
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NOx selective catalytic reduction (SCR) on self-supported V–W-doped TiO2 nanofibers†
Debora
Marani
 *,
Rafael Hubert
Silva
,
Apiwat
Dankeaw
,
Kion
Norrman
,
Rebecka Marie Larsen
Werchmeister
,
Davide
Ippolito
,
Mads
Gudik-Sørensen
,
Kent Kammer
Hansen
and
Vincenzo
Esposito
*,
Rafael Hubert
Silva
,
Apiwat
Dankeaw
,
Kion
Norrman
,
Rebecka Marie Larsen
Werchmeister
,
Davide
Ippolito
,
Mads
Gudik-Sørensen
,
Kent Kammer
Hansen
and
Vincenzo
Esposito
Department of Energy Conversion and Storage, Technical University of Denmark (DTU), Frederiksborgvej 399, Roskilde, DK-4000, Denmark. E-mail: debora.marani3@gmail.com
First published on 30th March 2017
Abstract
Electrospun V–W–TiO2 catalysts, resulting in a solid solution of V and W in the anatase phase, are prepared as nonwoven nanofibers for NOx selective catalytic reduction (SCR). Preliminary catalytic characterization indicates their superior NOx conversion efficiency to the-state-of-the-art material. A novel concept of a self-supported, ultra-compact, and lightweight nanofibrous SCR-reactor is defined.
Introduction
Despite the huge efforts being made to control the emissions of noxious waste into the atmosphere, air pollution is still a looming priority issue. Specifically, nitrogen oxides (NOx) among other pollutants represent a serious threat for human health and the environment.1,2 The most efficient and cost effective method to limit their emission into the atmosphere is the selective catalytic reduction (SCR) with NH3 used as a reducing agent.2–4 Commercial catalysts typically consist of active materials (e.g. V2O5–WO3 or V2O5–MoO3) dispersed via wet impregnation over a ceramic support (e.g. TiO2 anatase).2,5 The catalysts thus obtained are then wash-coated onto a highly porous honeycomb structure to ensure a highly exposed surface area and a fluid dynamic regime favourable for an efficient permeation of the reactant species.4,5In this system, the active material vanadium pentoxide (V2O5) is the component responsible for the catalytic abatement of NOx, and its amount is typically within the range of 0.5–3 wt%.6,7 To support and enhance the V2O5 activity, either tungsten trioxide (WO3) or molybdenum trioxide (MoO3), at concentrations in the range of 8–10 wt%, is used.2,8–11
To meet the upcoming and more stringent regulation, increasingly efficient systems are required. In this context, nanomaterials definitely offer unique opportunities.12–15 A promising strategy is to use 1-dimensional (1D) nanomaterials (e.g. nanotubes, nanofibers) with the merits of a high surface area-to-volume ratio associated with a high aspect ratio.13,14 Among others, ceramic nanofibers produced via electrospinning offer the further advantage of a fibrous membrane substrate characterized by wide open porosity and sufficient mechanical integrity.13–19 In such a structure, each single nanofiber might act as a nano-reactor arranged with the others into a widely interconnected nano-textured and highly porous network. Such a substrate conveniently assembled could be considered as a complete lightweight and compact SCR reactor unit. In this configuration, any other components (e.g. honeycombs) might not be required.
In addition, the electrospinning approach allows an easy control over specific morphological and compositional features.13,19
In this work, for the first time the NH3-SCR state-of-the-art catalyst (V2O5–WO3/TiO2) was prepared as V–W-doped titania (TiO2) electro-spun nanofibers. For the V–W co-doped catalysts the composition was fixed at V0.03W0.02Ti0.95O2, corresponding to a WO3 and V2O5 content of 5% and 3% in weight respectively. The ultimate aim was to prove the benefits for the NOx conversion efficiency deriving from both the nanofiber morphology and the doping approach.
Experimental
Materials
All materials were of reagent grade and used as received.Titanium tetraisopropoxide (97% purity, Sigma Aldrich, Denmark), tungsten ethoxide (5% w/v in ethanol, Alfa Aesar, Denmark,) and vanadium acetylacetonate (99.98% purity, Sigma Aldrich, Denmark) were used as precursors to V–W-doped titania (V–W–TiO2). Polyvinylpyrrolidone (PVP, Mw ∼ 1.3 M, Sigma Aldrich, Denmark) was used as a carrier polymer due to its good solubility in alcohols and its good compatibility with the other reagents. Glacial acetic acid (99.8%, Sigma Aldrich) served as a stabilizer to control the hydrolysis reaction of the alkoxides. In Table S2 (ESI†) the compositions developed are reported.
Preparation of spinning solution
Spinning solutions at different titanium tetraisopropoxide/acetic acid molar ratios (namely 2, 5, and 10) were prepared with the aim of optimizing the morphology of the resulting nanofibers.In a typical procedure to prepare the full composition, titanium tetraisopropoxide was first mixed with acetic acid (e.g. molar ratio of TTIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcH = 1:10) under magnetic stirring (20 °C) in a sealed container purged with an inert gas (e.g. argon). The required amounts of the other reagents were then added using adequate syringes. Vanadium acetylacetonate and PVP were previously dissolved in ethanol/acetic acid (1:1 in volume) solution (0.037 g ml−1) and pure ethanol (9.20% w/v), respectively. The concentration of PVP (∼0.03 g ml−1) is finally adjusted by adding an extra volume of pure ethanol. The solution was kept under magnetic stirring for 10 min, and finally loaded into a glass syringe equipped with a 21 gauge stainless steel needle.
:AcH = 1:10) under magnetic stirring (20 °C) in a sealed container purged with an inert gas (e.g. argon). The required amounts of the other reagents were then added using adequate syringes. Vanadium acetylacetonate and PVP were previously dissolved in ethanol/acetic acid (1:1 in volume) solution (0.037 g ml−1) and pure ethanol (9.20% w/v), respectively. The concentration of PVP (∼0.03 g ml−1) is finally adjusted by adding an extra volume of pure ethanol. The solution was kept under magnetic stirring for 10 min, and finally loaded into a glass syringe equipped with a 21 gauge stainless steel needle.
Electrospinning of the solution and calcination of the green nanofibers
RT Advanced (Linari Engineering) electrospinning equipment was used. The syringe loaded with the precursor solution was first installed inside of the chamber of the RT Advanced with fixed humidity (22% RH) and temperature (20 °C). The grounded substrate drum was placed 8 cm away from the needle and covered with an aluminium foil to collect the nanofibers. The electrospinning process was conducted in air with a voltage supply of 36 kV and a feeding rate of 1 ml h−1.The material resulted in a flexible green mat that was thermally treated in an oven at 500 °C for 3 h (heating rate 1 K min−1) to remove the organics and promote the formation of a crystallographic phase.
Characterization
TGA (NETZSCH, STA 409, CD) was conducted in air flux from room temperature to 700 °C with a heating rate of 2 °C min−1.BET (Brunauer–Emmett–Teller; Autosorb 1-MP, Quantachrome Instruments, Boynton Beach, FL, USA) analysis was conducted to measure the specific surface area (SSA) of the fibrous materials developed. The results are summarized in Table S2 (ESI†).
X-ray diffraction (XRD, Bruker D8, Germany) patterns were recorded at room temperature (Cu Kα radiation) at a scanning rate of 0.02° s−1 in the 2θ range from 20 to 90°.
The morphology of the nanofibers was analyzed by using field emission scanning electron microscopy (FESEM, Supra, Carl Zeiss, Germany) and transmission electron microscopy (TEM, JEM3000F, Oxford Instruments, UK).
The oxidation state of the elements involved was investigated using X-ray photoelectron spectroscopy (XPS, K-α, Thermo Electron Limited, Winsford, UK). The analyses were performed using a monochromatic Al-Kα X-ray source and a take-off angle of 90° from the surface plane. Atomic concentrations were determined from surface spectra (0–1300 eV, 100 eV detector pass energy, 3 scans) and were calculated by determining the relevant integral peak intensities using a Smart type background. High-resolution Ti2p, W4f, V2p, Au4f, and O1s XPS spectra were obtained using 50 eV detector pass energy and 10 scans. The binding energies were calculated by referencing to the Au4f7 peak at 84.0 eV.
A home-made lab-scale experimental setup was used for the SCR characterization. The core of the setup is a quartz reactor equipped with an inlet and outlet for feeding the reactant gases and measuring the resulting gases, respectively. The test setup is equipped with sensors for NOx and CO and NO and NH3 supply (equipped with mass flow controllers), gas analyzers (quadrupole mass spectrometer, Omnistar GSD 301, Pfeiffer Vacuum, Denmark) and a chemiluminescence detector (Model 42i High Level, Thermo Scientific, UK). The experiments were conducted on around 20 mg of titania-based nanofibers at gas concentrations such as [NO] = [NH3] = 1000 ppm, [O2] = 8% vol., [CO2] = 5% vol., and Ar balance (GHSV = 400000 h−1). The amount of material was intentionally kept as low as possible (20 g) with the aim of appreciating any differences in the NOx conversion efficiency (also at higher temperature) among the different materials.
Results and discussion
The nanofibers were produced starting from sol–gel solutions containing the precursors (alkoxides and salts) for all the elements of interest, and an adequate amount of the chelating agent (acetic acid) and of the carrier polymer (polyvinylpyrrolidone).20,21 The chemistry of the solution was optimized to obtain a stable jet resulting in continuous nanofibers.Morphological characterization of the V–W co-doped nanofibers (V–W–TiO2) prepared at different [Ac]/[Ti] ratios (2, 5 and 10) suggested that the highest ratio (e.g. 10) is the optimal composition to obtain the best morphology (Fig. S2, ESI†). At this content of acetic acid, defect-free continuous nanofibers characterized by a highly homogeneous distribution of their diameters are obtained. This finding was associated with the ability of the acetic acid to control the hydrolysis/condensation of the alkoxides. The [Ac]/[Ti] = 10 was identified as the optimal molar ratio to obtain homogeneous morphologies.
Based on this indication, only the complete series of samples (pure TiO2, V–TiO2, W–TiO2, and V–W–TiO2) at [Ac]/[Ti] = 10 was then prepared and further investigated.
Similar morphological features for all the titania-based samples (prepared at [Ac]/[Ti] = 10) were observed. For the sake of clarity in Fig. 1a–d only the results on the full composition material (V–W–TiO2, [Ac]/[Ti] = 10) are discussed. Fig. 1a and b show SEM micrographs at low and high magnifications respectively. The images show that the material has an extended nano-textured morphology, with nanofibers displaying high aspect ratios and a narrow diameter distribution (within the range of 80–120 nm). The formation of any dead-end pores (closed porosity) upon the electrospinning process is clearly excluded and a large open and all-interconnected porosity is instead obtained and associated with a wide pore volume. The volume fraction was estimated using two methods: (1) analysis of the SEM micrographs (via estimation of the mean intercept length)22 that provided values of ca. 80%; and (2) measurements of the relative density of the samples that resulted in values between 80 and 90%. Both methods provided an extremely high pore volume, definitely higher than the volume fraction usually measured for the nanoparticle catalyst (40–60%).23
 | ||
| Fig. 1 SEM micrographs of the V–W–TiO2 sample ([Ac]/[Ti] = 10) at (a) low and (b) high magnifications, (c) TEM micrograph of the V–W–TiO2 sample ([Ac]/[Ti] = 10) with the SAED as an inset, and (d) HT-TEM micrograph of the V–W–TiO2 sample ([Ac]/[Ti] = 10) with the fringe spacing attributed to the (101) plane of anatase. | ||
Both the high magnification SEM (Fig. 1b) and TEM (Fig. 1c) micrographs for the single ceramic nanofiber show a densely packed polycrystalline structure with a rough surface and a dimension of the crystallites around 25–35 nm. The selected-area electron diffraction (SAED) inset in Fig. 1c (TEM micrograph) suggests an anatase phase in the short range (nanoscale) for the co-doped material. This observation is supported by the HR-TEM image (Fig. 1d) that shows a well-defined organization of the atoms with a fringe spacing of 0.349 nm indexed to the (101) plane of the anatase phase.
The anatase arrangement of the atoms within the crystallographic lattice for the co-doped composition is further confirmed by the long range XRD pattern (Fig. 2a). An evident effect of the dopants on the phase of the titania-based nanofibers is identified as indicated in Fig. 2a. For the un-doped titania both anatase and rutile phases can be observed, whereas the addition of the dopants clearly inhibits the rutile phase formation. For the pure titania, the co-occurrence of the two phases can be attributed to a size-dependent effect, as also suggested by Barnard et al. Indeed, their study revealed that crystallites of around 20–30 nm in size obtained with a treatment at 500 °C result in a mixture of the two phases.24 For the doped materials, the annihilation of the oxygen vacancies due to the oxidation state of vanadium and tungsten (>4) inhibits the phase transformation.25 This is also verified for the V-doped titania sample, although the vanadium is known to promote the anatase–rutile transformation.24 The unusual effect of vanadium might be a combined result of the size-dependent and oxidation state effects.
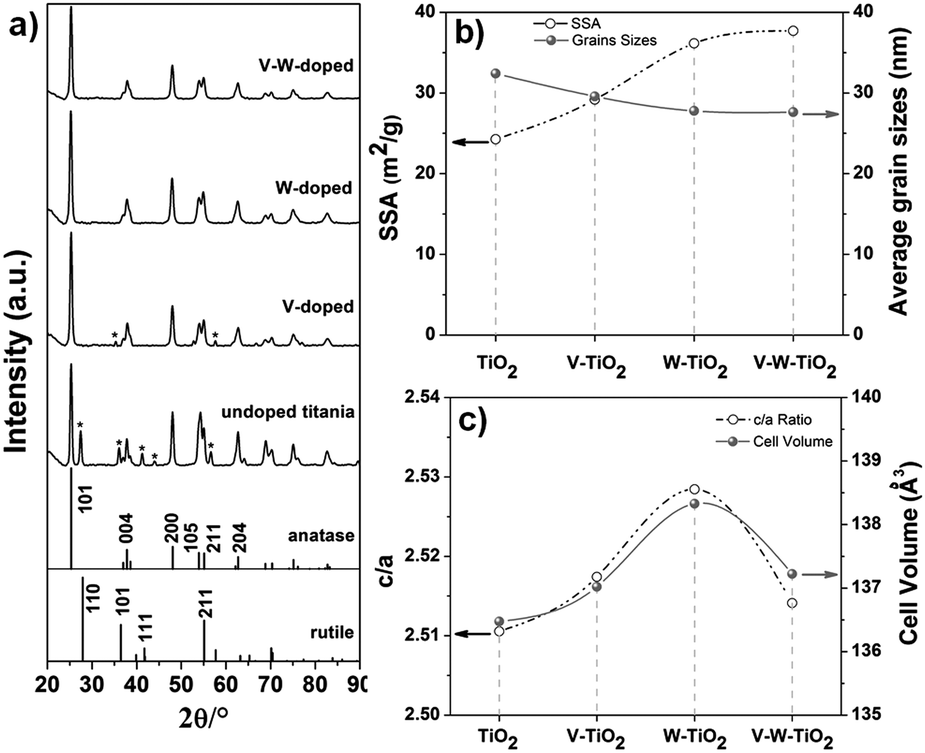 | ||
| Fig. 2 (a) XRD pattern, (b) SSA and grain sizes, and (c) lattice parameters (c/a and cell volume) for the complete series of samples prepared at [Ac]/[Ti] = 10. | ||
An effect on the grain sizes is also observed when the dopants are introduced. As indicated in Fig. 2b and reported in Table S3 (ESI†), a light decrease in the average grain sizes is achieved with the introduction of dopants into the anatase lattice (prepared at [Ac]/[Ti] = 10). The average grain size for the samples is calculated from the (101) plane of the anatase phase by applying the Debye–Scherrer equation (eqn (S1), ESI†). The slight reduction in the grain size corresponds to a slight increase of the SSA with a continuous increment of its values from pure titania to the V–W–TiO2 sample at fixed [Ac]/[Ti] (e.g. 10) (Fig. 2b and Table S2, ESI†). This effect is ascribed to the solute drag effect that limits the mass diffusion during the thermal treatment (calcination at low temperature, 500 °C).26,27 For the nanofibers prepared at optimized [Ac]/[Ti] (fixed at 10) and characterized by a homogeneous distribution of the nanofiber diameters, the main factor affecting the SSA can be recognised as the grain sizes.
By contrast, an effect of the inhomogeneous distribution of the nanofiber diameters can be identified as the main factor determining the SSA for the nanofibrous substrates prepared at a fixed composition (V–W–TiO2) but at different [Ac]/[Ti] molar ratios (e.g. 2, 5 and 10). As expected, the substrate with the highest level of inhomogeneity ([Ac]/[Ti] = 2) in the distribution of the nanofiber diameters (20–150 nm) also exhibits the highest value of SSA (Table S2, ESI†). As the molar ratio increases, the level of the inhomogeneity was reduced. At the optimized molar ratio ([Ac]/[Ti] = 10), the nanofibrous material exhibits an extremely homogeneous distribution of the nanofiber diameters (Fig. 1a and b) and also the lowest SSA (Table S2, ESI†).
Therefore, two major factors have been identified affecting the SSA: the homogeneity in the nanofiber diameters and the grain sizes. The first factor is crucial for samples characterized by inhomogeneity in the nanofiber distribution. When the distribution of the nanofiber diameters is optimized, the grain sizes play a major role in affecting the SSA.
The crystal parameters were also estimated considering a tetrahedral crystal structure for the TiO2 samples (a = b ≠ c and α = β = γ). The axes were determined by applying eqn (S2) (ESI†) and using the (101) and (200) crystal planes of the anatase phase. In Fig. 2c the parameters c/a and cell volume are plotted for the complete series of samples prepared at [Ac]/[Ti] = 10.
The introduction of the dopants into the anatase lattice clearly causes a distortion of the unit cell as indicated by the increase of the lattice parameter c/a (Fig. 2c). Specifically, the distortion resulted in an expansion of the cell with the three lattice axes and the cell volume increasing for the doped nanofibers compared to the un-doped TiO2 (see Fig. 2c, Fig. S3c and Table S3, ESI†). A larger elongation for the c-axis is observed compared to the a and b-axes. The expansion of the unit cell indicates that both V5+ and W6+ are substitutionally introduced into the anatase lattice, resulting in a solid solution. Indeed, Rodriguez-Talavera et al.28 have demonstrated that not only the ionic radii, which are similar to one another for the cations of interest (Table S1, ESI†), but also the ionic charge significantly affect the unit cell. The distortion of the unit cell is smaller for the co-doped material compared to the mono-doped ones, probably because of the different interactions of dopant ions with titanium ions. The distortion is likely due to the electrical field created between the dopant and the surrounding ions.28 In addition, the expansion of the unit cell likely occurs via formation of defects.28 This is also expected to affect the electronic structure with a resulting impact on the electrical properties of the materials.29,30 Specifically, the substitution of Ti4+ with Dn+ (D stands for dopant) ions at higher oxidation states (n > 4) introduces an excessive charge into the lattice that is compensated either by the formation of point defects (titanium vacancies) or by the delocalization of (n − 4) electrons around Dn+ sites.31 Yet in this last circumstance, the extra electrons thus introduced can be either promoted to the conduction bands, enhancing the conductivity properties of the materials, or transferred to the neighbouring sites, promoting their reduction (e.g. Ti4+ → Ti3+).32 The described charge compensation mechanisms compete with one another and the prevalence of one over the others essentially depends on the chemical environment.
The effect of V5+ and W6+ dopants on the defect chemistry of the anatase-based nanofibers was explored by XPS. Fig. 3 shows the titanium 2p (Fig. 3a), oxygen 1s (Fig. 3b), vanadium 2p (Fig. 3c) and tungsten 4f (Fig. 3d) spectra for the series of samples prepared at [Ac]/[Ti] = 10. Detailed information about the binding energy and surface composition is provided in Tables S4 and S5 (ESI†) respectively. The analysis of the surface composition provides a V2O5/WO3 molar ratio close to the nominal value (almost 1), indicating a formation of the phase without any segregation.
 | ||
| Fig. 3 X-ray photoelectron spectra of (a) Ti 2p, (b) O 1s, (c) V 2p, and (d) W 4f for the series of samples at [Ac]/[Ti] = 10. | ||
In Fig. 3a two peaks corresponding to Ti 2p3/2 and Ti 2p1/2 photo-electron lines for Ti4+ are observed for all the samples.33,34 Variations in the binding energy from pure titania to the full composition sample are observed and explained by the different electron densities around the titanium atoms (Table S1, ESI†). This is specifically the case for the W-doped material for which a shift of around 0.8 eV toward higher binding energy is produced for the Ti 2p3/2 signal. The observed shift is associated with a greater electronegativity of tungsten (2.36, Pauli scale, Table S1, ESI†) compared with titanium (1.54, Pauli scale, Table S1, ESI†). For the V-doped sample no evident shift toward the highest energy is observed because of the comparable values of electronegativity for the two elements (vanadium 1.63 on the Pauli scale, Table S1, ESI†). For the co-doped composition, the Ti 2p3/2 peak is observed to be only slightly shifted (0.2 eV) towards a higher binding energy. In this case the effect of tungsten on titanium electron density is attenuated by the presence of vanadium. No significant intensity for signals related to 2p photon–electron lines of Ti+3 can be detected. This result excludes for all the doped samples a charge compensation occurring via Ti4+ → Ti3+ reduction.
For all the samples a wide and asymmetric peak is observed in the O 1s region (Fig. 3b), which consists of two distinct bands corresponding to two chemical states.8,35 The main signal at low binding energy is assigned to the bulk oxygen bonded either to titanium or to any other cations in the crystal lattice (Ti–O–D), whereas the minor signal at higher energy corresponds to the chemisorbed oxygen (–OH).8,35 For simplicity, they are referred to as Oα and Oβ respectively.8
Depending on the chemical environment, a variation in the Oα amount is detected and estimated by considering the Oα/(Oα + Oβ) ratio. The corresponding values are listed in Table S5 (ESI†). Interestingly, upon the single addition of vanadium and tungsten the Oα/(Oα + Oβ) ratio decreases with respect to the pure titania nanofibers. Its initial amount is then restored in the co-doped material. This is likely the result of different charge compensation mechanisms occurring depending on the chemical environment. When V5+ and W6+ are individually introduced into the anatase lattice, the extra charge (electrons) might be compensated via formation of titanium vacancies (point defects). As a consequence of that, the number of –OH groups, bonded to titanium, decreases. By contrast, in co-doped material the transfer of electrons to neighbouring sites to favour their reduction might be the mechanism occurring. The analysis of the vanadium 2p spectra (Fig. 3c) definitely supports this assumption. Indeed, an increment of the V4+ signal with respect to the V5+ peak is identified in the XPS profile for the full composition sample compared with the V-doped material. Additionally, the reduction of vanadium from V5+ to V4+ is a convenient process as indicated by its standard reduction potential (E0 = 0.96 V). A higher concentration of V4+ corresponds to a larger amount of Brønsted proton acid, namely V4+-OH groups, consistent with the increment of the Oα/(Oα + Oβ) ratio. The analysis of the tungsten 4f spectra (Fig. 3d) excludes the reduction of W6+ to W5+, as only signals deriving from W6+ can be detected. This result is also supported by the standard reduction potential for the W6+/W5+ couple (E0 = −0.03 V).
The morphological features, the oxidation states and the chemistry defects observed for the developed materials provide some information for a clear understanding of their catalytic activity. Fig. 4a shows the NOx conversion efficiency for the nanofibers prepared at [Ac]/[Ti] = 10.
Preliminary experiments on the catalytic activity were conducted only under dry conditions and in the absence of SO2 with the aim of characterizing the intrinsic activity of the materials, when the catalysts work under the best conditions. In addition, the experiments were intentionally conducted on a small amount of materials (20 mg), again to appreciate any possible difference in the catalytic activity at the highest temperature. Indeed, in a typical experiment conducted on a larger amount of material (e.g. 100 mg), when the activation threshold temperature (Ta) for the process is exceeded, NOx conversion reaches the 100% value. Under these conditions, any differences in the conversion behaviour among the samples are not detectable. Only the high range of temperatures (250–500 °C) was investigated.
In Fig. 4a only the trends for V–TiO2 and V–W–TiO2 catalysts are plotted since pure titania and tungsten-doped materials exhibited a conversion efficiency lower than the detectable limit of our setup. This is likely due to the low amount of material used for the experiments.
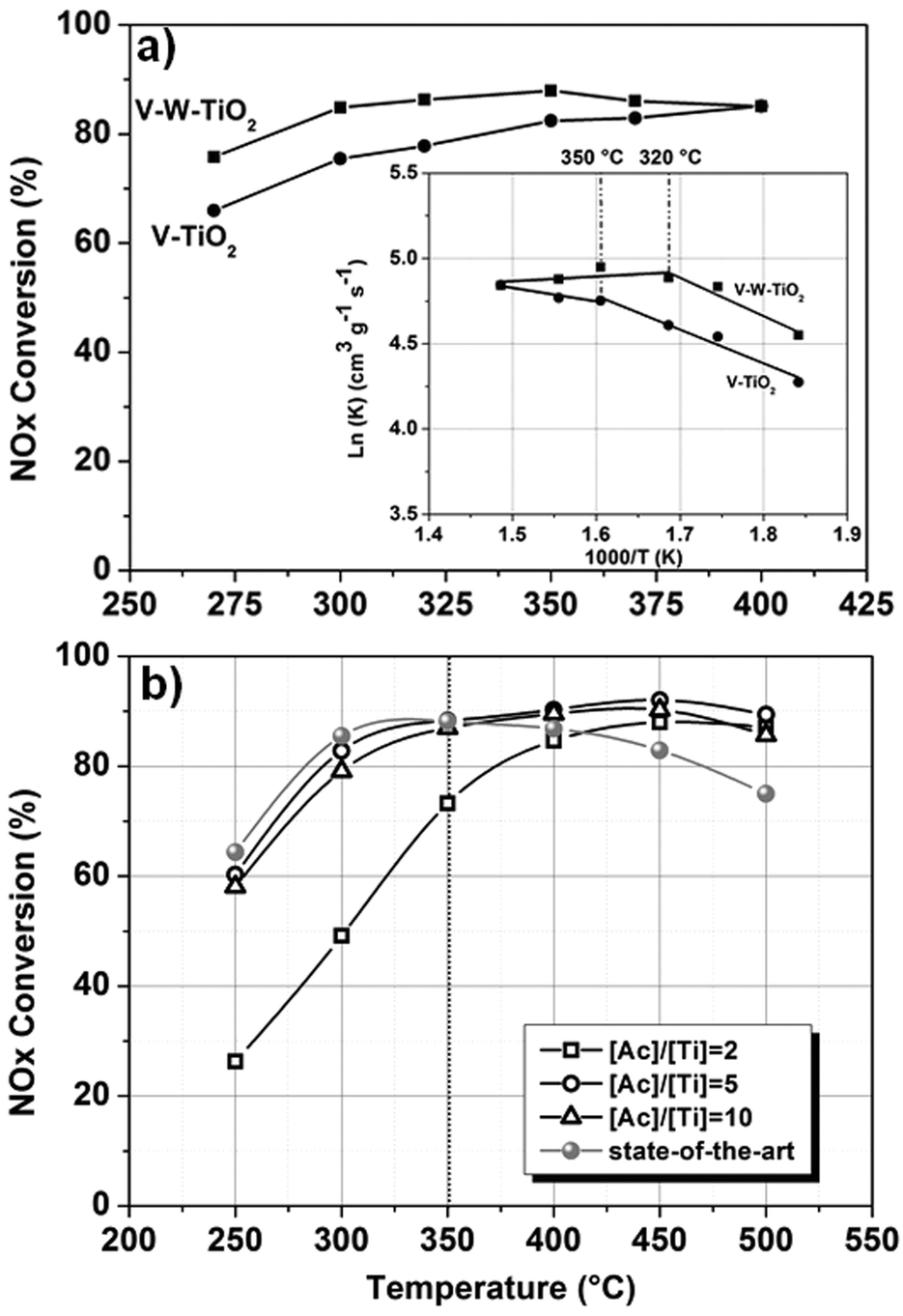 | ||
| Fig. 4 NOx conversion activity for the (a) complete series of nanofibers prepared at [Ac]/[Ti] = 10 along with their corresponding Arrhenius plots (inset), and for the (b) full composition materials prepared at different content [Ac]/[Ti] ratios. | ||
The superior performance of the full composition material over the V–TiO2 catalyst (Fig. 4a) is consistent with its higher concentration of Brønsted acids associated with the V4+-OH groups. These groups have been proven to be the active sites of the catalytic redox reaction, because of their higher mobility compared with the lattice oxygen.35,36
To further explore the effect of the doping on the conversion activity, the kinetics of the process were also investigated. The NH3-SCR reaction on V2O5–WO3–TiO2 is usually considered to be a first order reaction with respect to NO concentration.8,37 The activation energy (Ea) and the number of active sites can be estimated by the Arrhenius fitting of the data using the following equation for the rate constant (k):
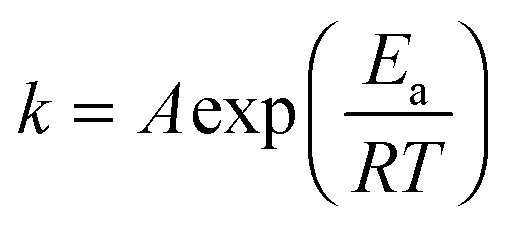 | (1) |
The values obtained are listed in Table S5 (ESI†). The Arrhenius plot is shown as an inset in Fig. 4a. For both materials two mechanisms of reaction are observed: the first mechanism at lower temperatures where a dependence of the reaction on temperature is observed and the second one at higher temperatures where instead the reaction is independent of the temperature with activation energy values (Ea) close to zero. In this region, the reaction is likely controlled by transport phenomena of reagents. This can be ascribed both to the morphology, characterized by a wide open and interconnected porosity, which favours a fluid dynamic regime and to the optimized dispersion of the active materials (introduced as dopants), which favours the exposure of the active sites towards the reagents. For the full composition material, the transition from the first to the second region occurs at 320 °C, whereas for the vanadium-doped material the transition is observed at 350 °C. This difference in the activation temperature (Ta) is likely the result of the different concentrations of OH groups.
The effects of the different morphologies and the SSA were also explored. Fig. 4b shows the NOx conversion efficiency for the full composition (V–W–TiO2) nanofibers prepared at different [Ac]/[Ti] values, namely 2, 5 and 10. Despite its high SSA (57.44 m2 g−1), the material prepared at [Ac]/[Ti] = 2 exhibits the lowest catalytic activity as a result of its poor morphological features (Fig. S2, ESI†). By contrast, the materials at [Ac]/[Ti] = 5 and 10, characterized by dissimilar values of SSA (59.52 m2 g−1 and 37.69 m2 g−1, respectively), exhibit comparable catalytic activity. The result confirms the predominant role of the morphology in the catalytic performances.
In Fig. 4b, a comparison with the-state-of-art nano-powder material (V–W–TiO2 catalyst, kindly provided by Haldor Topsøe A/S) is also shown. The state-of-the-art catalyst was tested under the same conditions and with the same set-up. Two temperature ranges with two different trends can be clearly observed. The first region is observed from 250 to 350 °C where the NOx conversion efficiencies of the state-of-the-art catalyst and of two best performing fibrous catalysts ([Ac]/[Ti] = 5 and 10) are comparable (within the experimental errors). When the temperature of 350 °C is exceeded, while the performance of the state-of-the-art material gradually declines, for the nano-fibrous catalysts ([Ac]/[Ti] = 5 and 10), a stable and high level of conversion is maintained. As indicated by the kinetic investigation, at these temperatures the reaction might be controlled by the transport phenomena of the gaseous reagents. The nanofibrous morphology is likely a major factor in enhancing the catalytic activity, supporting a favourable fluid-dynamic regime and efficient exposure of the active materials towards the reagents. The superior performances might also be associated with the doping approach that enables a high dispersion and immobilization of the supported active phase (V2O5).
Conclusions
Electro-spun V–W doped-titania materials were prepared and tested for the first time for their de-NOx activity in an SCR process. The purpose was to demonstrate the potential and the benefits arising from the nanofiber (1D) morphologies and doping approach. The effect of the dopants on the morphological features, crystallographic phase and chemical environment was investigated and used to explain the NOx conversion activity. Tungsten was observed to increase the concentration of Brønsted acid sites (V4+-OH) and was demonstrated to exert its role by favoring the reduction of the V5+ to V4+. The nano-fibrous morphology and the doping approach were proven to be main factors impacting the NOx efficiency conversion, enabling an effective exposure of the supported active material (V2O5) and a favourable fluid-dynamic regime of the reagents. The materials developed exhibit superior catalytic performances to the-state-of-the-art material.Acknowledgements
Financial support from the European Commission within the frame of FP7 with the project BLUESHIP (ref. no. 605102) is gratefully acknowledged. Haldor Topsøe A/S is also gratefully acknowledged for providing the state-of-the-art catalyst.Notes and references
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155 CrossRef CAS PubMed.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1 CrossRef CAS.
- P. Forzatti, I. Nova, E. Tronconi, A. Kustov and J. R. Thøgersenb, Catal. Today, 2012, 184, 153 CrossRef CAS.
- B. Liu, J. Du, X. X. Lv, Y. Qiu and C. Tao, Catal. Sci. Technol., 2015, 5, 1241–1250 CAS.
- H. Li, D. Zhang, P. Maitarad, L. Shi, R. Gao, J. Zhang and W. Cao, Chem. Commun., 2012, 48, 10645 RSC.
- S. B. Kristensen, A. J. Kunov-Kruse, A. Riisager, S. B. Rasmussen and R. Fehrmann, J. Catal., 2011, 284, 60 CrossRef CAS.
- F. Liu, Y. Yu and H. He, Chem. Commun., 2014, 50, 8445 RSC.
- G. Dong, Y. Bai, Y.-F. Zhang and Y. Zhao, New J. Chem., 2015, 39, 3588 RSC.
- C. Z. Wang, S. J. Yang, H. Z. Chang, Y. Peng and J. H. Li, Chem. Eng. J., 2013, 225, 520 CrossRef CAS.
- G. T. Went, L. J. Leu and A. T. Bell, J. Catal., 1992, 134, 479 CrossRef CAS.
- L. J. Alemany, F. Berti, G. Busca, G. Ramis, D. Robba and G. P. Toledo, Appl. Catal., B, 1996, 10, 299 CrossRef CAS.
- M. M. Khin, A. S. Nair, V. J. Babu, R. Murugana and S. A. Ramakrishna, Energy Environ. Sci., 2012, 5, 8075 CAS.
- S. Zhan, M. Qiu, S. Yang, D. Zhu, H. Yu and Y. Li, J. Mater. Chem. A, 2014, 2, 20486 CAS.
- A. O. Ogunlaja, P. E. Kleyi, R. S. Welmsley and Z. R. Tshentu, Catalysis, 2016, 28, 144 Search PubMed.
- L. J. Alemany, L. Lietti, N. Ferlazzo, P. Forzatti, G. Busca, E. Giamello and F. Bregani, J. Catal., 1995, 155, 117 CrossRef CAS.
- S. S. Reddy Putluru, L. Schil, D. Gardini, S. Mossin, J. B. Wagner, A. D. Jensen and V. Fehrmann, J. Mater. Sci., 2014, 49, 2705 CrossRef.
- Y. Dai, W. Liu, E. Formo, V. Sun and Y. Xia, Polym. Adv. Technol., 2011, 22, 326 CrossRef CAS.
- H. Wu, W. Pan, D. Lin and H. Li, J. Adv. Ceram., 2012, 1, 2 CrossRef CAS.
- L. Shahreen, G. G. Chase, A. J. Turinske, S. A. Nelson and N. Stojilovic, Chem. Eng. J., 2013, 225, 340 CrossRef CAS.
- D. Li and Y. Xia, Nano Lett., 2003, 3, 555 CrossRef CAS.
- D. Marani, C. Trakanprapai, S. Licoccia, E. Traversa and M. Miyayama, Mater. Res. Soc. Symp. Proc., 2009, 1126, 99 Search PubMed.
- M. M. Tomadakis and T. J. Robertson, J. Compos. Mater., 2005, 39, 163 CrossRef.
- P. Serp and B. Machado, Nanostructured carbon materials for catalysis, Royal Society of Chemistry, Cambridge CB4 0WF, UK, 2013 Search PubMed.
- S. Barnard and H. Xu, ACS Nano, 2008, 11, 2237 CrossRef PubMed.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855 CrossRef CAS.
- V. Esposito, D. W. Ni, Z. He, W. Zang, A. S. Prasad, J. A. Glasscock, C. Chatzichristodoulou, S. Ramousse and A. Kaiser, Acta Mater., 2013, 61, 6290 CrossRef CAS.
- D. Z. de Florio, V. Esposito, E. Traversa, R. Muccillo and F. C. Fonseca, J. Therm. Anal. Calorim., 2009, 97, 143 CrossRef CAS.
- R. Rodriguez-Talavera, S. Vargas, R. Arroyo-Murrillo, R. Montiel-Campas and E. H. Poniatowski, J. Mater. Res., 1997, 12, 439–443 CrossRef CAS.
- D. S. Bhachu, S. Sathasivam, G. Sankar, D. O. Scanlon, G. Cibin, C. J. Carmalt, I. P. Parkin, G. W. Watson, S. M. Bawaked, A. Y. Obaid, S. Al-Thabaiti and S. N. Basahel, Adv. Funct. Mater., 2014, 24, 5075 CrossRef CAS.
- L. R. Sheppard, T. Bak and J. Nowotny, J. Phys. Chem. B, 2006, 110, 22447 CrossRef CAS PubMed.
- Y. Liu, J. M. Szeifert, J. M. Feckl, B. Mandlmeier, J. Rathousky, O. Hayden, D. Fattakhova-Rohlfi and T. Bein, ACS Nano, 2010, 4, 5373 CrossRef CAS PubMed.
- C. R. A. Catlow, A. A. Sokol and A. Walsh, Chem. Commun., 2011, 47, 3386 RSC.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST Standard Reference Database 20, Version 4.1, (web version, http://srdata.nist.gov/xps/), 2012 Search PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887 CrossRef CAS.
- W. Cha, S.-T. Yun and J. Jurng, Phys. Chem. Chem. Phys., 2014, 16, 17900 RSC.
- N.-Y. Topsøe, Science, 1994, 265, 1217 Search PubMed.
- S. Roy, M. S. Hedge and G. Madras, Appl. Energy, 2009, 86, 2283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characteristic properties of the elements investigated, details on the experimental procedures, thermogravimetric analysis, scanning electron microscopy micrographs for co-doped materials prepared at different [Ac]/[Ti] ratios, details on the X-ray photon electron analysis (e.g. binding energy), and details on the kinetic analysis of the catalytic process. See DOI: 10.1039/c6nj03205b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |