducted within the whole investigated potential range. Electrodes obtained in such a manner are electroactive in the supporting electrolyte solution. The characteristics of their voltammograms are similar to those gained during electropolymerization over the entire short range of potentials (Fig. 4 and 7 dashed black line).
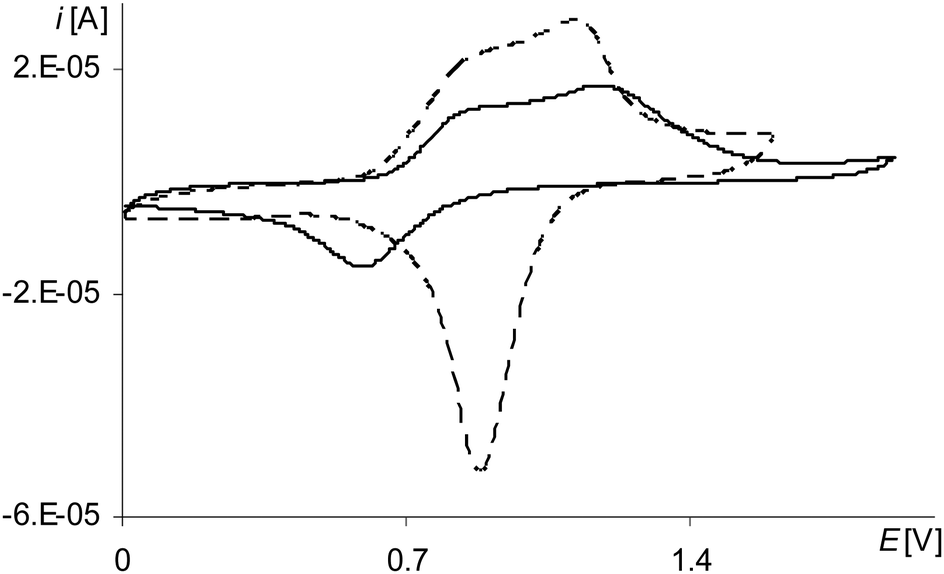 | ||
| Fig. 4 Cyclic voltammograms of poly[Ni(salcn)] in TBAH/CH2Cl2, 2nd scan, v = 0.05 V s−1, vs. Ag/AgCl; solid line – after electropolymerization: 3 scans, 0–2.1 V, v = 0.05 V s−1; dashed line – after electropolymerization: 10 scans, 0–1.6 V, v = 0.05 V s−1. | ||
The results of FTIR ATR studies indicate that the subsequent reactions are dimerization and polymerization.
The further peaks on the polymerization curves may have originated both from the oxidation of the complex solution as well as from the oxidation of the film.
Curves for all of the investigated complexes and their films show differences regarding the types of substituents in phenolate moieties. This phenomenon indicates that the oxidation process is based on the ligand.19,47 In the oxidation process based on the central metal ion it seems that such effect should be much lower.47
Analysis of the peaks was carried out based on a study of modified electrode electroactivity in a solution of the same supporting electrolyte as that in which electropolymerization was carried out.
The shape of most of the film peaks is sharp which indicates the advantage of the surface character of the electrode processes. Typical superficial processes are characterized by sharp peaks and high symmetry against the current–voltage axis.48 This is a consequence of such properties of the film (low thickness and/or high charge transfer rate) which enables the redox process along the whole length of polymer chains.48 The sharpest shape of peaks was observed for polymers of the [Ni(salcn(Bu))] complex. The milder character of polymer peaks of other complexes, especially [Ni(salcn)], indicates that redox conductivity in these films is partly limited and does not allow the complete redox process to occur in the outer layers of the films.
The last peak in the first polymerization cycle (Fig. 3, gray line 4a) is associated with the oxidation of complex solution because it is not observed on the cyclic voltammogram of poly[Ni(salcn)] (Fig. 4, dashed black line)). By analogy to unpolymerized complexes it indicates the oxidation of the Ni(II)-phenoxyl radical complex to the Ni(II)-bis-phenoxyl radical complex (II oxidation step). The higher potential as compared to the oxidation potential of [Ni(salcn(2Bu))]˙+ and [Ni(salcn(BuOMe))]˙+23 is the result of the lack of substituents in the phenolate moiety, which can influence the charge density of active centres and hence the value of the oxidation potential. This peak is absent in the subsequent polymerization curves (Fig. 3, 2nd–10th scan), the other peaks grow thus indicating the increase in the weight of the polymer. The increase of the peaks is accompanied by shifting of the potential of the first anodic peak (Fig. 3, 2nd–10th scan, 1a) to the right. This fact indicates that the increase in the polymer film thickness enhances difficulties in the charge transfer to the extent that after the 20th polymerization cycle the peak currents decrease or even disappear.
The reason of the fast decay of film activity is the polymer net structure that was evidenced by the FTIR ATR investigation results.
During the electropolymerization of [Ni(salcn)] within the long potential range, there is a third anodic peak (Fig. 5, the 1st scan – black line 6a; Table 2, Epa(IIIstep)). This peak is not visible from the second polymerization cycle. Because it does not occur on the cyclic voltammogram of poly[Ni(salcn)] (Fig. 4, the solid black line), hence it indicates that the complex solution is the sole source of the signal of this step of electropolymerization process. By analogy to its unpolymerized counterparts it corresponds to the III oxidation step of the complex solution. Due to the lack of the substituent within the phenolate moieties, for this step also, the potential peak value (Table 2, Epa(IIstep)) is higher than the potential value for unpolymerized complexes.23 The absence of this peak during the process carried out on the modified electrode (the second polymerization cycle) indicates the participation of [Ni(salcn)]2+ in the subsequent reaction with the film.
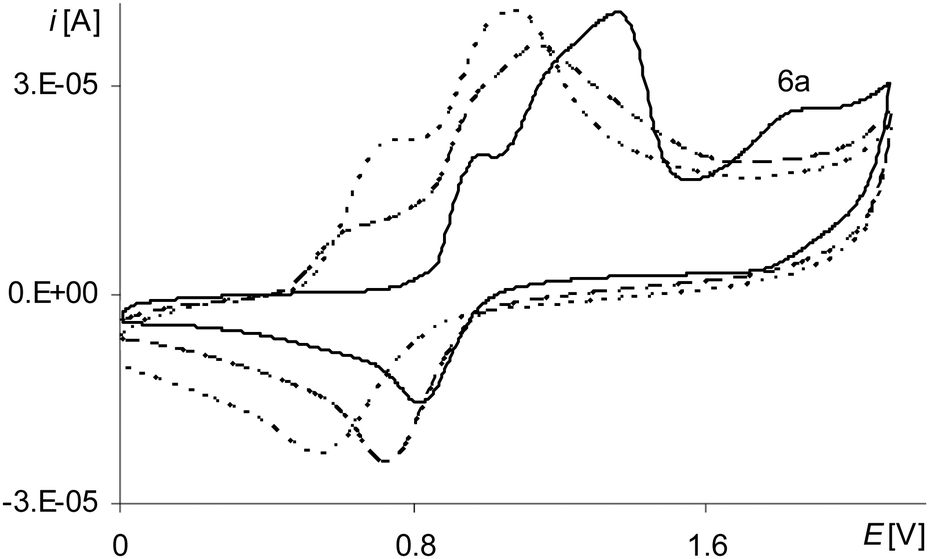 | ||
| Fig. 5 Cyclic voltammograms of [Ni(salcn)] in TBAH/CH2Cl2, 0–2.1 V, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; solid line – 1st scan, dashed line – 2nd scan, dotted line – 3rd scan. | ||
 | ||
| Fig. 6 Cyclic voltammograms of [Ni(salcn(Me))] in TBAH/CH2Cl2, 0–1.6 V, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; gray line – 1st scan, black lines – 2nd–10th scan. | ||
The peak of the third electropolymerization step of the process (Fig. 8, the gray line 6a; Table 2Epa(IIIstep), Fig. S3, ESI†), analogously as for [Ni(salcn)], reflects only the oxidation reaction of the complex solution and it is observed also only in the first cycle. However, unlike the case of [Ni(salcn)] at the first electropolymerization cycle for [Ni(salcn(Me))] at a slightly lower potential (Fig. 8, the gray line 5a, Fig. S2, ESI†), there is inflection that in subsequent cycles changes into a very small peak (Fig. 8, the dotted and solid black lines, 5a). The presence of the corresponding peak at the same potential value on the voltammogram of poly[Ni(salcn)] (Fig. 7, solid black line) indicates the third step of film oxidation. The fact that the third oxidation step of the film takes place also during the electropolymerization process of this complex, contrary to [Ni(salcn)], is probably connected with the steric effect of methyl substituents affecting the film structure and hence the mass transfer for polymer films. The presence of this peak may indicate that the mass transfer into the o-substituted polymer is easier than into the polymer film with free active positions located on the phenolate moieties.
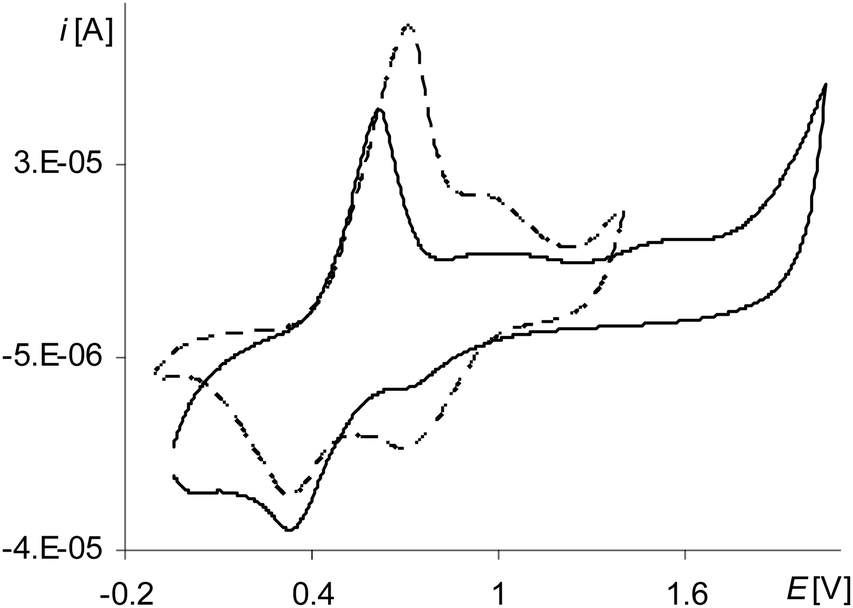 | ||
| Fig. 7 Cyclic voltammograms of poly[Ni(salcn(Me))] in TBAH/CH2Cl2, 2nd scan, v = 0.05 V s−1, vs. Ag/AgCl; solid black line – after electropolymerization: 3 scans, 0–2.1 V, v = 0.05 V s−1; dashed black line – after electropolymerization: 10 scans, 0–1.6 V, v = 0.05 V s−1. | ||
However, the process of the third oxidation step of the poly[Ni(salcn(Me)) film is restricted. The low value of the peak current observed for this step (Fig. 8, 5a) indicates that the process is harder than the oxidation of the film in the initial two steps (Fig. 6, 1a, 3a). This process also depends on the solvent in which the modified electrode is working. The peaks of the third oxidation step of the poly[Ni(salcn(Me))] film occur at polymerization curves up to only the first 3–5 cycles, depending on the speed of the process, while at the cyclic voltammogram of poly[Ni(salcn(Me))] up to the 6–7 ones. This shows that the reason probably lies in the kinetics restrictions, arising from the transfer of the bigger amount of mass into the film during the electropolymerization process than during the electroactivity study.
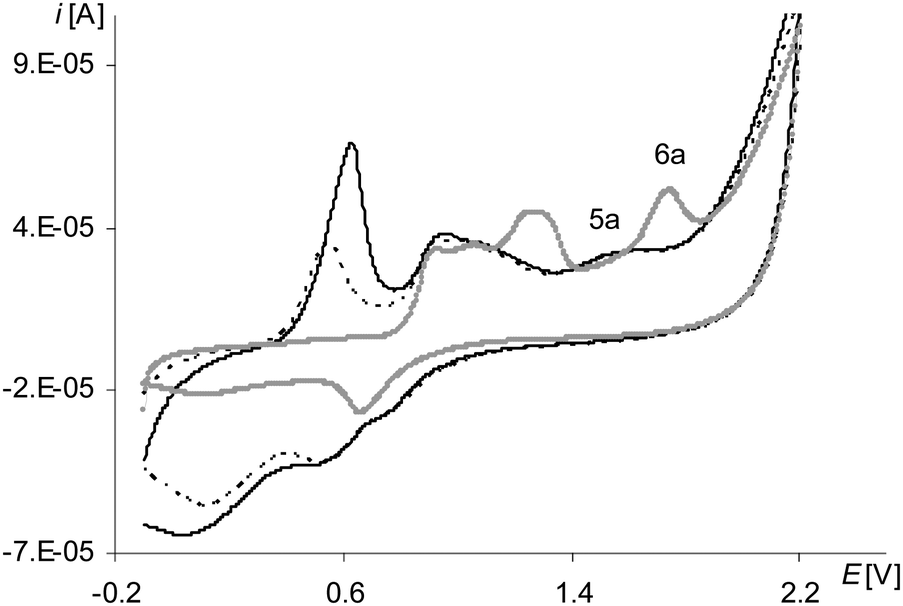 | ||
| Fig. 8 Cyclic voltammograms of [Ni(salcn(Me))] in TBAH/CH2Cl2, 0–2.1 V, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; gray line – 1st scan, dotted line – 2nd scan, solid black line – 3rd scan. | ||
The presence and growth of the peak in all polymerization cycles (Fig. 9, black lines, 2a) that in the first cycle was the signal of the oxidation of the complex solution (Fig. 9, gray line 2a) as well as the presence of the peak on the cyclic voltammogram of poly[Ni(salcn(Bu))] at the corresponding potential (Fig. 10, black lines) indicate the origin of the first anodic peak of polymerization both from the Ni(II)-phenoxyl radical complex and from the film.
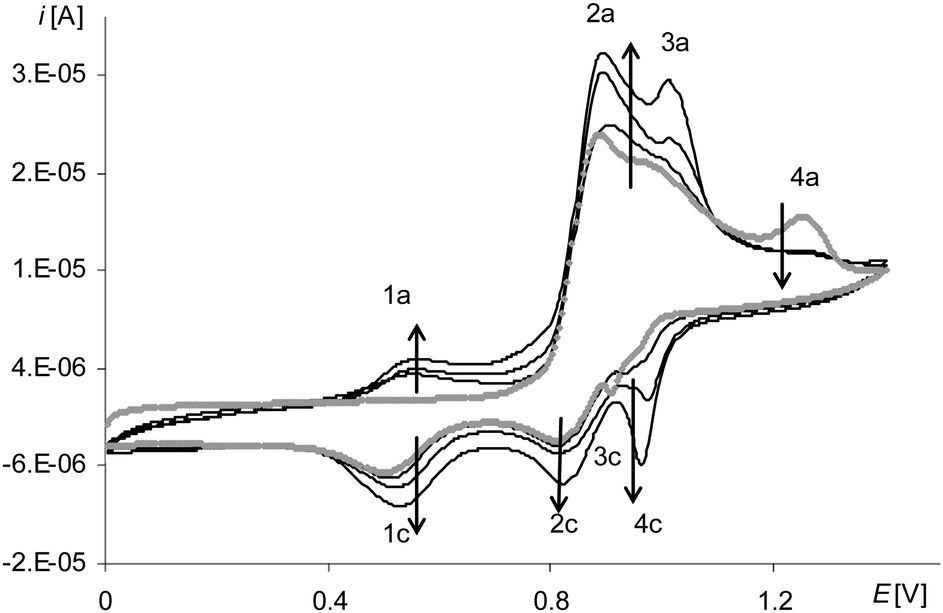 | ||
| Fig. 9 Cyclic voltammograms of [Ni(salcn(Bu))] in TBAH/CH2Cl2, 0–1.6 V, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; gray line – 1st scan, black lines – 2nd, 10th, 30th scan. | ||
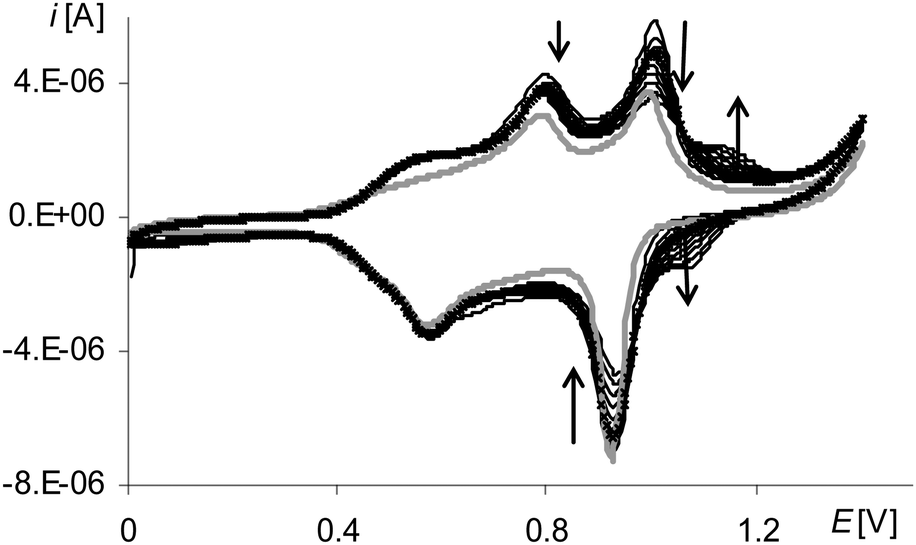 | ||
| Fig. 10 Cyclic voltammograms of poly[Ni(salcn(Bu))] in TBAH/CH2Cl2, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; black lines – 1st–10th scan after electropolymerization: 10 scans, 0–1.6 V, v = 0.05 V s−1; gray line – 10th scan after electropolymerization: 3 scans, 0–2.1 V, v = 0.05 V s−1. | ||
After the oxidation step of the complex solution to the Ni(II)-phenoxyl radical on the anodic curve, one can see a lower, slightly formed peak (Fig. 9, gray line 3a). On the cathodic part of the curve at the highest potential, there is very little visible inflection (Fig. 9, gray line 4c), followed by a small peak (Fig. 9, gray line 3c). In the next cycle, within the potential range of 4c (Fig. 9) there are inflections observed that, in the electropolymerization process, change into one sharply-outlined peak (Fig. 9, 30th scan 4c). Also in this potential range, the corresponding oxidation peak, observed from the first cycle, increases (Fig. 9, 3a), which clearly indicates that the finally formed redox system 3a/4c (Fig. 9) originates from the film. However, the method of forming of a cathodic peak of the redox system indicates certain heterogeneity of the film at the initial stage of its formation. One of the factors affecting this lack of homogeneity can be adsorption of the complex on the electrode surface. For such a step of the electrode process a cathodic peak 3c (Fig. 9, gray line) is observed during the first cycle, that is characteristic of substrate adsorption.50 The shape of the corresponding peak 3a (Fig. 9, gray line) points out that it is complicated, hence it can also comprise the signal originating from the oxidation of the adsorbed substrate. The higher redox potential value of the adsorbed complex is connected with the necessity of overcoming the adsorption forces.
Another redox system (Fig. 9, 1a/1c), for which the oxidation peak appears only in the second cycle, is derived from the film.
In contrast, the last peak on the first anodic polymerization curve (Fig. 9, gray line 4a, Fig. S1, ESI†), by analogy with other complexes, corresponds to the oxidation of the Ni(II)-phenoxyl radical to the Ni(II)-bis-phenoxyl radical. Its presence, though marked to a small extent but observed until the end of the polymerization process, indicates the smallest, among all the analyzed radicals, reactivity of [Ni(salcn(Bu))]+.
The electrode after the electropolymerization process is covered, as the previously presented ones, with a yellow film.
The polymerization process of the [Ni(salcn(Bu))] complex in the short potential range also differs from the other two complexes. A small increase in peak current is seen during the progression of the process. The confirmation of such behaviour is a small increase in the film as well as much lower (∼6 times) peak currents at the voltammogram of poly[Ni(salcn(Bu))] (Fig. 10, black lines). In addition, on the voltammogram of poly[Ni(salcn(Bu))] after repeated scanning processes (regardless of the thickness of the polymer) a new redox system (Fig. 10, black lines, last redox couple) is observed. This can suggest the presence of some species which activate during the scanning of the modified electrode.
In order to analyze the electropolymerization mechanism, the process was carried out in limited potential ranges, successively to the first peak potential (Fig. 9, gray line, 2a) and a second peak potential (Fig. 9, gray line 3a) within the first electropolymerization curve.
During the process carried out to the first peak potential, on the electropolymerization curve there is a redox system 1a/1c (Fig. 11A, black lines) that originated from the film and an anodic peak 2a (Fig. 11A, black lines) that within the unlimited potential range corresponds to the oxidation of the complex solution and the film. In the range limited to the first peak potential, the anodic peak 2a (Fig. 11A, black line) does not increase during successive cycles, indicating the oxidation only of complex solution. After this process, the electrode is covered with a gray unstable film (not visible after washing the electrode) and on the voltammogram in this way modified the electrode (Fig. 11B, black line), no peak is observed. The result of the experiment shows that in the range limited to a first peak potential, the electropolymerization process does not proceed and the redox system 1a/1c (Fig. 11A, black line), even though it originated from the film, is not a response connected with the structure of the neat polymer.
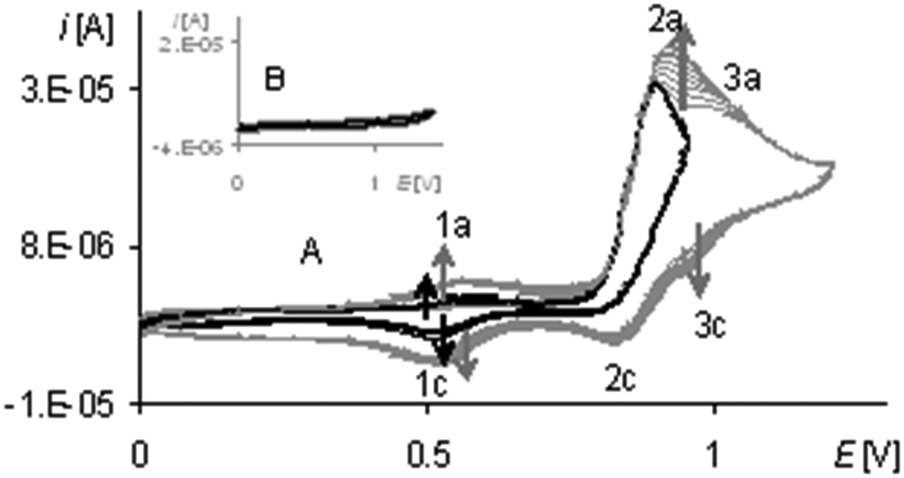 | ||
| Fig. 11 (A) Cyclic voltammograms of [Ni(salcn(Bu))] in TBAH/CH2Cl2, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; gray lines – 1–10th scan, 0–1.2 V; black lines – 1–10th scan, 0–0.9 V. (B) Cyclic voltammetry of poly[Ni(salcn(Bu))] in TBAH/CH2Cl2, 1st scan, v = 0.05 V s−1, vs. Ag/AgCl; gray line – after electropolymerization: 10 scans, 0–1.2 V, v = 0.05 V s−1; black line – after electropolymerization: 10 scans, 0–0.9 V, v = 0.05 V s−1. | ||
Even after the electropolymerization process performed within the range limited to the second peak potential, observed on the first polymerization curve (Fig. 9, gray line 3a) a gray film is observed, which is also not visible after washing the electrode.
On the basis of the measurements carried out in the limited potential ranges it can be concluded that the preparation of the polymer poly[Ni(salcn(Bu))], in contrast to others, is possible only at the potential of the last peak (on the first polymerization curve (Fig. 9, gray line 4a), and thus at the potential of Ni(II)-bis-phenoxyl radical formation. These investigations indicate that the poly[Ni(salcn(Bu))] film may comprise in its structure species that are weakly bound to the electrode surface. Such hypothesis may be justified taking into account the formation of a new redox system during the scanning of the modified electrode within the whole short potential range (Fig. 10, black lines, last redox coupe). In the polymer film obtained from the solution consisting of only the monomer and supporting electrolyte, the species that may be present in the film may be dimers or oligomers.
The reason of the presence of such species within the structure of poly[Ni(salcn(Bu))] is probably connected with the nature of the monomer in which the biggest substituents, among all of the investigated compounds, are attached.
It is generally known that tert-Bu substituents in o-, m- and p-positions in phenol moieties stabilize phenoxyl radicals.51 In non-polymerizing [Ni(salcn(2Bu))] molecules23tert-Bu substituents stabilize the Ni(II)-phenoxyl radicals so much that they exist in the solution for a period of about 5 min. Therefore, it seems to be reasonable, in the [Ni(salcn(Bu))] molecule, although containing two times less tert-Bu substituents, that it shows the stabilizing effect of Ni(II)-phenoxyl radicals as well as macro-Ni(II)-phenoxyl radicals. The latter impede the formation of the polymer so that it occurs only after the step of creating a more reactive species of Ni(II)-bis-phenoxyl radicals. The consequence of this stabilization may be the presence of additional dimers and oligomers in the film structure, affecting the heterogeneity of the film. Hence the low yield of the process of electropolymerization is observed, a measure in which there are much lower peaks on the voltammogram of poly[Ni(salcn(Bu))] than the ones noticed on the electropolymerization curve. Unbound polymer oligomers and dimers are active within the range of the peaks potential 1a/1c and 2a/2c (Fig. 11B, gray lines), but they are so weakly adsorbed on the electrode surface that the film containing the polymer is destroyed after rinsing of the electrode (Fig. 11A, gray lines).
The result of the experiment carried out in each of the short potential range variant shows that the film structure of poly[Ni(salcn(Bu))], apart from the polymer, consists of the complexes adsorbed on the electrode surface. There can be also some oligomers present that may cause the lack of identity of the polymer film. The FTIR ATR studies indicate the presence of the complex in the film.
The process of electropolymerization of [Ni(salcn(Bu))] within the long potential range is different from the discussed process for other complexes. This difference is that the peak of the third stage of complex solution oxidation (Fig. 12, gray and black lines, 6a) and the peak of the third stage of film oxidation (Fig. 12, gray and black lines, 5a) were observed until the last cycle of the process, regardless of the number of electropolymerization cycles. In analogy to their non-polymerized counterpart ([Ni(salcn(2Bu))]) (1.73 V in TBAP/CH2Cl2)23), the abovementioned peaks (Table 2) correspond to the presence of Ni(II)-phenoxonium cations in the solution of the complex and in the film.
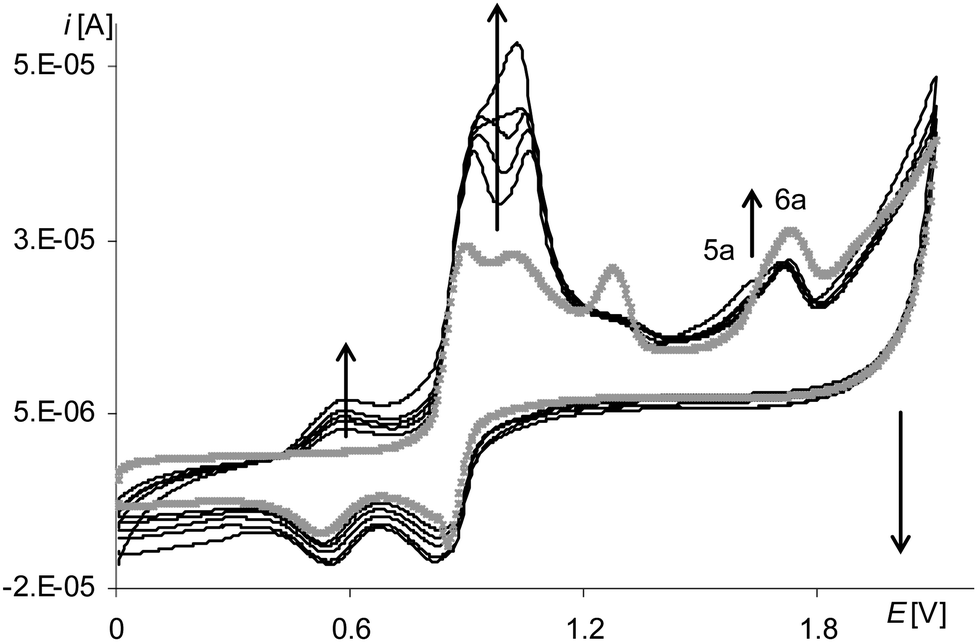 | ||
| Fig. 12 Cyclic voltammograms of [Ni(salcn(Bu))] in TBAH/CH2Cl2, 0–2.1 V, v = 0.05 V s−1, vs. Ag/AgCl; T = 293 K; gray line – 1st scan, black lines – 5–10th scan. | ||
Such a result indicates that the three-step oxidation of the film is inhibited, neither during the electropolymerization, nor during the electroactivity test, as it was in the case of [Ni(salcn(Me)]. In contrast to the other two complexes, the oxidation of the complex solution during electropolymerization is also not inhibited.
After the electropolymerization process in the long potential range, difference between peak currents in the cyclic voltammograms of poly[Ni(salcn(Bu))] and electropolymerization curves becomes comparable with those differences for other polymers. This indicates an increase in electropolymerization process efficiency, whose only reason in a solution with unchanged concentration can only be the presence of more reactive forms of Ni(II)-phenoxonium cations. Furthermore, after such electropolymerization processes, on the voltammograms of poly[Ni(salcn(Bu))] the formation of the additional redox system is not observed (Fig. 10, gray line). Such a system is shown in the voltammograms of poly[Ni(salcn(Bu))] after the electropolymerization process was carried out in a short potential range (Fig. 10, black lines, last redox couple). This fact may suggest that in the film formed in the range covering the higher potential values, some of the oligomers might be incorporated into the film structure. On the other hand the polymer film obtained by electropolymerization in a short potential range is not fully formed and further modification of the process occurs during the scanning of the modified electrode in a solution of the supporting electrolyte.
Conclusions
The anodic electropolymerization method applied to [Ni(salcn)], [Ni(salcn(Me))] and [Ni(salcn(Bu))] complexes yields three yellow-coloured electroactive polymer films.In these polymers FTIR and FTIR ATR measurements showed the existence of phenyl–phenyl type bonds.
The poly[Ni(salcn(Me))] and poly[Ni(salcn(Bu))] films form such bonds through the only active and free 5 position. Whereas the poly[Ni(salcn)] film consists of 1,2,4 and 1,2,3,5 chains and can also incorporate 1,2,3 tri-substituted aromatic rings. In all of the investigated films the presence of the free complex, supporting the electrolyte and solvent molecules, has been detected.
On the basis of cyclic voltammetry, the analyses of the electropolymerization mechanisms for the complexes and the redox processes occurring in the modified electrode films have been performed. Three-step processes of the complex oxidation were found. First the oxidation proceeds through Ni(II)-phenoxyl radical form, then the Ni(II)-bis-phenoxyl radical and finally Ni(II)-phenoxonium cation form. The obtained species differ in oxidation potential and stability of various forms depending on the nature of the substituent in the phenolate moiety. It has been shown that the step after which the follow-up reactions occurs (dimerization and polymerization) also depends on the nature of the substituent affecting the reactivity of the radicals.
Oxidized species derived from [Ni(salcn(Bu))] seem to show the least activity, due to the stabilizing effect of the most bulky substituents, which allows us to observe the second and the third oxidation step of the complex until the end of the whole electropolymerization process. Also for this reason, the preparation of poly[Ni(salcn(Bu))], in contrast to obtaining the polymers of other complexes, takes place only at the potential of the second oxidation step of the complex. Furthermore the electropolymerization of [Ni(salcn(Bu))] is the least efficient.
[Ni(salcn)] and [Ni(salcn(Me))] complexes polymerize after the first oxidation step, while the [Ni(salcn(Bu))] complex at the same potential forms only oligomers weakly bound to the electrode surface. For the [Ni(salcn(Bu))] complex the additional step of electropolymerization process which is the adsorption of the substrate to the electrode surface was evidenced. For this reason poly[Ni(salcn(Bu))] films have the least regular structures.
The heterogeneity of poly[Ni(salcn(Bu))] structures is partially reduced by the electropolymerization process carried out to the potential value corresponding to the third oxidation step of the complex. This may probably influence the inclusion of oligomers in the polymer structure. Therefore, poly[Ni(salcn(Bu))] films obtained during the process of electropolymerization carried out in a short potential range (0 to 1.6 V) undergo modification in an electrolyte solution. The films formed in a long potential range (0 to 2.1 V) are stable.
References
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420 CrossRef CAS; T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef PubMed; Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603 CrossRef PubMed; S. Trevin, F. Bedioui, M. G. G. Villegas and C. Bied-Charreton, J. Mater. Chem., 1997, 7, 923 RSC; A. Szczepanik and A. Sobkowiak, Catal. Lett., 2008, 126, 261 CrossRef; D. Naróg, U. Lechowicz, T. Pietryga and A. Sobkowiak, J. Mol. Catal. A: Chem., 2004, 212, 25 CrossRef; P. Seliger, N. Sołtys, G. Andrijewski and M. Siwy, New J. Chem., 2012, 36, 2607 RSC; N. Sołtys, P. Seliger, G. Andrijewski and M. Siwy, RSC Adv., 2013, 3, 25351 RSC; T. Matsushita, D. T. Sawyer and A. Sobkowiak, J. Mol. Catal. A: Chem., 1999, 137, 127 CrossRef; L. Nowak, G. Andrijewski, D. Tomczyk and T. Błaszczyk, Pol. J. Chem., 2009, 83, 2195 Search PubMed; D. Tomczyk, G. Andrijewski, L. Nowak, P. Urbaniak and D. Sroczyński, Inorg. Chim. Acta, 2012, 390, 70 CrossRef.
- L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85 RSC.
- E. Szłyk, A. Surdykowski, M. Barwiolek and E. Larsen, Polyhedron, 2002, 21, 2711 CrossRef CAS; E. Szłyk, M. Barwiołek, R. Kruszynski and T. J. Bartczak, Inorg. Chim. Acta, 2005, 358, 3642 CrossRef; M. Barwiołek, E. Szłyk, A. Surdykowski and A. Wojtczak, Dalton Trans., 2013, 42, 11476 RSC; M. K. Bera, Ch. Chakraborty and S. Malik, J. Chem., 2015, 39, 9207 Search PubMed; A. Choudhary, B. Das and S. Ray, Dalton Trans., 2015, 44, 3753 RSC; M. Mohammadikish, CrystEngComm, 2014, 16, 8020 RSC.
- C. Gosden, K. P. Hely and D. Pletcher, J. Chem. Soc., Dalton Trans., 1978, 972 RSC; C. Gosden, J. B. Kerr, D. Pletcher and R. Rosas, J. Electroanal. Chem. Interfacial Electrochem., 1981, 117, 101 CrossRef CAS; A. Gogoi, A. Dewan, G. Borah and U. Bora, New J. Chem., 2015, 39, 3341 RSC; E. Szłyk, A. Wojtczak, E. Larsen, A. Surdykowski and J. Neumann, Inorg. Chim. Acta, 1999, 293, 239 CrossRef; E. Dunach, M. J. Medeiros and S. Olivero, New J. Chem., 2006, 30, 1534 RSC; P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC.
- C. Gosden and D. Pletcher, J. Organomet. Chem., 1980, 186, 401 CrossRef CAS; F. Azevedo, C. Freire and B. de Castro, Polyhedron, 2002, 21, 1695 CrossRef; P. W. Raess, M. S. Mubarak, M. A. Ischay, M. P. Foley, T. B. Jennermann, K. Raghavachari and D. G. Peters, J. Electroanal. Chem., 2007, 603, 124 CrossRef; S. Özalp-Yaman, V. T. Kasumov and A. M. Önal, Polyhedron, 2005, 24, 1821 CrossRef.
- S. Bhattacharjee, K.-E. Jeong, S.-Y. Jeong and W.-S. Ahn, New J. Chem., 2010, 34, 156 RSC; M. Martins, C. Freire and A. R. Hillman, Chem. Commun., 2003, 434 RSC; Z. Li, S. Wu, H. Ding, H. Lu, J. Liu, Q. Huo, J. Guan and Q. Kann, New J. Chem., 2013, 37, 4220 RSC; M. F. S. Teixeira and T. R. L. Dadamos, Procedia Chem., 2009, 1, 297 CrossRef CAS; H. Yoon, T. R. Wagler, K. J. O'Connor and C. J. Burrows, J. Am. Chem. Soc., 1990, 112, 4568 CrossRef; R. Zhang, L. Liu, Y. Li, W. Wang and R. Li, Int. J. Electrochem. Sci., 2015, 10, 2355 Search PubMed.
- F. Azevedo, M. A. A. F. de C. T. Carrondo, B. de Castro, M. Convery, D. Domingues, C. Freire, M. T. Duarte, K. Nielsen and I. C. Santos, Inorg. Chim. Acta, 1994, 219, 43 CrossRef CAS.
- B. de Castro and C. Freire, Inorg. Chem., 1990, 29, 5113 CrossRef CAS.
- O. Rotthaus, O. Jarjayes, F. Thomas, C. Philouze, C. P. D. Valle, E. Saint-Aman and J.-L. Pierre, Chem. – Eur. J., 2006, 12, 2293 CrossRef CAS PubMed; O. Rotthaus, O. Jarjayes, C. P. D. Valle, C. Philouze and F. Thomas, Chem. Commun., 2007, 4462 RSC.
- O. Rotthaus, F. Thomas, O. Jarjayes, C. Philouze, E. Saint-Aman and J.-L. Pierre, Chem. – Eur. J., 2006, 12, 6953 CrossRef CAS PubMed.
- Y. Shimazaki, T. Yajima, F. Tani, S. Karasawa, K. Fukus, Y. Naruta and O. Yamauchi, J. Am. Chem. Soc., 2007, 129, 2559 CrossRef CAS PubMed.
- Y. Shimazaki, T. D. Stack and T. Storr, Inorg. Chem., 2009, 48, 8383 CrossRef CAS PubMed; Y. Shimazaki, N. Arai, T. J. Dunn, T. Yajima, F. Tani, C. F. Ramogida and T. Storr, Dalton Trans., 2011, 40, 2469 RSC.
- L. Chiang, R. M. Clarke, K. Herasymchuk, M. Sutherland, K. E. Prosser, Y. Shimazaki and T. Storr, Eur. J. Inorg. Chem., 2016, 49 CrossRef CAS; F. Thomas, Dalton Trans., 2016, 45, 10866 RSC.
- K. A. Goldsby, J. K. Blaho and L. A. Hoferkamp, Polyhedron, 1989, 8, 113 CrossRef CAS.
- L. A. Hoferkamp and K. A. Goldsby, Chem. Mater., 1989, 1, 348 CrossRef CAS.
- P. Audebert, P. Capdevielle and M. Maumy, Synth. Met., 1991, 41, 3049 CrossRef CAS; P.-H. Aubert, P. Audebert, P. Capdevielle, M. Maumy and M. Roche, New J. Chem., 1999, 23, 297 RSC.
- P. Audebert, P. Hapiot, P. Capdevielle and M. Maumy, J. Electroanal. Chem., 1992, 338, 269 CrossRef CAS; M. Maumy, P. Capdevielle, P.-H. Aubert, P. Audebert and M. Roche, New J. Chem., 1997, 21, 621 Search PubMed.
- C. E. Dahm, D. G. Peters and J. Simonet, J. Electroanal. Chem., 1996, 410, 163 CrossRef.
- M. Vilas-Boas, C. Freire, B. de Castro, P. A. Christensen and A. R. Hillman, Inorg. Chem., 1997, 36, 4919 CrossRef CAS.
- J. Tedim, S. Patrício, J. Fonseca, A. L. Magalhães, C. Moura, A. R. Hillman and C. Freire, Synth. Met., 2011, 161, 680 CrossRef CAS.
- T. Y. Rodyagina, P. V. Gaman'kov, E. A. Dmitrieva, I. A. Chepurnaya, S. V. Vasil'eva and A. M. Timonov, Russ. J. Electrochem., 2005, 41, 1239 CrossRef.
- O. V. Levin, M. P. Karushev, A. M. Timonov, E. V. Alekseeva, S. Zhang and V. V. Malev, Electrochim. Acta, 2013, 109, 153 CrossRef CAS.
- D. Tomczyk, L. Nowak, W. Bukowski, K. Bester, P. Urbaniak, G. Andrijewski and B. Olejniczak, Electrochim. Acta, 2014, 121, 64 CrossRef CAS.
- L. Nowak, G. Andrijewski, D. Tomczyk and N. Cichomski, Pol. J. Chem., 2007, 81, 493 CAS.
- H. H. Freedman, J. Am. Chem. Soc., 1961, 83, 2900 CrossRef CAS.
- O. Signorini, E. R. Dockal, G. Castellano and G. Oliva, Polyhedron, 1996, 15, 245 CrossRef CAS.
- J. R. Zamian and E. R. Dockal, Transition Met. Chem., 1996, 21, 370 CrossRef CAS.
- G. A. Kolawole and K. S. Patel, J. Coord. Chem., 1986, 14, 235 CrossRef CAS; M. Lalia-Kantouri, M. Hartophylles, P. D. Jannakoudakis and G. P. Voutsas, Z. Anorg. Allg. Chem., 1995, 621, 645 CrossRef.
- G. C. Percy and D. A. Thornton, J. Inorg. Nucl. Chem., 1973, 35, 2319 CrossRef CAS.
- G. Condorelli, I. Fragalá, S. Giuffrida and A. Cassol, Z. Anorg. Allg. Chem., 1975, 412, 251 CrossRef CAS; F. A. Bottino, P. Finocchiaro and E. Libertini, J. Coord. Chem., 1988, 16, 341 CrossRef.
- V. T. Kasumov and F. Köksal, Spectrochim. Acta, Part A, 2005, 61, 225 CrossRef PubMed.
- G. L. Estiú, A. H. Jubert, J. Costamagna and J. Vargas, THEOCHEM, 1996, 367, 97 CrossRef.
- H. B. Gray and C. J. Ballhausen, J. Am. Chem. Soc., 1963, 85, 260 CrossRef CAS.
- V. R. de Souza, A. M. da Costa Ferreira and H. E. Toma, Dalton Trans., 2003, 458 RSC; T. Kizaki, T. Matsumoto and S. Ogo, Dalton Trans., 2010, 39, 1339 RSC , ESI.†.
- Ch. Zafiu, G. Trettenhahn, D. Pum, U. B. Sleytr and W. Kautek, Phys. Chem. Chem. Phys., 2011, 13, 13232 RSC; S. López-Romero, J. Castillo-Mendoza, J. Chávez-Ramírez and J. Muñoz-García, Rev. Mater., 2005, 10, 413 Search PubMed.
- J. Mohan, Organic Spectroscopy: Principles and Applications, CRC Press, Alpha Science, 2nd edn, 2004, pp. 81–82 Search PubMed.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons, 2004, p. 74 Search PubMed.
- J. Mohan, Organic Spectroscopy: Principles and Applications, CRC Press, Alpha Science, 2nd edn, 2004, pp. 78–79, 95 Search PubMed.
- NIST Chemistry WebBook CH2Cl2, http://webbook.nist.gov.
- NIST Chemistry WebBook benzene diphenyl, http://webbook.nist.gov.
- Y. Li, H. Xu, X. Tao, K. Qian, S. Fu, Y. Shen and S. Ding, J. Mater. Chem., 2011, 21, 1810 RSC.
- J. Mohan, Organic Spectroscopy: Principles and Applications, CRC Press, Alpha Science, 2nd edn, 2004, pp. 79–80 Search PubMed; NIST Chemistry WebBook cyclohexane, http://webbook.nist.gov.
- O. Signorini, E. R. Dockal, G. Castellano and G. Oliva, Polyhedron, 1996, 15, 245 CrossRef CAS; J. R. Zamian and E. R. Dockal, Transition Met. Chem., 1996, 21, 370 CrossRef; X. Wang, X. M. Zhang and H. X. Liu, Polyhedron, 1995, 14, 293 CrossRef; G. C. Percy and D. A. Thornton, J. Inorg. Nucl. Chem., 1973, 35, 2319 CrossRef.
- J. A. Faniran, K. S. Patel and J. C. Bailar Jr., J. Inorg. Nucl. Chem., 1974, 36, 1547 CrossRef CAS; A. Seminara, S. Giuffrida, A. Musumeci and I. Fragalá, Inorg. Chim. Acta, 1984, 95, 201 CrossRef; Ch.-W. Ho, W.-Chi. Cheng, M.-Ch. Cheng, S.-M. Peng, K.-F. Cheng and Ch.-M. Che, J. Chem. Soc., Dalton Trans., 1996, 405 RSC.
- D. Talbi and G. S. Handler, J. Mol. Spectrosc., 2012, 275, 21 CrossRef CAS.
- J. R. Dyer, Application of Absorption Spectroscopy of Organic Compounds, Prentice-Hall, Englewood Cliffs, New York, 1965 Search PubMed, Fig. 2.28.
- M. Vilas-Boas, I. C. Santos, M. J. Henderson, C. Freire, A. R. Hillman and E. Vieil, Langmuir, 2003, 19, 7460 CrossRef CAS.
- P. T. Kissinger and W. R. Heineman, Laboratory Techniques in Electroanalytical Chemistry, Marcel Dekker, 2nd edn, 1996, pp. 420–421 Search PubMed.
- C. G. Zoski, Handbook of Electrochemistry, Elsevier Science, 1st edn, 2006, p. 183 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods. Fundamentals and applications, John Wiley & Sons, New York, 2nd edn, 2001, p. 595 Search PubMed.
- R. D. Webster, Electrochem. Commun., 2003, 5, 6 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1. Comparison of the FTIR ATR bands of poly[Ni(salcn)] films (electrosynthesized in Bu4NPF6 or Bu4NClO4) with the FTIR bands of the [Ni(salcn)] complex; Fig. S1. Cyclic voltammograms of complexes in TBAH/CH2Cl2, 0.1–1.6 V, v = 0.05 V s−1, 1st scans vs. Ag/AgCl and Fig. S2. Cyclic voltammograms of complexes in TBAH/CH2Cl2, 0.1–2.1 V, v = 0.05 V s−1, 1st scans vs. Ag/AgCl. See DOI: 10.1039/c6nj03635j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |