
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and optimization of a reactive oxygen species responsive cellular delivery system†
Ana M.
Perez-Lopez‡
 ,
Elsa
Valero‡
and
Mark
Bradley
*
,
Elsa
Valero‡
and
Mark
Bradley
*
School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3JJ, UK. E-mail: mark.bradley@ed.ac.uk
First published on 1st February 2017
Abstract
Reactive oxygen species play numerous roles in a number of pathological processes. Monitoring H2O2 is a powerful tool for imaging and therapy of diseases wherein oxidative stress is involved. In particular, we report a specific application of functional microspheres as sensors of H2O2. Reactive oxygen species responsive delivery systems were developed to detect in vitro peroxides thanks to the presence of a boronic ester which is readily cleaved with H2O2. This ROS-sensitive cleavable linker underwent a 1,6-elimination to disrupt fluorescence resonance energy transfer by coupled near-infrared fluorophores such as Cy5.5/Cy7. This technology would allow real-time monitoring of therapeutic regimes (and their success), as well as optical detection of inflammation.
1. Introduction
In healthy cells and tissues, there exists a delicate balance between the generation of reactive oxygen species and radical generation and a cell's antioxidant defences1,2 with both important for normal metabolism, and regulation of the cellular function.3,4 Reactive oxygen species (ROS) cover a number of oxygen-based metabolites including superoxide anion radicals (O2˙−), hydrogen peroxide (H2O2) and hydroxyl radicals (˙OH). These species have been demonstrated to cause damage to various cellular components, effecting cellular proliferation and survival,5 as well as being found in many hypoxic tissues. Perhaps counter intuitively compounds such as hydrogen peroxide are potentially more damaging to DNA than hydroxyl radicals, since their lower reactivity provides a sufficient time for hydrogen peroxide to travel into the nucleus of a cell from its site of generation.6H2O2 imbalance has potential as a trigger for tissue directed delivery,7–10 and has been extensively targeted with the synthesis of switch-on optical imaging probes.11–14 Tsien developed a strategy whereby H2O2 produced in the proximity of a cancer reacted with a so-called Activatable Cell Penetrating Peptide (ACPP) construct, inducing delivery of a labelled polyarginine scaffold into cells and with application in an in vivo inflammation model.15
The uptake of nano and microspheres (0.2–5 μm) has been successfully established in a broad range of cell types, including adherent and suspension cultures of primary cells. The broad applicability of this approach has been demonstrated with the “loading” of stem cells, giving rise to viable chimeras thus demonstrating their remarkable cellular biocompatibility.16–18
Here, ROS-cleavable linkers were designed, synthesised, loaded with a cargo and attached to microspheres, such that when these “carriers” entered cells undergoing oxidative stress cargo release was triggered. To allow evaluation of the cleavage of the ROS-sensitive linker, FRET-based probes were synthesised, as shown in Fig. 1,19 where cleavage of the ROS-cleavable linker by H2O2 perturbs the FRET system (consisting of the well-known FRET pair 5(6)-carboxyfluorescein (λex/em 488/528 nm)/methyl red (λAbs 480 nm) or the NIR cyanine dyes Cy5.5 (λex/em 675/694 nm)/Cy7 (λex/em 743/767 nm)).20–22 As a second example a cell carrier consisting of a polycationic lysine-like peptide,23 which is resistant to proteolysis, non-toxic in vivo and has demonstrated a highly efficient cell entry profile24–34 was used to confirm the versatility of a ROS-sensitive linker.
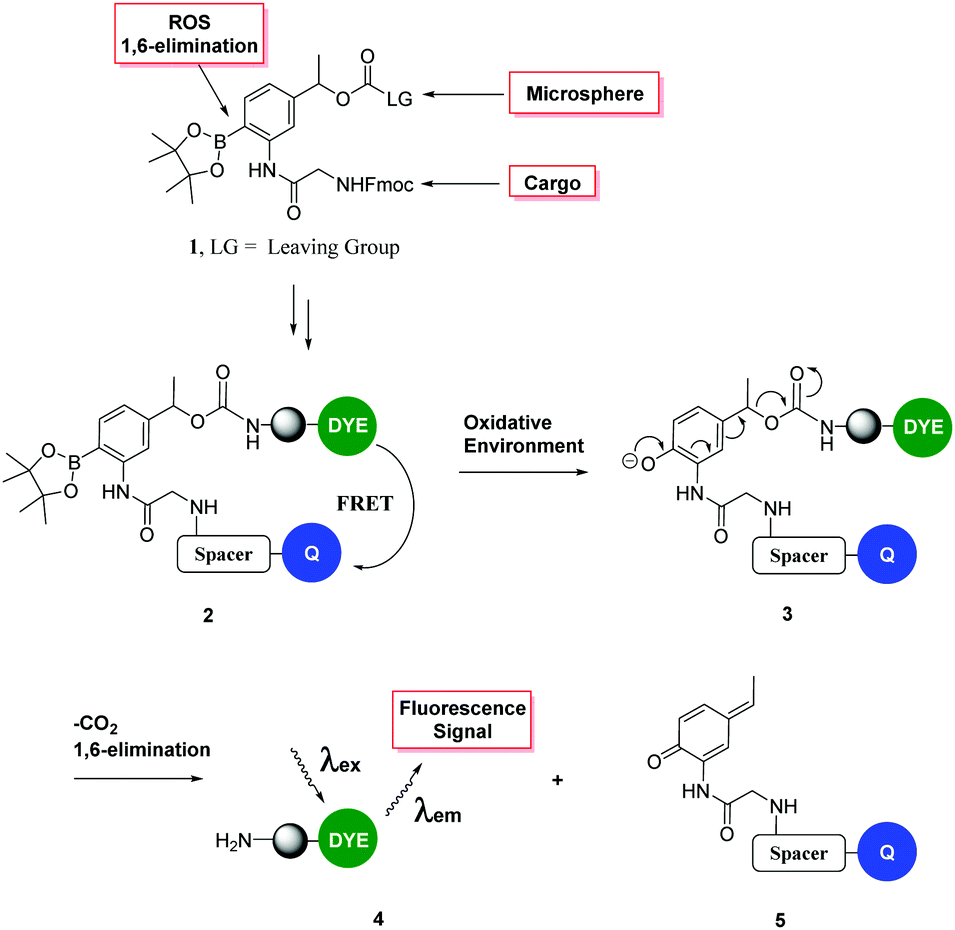 | ||
| Fig. 1 Fluorescence “turn on” ROS-responsive cleavable linker. Upper: Compound 1 shows the three major elements of the linker – namely (i) its ability to attach to a variety of cargos via Fmoc chemistry; (ii) a leaving group to allow conjugation to a cellular carrier and (iii) cyclic boronate to allow peroxide mediated rearrangement to the phenol with subsequent 1,6-elimination.19 The lower figure shows the mechanism of cleavage and the designed FRET system. | ||
This safety catch linker cleaves via the well-known oxidative rearrangement, which generates a phenol/phenolate which undergoes a 1,6-elimination reaction (as we described in 1997)35 to liberate a fluorescent label or cargo.36
2. Experimental
2.1 Chemicals and apparatus
All solvents and reagents were obtained from commercial suppliers and used without purification, unless otherwise stated. Microwave reactions were carried out using a Biotage Initiator instrument in sealed heavy-walled Pyrex tubes (Biotage AB, size 5 mL or 20 mL). Microsphere couplings were carried out on an Eppendorf ThermoMixer® comfort and washed with centrifugation in a Heraeus Pico 17 centrifuge.Thin Layer Chromatography (TLC) was carried out using Merck silica gel 60 F254 (0.25 mm) foil-backed plates with visualisation by ultraviolet light. Flash chromatography was carried out using Silica 60 Å, particle size 35–70 micron under positive pressure.
High pressure liquid chromatography (HPLC) analyses were performed using the following eluents: (A) H2O + 0.1% formic acid and (B) MeOH + 0.1% formic acid. Analytical HPLC was performed on an Agilent 1100 analytical system with a Supelco Discovery® C18, 5 μm, 5 cm column coupled to a Polymer laboratories 100 ES evaporative light scattering detector (ELSD). HPLC grade eluents were employed, at a flow rate of 1 mL min−1. The following method was used: eluents A and B, 5 to 95% B over 8 min, then 95% B for 1 min. Semi-preparative reverse phase-HPLC purifications were performed on an Agilent Technologies HP1100 Chemstation eluting on an Eclipse XDB-C18 column (9.4 × 250 mm, 5 μm), at a flow rate of 2 mL min−1. Detection was carried out at 254 nm. The following gradient was used: 5 to 50% A over 6 min, 50 to 100% B for 30 min.
High resolution mass spectrometry was measured in a Bruker MicroToF 2. Matrix assisted laser desorption ionisation-time of flight (MALDI-TOF) mass spectra were recorded using a Bruker Ultraflex Extreme MALDI TOFTOF instrument and analysed with the Bruker Daltonics flexAnalysis software. For peptide analysis, sinapic acid was used as a matrix and the positive ion mass spectra were recorded. The low resolution electrospray mass spectra (ES/MS) were recorded on an Agilent Technologies LC/MSD 1100 Quadrupole Mass Spectrometer (QMS) with an electrospray ion source.
Nuclear magnetic resonance experiments were run in a Bruker Avance NMR Spectrometer 400 MHz or 500 MHz for small molecular weight compounds. Chemical shifts are reported as δ in units of parts per million (ppm) relative to the indicated solvent. Multiplicities in 1H NMR are reported as follows: s (singlet), d (doublet), t (triplet), q (quadruplet), dd (doublet of doublets), ddd (doublet of doublet of doublets), m (multiplet). Coupling constants are reported as a J value in Hertz (Hz). The number of protons (n) for a given resonance is indicated as nH, and is based on spectral integration values.
Dynamic Light Scattering (DLS) and zeta potential were measured on a Zetasizer Nano ZS ZEN 3500 in molecular biology grade water in a disposable sizing cuvette for size measurements or clear disposable zeta cuvettes for zeta potential measurements.
Fluorescence spectrophotometry was performed on a Synergy™ HT Multi-Mode Microplate Reader from BioTek. Reader control is via BioTek's Gen5™ Data Analysis Software.
Flow cytometry was performed on a BD FACS Aria® System using the BD FACS Diva® software. The data were analysed using the software Flowjo® 7.5.
2.2 Synthesis of ROS-cleavable linkers 1a/1b
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under N2. Pyridine (anhydrous) (0.12 mL, 1.48 mmol) was added followed by 4-nitrophenyl chloroformate (0.16 g, 0.81 mmol) and the reaction was allowed to stir at room temperature for 12 h. The reaction was diluted with EtOAc and washed with 1 M HCl followed by saturated NaHCO3. The organic layer was dried over MgSO4, filtered and concentrated. Compound 1b was purified on a silica gel column eluting with 5% EtOAc in hexane. Yield: 60%. HRMS: calcd for C38H38N3O10B1 707.264480, found 707.264750. 1H NMR (500 MHz, MeOD) δ 8.00 (s, 1H), 7.80 (t, J = 8.3, 3H), 7.70 (d, J = 7.3, 3H), 7.59 (d, J = 8.3, 1H), 7.44–7.27 (m, 6H), 7.12 (d, J = 8.0, 1H), 5.84 (dd, J = 12.8, 6.2, 1H), 4.46 (d, J = 6.7, 2H), 4.27 (t, J = 6.6, 1H), 3.99 (s, 2H), 1.52 (d, J = 7.6, 3H), 1.27 (s, 12H). 13C NMR (126 MHz, MeOD) δ 171.93, 169.25, 143.82, 142.43, 141.24, 135.36, 132.39, 127.43, 126.80, 124.80, 119.58, 83.26, 74.45, 71.76, 66.90, 46.98, 35.00, 34.52, 28.98, 28.45, 26.44, 26.34, 25.86, 25.84, 23.96, 23.94, 23.65, 21.06.
:1) under N2. Pyridine (anhydrous) (0.12 mL, 1.48 mmol) was added followed by 4-nitrophenyl chloroformate (0.16 g, 0.81 mmol) and the reaction was allowed to stir at room temperature for 12 h. The reaction was diluted with EtOAc and washed with 1 M HCl followed by saturated NaHCO3. The organic layer was dried over MgSO4, filtered and concentrated. Compound 1b was purified on a silica gel column eluting with 5% EtOAc in hexane. Yield: 60%. HRMS: calcd for C38H38N3O10B1 707.264480, found 707.264750. 1H NMR (500 MHz, MeOD) δ 8.00 (s, 1H), 7.80 (t, J = 8.3, 3H), 7.70 (d, J = 7.3, 3H), 7.59 (d, J = 8.3, 1H), 7.44–7.27 (m, 6H), 7.12 (d, J = 8.0, 1H), 5.84 (dd, J = 12.8, 6.2, 1H), 4.46 (d, J = 6.7, 2H), 4.27 (t, J = 6.6, 1H), 3.99 (s, 2H), 1.52 (d, J = 7.6, 3H), 1.27 (s, 12H). 13C NMR (126 MHz, MeOD) δ 171.93, 169.25, 143.82, 142.43, 141.24, 135.36, 132.39, 127.43, 126.80, 124.80, 119.58, 83.26, 74.45, 71.76, 66.90, 46.98, 35.00, 34.52, 28.98, 28.45, 26.44, 26.34, 25.86, 25.84, 23.96, 23.94, 23.65, 21.06.
2.3 Synthesis of ROS-responsive delivery systems
37–39 The colloidal stability of the microspheres was measured by Phase Analysis Light Scattering in the Zetasizer Nano ZS. It was observed that the values of zeta potential change significantly after each step.
2.4 ROS-cleavable linker detection assay
ROS cleavable linker 1b (1.4 mg) was dissolved in 2 mL of 2 M hydrogen peroxide solution (1 mM), and was analysed by HPLC and LCMS to determine the cleavable products after 5 min. ROS cleavable linker 1b was dissolved in 2 mL of water at 1 mM as a control.2.5 Sensitivity test
ROS-responsive cell penetrating peptide probes 22 and 23 (20 μM) were incubated for 10 min with H2O2 (0–40 mM) in PBS (1 mL) and fluorescence emission at λex/em 485/528 nm for FAM/MR and 590/645 nm for Cy5.5/Cy7 was monitored (n = 3) in a BioTek microplate reader. Error is ± standard deviation.ROS-responsive microspheres probe 15 and Cy5.5 labelled-microspheres (positive control) (85 μg mL−1) were incubated with H2O2 (40 mM) in PBS (1 mL) and fluorescence emission at λex/em 590/645 nm for Cy5.5/Cy7 was monitored (n = 3) using a BioTek microplate reader. Also, the fluorescence was measured at λex/em 482/690 nm, using a BD FACSAria flow cytometer. Fluorescence was evaluated as the mean fluorescence intensity (MFI) with 1000 events per sample plotted in two-dimensional dot plots based on forward and side scattering (SSC-H vs. FSC-H) to gate microsphere populations. The data were analysed using the software Flowjo® 7.5. Error is ± standard deviation.
2.6 Cellular assays
Cell experiments were performed in a Heracell 150 incubator and carried out in a Herasafe KS 18 class II negative-flow cabinet from Heraeus. RAW 264.7 (mouse leukaemic monocyte macrophage cell line) was from The European Collection of Animal Cell Cultures (ECACC). Cells were cultured in DMEM containing high glucose (4.5 mg mL−1) supplemented with 4 mM glutamine, 100 units per mL penicillin, 50 mg mL−1 streptomycin, and 10% FBS. The cells were maintained in a humid chamber at 37 °C in an atmosphere of 5% CO2 in T75 cell culture flasks until 70% confluency. RAW 264.7 macrophage cells in their complete growth medium were transferred (100000 cells per well) to 24-well plates. To activate H2O2 production, PbCrO4 was added (150 μM) and incubated for 20 min. After the removal of media, ROS-responsive cell penetrating peptide probes 22–23 and CPPM positive control (5 μM) or ROS-responsive microspheres probe 15 and Cy5.5 labelled-microspheres positive control (85 μg mL−1) were incubated overnight. Then, cells were washed twice with PBS and detached with trypsin/EDTA, harvested with 2% fetal bovine serum (FBS) in PBS (supplemented with Trypan Blue (0.04%) for FITC), analysing using a BD FACSAria flow cytometer. Cellular fluorescence of cells was analysed in a Zeiss Axiovert 200M pseudo confocal microscope with a 100 W Hg lamp or in a Leica SP5 Confocal (GFP and RFP channel).
3. Results and discussion
The ROS-cleavable linker was synthesised in 4 steps following the approach shown in Scheme 1.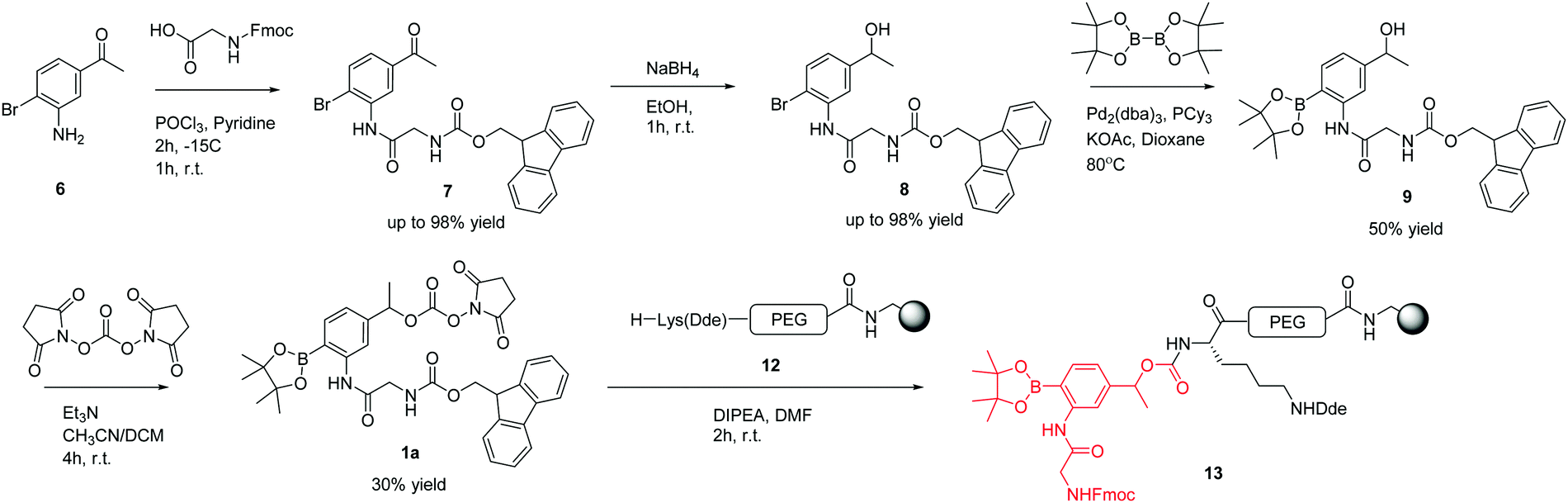 | ||
| Scheme 1 Successful synthetic route towards the ROS-cleavable linker. | ||
Compound 7 was synthesised from 3-amino-4-bromoacetophenone 6 by the amide coupling of Fmoc-Gly-OH. Presumably due to the poor nucleophilic behaviour and steric bulk of the bromoaniline, compound 7 was only obtained using the acid chloride of Fmoc-Gly-OH, generated using phosphoryl chloride (5.8% w/v) in pyridine for 2 h at −15 °C.
Following successful amide bond formation, the ketone was simply reduced by NaBH4 to give alcohol 8. Palladium mediated conversion of the bromide to the boronic ester was problematic and took extensive optimisation (Table 1). The use of the ligand tricyclohexylphosphine and the palladium catalyst Pd2(dba)3 gave the desired product in 50% yield.
| Reagents | Ratios (8)a | Solvent/Conc. of substrate (8) | Conditions | Yield (%) |
|---|---|---|---|---|
|
a Bis(pinacolato)diboron was used at 1:1 equivalents according to compound 8 in all cases.
|
||||
| PdCl2(dppf) | 0.03 | Dioxane/0.1 M | 24 h 80 °C | 14 |
| KOAc | 1.5 | |||
| PdCl2(dppf) | 0.03 | Dioxane/0.1 M | 48 h 80 °C | 19 |
| KOAc | 1.5 | |||
| PdCl2(dppf) | 0.03 | Dioxane/0.1 M | 3 h 100 °C | 14 |
| Et3N | 1.5 | |||
| Pd2(dba)3 | 0.03 | Dioxane/H2O (1:3)/0.2 M |
3 h 80 °C microwave heating | — |
| PCy3 | 0.07 | |||
| KOAc | 1.5 | |||
| Pd2(dba)3 | 0.03 | DMF/H2O (1:3)/0.2 M |
3 h 80 °C microwave heating | — |
| PCy3 | 0.07 | |||
| KOAc | 1.5 | |||
| Pd2(dba)3 | 0.03 | Dioxane/0.2 M | 6 h 80 °C | 31 |
| PCy3 | 0.07 | |||
| KOAc | 1.5 | |||
| Pd2(dba)3 | 0.03 | Dioxane bubbled/0.2 M | 6 h 80 °C | 50 |
| PCy3 | 0.07 | |||
| KOAc | 1.5 | |||
The next step was the activation of the secondary alcohol group. Several strategies were tried, including triphosgene bis(trichloromethyl) carbonate (BTC), 4-nitrophenyl chloroformate, 1,1′-carbonyldiimidazole (CDI), and N,N′-disuccinimidyl carbonate (DSC) (Table 2).
| Reagents | Ratios (9) | Solvent/Conc. of substrate (9) | Conditions | Yield (%) |
|---|---|---|---|---|
| BTC | 2 | DCM/0.1 M | 1 h 0 °C 6 h r.t. | — |
| TEA | 1 | |||
| BTC | 2.3 | DCM/0.1 M | 2 h | — |
| Pyridine | 13.5 | |||
| BTC | 0.1 | DMF/CH3CN (1:1)/0.1 M |
24 h | — |
| CDI | 4 | DMF/0.2 M | 24 h | — |
| DMAP | 0.5 | |||
| 4NO2PhCOCl | 1.1 | THF/0.1 M | 2 h | — |
| TEA | 2 | |||
| 4NO2PhCOCl | 2 | DCM/0.8 M | 3 h | 10 |
| Pyridine | 3 | |||
| DSC | 1 | CH3CN/DCM (1:1)/0.4 M |
4 h | 30 (1a) |
| Et3N | 2 | |||
| 4NO2PhCOCl | 2 | DCM/THF (7:3)/0.4 M |
o/n | 60 (1b) |
| Pyridine | 3 | |||
To test whether the linker was cleaved by H2O2, linker 1b (3-[Fmoc-Gly]amino-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenylethyl 4-nitrophenyl ester carbonic acid) was treated with hydrogen peroxide and analysed by HPLC/LCMS. This clearly showed (Fig. 2) the generation of the phenol derivative (2-[Fmoc-Gly]amino-4-hydroxyethylphenol), a product of the 1,6-elimination reaction generated by the oxidation of the boronic acid in the classical boronic acid to phenol rearrangement.
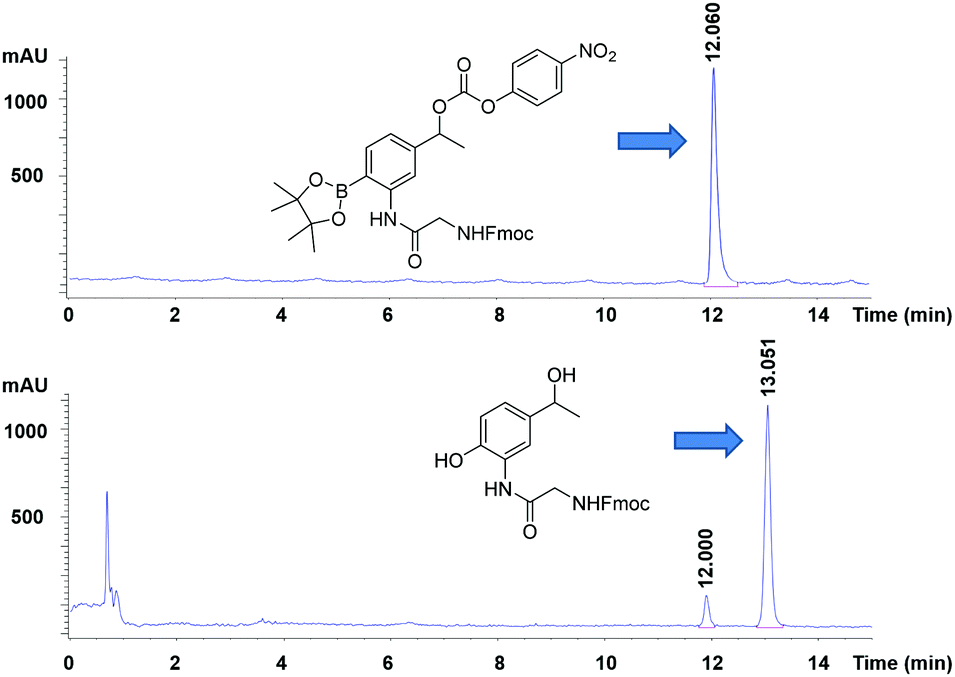 | ||
| Fig. 2 HPLC chromatograms (detection at 254 nm) of (upper) the linker (1b), (lower) its cleavage products following treatment with peroxide. | ||
Using 4-nitrophenyl chloroformate or N,N′-disuccinimidyl carbonate, the linker was successfully activated to give the 4-nitrophenyl and succinimidyl carbonates respectively. However, 4-nitrophenol was not a good enough leaving group to enable coupling to the amino derivatised microspheres, whereas the N-hydroxysuccinimide was. This then allowed solid-phase orthogonal chemistry (based on Dde and Fmoc protecting group chemistry)36 to allow coupling of peptide cargos/dyes to the microspheres (Scheme 2).
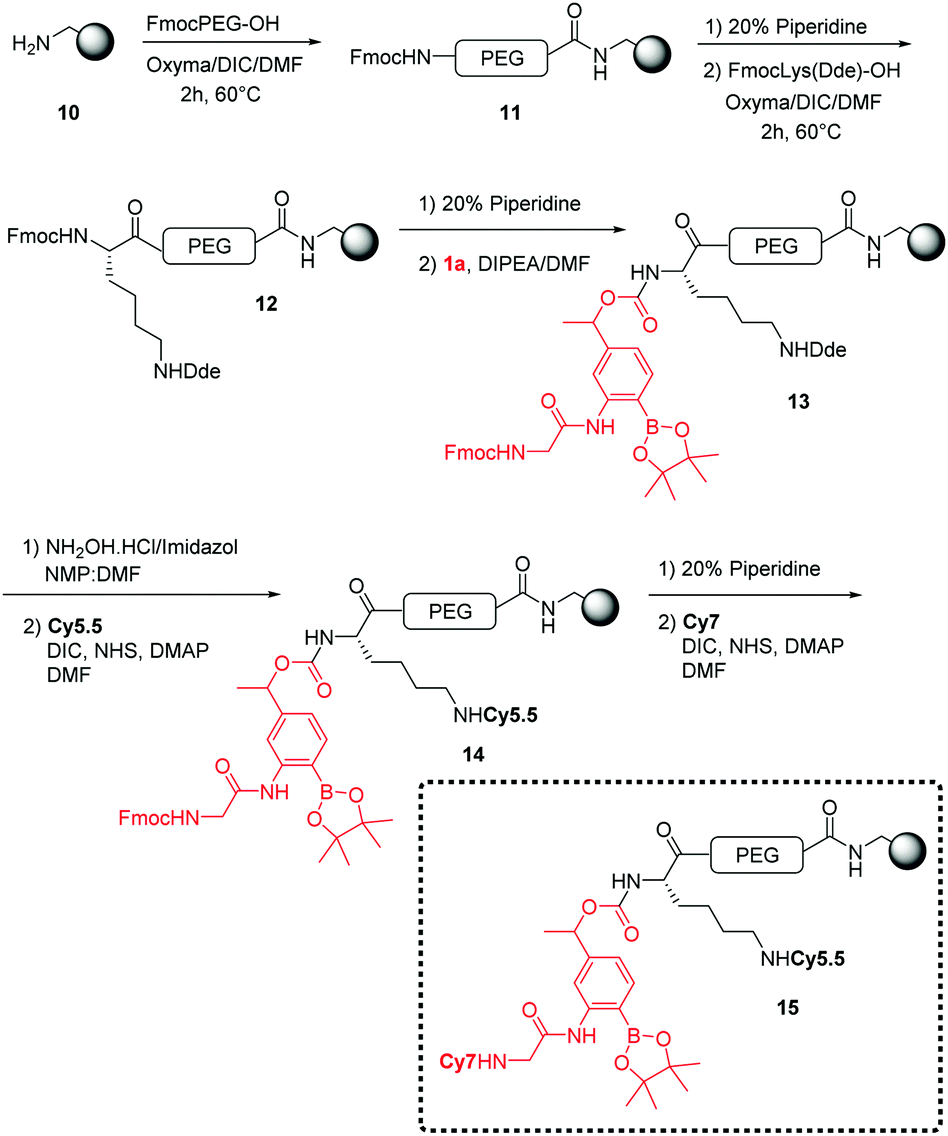 | ||
| Scheme 2 General synthesis for the generation of ROS-responsive microspheres (15). | ||
The polymeric microspheres were synthesized by an emulsifier-free emulsion polymerisation16 where vinylbenzylamine hydrochloride salt (VBAH) acts as the emulsifier and is copolymerized with styrene along with p-divinylbenzene (DVB) as a cross-linking agent (see ESI†). A Cy5.5/Cy7 FRET pair was used to allow evaluation of cellular-uptake efficiency and monitoring of ROS-production. The dual-labelled 200 nm microspheres (loading 0.2 mmol g−1) were prepared using an orthogonal Fmoc/Dde-based strategy. Fmoc-PEG-OH was coupled onto the microspheres followed by coupling of Fmoc-Lys(Dde)-OH and attachment of the linker 1a.18 The Dde protecting group was selectively removed36 followed by the addition of Cy5.5. The dye Cy7 was coupled following Fmoc cleavage (see Scheme 2).
To test the efficiency of ROS-responsive microspheres (15) for the monitoring of oxidative species in solution, a flow cytometry assay was performed on the beads with probe 15 incubated with H2O2 (see Fig. 3). This showed a >3-fold Cy5.5-fluorescence increase and confirms Cy5.5 fluorescence quenching (Cy5.5/Cy7 FRET) with disruption of this FRET pair through cleavage of the ROS-linker.
 | ||
| Fig. 3 (a) Design of ROS-responsive polymeric microspheres (15) as a FRET-based probe attached to the ROS-cleavable linker (1a), which is cleaved by oxidative conditions perturbing the FRET system (consisting of NIR cyanine dyes Cy5.5 (λex/λem 675/694 nm)/Cy7 (λex/λem 743/767 nm)). (b) Flow cytometry analysis of the fluorescence intensity. The ROS-responsive polymeric microspheres (15) showed a > than 3-fold fluorescence increase after 10 min incubation with H2O2 (40 mM). (c) Histograms of microspheres probe 15 before (dark grey) and after (light grey) the addition of H2O2 (λex/λem 482/690 nm), using a BD FACSAria flow cytometer. Fluorescence was evaluated as the mean fluorescence intensity (MFI) with 1000 events per sample plotted in two-dimensional dot plots based on forward and side scattering (SSC-H vs. FSC-H) to gate microsphere populations. The data were analysed using the software Flowjo® 7.5. | ||
Another approach to confirm the efficiency of the ROS-sensitive linker was designed using a cell penetrating peptide as a carrier system. This consisted of the polycationic N-alkylglycine peptide24–34 connected to the ROS-cleavable linker 1b (which showed high coupling efficiency to the Rink amide resin than 1a), but with cellular uptake blocked due to the interaction with a polyanionic chain (L-Glu9) analogous to the system of Tsein (for synthesis details, see the ESI†). The ROS-responsive cell penetrating peptides 22–23 were synthesized on a Rink amide-functionalised aminomethyl polystyrene resin using an Fmoc/tBu-based strategy and labeled with the fluorophores 5(6)-carboxyfluorescein or Cy5.5 and the corresponding quenchers methyl red and Cy7 (see Scheme 3).23,24 After deprotection and cleavage from the resin with TFA/TIS, the ROS-responsive Activatable Cell Penetrating Peptides were purified by preparative HPLC and analysed with structures confirmed by MALDI-TOF MS.
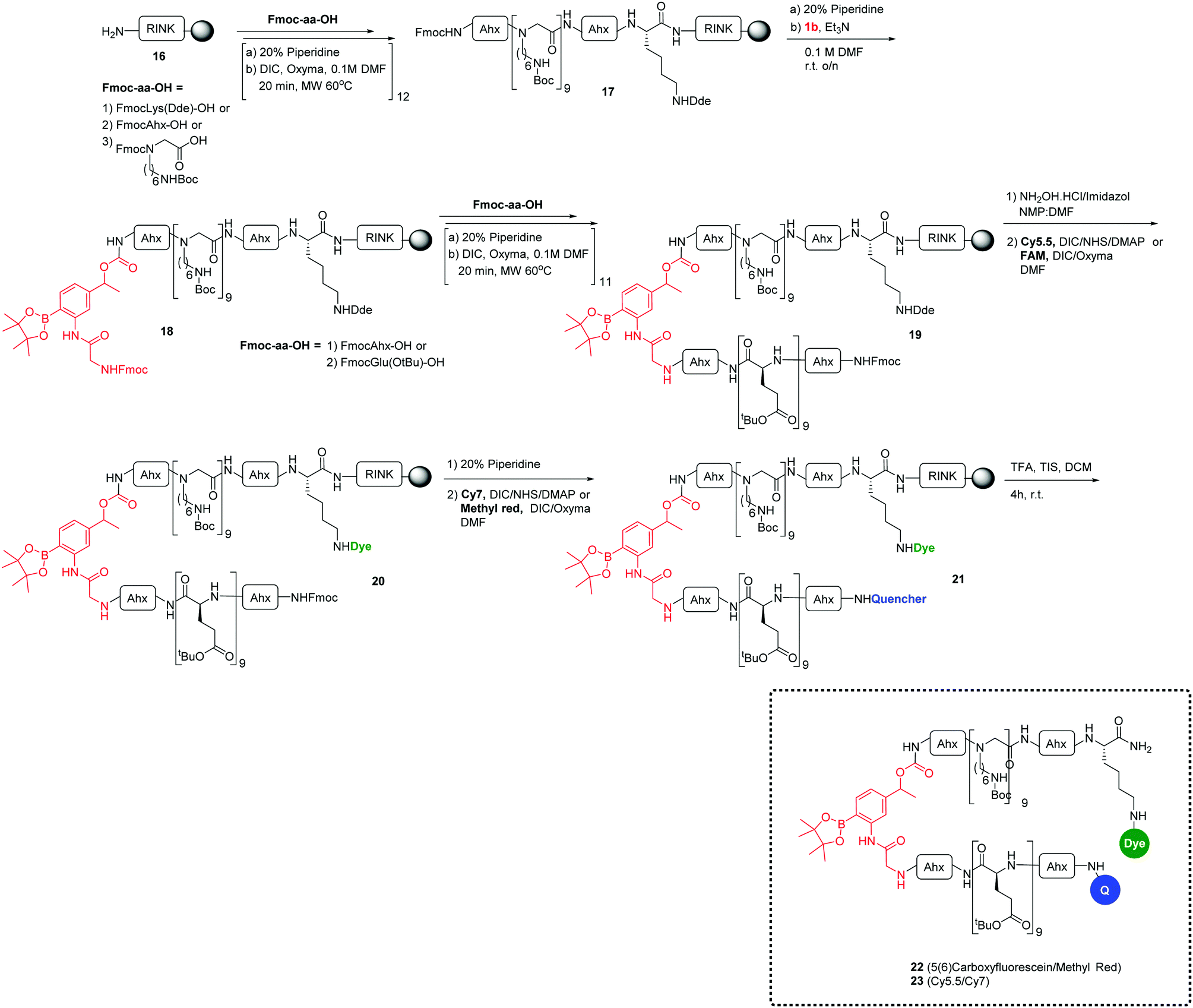 | ||
| Scheme 3 General synthesis of ROS-responsive cell penetrating peptides (22 and 23). | ||
The efficiency of the release of the cargos was evaluated in solution assays with monitoring of the fluorescence increase when probes 22 and 23 were incubated with increasing concentrations of H2O2 (Fig. 4b) and showed a 6-fold fluorescence increase for the peptide 22 carrying the FAM/MR pair and a 15-fold fluorescence increment for the peptide 23 which contained the Cy5.5/Cy7 couple.
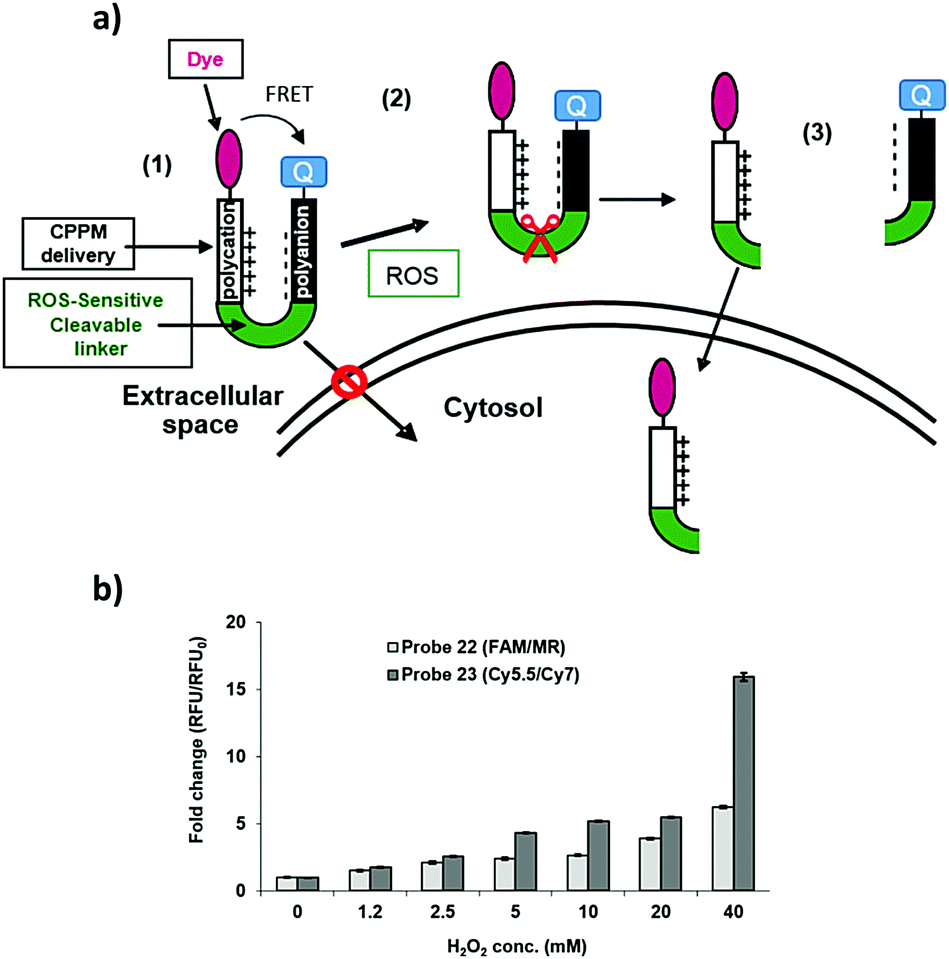 | ||
| Fig. 4 (a) ROS-responsive cell penetrating probes (22 and 23) – the reactive oxygen species – the activatable cell penetrating strategy. (a) (1) Blocked cellular uptake of the delivery system due to neutralisation/interaction with the polyanionic chain; (2) selective extracellular cleavage of the linker by the reactive oxygen species and (3) the liberated polycationic peptide linked to the dye can enter the cell. (b) Analysis of the cell penetrating peptides 22 and 23 (at 20 μM) with differing concentrations of hydrogen peroxide (measured 10 min after addition of H2O2): peptide 22, λex/em 485/528 nm or 23, λex/em 590/645 nm (Cy5.5 λex/em 675/694 nm and Cy7 λex/em 743/767 nm). | ||
RAW 264.7 macrophage cells (mouse leukemic macrophage) are a cell line established from a tumor induced by Abelson murine leukemia virus. They are capable of producing H2O2 upon stimulation with various inorganic/organic molecules.40,41
Probes 15 and 23 were incubated in either oxidative or non-oxidative environments, with RAW macrophage cells and cell fluorescence analysed by flow cytometry and fluorescence microscopy. It has been shown that PbCrO4 can be phagocytized by cells and can accumulate within vacuoles in the cytoplasm. Cr(VI) compounds undergo a complex metabolic reducing pathway, which generates a variety of reactive forms of chromium and ROS, and generate hydroxyl radicals (˙OH) from H2O2via a Fenton-like reaction. Also, during the cellular Cr(VI) reduction process, molecular oxygen is consumed to generate superoxide anion radicals (O2˙−) and H2O2.42
When RAW cells were treated with the ROS-responsive microspheres (probe 15) (85 μg mL−1) and were stimulated with PbCrO4 (150 μM) the flow cytometric histograms showed >20-fold more fluorescence for ROS-production stimulated cells (Fig. 5a). Semi-confocal microscopy confirmed that the fluorescence signal was far greater after linker cleavage (Fig. 5c and d), confirming the results obtained by flow cytometry.
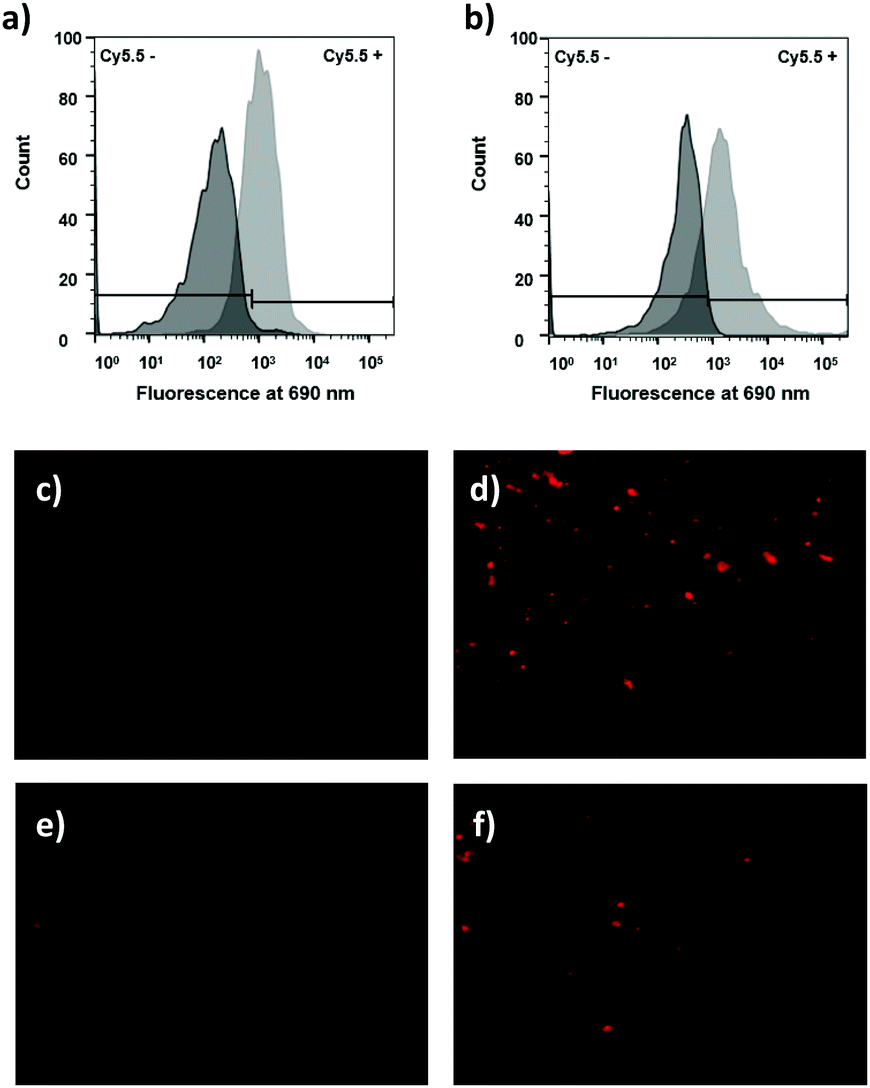 | ||
| Fig. 5 Assessment of the ROS-sensitive linker in probes 15 and 23 in live RAW cells showing cleavage by H2O2, as demonstrated by FACS analysis and microscopy. Histograms of RAW cells incubated with (a) ROS-sensitive microspheres probe 15 (85 μg mL−1) (dark grey) and probe 15 (85 μg mL−1) + PbCrO4 (150 μM for 20 min) (light grey) or incubated with (b) ROS-sensitive cell penetrating peptide probe 23 (5 μM) (dark grey) and probe 23 (5 μM) + PbCrO4 (150 μM for 20 min) (light grey). (c–f) Zeiss Axiovert 200M pseudo confocal microscope images of RAW cells analysed under a Cy5 filter (λex/λem 640/690 nm), with a ×20 objective (c) after incubation with 15 (85 μg mL−1) and (d) after incubation with 15 (85 μg mL−1) + PbCrO4 (150 μM for 20 min); (e) after incubation with 23 (5 μM) and (f) after incubation with 23 (5 μM) + PbCrO4 (150 μM for 20 min). Red fluorescence dots are from Cy5.5 “turned on” by hydrogen peroxide cleavage of the ROS-sensitive linker in the ROS-sensitive microspheres probe 15 (upper) and the ROS-sensitive cell penetrating peptide probe 23 (lower) inside the cytoplasm of RAW macrophage cells. | ||
It was explored whether the ROS-responsive cell penetrating peptide (probe 23) (5 μM) could be used to detect H2O2 in the cellular environment as observed previously using the ROS-responsive microspheres. Probe 23 was incubated with RAW cells with/without PbCrO4 and analysed by flow cytometry and fluorescence microscopy. In this case, the flow cytometric histograms showed a 6-fold fluorescence increment for cells treated with PbCrO4 (Fig. 5b), corroborating the successful cleavage of the ROS-sensitive linker when the H2O2 level increased.
4. Conclusions
In conclusion an efficient route towards synthesizing a ROS-cleavable linker that is sensitive to H2O2 has been developed. This linker allowed the design of ROS-responsive delivery systems able to liberate cargos selectively into the cellular cytoplasm in an oxidative environment.The synthesis of a ROS-responsive polymer delivery system that detects ROS inside cells allowed the release of the cargos in the cellular cytoplasm. Cell penetrating peptide mimetics were designed with penetrating properties disrupted through a hairpin structure with a FRET pair, and the cleavage of the linker permits the internalization of labelled-peptides. In live cells, fluorescence increases were detected by flow cytometry, confirming the potential of these ROS-responsive delivery systems for imaging of the oxidative environment. Based on these studies, phagocytic cells (macrophages) which generate high levels of H2O2 could be monitored thanks to the efficient cleavage of this ROS-sensitive linker. Importantly, a similar targeting mechanism could be further used for directed delivery of therapeutic cargos.
Acknowledgements
The authors would like to thank the Caja Madrid and Ramon Areces Foundations for funding AMPL. EV acknowledges European Union Seventh Framework Programme FP7 2012 for financial support. The research leading to these results has received funding from the European Union Seventh Framework Programme FP7 2012 under grant agreement no. 327903 (E. V.).Notes and references
- G. S. Devi, M. H. Prasad, D. Raghu, D. N. Rao and P. P. Reddy, Clin. Chim. Acta, 2000, 293, 53–62 CrossRef CAS.
- G. Poli, F. Biasi and E. Chiarpotto, Curr. Med. Chem., 2004, 11, 1163–1182 CrossRef CAS PubMed.
- T. Finkel, J. Cell Biol., 2011, 194, 7–15 CrossRef CAS PubMed.
- J. Chandra, A. Samali and S. Orrenius, Free Radical Biol. Med., 2000, 29, 323–333 CrossRef CAS PubMed.
- W. Droge, Physiol. Rev., 2002, 82, 47–95 CrossRef CAS PubMed.
- L. Pecorino, Molecular Biology of Cancer: Mechanisms, Targets, and Therapeutics, 3rd edn, Oxford, 2012 Search PubMed.
- C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef CAS PubMed.
- J. Liu, Y. Pang, Z. Zhu, D. Wang, C. Li, W. Huang, X. Zhu and D. Yan, Biomacromolecules, 2013, 14, 1627–1636 CrossRef CAS PubMed.
- Y. Kuang, K. Balakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- E. J. Kim, S. Bhuniya, H. Lee, H. Min Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- G. C. Van de Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316–21321 CrossRef CAS PubMed.
- G. C. Van de Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783–1795 CrossRef CAS PubMed.
- D. Lee, S. Khaja, J. C. Velasquez-Castano, M. Dasari, C. Sun, J. Petros, W. R. Taylor and N. Murthy, Nat. Mater., 2007, 6, 765–769 CrossRef CAS PubMed.
- N. Karton-Lifshin, E. Segal, L. Omer, M. Portnoy, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2011, 133, 10960–10965 CrossRef CAS PubMed.
- R. Weinstain, E. N. Savariar, C. N. Felsen and R. Y. Tsien, J. Am. Chem. Soc., 2014, 136, 874–877 CrossRef CAS PubMed.
- R. Sanchez-Martin, M. Muzerelle, N. Chitkul, S. E. How, S. Mittoo and M. Bradley, ChemBioChem, 2005, 6, 1341–1345 CrossRef CAS PubMed.
- R. M. Sanchez-Martin, M. Cuttle, S. Mittoo and M. Bradley, Angew. Chem., Int. Ed., 2006, 45, 5472–5474 CrossRef CAS PubMed.
- J. M. Cardenas-Maestre, A. M. Perez-Lopez, M. Bradley and R. M. Sanchez-Martin, Macromol. Biosci., 2014, 14, 923–928 CrossRef CAS PubMed.
- C. Chung, D. Srikun, C. S. Lim, C. J. Chang and B. R. Cho, Chem. Commun., 2011, 47, 9618–9620 RSC.
- V. V. Didenko, Biotechniques, 2011, 31, 1106–1121 Search PubMed.
- A. Solinas, L. J. Brown, C. McKenn, J. M. Mellor, J. T. G. Nicol, N. Thelwell and T. Brown, Nucleic Acids Res., 2011, 29, e96 CrossRef.
- S. Zhang, V. Metelev, D. Tabatadze, P. C. Zamecnik and A. Bogdanov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4156–4161 CrossRef CAS PubMed.
- T. Jong, A. M. Pérez-López, E. M. V. Johansson, A. Lilienkampf and M. Bradley, Bioconjugate Chem., 2015, 26, 1759–1765 CrossRef CAS PubMed.
- M. A. Fara, J. J. Diaz-Mochon and M. Bradley, Tetrahedron Lett., 2006, 47, 1011–1014 CrossRef CAS.
- A. M. Pérez-López, M. L. Soria-Gila, E. R. Marsden, A. Lilienkampf and M. Bradley, PLoS One, 2016, 11, e0153209 Search PubMed.
- I. Peretto, R. Sanchez-Martin, X. Wang, J. Ellard, S. Mittoo and M. Bradley, Chem. Commun., 2003, 2312–2313 RSC.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS PubMed.
- A. Unciti-Broceta, F. Diezmann, C. Y. Ou-Yang, M. A. Fara and M. Bradley, Bioorg. Med. Chem., 2009, 3, 959–966 CrossRef PubMed.
- K. Dhaliwal, L. Alexander, G. Escher, A. Unciti-Broceta, M. Jansen, N. Mcdonald, J. M. Cardenas-Maestre, R. Sanchez-Martin, J. Simpson, C. Haslett and M. Bradley, Faraday Discuss., 2011, 149, 107–114 RSC.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg, C. K. Marlowe, D. C. Spellmeyer, R. Tan, A. D. Frankel, D. V. Santi, F. D. Cohen and P. A. Bartlett, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS.
- R. N. Zuckermann and T. Kodadek, Curr. Opin. Mol. Ther., 2009, 11, 299–307 CAS.
- J. T. Nguyen, C. W. Turck, F. E. Cohen, R. N. Zuckermann and W. A. Lim, Science, 1998, 282, 2088–2092 CrossRef CAS PubMed.
- B. Chitkula, B. Atrasha and M. Bradley, Tetrahedron Lett., 2001, 42, 6211–6214 CrossRef.
- J. J. Díaz-Mochón, L. Bialy and M. Bradley, Org. Lett., 2004, 6, 1127–1129 CrossRef PubMed.
- R. M. Sanchez-Martin, L. Alexander, M. Muzerelle, J. M. Cardenas-Maestre, A. Tsakiridis, J. M. Brickman and M. Bradley, ChemBioChem, 2009, 10, 1453–1456 CrossRef CAS PubMed.
- F. Thielbeer, S. V. Chankeshwara and M. Bradley, Biomacromolecules, 2011, 12, 4386–4391 CrossRef CAS PubMed.
- A. Unciti-Broceta, E. M. V. Johansson, R. M. Yusop, R. M. Sánchez-Martín and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS PubMed.
- S. Y. Park, G. E. Ji, Y. T. Ko, H. K. Jung, Z. Ustunol and J. J. Pestka, Int. J. Food Microbiol., 1999, 46, 231–241 CrossRef CAS PubMed.
- P. Ping Wu, Z. Cai, J. Chen, H. Zhang and C. Cai, Biosens. Bioelectron., 2011, 26, 4012–4017 CrossRef PubMed.
- S. S. Leonard, J. R. Roberts, J. M. Antonini, V. Castranova and X. Shi, Mol. Cell. Biochem., 2004, 255, 171–179 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj02985j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |