
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, metal binding and spectral properties of novel bis-1,3-diketone calix[4]arenes†
Sergey N.
Podyachev
 *a,
Svetlana N.
Sudakova
a,
Gulnaz Sh.
Gimazetdinova
b,
Nataliya A.
Shamsutdinova
a,
Victor V.
Syakaev
a,
Tatjiana A.
Barsukova
a,
Nobuhiko
Iki
c,
Dmitry V.
Lapaev
d and
Asiya R.
Mustafina
a
*a,
Svetlana N.
Sudakova
a,
Gulnaz Sh.
Gimazetdinova
b,
Nataliya A.
Shamsutdinova
a,
Victor V.
Syakaev
a,
Tatjiana A.
Barsukova
a,
Nobuhiko
Iki
c,
Dmitry V.
Lapaev
d and
Asiya R.
Mustafina
a
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, Arbuzov str., 8, 420088, Kazan, Russia. E-mail: spodyachev@iopc.ru; Fax: +7-843-273-1872
bKazan National Research Technological University, K. Marks str., 68, 420015, Kazan, Russia
cGraduate School of Environmental Studies, Tohoku University, 6-6-07 Aramaki-Aoba, Aoba-ku, Sendai 980-8579, Japan
dZavoisky Physical-Technical Institute of Kazan Scientific Center of Russian Academy of Sciences, 420029 Kazan, Russia
First published on 19th January 2017
Abstract
New bis-1,3-diketone derivatives of calix[4]arene (3–5) have been synthesized with good yields by the addition of a sodium salt of acetylacetone, 1-benzoylacetone and dibenzoylmethane to 5,17-bis-(bromomethyl)-25,26,27,28-tetrahydroxycalix[4]arenes. The structural properties of the obtained compounds and their complexes have been established by means of IR, UV-Vis, NMR spectroscopy and quantum-chemical calculations. The complex ability of bis-1,3-diketones towards Al3+, Ni2+, Cu2+ and lanthanide ions (Nd3+, Eu3+, Tb3+) has been investigated by using a liquid–liquid extraction method. The UV-Vis data indicate thermodynamically favorable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex formation of the ligands with Tb3+ in alkaline DMF, although the time required for the equilibration reveals the difference between calix[4]arenes bearing acetylaceton-, benzoylaceton- and dibenzoylmethane-substituents. The steric hindrance effect on keto–enol transformation is the reason for the difference. The ligand-centered emissions of Gd3+ complexes with benzoylaceton- and acetylaceton-substituted calix[4]arenes reveal them both as more convenient antennae for red and infra-red than for green lanthanide luminescence. Indeed, the benzoylaceton-substituted counterpart sensitizes Yb3+-centered luminescence to a good extent. Nevertheless, the luminescence of Tb3+ is sensitized by the acetylaceton-substituted counterpart to a better extent than that of Yb3+, while only poor red Eu3+ emission is observed under sensitization by both the ligands.
:1 complex formation of the ligands with Tb3+ in alkaline DMF, although the time required for the equilibration reveals the difference between calix[4]arenes bearing acetylaceton-, benzoylaceton- and dibenzoylmethane-substituents. The steric hindrance effect on keto–enol transformation is the reason for the difference. The ligand-centered emissions of Gd3+ complexes with benzoylaceton- and acetylaceton-substituted calix[4]arenes reveal them both as more convenient antennae for red and infra-red than for green lanthanide luminescence. Indeed, the benzoylaceton-substituted counterpart sensitizes Yb3+-centered luminescence to a good extent. Nevertheless, the luminescence of Tb3+ is sensitized by the acetylaceton-substituted counterpart to a better extent than that of Yb3+, while only poor red Eu3+ emission is observed under sensitization by both the ligands.
1. Introduction
Among various functional building blocks, 1,3-diketones have found wide applications as key reagents for the design of a variety of organic compounds1,2 and the synthesis of some drug compounds.3 Moreover, 1,3-diketone derivatives are known as invaluable chelating ligands for many transition metals in materials chemistry3–6 and can be potentially used in the synthesis of extractants6 and luminescent materials.7–11 Luminescent lanthanide complexes attract much attention due to their usage in optical communications and solar energy conversion,12–15 as well as in fluoroimmunoassay, bio-medical diagnostics and therapy.16–20 It has been well demonstrated that 1,3-diketones are ideal candidates for the sensitization of visible and near infrared (NIR) luminescence emitted by lanthanide ions due to the significant antenna effect. The introduction of bulky and fluoro-aliphatic substituents into 1,3-diketones can significantly improve lanthanide luminescence.11,21 Therefore, the design and synthesis of new 1,3-diketone ligands bearing various substituents seems to be an exciting topic of current investigations.Anchoring of functional groups on the suitable molecular platforms is worth noting as a promising strategy for preparing more advanced ligands with improved binding efficiency and selectivity towards metal ions. This tendency has become a challenging factor for the synthesis of poly-1,3-diketones.5,22,23
Recently, we have shown24,25 that calix[4]arenes and calix[4]resorcines serve as very promising platforms for embedding four 1,3-diketone moieties. The synthesized compounds appeared to be a rather effective antennae for Tb(III)-centered luminescence. The hydrophilic colloids prepared on the basis of Tb(III) and Gd(III) tetra-1,3-diketonate complexes were found to be promising candidates for biomedical applications as contrast agents due to their high photophysical24 and magnetic relaxation26 parameters correspondingly.
Despite the presence of four 1,3-diketone groups at the upper rim of the above mentioned calix[4]arenes, the coordination with metal ions was realized only by means of two chelating groups of one molecule, which favors further formation of a ternary complex. This fact is obviously the main reason for the luminescence response and realization of a sensing function of polyelectrolyte-coated colloids based on Tb(III) complexes of the calix[4]resorcinarene cavitand towards some of substrates in aqueous solutions.27 From this point of view, the synthesis of bis-1,3-diketone derivatives based on the calix[4]arene platform is an attractive direction in the design of lanthanide complexes for bio- and chemosensing. It is also worth noting that embedding aromatic substituents to ligands is a widely applied route to enhance the antenna-effect of the ligand and/or to make the ligand more suitable for sensitizing red or near infra-red lanthanide-centered luminescence.
Thus, herein, we report the synthesis of new bis-1,3-diketone derivatives of calix[4]arene bearing methyl and phenyl terminal substituents in 1,3-diketone fragments. The lipophilicity of calix[4]arene derivatives is the prerequisite for studying them in liquid–liquid extraction of some metal ions (Al3+, Ni2+, Cu2+, Nd3+, Eu3+, and Tb3+). The spectroscopic properties of the new calix[4]arene derivatives and the luminescence behaviour of their Tb3+, Gd3+, Eu3+ and Yb3+ complexes have been considered and compared with the previously obtained data for the tetra-1,3-acetylacetonyl analogue with an aim to highlight structure impact in complex ability and the antenna effect of novel compounds.
2. Results and discussion
2.1. Synthesis and characterization of bis-1,3-diketone calix[4]arenes 3–5
The synthetic procedures for the series of bis-1,3-diketones are summarized in Scheme 1. The halogenmethylated derivatives of calix[4]arenes can be successfully applied for the anchoring of 1,3-diketone groups at the upper rim of calix[4]arenes. In the case of tetra-hydroxy substituted calix[4]arenes, the halogenmethylation is carried out in the presence of an excess of alkyl chloromethyl ether and tin tetrachloride.28 The calix[4]arenes can be also chloromethylated by using paraformaldehyde/HCl in a dioxane/H3PO4/AcOH mixture.29–31 The synthesis of tetrahydroxytetrakis(halogenmethyl)calix[4]arene was previously performed by the interaction of 1 with an excess of paraformaldehyde in the presence of Zn and HBr in glacial acetic acid.32 It was recently demonstrated that this reaction can be also applied for obtaining bis-halogen derivatives of the calix[4]arene 2.33 We have successfully synthesized compound 2 by using a slightly modified literature procedure. The reaction was carried out at room temperature for 5 days in the presence of 3 equivalents of paraformaldehyde. The spectral parameters of the obtained product 2 are in full agreement with the literature data.33 | ||
| Scheme 1 Synthetic routes and structural formulae of the investigated compounds 1–8. A similar numbering system of atoms is used in Table 1. | ||
The synthesis of bis-1,3-diketone derivatives was fulfilled by the addition of a sodium salt of the corresponding 1,3-diketone to the 5,17-bis-(bromomethyl)-25,26,27,28-tetrahydroxycalix[4]arene 2 in anhydrous dioxane under vigorous stirring. It should be noted that in all cases, the addition of sodium salt was accompanied by an intense pink-violet color which gradually changed to brownish yellow. A similar phenomenon was previously reported for tetra-substituted analogue 6.25 This color can be obviously explained by the formation of intermediate quinone structures in the reaction mixture after addition of a basic salt to compound 2.34,35 A high reactability of this intermediate brought about the target bis-1,3-diketones 3–5 with good yields (52–60%) even without heating. The obtained novel compounds were characterized by IR, 1H NMR, 13C NMR and MALDI-MS techniques.
The presence of a more broadened absorption band at ∼1600 cm−1 in the IR spectrum of compound 3 in comparison with 4 and 5 is probably caused by a considerable amount of the enol form in the former compound. The strong hydrogen bonding of the lower rim hydroxyl groups for 3–5 is evident from broadened and intensive bands ν(OH) at ∼3160–3193 cm−1 that are substantially lower than ν(OH)free ∼3500–3600 cm−1.36
Assignment of the signals in the NMR spectra of 3–5 (Table 1) was accomplished by means of 2D COSY, 1H–13C HSQC and 1H–13C HMBC experiments. It is well known that 1,3-diketones can exist as keto–enol tautomers.37 In the case of the bis-1,3-diketones, three main equilibrium forms can be realized (Scheme 2). According to 1H NMR data, the content of the enol form in CDCl3 solution amounts to 39% for 3 (C3 = 30 mM), which leads to the appearance of an additional set of signals in the NMR spectra of the compound. However, it is difficult to estimate the contribution of each form because of overlapping of the signals. In the case of 4 and 5, the enol form content is negligible (<0.1% for C3,4 = 30 mM).
| Atom | Compound | |||||
|---|---|---|---|---|---|---|
| 3 | 4 | 5 | ||||
| 1H | 13C | 1H | 13C | 1H | 13C | |
| a Numbering according to Scheme 1. b The assignment for the enolic forms is given in parentheses. | ||||||
| 1 | (16.80–16.83(OH)) | 203.6 (192.0) | (17.05(OH)) | 203.3 | (17.1–17.3(OH)) | 195.7 |
| 2 | 3.89 | 70.3 (108.5) | 4.69 | 65.0 | 5.40 t, 3J = 6.7 | 59.2 |
| 3 | 196.2 | |||||
| 4 | 2.91 (3.43) | 34.2 (32.08) | 3.10 | 34.2 | 3.19 d, 3J = 6.7 | 44.6 |
| 5 | 131.9 (135.1) | 132.2 | 132.8 | |||
| 5′ | 6.77 t, 3J = 7.3 | 129.4 | 6.74 t, 3J = 7.6 | 122.4 | 6.74 tr, 3J = 7.5 | 122.4 |
| 6 | 6.76 (6.71) | 128.4 (122.4) | 6.83 | 129.5 | 6.87 | 129.7 |
| 6′ | 7.03 d, 3J = 7.3 | 129.2 | 7.07 d, 3J = 7.6 | 129.2 | 7.07 d, 3J = 7.5 | 129.2 |
| 7 | 128.0 (127.9) | 128.5 | 128.5 | |||
| 7′ | 122.4 | 128.4 | ||||
| 8 | 10.0–10.3 | 149.0 (147.4) | 10.2 (OH) | 149.0 | 10.07 (OH) | 147.5 |
| 8′ | 147.7 | 147.5 | 10.20 (OH) | 149.0 | ||
| 9 | 3.41 eq. | 31.9 | 3.37 eq. | 31.8 | 3.33 eq. d, 2J = 13.9 | 31.7 |
| 4.17 ax. | 4.11 ax | 4.07 ax. d, 2J = 13.9 | ||||
| 10 | 2.10 (2.00) | 2.10 | 28.82 | |||
| 11 | 136.6 | 136.1 | ||||
| 12 | 7.78 | 128.7 | 7.77 d, 3J = 7.6 | 128.6 | ||
| 13 | 7.22 | 128.8 | 7.20 t, 3J = 7.6 | 128.9 | ||
| 14 | 7.34 | 133.8 | 7.33 t, 3J = 7.6 | 133.6 | ||
 | ||
| Scheme 2 Keto–enol tautomerization of bis-1,3-diketone calix[4]arenes. | ||
Hydrogen atoms in the enol fragments participate in the formation of strong hydrogen bonds (δ1H(OH) = 16.80–16.83 ppm for 3). It is interesting to note that the amount of the enol form realized in compound 3 is almost the same as in tetra-1,3-diketone 6 (40%).25 This fact testifies a weak mutual influence of 1,3-diketone groups on the keto–enol tautomerism in the investigated calix[4]arene derivatives. It can be also supposed that the substitutents in these compounds are turned out from the calix[4]arene cavity.
For all synthesized compounds 3–5 only a single peak of methylene-bridged carbon atoms is detected, which indicates a cone or a 1,3-alternate isomer of calix[4]arenes. According to the “de Mendoza rule”,38 the determined values of chemical shifts for these atoms ((δ13C(9) = 31.9 ppm for 3, 31.8 ppm for 4 and 31.7 ppm for 5) testify undoubtedly the cone isomer form for all investigated calix[4]arenes.
The chemical shifts for hydroxyl protons localized at the low rim of calix[4]arene molecules are not practically changed on going from parent calix[4]arene 1 (10.19 ppm)39 to the calix[4]arenes 3–5 (δ1H(8) = 10.0–10.3 ppm). These data indicate the maintenance of the initial cone conformation for calix[4]arene derivatives 3–5, stabilized by a circular hydrogen bond between hydroxyl groups similar to the calix[4]arene 1.40
2.2. Extraction of metal ions by bis-1,3-diketones 3–5
As it was mentioned above, 1,3-diketones are invaluable chelating ligands for complex formation with various transition metal ions and thus can extract metal ions from an aqueous phase to an immiscible organic phase. However, the application of more simple representatives of 1,3-diketones, such as acetylacetone or 1-benzoylacetone, is rather complicated due to the fact that their metal complexes are too hydrophilic to allow extraction into a nonpolar organic phase.6 Therefore, fixing of 1,3-diketone chelating groups on the calix[4]arene platform should be a very promising way to gain hydrophobicity and water insolubility, which can contribute to further development of extracting agents.The extractability of new bis-1,3-diketone calix[4]arenes 3–5 towards some industrially critical metal ions such as Al3+, Ni2+, Cu2+ and rare-earth ions (Nd3+, Eu3+, Tb3+) was estimated by the competitive extraction of the cations from their aqueous mixture into chloroform. The extraction was carried out during 24 hours to reach equilibrium conditions. The results shown in Fig. 1 demonstrate that the extraction percentage for metal ions goes up on going to higher pH, which can be explained by easier ionization of 1,3-diketone groups under alkaline conditions and hereby testifies that these cations are extracted due to the ion-exchange process.
 | ||
| Fig. 1 Effect of pH on the extraction percentage for different metal ions with (a) 3, (b) 4 and (c) 5. [Metal ion] = 0.1 mM; [L3–5] = 0.5 mM. | ||
In the case of bis-1,3-acetylacetonyl derivative 3, notable extraction of Al3+ and Cu2+ was detected after pH 4.0, but Nd3+, Eu3+ and Tb3+ metal ions were extracted by this compound only at pH > 6. An almost quantitative extraction (>90%) for the former metal ions was already observed at pH 6, while the extraction of lanthanide ions reached a maximum at pH 8. At the same time, a quite low extractability of the derivative 3 towards Ni2+ ions (E < 11%) was observed in the whole range of pH values. In the case of bis-1,3-diketones 4 and 5, the extraction of Al3+ and Cu2+ was detected only after pH 5 and 6, which is obviously due to the lower acidity of these compounds in comparison with 3. The results obtained earlier for the acidity of tris(1,3-diketones) having similar substitutents and linked by the mesitylene spacer support this assumption.41 However, the extraction of lanthanide ions by compounds 4 and 5 also begins after pH 6 and becomes quantitative at pH 8, as well as for 3. The extraction efficiency for the investigated bis-1,3-diketones 3–5 remains the same and depends only slightly on the nature of the lanthanide ion.
It is worth noting that the presence of hydroxyl groups at the low rim of calix[4]arene molecules makes their participation in the coordination of metal ions possible. It was established, however, that the unmodified “classical” p-tert-butylcalix[4]arene only scarcely extracts transition metal ions (E% for Ni2+ and Cu2+ ∼1%) under similar experimental conditions.42 Therefore, a main role in the binding of metal ions by compounds 3–5 belongs to 1,3-diketone groups fixed at the upper rim of the calix[4]arene backbone.
Thus, we have shown here that bis-1,3-diketone calix[4]arenes 3–5 can act as rather effective pH-dependent extraction agents. The selective extraction of Cu2+versus Ni2+ can be utilized for analytical purposes. The lack of selectivity in the extraction of Nd3+, Eu3+, and Tb3+ reveals 3–5 as group extractants for lanthanide ions.
2.3. Electronic absorption spectroscopy
UV-Vis spectroscopy is a powerful tool to study complex formation of 1,3-diketones with lanthanide ions. This method is of particular importance for 1,3-diketone derivatives of calix[4]arenes and calix[4]resorcinarenes, since phenol and resorcinol moieties significantly contribute to the spectral behavior of their derivatives.24,25 The UV-Vis absorption spectra of the calix[4]arenes 1, 3–6, model compound 7 as well as phenol 8 recorded in DMF are shown in Fig. 2 and 3. Spectroscopic data are summarized in Table 2. | ||
| Fig. 2 UV-vis spectra of compounds 1, and 6–8 in DMF solution (C1 = 0.1 mM, C6 = 0.05 mM, C7 = 0.2 mM, C8 = 0.4 mM). | ||
 | ||
| Fig. 3 UV-Vis spectra of L (3 (a), 4 (b) and 5 (c)) (CL = 0.1 mM) in DMF (L); L in alkalized DMF (CTEA = 0.6 mM) (L-TEA (1:6)); L with Tb(NO3)3 (CTb3+ = 0.1 mM) (L-Tb (1:1)), and L with Tb(NO3)3 in alkalized DMF (L-Tb-TEA (1:1:6)). The spectra were recorded under equilibrium conditions. | ||
| Compound | λ max (nm) | ε max (103 M−1 cm−1) | Compound | λ max (nm) | ε max (103 M−1 cm−1) |
|---|---|---|---|---|---|
| 1 | 275 | 9.22 | 6 | 292 | 20.81 |
| 282 | 8.83 | ||||
| 3 | 288 | 13.42 | 7 | 282 | 3.71 |
| 4 | 282 | 13.25 | 8 | 274 | 2.28 |
| 280 | 1.86 | ||||
| 5 | 283 | 13.33 |
The absorption spectra of the investigated compounds demonstrate rather high values of the molar extinction coefficients (εmax = 8.83 × 103 M−1 cm−1 to 20.81 × 103 M−1 cm−1) with λmax in the range of 275 nm to 292 nm (Table 2) typical for π–π* electron transitions. Additionally, it is worth noting the shoulders at 310–340 nm in the spectra of the calix[4]arenes 3–5. A comparison of UV–Vis spectra for the model compounds 7 and 8 and the calix[4]arene molecules indicates that the absorptions of phenol and 1,3-dikenone fragments incorporated on the calix[4]arene platform are practically independent and additive. This tendency can be exemplified by the εmax values of phenol 8 (εmax = 2.28 × 103 M−1 cm−1 at 274 nm) and calix[4]arene 1 (εmax = 9.22 × 103 M−1 cm−1 at 275 nm). Similarly to this case, the absorption of bis-1,3-diketone 3 (εmax = 13.42 × 103 M−1 cm−1) can be approximated as the sum of the absorptions of its structural blocks (εcalc = 2(ε7 + ε8) = 11.98 × 103 M−1 cm−1). Such additivity indicates the lack of any conjugation between the chromophoric units of 3. However, a rather noticeable bathochromic shift (∼10 nm) is observed for 6 in comparison with 7. The εmax value for 6 (εmax (292 nm) = 20.81 × 103 M−1 cm−1) also deviates from εcalc (εcalc = 4ε7 = 14.84 × 103 M−1 cm−1). This result points to an impact of intramolecular interactions between four 1,3-diketone substitutents fixed on the macrocyclic platform on the spatial structure and spectral behavior of 6.
UV-Vis spectroscopy was applied to reveal the complex formation properties of ligands 3–5. The spectral changes of 3 under its complex formation with Tb(III) (Fig. 3a) are similar to those previously reported for its tetra-1,3-diketone counterpart 6.25 The spectral changes of 4 (Fig. 3b) are much less pronounced than those of 3 immediately after sample preparation, and tend to increase with time on approaching the equilibrium conditions in three days. A new intensive maximum at ∼320 nm appears in the spectrum of 4 when equilibrium conditions are attained. The time dependence for ligand 5 is more pronounced than that for 4 (Fig. S1, ESI†). In particular, insignificant spectral changes of 5 are observed within one day after the admixture of Tb3+ and TEA. Moreover, four days of storage of the solutions is not obviously enough for the achievement of equilibrium conditions for 5 (Fig. S1, ESI†). Moreover, the appearance of a new absorption band at about 350 nm in alkalized DMF solutions of calix[4]arene 5 and Tb3+ salt (Fig. 3c) is another difference between 5 and 4. The reasons for the time consuming complex formation are worth discussing.
Literature data highlight keto–enol tautomeric transformation as the most time-consuming step in complex formation, which is greatly affected by the substituent effect37,43,44 and nature of the solvent.37,45 It is worth noting that the α-substituent effect (α-position is designated in Scheme 2) is well documented in the literature,37,46 although no significant retardation of keto–enol transformation is observed for 3 (Fig. S1, ESI†) and its tetrakis-1,3-diketone counterpart,25 where acetylacetone moieties are fixed at the calix[4]arene backbone through α-substitution by a methylene linker. Nevertheless, in the case of the tetrakis-[(1,3-acetylaceton-3-yl)] derivative of calix[4]resorcine cavitand, which possesses a rather more rigid conformational structure, time dependent complex formation was observed.24
Some differences between keto–enol tautomeric transformations under substitution of methyl-groups of acetylacetone to phenyl moieties in dibenzoylmethane are highlighted in the literature,47 although they cannot explain the above mentioned difference in the complex formation for 3 and 5. Thus, the observed tendency points to the impact of both α- and β-substituents on the keto–enol tautomerism of 1,3-diketone derivatives of calix[4]arenes 3–5 under alkaline conditions. The above mentioned 1H NMR data also reveal the insignificant contribution of the enolic form for 4 and 5 in CDCl3, while both forms of 3 are in equilibrium under similar conditions.
The stabilization of the enolic form in non-polar solvents is well documented.37 On the other hand, the hydration is worth noting as another factor shifting the keto–enol equilibrium.41 These factors explain the lack of a detectable difference in the complex formation behavior of compounds 3–5 at the water–CHCl3 interface after 24 hour exposition (Fig. 1). This result indicates that the difference in keto–enol tautomerization for 3–5 is negligible in the extraction process conditions due to a greater rate of keto–enol transformation in the biphasic system.
In order to evaluate the stoichiometry of complex formation, a Job plot analysis of the spectral measurements with a varied L:Tb3+ molar ratio and a constant L:TEA molar ratio in DMF solutions has been performed. It can be clearly seen from Fig. 4 that in all cases, the long-term storage of DMF solutions containing Tb3+ complexes of the investigated ligands 3, 4 and 5 results in the maximum in the Job's plot being at 0.5 molar ratio. This fact apparently indicates that the Tb3+ ion preferably forms complexes of 1:1 stoichiometry with these compounds. It should be also noticed that at the initial stage, complexes of n:1 (n > 1) stoichiometry are obviously accumulated, which is well demonstrated in Fig. 4b for the ligand 4 after one day of solution storage (maximal L:Tb3+ ratio at 0.67). The observed tendency points to the transformation of kinetically favorable complexes of n:1 stoichiometry into more thermodynamically stable 1:1 complexes after more prolonged time of storage (3 days).
 | ||
| Fig. 4 The Job plot profiles of DMF solutions at varied L:Tb3+ molar ratios: (a) λ = 320 nm, [Tb3+] + [3] = 0.1 mM, L:TEA (1:6) and time of solution storage 1 day; (b) λ = 320 nm, [Tb3+] + [4] = 0.2 mM, L:TEA (1:6) and times of solution storage 1 day and 5 days; (c) λ = 340 nm, [Tb3+] + [5] = 0.2 mM, L:TEA (1:10) and time of solution storage 5 days. | ||
To confirm the assumed stoichiometry of the thermodynamically stable complexes we have accomplished a spectral titration experiment for compounds 3–5 with 1:1 molar ratio of Tb:L and varied L:TEA molar ratio in DMF solutions (Fig. 5). The obtained data testify that the addition of an excess of TEA leads to the deprotonation of two diketone groups from the molecule of bis-1,3-diketone 3 (Fig. 5a). Only one proton is eliminated in Tb(III)-containing alkaline solutions of 4 and 5 after one day of storage, while storage for four days at least is required for the second proton elimination (Fig. 5b and c). The results indicate 1:1 complex formation of Tb3+via two deprotonated 1,3-diketone groups. Additionally, quantum-chemical calculations have been accomplished to confirm the probability of this coordination mode.
 | ||
| Fig. 5 UV-Vis spectra and ΔA (λ = 320 nm) of the DMF solutions of 3 (a), 4 (b) and 5 (c) with Tb(NO3)3 ([3] = [4] = [5] = [Tb3+] = 0.1 mM) at varied TEA:L molar ratios and time of solution storage 1 day (1) and 4 days (2). | ||
2.4. The quantum-chemical calculations
Our previous report25 highlighted that only two 1,3-diketonate groups of 6 participate in the coordination of one lanthanide ion. Moreover, efficient coordination of lanthanide ions can be realized for all possible conformations of the calix[4]arene backbone. This fact is of particular importance under alkaline conditions, where deprotonation of the phenolic lower rim is the reason for conformation shift from cone to 1,2- or 1,3-alternates. Taking into account these tendencies, the efficiency of coordination of bis-1,3-diketone-derivatives 3–5 with lanthanide ions also can be realized in different conformations of the calix[4]arene backbone, including cone, partial cone and 1,3-alternate.Relative heat of formations for conformers of calix[4]arene 3 and their complexes [Tb3+Ln−] were obtained using MOPAC 2012 (Table 3). The structures obtained after SPARKLE/PM7 optimizations are presented in Fig. 6. The theoretically predicted stability order for 3 is cone > partial cone > 1,3-alternate (Table 3), which correlates with the number of hydrogen bonds between the OH groups at the calix[4]arene lower rim. This tendency is in agreement with the previous reports about the same stability order for conformers of calix[4]arene without any upper-rim substituents.48,49 The same tendency is revealed for Tb3+ complexes of 3, where the most energy gain is in cone conformation. In particular, the cone conformer of 3 is most favorable for both [Tb3+L2−] and [Tb3+L3−], where two 1,3-diketone groups are deprotonated. It is also worth noting that deprotonation of the phenolic lower rim of calix[4]arene 3 insignificantly influences the energy gap between the conformers (Table 3).
| Compound |
|
||
|---|---|---|---|
| Cone | Partial cone | 1,3-Alternate | |
| L | 0 | 17.38 | 22.48 |
| [Tb3+L2−] | 0 | 13.47 | 27.33 |
| [Tb3+L3−] | 0 | 13.69 | 29.37 |

 | ||
| Fig. 6 Sparkle/PM7 model optimized structures of three conformers of 3 and their Tb3+ complexes. Hydrogen atoms are omitted for clarity with the exception of the phenolic hydrogens. | ||
2.5. Photoluminescence spectroscopy
UV-Vis spectral data reveal similarity in complex formation for bis-(3) and tetrakis-1,3-diketone 6, which is confirmed by the similarity in the spectra of Tb3+-centered steady-state luminescence for the Tb3+ complexes of 3 and 6 (Fig. 7a, corresponding excitation spectra are shown in Fig. S2a, ESI†). At the same time, the sensitizing effects of ligands 4 and 5 on Tb3+-centered luminescence are insignificant. The steady state spectra point to some difference in the antenna-effects of ligands 3 and 6 on Tb3+-centered luminescence.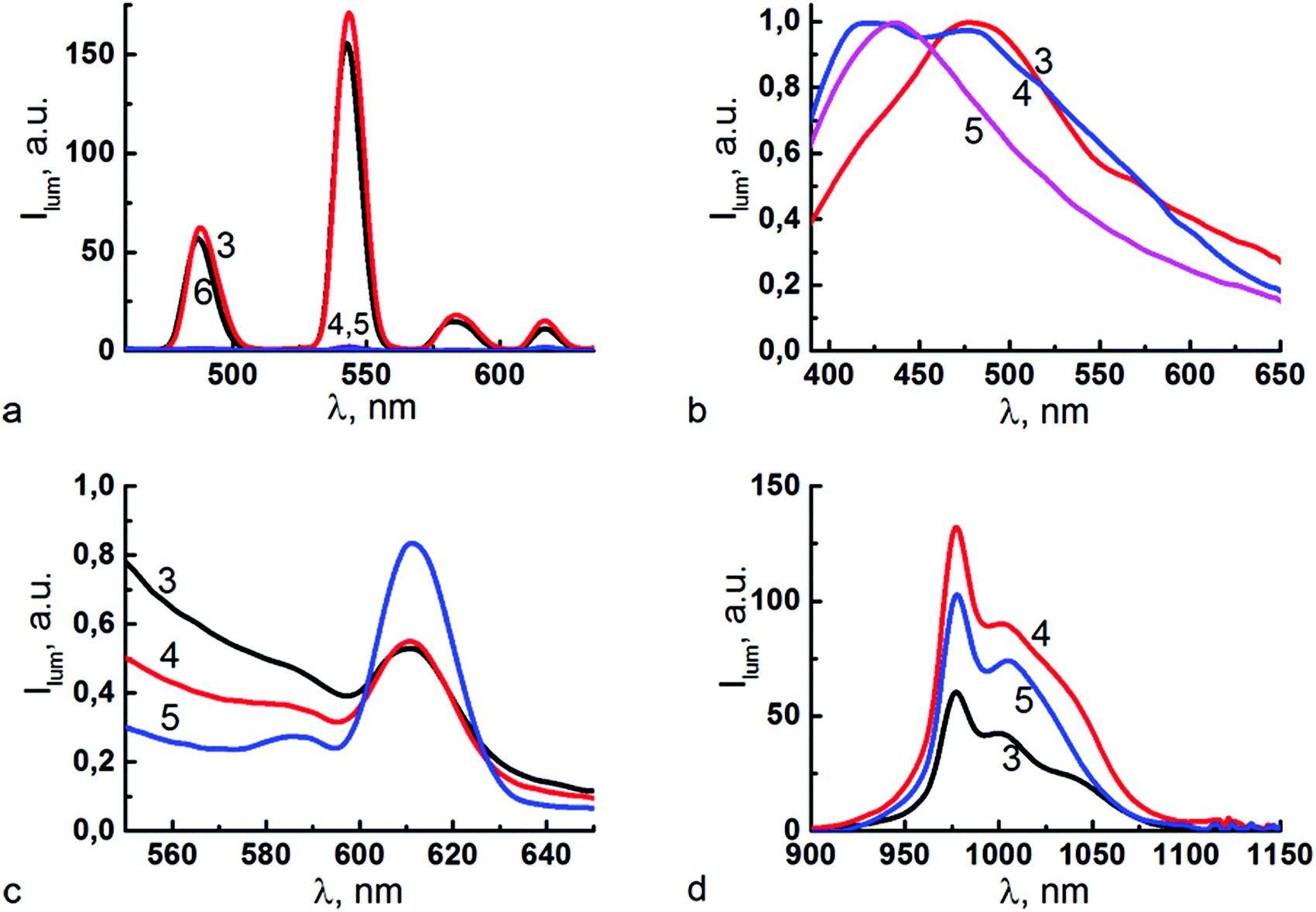 | ||
| Fig. 7 Luminescence spectra of alkalized DMF solutions of ligands L = 3–5 with Ln3+ = Tb3+ (a); Gd3+ (b), Eu3+ (c) and Yb3+ (d). λex = (a) 327 nm (3), 340 nm (4 and 5) and 330 nm (6); (b) 337 nm (3–5); (c) 345 nm (3), 340 nm (4 and 5); (d) 330 nm (3), 370 nm (4 and 5). (a and c) CL = CLn3+ = 0.1 mM, CTEA = 0.4 mM. (b) CL = CGd3+ = 1 mM, CTEA = 4 mM. (d) CL = CYb3+ = 0.2 mM, CTEA = 0.8 mM. Ligands are designated in the spectra by corresponding numbers. Luminescence spectra of Gd(III) complexes were recorded with 20 μs delay time. | ||
The time-resolved luminescence measurements (see Fig. S3 in the ESI†) reveal the longer excited state lifetime for the Tb3+-centered luminescence in complexes with ligand 3 (0.127 ± 0.004 ms) than that for its tetra-1,3-diketone counterpart 6 (0.053 ± 0.002 ms). This fact confirms coordination of Tb3+ ions by 1,3-diketonate groups of 3 as the dominant coordination mode under alkaline conditions (see Fig. 6). The longer lifetime of the Tb(III) complex with 3 compared with 6 points to either less non-radiative de-activation or less back transfer, or both. The interfering effect of 1,3-diketone groups of 6 not participating in coordination with Tb3+ ions can be assumed as a reason for more energy stock from the excited Tb3+ state to the vibrational levels of the ligand in comparison with 3. It is worth noting that the above mentioned peculiarity in the spectral behavior of 6, which is a bathochromic shift and adsorption non-additivity (see Section 2.3), correlates with the interfering effect of four 1,3-diketone groups embedded to the calix[4]arene backbone.
Literature results highlight the effect of aromatic or aliphatic substituents in 1,3-diketones on both the energy of singlet and triplet excited state levels and the intersystem crossing, which in turn affects their ability to sensitize lanthanide-centered luminescence.50 Dibenzoylmethane is well documented for its good antenna effect on near infra-red lanthanide-centered luminescence.51 Thus, Tb3+-, Eu3+- and Yb3+-centered luminescence measured for 1:1 complexes of ligands 3, 4, 5 and 6 is presented in Fig. 7 along with ligand-centered luminescence of the Gd3+ complex with 3–5. The corresponding excitation spectra are presented in Fig. S2 in the ESI.† The luminescence spectra of Gd(III) complexes are commonly applied in measuring the triplet level energy of the ligands in their lanthanide complexes due to Gd(III)-enhanced intersystem crossing.52 The luminescence spectra of Gd(III) complexes presented in Fig. 7b were recorded with a pronounced (20 μs) delay time in order to minimize the contribution of singlet level derived emission. Fig. S3 in the ESI† represents the ligand-centered luminescence for Gd(III) complexes recorded with the delay time varying within 1–20 μs.
The measured spectra indicate that Tb3+-centered luminescence is the highest for the complexes with 3versus those with 4 and 5 (Fig. 7a). The Eu3+-centered luminescence is very weak for all measured complexes (Fig. 7c). The Yb3+-centered luminescence is the highest for the complexes with 4versus those with 5 and 3. It is worth noting that the different kinetic conditions designated in the previous section were taken into account in the measurements of lanthanide-centered luminescence. Nevertheless, storage for three days is enough for the completed complex formation with lanthanides for 4, while the complex formation is still incomplete for 5 under the same conditions. Thus, the data obtained for 5 is greatly affected by the kinetic retardation of complex formation, while the kinetic effect is negligible for 4.
The luminescence spectra presented in Fig. 7a indicate that ligand 4 provides a worse antenna effect on Tb3+-centered luminescence than 3 and 6. The opposite tendency is observed for Yb3+-centered luminescence (Fig. 7d), where ligand 4 sensitizes Yb3+ luminescence to a better extent than 3, while rather weak emission bands at 614 nm are observed for Eu3+-centered luminescence for all ligands. The spectral pattern of Yb3+ complexes (Fig. 7d) reveals emission at 976 nm, which corresponds to the 0-phonon component of the 2F5/2 → 2F7/2 transition with additional transitions at lower energy assigned to transitions to other crystal field sublevels of the ground state and/or to vibronic contribution. Thus, ligand 4 can be considered as an antenna for Yb3+-centered NIR-luminescence, but not for green Tb3+- and red Eu3+-centered luminescence.
Since a ligand-to-metal energy transfer is of great impact in lanthanide-centered luminescence, the ligand-centered emission of Gd3+ complexes with ligands 3 and 4 (Fig. 7b) should correlate with the experimentally observed sensitizing effects of the ligands on the emission of their lanthanide complexes (Fig. 7a, c and d). The data (Fig. 7b) reveal similar bands at 480–490 nm, while the emission of the Gd(III) complex with 5 is characterized by a band at shorter wavelengths (∼420 nm). A shoulder at the shorter wavelengths is also revealed for the Gd(III) complex with 4. The emission at 480–500 nm tends to enhance under the longer delay time (Fig. S3 in the ESI†). The decay measurements at 475–500 nm reveal rather long excited state lifetimes (3.5–4.0 μs), which confirms the triplet derived emission at these wavelengths. The phosphorescence emission at 480–500 nm is in good confirmation with the literature results on triplet level energies measured at 77 K for benzoyl-substituted 1,3-diketonate Gd(III) complexes.53 The emission at 400–430 nm characterized by the excited lifetime in the range of 3.2–3.3 μs should be assigned to 1,3-diketone derived emission.52 Thus, the data presented in Fig. 7b indicate the great contribution of the 1,3-diketone form for the complexes with ligand 5, and the small or negligible contributions of its enol forms complexed with Gd(III). This spectral behavior of the complex with 5 is in good agreement with the above mentioned incomplete complex formation of this ligand. It is also worth noting that the spectral behavior of Gd(III) complexes with 3 and 4 reveals them both as better antennae for red Eu3+- and infra-red Yb3+-centered luminescence versus green Tb3+-centered emission. Thus, the poor Eu3+-centered luminescence (Fig. 7c) points to efficient quenching processes peculiar for Eu(III) complexes.54 Moreover, both Tb3+- and Yb3+-centered luminescence (Fig. 7a and d) reveal the difference between ligands 3 and 4, which indicates that further studies are required to reveal the reasons for the difference.
3. Conclusions
The present work represents the synthesis of novel bis-1,3-diketone derivatives of calix[4]arene with (acetylaceton-3-yl)methyl-, (benzoylaceton-3-yl)methyl- and (dibenzoylmethan-3-yl)methyl-moieties as promising ligands for complex formation with lanthanide ions. The pH-dependent extraction of metal ions at the water–CHCl3 interface indicates efficient complex formation of differently substituted ligands with metal ions, including lanthanides under the equilibrium conditions, while no difference in complex formation with lanthanides is revealed for differently substituted ligands under these conditions.The great difference between the ligands is revealed from their complex formation with Tb(III) ions in alkaline DMF solutions. The difference results from the retardation of keto–enol transformation in the series of studied calix[4]arenes, which results from bulky β-substituents for the α-substituted 1,3-diketones. In particular, efficient complex formation with Tb3+ ions with quick equilibration is observed for the bis-(acetylaceton-3-yl)methyl-acetylacetonate derivative of calix[4]arene in DMF. The substitution of one or two methyl groups in 1,3-diketone fragments by phenyl ones significantly increases the time for the establishment of equilibrium conditions. The Job plot analysis revealed 1:1 complex stoichiometry accompanied by the deprotonation of both the 1,3-diketone groups for the ligands under the equilibrium conditions. The quantum-chemical calculations (Sparkle/PM7) point to the cone conformation of calix[4]arene as the most stable one for both free ligand 3 and its deprotonated forms in the Tb3+complexes.
The photophysical properties of the complexes with Tb3+, Eu3+, Yb3+ and Gd3+ were analyzed with the aim of revealing sensitizing effects of the ligands. The calix[4]arene disubstituted by (acetylaceton-3-yl)methyl-moieties is highlighted for its better antenna effect on Tb3+ centered luminescence than the tetra-substituted counterpart. The ligand-centered emission of Gd3+ complexes with (benzoylaceton-3-yl)methyl- and (acetylaceton-3-yl)methyl-substituted calix[4]arenes reveals them both as more convenient antennae for red and infra-red than for green lanthanide luminescence, while the data on the (dibenzoylmethan-3-yl)methyl-substituted ligand confirm its incomplete complex formation with lanthanides. Nevertheless, Yb3+-centered luminescence is sensitized to a better extent by a benzoyl-substituted ligand versus its acetyl-substituted counterpart, while Tb3+ luminescence is sensitized by an acetyl-substituted ligand, not by its benzoyl-substituted counterpart. Moreover, only poor red Eu3+ emission is observed under sensitization by both ligands. Thus, the observed tendency points to the importance of quenching mechanisms along with ligand-to-metal energy transfer on lanthanide-centered luminescence in the complexes with (benzoylaceton-3-yl)methyl- and (acetylaceton-3-yl)methyl-substituted calix[4]arenes.
4. Experimental section
4.1. Reagents and materials
Acetylacetone was distilled before use. 1,4-Dioxane and toluene were purified by distillation over metallic sodium. N,N-Dimethylformamide (DMF) (Acros Organics) was distilled over P2O5. CDCl3 (99.8% isotopic purity) from Aldrich was used for NMR spectroscopy. Terbium nitrate (Tb(NO3)3·xH2O) (Alfa Aesar), europium nitrate Eu(NO3)3·6H2O and triethylamine (TEA) (Acros Organics), neodymium(III) nitrate hexahydrate (Nd(NO3)3·6H2O) (Aldrich), copper(II) chloride dehydrate (CuCl22H2O), nickel(II) chloride hexahydrate (NiCl26H2O) and aluminum(III) chloride hexahydrate (AlCl36H2O) (Fluca) were used as commercially received without further purification. Stock solutions of metal ions were prepared by dissolving salts in deionized water. All these solutions were standardized by titration using EDTA solution. HI-7004L/C pH 4.01 buffer solution (HANNA), Mes (2-(N-morpholino)ethanesulfonicacid (Sigma-Aldrich) and Tris (tris(hydroxymethyl)aminomethane) (Sigma-Aldrich) were used for pH 4.0, pH 5.0–7.0 and pH 8.0, respectively.4.2. Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)), 1605 (ν(CO) and ν(CC)), 1463 (ν(Ph)), 1377, 1358, 1245, 1225, 1150 (νas(CCC)), 953 (νs(CCC)). Anal. calcd for C40H40O8 (648.74): C, 74.06; H, 6.21. Found: C, 73.88; H, 6.15. Mass spectrum (MALDI-TOF): m/z = 649.3 [M + H]+, 671.2 [M + Na]+.
O)), 1596, 1580 (ν(CO) and ν(CC)), 1464 (ν(Ph)), 1377, 1358, 1247, 1222, 1152 (νas(CCC)), 1077, 970 (νs(CCC)). Anal. calcd for C50H44O8 (772.88): C, 77.70; H, 5.74. Found: C, 77.88; H, 6.02. Mass spectrum (MALDI-TOF): m/z = 773.3 [M + H]+, 795.3 [M + Na]+.
O)), 1596, 1580 (ν(CO) and ν(CC)), 1465, 1449 (ν(Ph)), 1377, 1340, 1268, 1229, 1199, 1180 (νas(CCC)), 1077, 942 (νs(CCC)). Anal. calcd for C60H48O8 (897.02): C, 80.34; H, 5.39. Found: C, 80.48; H, 5.15. Mass spectrum (MALDI-TOF): m/z = 897.3 [M + H]+, 919.3 [M + Na]+.
O)), 1605 (ν(CO) and ν(CC)), 1463 (ν(Ph)), 1377, 1358, 1245, 1225, 1150 (νas(CCC)), 953 (νs(CCC)). Anal. calcd for C40H40O8 (648.74): C, 74.06; H, 6.21. Found: C, 73.88; H, 6.15. Mass spectrum (MALDI-TOF): m/z = 649.3 [M + H]+, 671.2 [M + Na]+.
O)), 1596, 1580 (ν(CO) and ν(CC)), 1464 (ν(Ph)), 1377, 1358, 1247, 1222, 1152 (νas(CCC)), 1077, 970 (νs(CCC)). Anal. calcd for C50H44O8 (772.88): C, 77.70; H, 5.74. Found: C, 77.88; H, 6.02. Mass spectrum (MALDI-TOF): m/z = 773.3 [M + H]+, 795.3 [M + Na]+.
O)), 1596, 1580 (ν(CO) and ν(CC)), 1465, 1449 (ν(Ph)), 1377, 1340, 1268, 1229, 1199, 1180 (νas(CCC)), 1077, 942 (νs(CCC)). Anal. calcd for C60H48O8 (897.02): C, 80.34; H, 5.39. Found: C, 80.48; H, 5.15. Mass spectrum (MALDI-TOF): m/z = 897.3 [M + H]+, 919.3 [M + Na]+.
4.3. Methods
UV-VIS absorption spectra have been recorded on a Lambda 35 spectrophotometer (Perkin-Elmer) in 10 mm quartz cuvettes. The Job plotting and spectrometric titration were done from monitoring of absorbance (Aλ).
The steady-state luminescence spectra have been recorded on a spectrofluorometer FL3-221-NIR (Jobin Yvon) with a SPEX FL-1042 phosphorimeter in 10 mm quartz cuvettes. Excitation of the samples was performed at the wavelength values designated in the figure captions and emission detected at 545 nm for Tb(III) with 6/6 nm excitation and emission slits. Time-resolved measurements have been performed on a spectrofluorometer FL3-221-NIR (Jobin Yvon) using the following parameters: time per flash 49.00 ms, flash count 200 ms, initial delay 0.02 ms and sample window 2 ms. The excitation and luminescence spectra of the Gd(III) and Eu(III) complexes were measured with 14/14 nm excitation and emission slits at the wavelength values designated in the figure captions. The Yb(III)-centered excitation and luminescence spectra were measured with 6/6 nm excitation and emission slits at the wavelength values designated in the figure captions.
The time-resolved luminescence spectra and the decay curves were recorded using an optical spectrometer based on an MDR-23 grating monochromator (LOMO, Saint Petersburg, Russia) coupled to a FEU-100 photomultiplier tube.56 The luminescence was excited by an LGI-21 pulsed nitrogen laser (337 nm wavelength, 2.1 mW laser pulse average output power, 10 ns pulse duration, 100 Hz repetition rate). The average output power of the laser near the samples was 1.7 mW. The exposed surface areas of the samples were 7 mm2.
All measurements were performed at room temperature under aerated conditions.
Acknowledgements
This work is supported by the Russian Fund for basic Research (grant 16-03-00007 A). N. Shamsutdinova and G. Gimazetdinova thank the President of Russian Federation grant for young scientists (MK-4456.2015.3) for financial support.Notes and references
- A. V. Kel'in and A. Maioli, Curr. Org. Chem., 2003, 7, 1855–1886 CrossRef.
- E. A. Shokova, J. K. Kim and V. V. Kovalev, Russ. J. Org. Chem., 2015, 51(6), 755–830 CrossRef CAS.
- J. Pradhan and A. Goyal, Int. J. Pharm. Res. Allied Sci., 2015, 4(2), 1–18 Search PubMed.
- G. G. Condorelli, G. Malandrino and I. L. Fragala, Coord. Chem. Rev., 2007, 251, 1931–1950 CrossRef CAS.
- P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2009, 253, 1099–1201 CrossRef CAS.
- K. Binnemans, K. A. Gschneidner Jr., J. C. G. Bünzli and V. K. Pecharsky, Handbook on the Physics and Chemistry of Rare Earths, Amsterdam, 2005, ch. 225, vol. 35, pp. 107–272 Search PubMed.
- M. L. P. Reddy, V. Divya and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC.
- M. A. Katkova, A. G. Vitukhnovskii and M. N. Bochkarev, Russ. Chem. Rev., 2005, 74(12), 1089–1109 CrossRef CAS.
- G. F. de Sá, O. L. Malta, C. de Mello Donegá, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva Jr., Coord. Chem. Rev., 2000, 196, 165–195 CrossRef.
- J.-C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- J.-C. G. Bunzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 2347–2356 CrossRef CAS PubMed.
- X. H. Wang, H. J. Chang, J. Xie, B. Z. Zhao, B. Liu, S. L. Xua, W. B. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273, 201–212 CrossRef.
- J.-C. G. Bunzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- S. V. Eliseeva and J.-C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- T. Nishioka, J. L. Yuan, Y. J. Yamamoto, K. Sumitomo, Z. Wang, K. Hashino, C. Hosoya, K. Ikawa, G. L. Wang and K. Matsumoto, Inorg. Chem., 2006, 45(10), 4088–4096 CrossRef CAS PubMed.
- M. Shi, C. Ding, J. Dong, H. Wang, Y. Tian and Z. Hu, Phys. Chem. Chem. Phys., 2009, 11, 5119–5123 RSC.
- J.-C. G. Bunzli, Coord. Chem. Rev., 2015, 293, 19–47 CrossRef.
- C.-J. Xu, B.-G. Li, J.-T. Wan and Z.-Y. Bu, Spectrochim. Acta, Part A, 2011, 82, 159–163 CrossRef CAS PubMed.
- G. Aromí, P. Gamez and J. Reedijk, Coord. Chem. Rev., 2008, 252, 964–989 CrossRef.
- J. K. Clegg, S. S. Iremonger, M. J. Hayter, P. D. Southon, R. B. Macquart, M. B. Duriska, P. Jensen, P. Turner, K. A. Jolliffe, C. J. Kepert, G. V. Meehan and L. F. Lindoy, Angew. Chem., Int. Ed., 2010, 49, 1075–1078 CrossRef CAS PubMed.
- N. A. Shamsutdinova, S. N. Podyachev, S. N. Sudakova, A. R. Mustafina, R. R. Zairov, V. A. Burilov, I. R. Nizameev, I. K. Rizvanov, V. V. Syakaev, B. M. Gabidullin, S. A. Katsuba, A. T. Gubaidullin, G. M. Safiullin and W. Dehaen, New J. Chem., 2014, 38, 4130–4140 RSC.
- R. Zairov, N. Shamsutdinova, S. Podyachev, S. Sudakova, G. Gimazetdinova, I. Rizvanov, V. Syakaev, V. Babaev, R. Amirov and A. Mustafina, Tetrahedron, 2016, 72, 2447–2455 CrossRef CAS.
- N. A. Shamsutdinova, A. T. Gubaidullin, B. M. Odintsov, R. J. Larsen, V. D. Schepkin, I. R. Nizameev, R. R. Amirov, R. R. Zairov, S. N. Sudakova, S. N. Podyachev, A. R. Mustafina and A. S. Stepanov, ChemistrySelect, 2016, 1, 1377–1383 CrossRef CAS.
- N. Shamsutdinova, R. Zairov, A. Mustafina, S. Podyachev, S. Sudakova, I. Nizameev, M. Kadirov and R. Amirov, Colloids Surf., A, 2015, 482, 231–240 CrossRef CAS.
- R. V. Rodik, A. S. Klymchenko, N. Jain, S. I. Miroshnichenko, L. Richert, V. I. Kalchenko and Y. Mély, Chem. – Eur. J., 2011, 17, 5526–5538 CrossRef CAS PubMed.
- A. Ikeda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 3102–3110 CrossRef CAS.
- M. H. Düker, R. Gómez, C. M. L. Vande Velde and V. A. Azov, Tetrahedron Lett., 2011, 52, 2881–2884 CrossRef.
- M. Strobel, K. Kita-Tokarczyk, A. Taubert, C. Vebert, P. A. Heiney, M. Chami and W. Meier, Adv. Funct. Mater., 2006, 16, 252–259 CrossRef CAS.
- T.-D. Guo, Q.-Y. Zheng, L.-M. Yang and Z.-T. Huang, J. Inclusion Phenom. Macrocyclic Chem., 2000, 36, 327–333 CrossRef CAS.
- L. O'Toole, J. McGinley and B. S. Creaven, Tetrahedron, 2013, 69, 7220–7226 CrossRef.
- C. D. Gutsche and K. C. Nam, J. Am. Chem. Soc., 1988, 110, 6153–6162 CrossRef CAS PubMed.
- M. Almi, A. Arduini, A. Casnati, A. Pochini and R. Ungaro, Tetrahedron, 1989, 45, 2177–2182 CrossRef CAS.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Springer Netherlands, 1980 Search PubMed.
- J. Toullec, The Chemistry of Enols, Wiley, New York, 1990, vol. 6, pp. 323–398 Search PubMed.
- C. Jaime, J. De Mendoza, P. Prados, P. M. Nieto and C. Sanchez, J. Org. Chem., 1991, 56, 3372–3376 CrossRef CAS.
- C. D. Gutsche, J. A. Levine and P. K. Sujeeth, J. Org. Chem., 1985, 50, 5802–5806 CrossRef CAS.
- J. Lang, V. Deckerová, J. Czernek and P. Lhoták, J. Chem. Phys., 2005, 122, 044506 CrossRef PubMed.
- S. N. Podyachev, S. N. Sudakova, A. K. Galiev, A. R. Mustafina, V. V. Syakaev, R. R. Shagidullin, I. Bauer and A. I. Konovalov, Russ. Chem. Bull., 2006, 55, 2000–2007 CrossRef CAS.
- N. Iki, N. Morohashi, F. Narumi and S. Miyano, Bull. Agric. Chem. Soc. Jpn., 1998, 71, 1597–1603 CrossRef CAS.
- A. V. Kel'in, Curr. Org. Chem., 2003, 7, 1691–1711 CrossRef.
- Q. T. H. Le, S. Umetani, M. Suzuki and M. Matsui, J. Chem. Soc., Dalton Trans., 1997, 643–647 RSC.
- D. J. Sardella, D. H. Heinert and B. L. Shapiro, J. Org. Chem., 1969, 34, 2817–2821 CrossRef CAS.
- V. Bertolasi, V. Ferretti, P. Gilli, X. Yao and C.-J. Li, New J. Chem., 2008, 32, 694–704 RSC.
- Y. Suwaa, M. Yamaji and J. of Photochemistry, J. Photochem. Photobiol., A, 2016, 316, 69–74 CrossRef.
- J. Hong and S. Ham, Tetrahedron Lett., 2008, 49, 2393–2396 CrossRef CAS.
- R. J. Bernardino and B. J. Costa Cabral, J. Phys. Chem. A, 1999, 103, 9080–9085 CrossRef CAS.
- P. Gacoin, J. Chem. Phys., 1972, 57, 1418–1425 CrossRef CAS.
- X.-Y. Chen and X. Yang, Bradley J. Holliday, Inorg. Chem., 2010, 49, 2583–2585 CrossRef CAS PubMed.
- R. E. Whan and G. A. Crosby, J. Mol. Spectrosc., 1962, 8, 315–327 CrossRef CAS.
- S. Biju, Y. K. Eom, J.-C. G. Bünzli and H. K. Kim, J. Mater. Chem. C, 2013, 1, 6915–7118 RSC.
- D. D'Alessio, S. Muzzioli, B. W. Skelton, S. Stagni, M. Massi and M. I. Ogden, Dalton Trans., 2012, 41, 4736 RSC.
- S. N. Podyachev, S. V. Bukharov, I. A. Litvinov, V. I. Morozov, A. T. Gubaidullin, G. N. Nugumanova and N. A. Mukmeneva, Russ. J. Gen. Chem., 2004, 74, 1775–1781 CrossRef.
- D. V. Lapaev, V. G. Nikiforov, G. M. Safiullin, I. G. Galyaviev, V. I. Dzabarov, A. A. Knyazev, V. S. Lobkov and Y. G. Galyametdinov, J. Struct. Chem., 2009, 50, 775–781 CrossRef CAS.
- J. J. P. Stewart, MOPAC, 2012 Search PubMed.
- J. D. L. Dutra, M. A. M. Filho, G. B. Rocha, R. O. Freire, A. M. Simas and J. J. P. Stewart, J. Chem. Theory Comput., 2013, 9, 3333–3341 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj03381d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |