DOI:
10.1039/C6NJ01703G
(Paper)
New J. Chem., 2017,
41, 1566-1573
Synthesis and evaluation of benzothiazole-triazole and benzothiadiazole-triazole scaffolds as potential molecular probes for amyloid-β aggregation†
Received
31st May 2016
, Accepted 5th January 2017
First published on 12th January 2017
Abstract
Small-molecule ligands that bind to misfolded protein aggregates are essential tools for the study and detection of pathological hallmarks in neurodegenerative disorders, such as Alzheimer's disease (AD). In the present study, three compounds (one benzothiazole-triazole, L1, and two benzothiadiazole-triazoles, L2 and L3) were synthesized via a modular approach (azide–alkyne cycloaddition) and evaluated as potential ligands for amyloid-β (Aβ) aggregates. The binding to amyloid-like fibrils, generated from recombinant Aβ1–42, were studied and the binding specificity to amyloid deposits was evaluated in brain sections from transgenic mice with AD pathology. All three derivatives showed significant reduced emission in the presence of recombinant Aβ1–42 amyloid fibrils. In addition, the observed binding to Aβ deposits in tissue sections suggests that the benzothiazole-triazole and benzothiadiazole-triazole structures are promising molecular scaffolds that can be modified for binding to specific protein aggregates.
1. Introduction
One of the common pathological hallmarks of neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's and Huntington's disease, and the infectious prion diseases are the formation of misfolded protein aggregates in the brain.1–4 Thus, development of new molecular probes for the detection of protein aggregates is of great importance in order to understand the pathology of these diseases.5–8 For example, probes that can be prepared via a modular synthetic approach to generate structures with diversity are desirable. To date, the most commonly used ligands for studying protein aggregation are thioflavin T (ThT) and Congo red (CR) (Fig. 1). ThT, which is a benzothiazole dye, is used due to its impressive increase in fluorescence intensity (ca. 1000-fold) in the presence of protein fibrils, a phenomenon that is due to conformational rigidity upon binding.9–11 However, the practical use of ThT has been debated since it has limitations; e.g. it is not able to distinguish between prefibrillar and fibrillar aggregates and some protein aggregates do not affect its fluorescence, leading to false negatives.12,13 In addition, CR, (an aromatic azo dye) is commonly used for staining protein aggregates in histology samples.14–16 However, CR staining can give misleading results due to the thickness of the sample and the orientation of the amyloid deposits.13 Hence, CR staining usually requires experience and sample control in order to generate reliable results.17 Although both ThT and CR are useful and well-established dyes for amyloid detection in tissue there is a need for alternative ligands, e.g. compounds that could be generated via a modular approach, in order to study different protein aggregates and their morphologies. Moreover, the use of nuclear imaging techniques, such as positron emission tomography (PET) and single photon emission computed tomography (SPECT) for the in vivo detection of protein aggregates (such as Aβ aggregates in AD) has increased interest in the development of radiolabelled binding ligands.18 For example, Pittsburgh compound B, [11C]PIB, and Amyvid™ (florbetapir) (Fig. 1) are widely used in vivo as indicators for Aβ-deposits in brain tissue.19–21
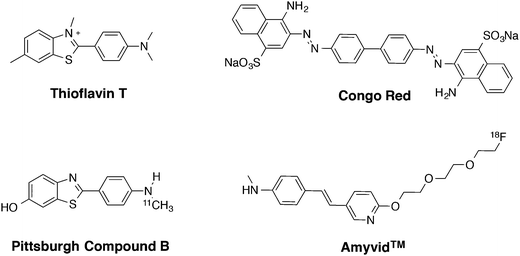 |
| | Fig. 1 Chemical structures of known amyloid-β ligands; thioflavin T (ThT), Congo red (CR), Pittsburgh compound B ([11C]PIB) and Amyvid™ (florbetapir). | |
In our search for small molecular scaffolds that bind to protein aggregates, we are particularly interested in synthetic pathways that involve a modular approach (e.g. click chemistry) that could generate diverse derivatives with specific binding properties. Initially for this study, we investigated if the combination of a 1,2,3-triazole moiety and two well-known chromophores, benzothiazole and benzothiadiazole, could afford ligands that exhibit amyloid binding properties in histology samples. Our approach was to use a small azide (3-azidopropan-1-ol) when forming the triazole ring in order to generate model compounds that could be used as a proof-of-concept for amyloid detection.22,23 Hence, in this paper we report the synthesis and evaluation of three new compounds (one benzothiazole-triazole, L1, and two benzothiadiazole-triazoles, L2 and L3, Fig. 2) as potential ligand scaffolds for amyloid-β aggregates in mouse brain sections.
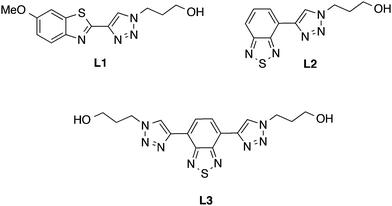 |
| | Fig. 2 Chemical structures of the benzothiazole-triazole (L1) and the benzothiadiazole-triazole derivatives (L2 and L3). | |
2. Results and discussion
2.1. Synthesis
The target compounds (L1–L3) were synthesized as presented in Schemes 1 and 2. The benzothiazole-triazole derivative (L1, Scheme 1) was prepared by using commercially available 2-amino-6-methoxy-benzothiazole (1) as starting material. First, the amino functionality in the 2-position of 1 was replaced with iodine via diazotization-Sandmeyer iodination using KI, NaNO2 and p-toluenesulfonic acid monohydrate in H2O/acetonitrile to afford compound 2.24 Subsequently, TMS-acetylene was introduced in the 2-position of the iodinated compound (2) by using a palladium-mediated Sonogashira coupling under inert conditions with ethynyltrimethylsilane, PdCl2(PPh3)2, and CuI in dichloromethane to give compound 3. Finally, deprotection of the TMS-group and the subsequent azide–alkyne Huisgen cycloaddition reaction (click chemistry) was performed simultaneously by mixing compound 3 with TBAF (1 M in THF), 3-azidopropan-1-ol, CuSO4·H2O and L-sodium ascorbate in H2O/THF to give compound L1 in 55% yield.25,26
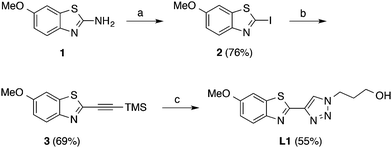 |
| | Scheme 1 Synthesis of the benzothiazole-triazole derivative, L1. Reagents and conditions: (a) KI, NaNO2, p-toluenesulfonic acid monohydrate, H2O, acetonitrile, 0 °C to rt, overnight. (b) Ethynyltrimethylsilane, PdCl2(PPh3)2, CuI, dichloromethane, reflux, 3 h. (c) TBAF (1 M in THF), 3-azidopropan-1-ol, CuSO4·H2O, L-sodium ascorbate, H2O, THF, rt, 3 days. | |
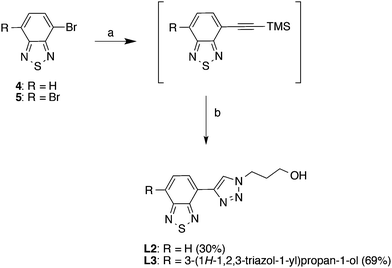 |
| | Scheme 2 Synthesis of the benzothiadiazole-triazole derivatives, L2 and L3. Reagents and conditions: (a) ethynyltrimethylsilane, PdCl2(PPh3)2, PPh3, CuI, NEt3, reflux, 4–18 h; (b) TBAF (1 M in THF), 3-azidopropan-1-ol, CuSO4·H2O, L-sodium ascorbate, H2O, THF, rt, 3 days. | |
The benzothiadiazole-triazole derivatives (L2 and L3) (Scheme 2) were synthesized using the same procedure as above. However, the syntheses were regulated using different equivalents of the reagents in order to generate the mono- or disubstituted products. First, the starting materials, 4-bromo-2,1,3-benzothiadiazole (4) and 4,7-dibromo-2,1,3-benzothiadiazole (5), were synthesized using previous reported protocols (ESI,† Scheme S1).27–29 Subsequently, TMS-acetylene was introduced by a palladium-mediated Sonogashira reaction in the presence of ethynyltrimethylsilane, PdCl2(PPh3)2, PPh3 and CuI, in triethylamine. Notably, the crude products were immediately carried forward in the next reaction step due to limited compound stability. Next, deprotection of the TMS-group and subsequent click reaction was performed simultaneously by mixing the crude product from the previous reaction step with TBAF (1 M in THF), 3-azidopropan-1-ol, CuSO4·H2O and L-sodium ascorbate in H2O/THF, affording L2 and L3 in 30% and 69% yield, respectively (over three steps).30
2.2. Photophysical measurements
2.2.1. Photophysical characterization of compound L1–L3.
Absorption and emission profiles for L1–L3 were determined in 5% DMSO in phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4) (Table 1) (for spectra see ESI,† Fig. S1). The molar extinction coefficients for the ligands were similar, ranging from 8000–11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 [M−1 cm−1]. The benzothiazole-triazole derivative (L1) showed a high-energy absorption band at 313 nm and emission maxima at 391 nm, whereas the absorption and emission maxima for the benzothiadiazole-triazole derivatives (L2–L3) were more redshifted (L2: λabs = 353 nm, λem = 494 nm; L3: λabs = 387 nm, λem = 531 nm). These results were expected since the benzothiadiazole fragment is well known for its interesting photophysical properties, such as the strong electron-withdrawing capacity that facilitates intramolecular charge transfer (ICT), large Stoke's shifts, high signal-to-noise ratio in cells and red-shifted optical profiles.31 In particular, the latter is beneficial in protein binding studies and tissue staining experiments due to the use of lower energies for excitation, and resulting lower autofluorescence signals and reduced light scattering. Further, quantum yield measurements for the ligands (in 5% DMSO in PBS buffer, pH 7.4) afforded moderate to high ϕF values (0.67, 0.24 and 0.31 for L1, L2 and L3, respectively) (Table 1). The pH dependence (pH 2–12) of the fluorophores was also investigated, and the fluorescent intensities and emission maxima was unchanged in a wide range (pH 4–12, see ESI,† Fig. S2). However, a reduced intensity was observed at pH 2, likely due to protonation of the benzothia(dia)zole fragments.32 Despite the blue-shifted absorption and emission maxima for L1 we further investigate all three compounds (L1–L3) for their amyloid binding potential in solution and in AD brain tissue.
000 [M−1 cm−1]. The benzothiazole-triazole derivative (L1) showed a high-energy absorption band at 313 nm and emission maxima at 391 nm, whereas the absorption and emission maxima for the benzothiadiazole-triazole derivatives (L2–L3) were more redshifted (L2: λabs = 353 nm, λem = 494 nm; L3: λabs = 387 nm, λem = 531 nm). These results were expected since the benzothiadiazole fragment is well known for its interesting photophysical properties, such as the strong electron-withdrawing capacity that facilitates intramolecular charge transfer (ICT), large Stoke's shifts, high signal-to-noise ratio in cells and red-shifted optical profiles.31 In particular, the latter is beneficial in protein binding studies and tissue staining experiments due to the use of lower energies for excitation, and resulting lower autofluorescence signals and reduced light scattering. Further, quantum yield measurements for the ligands (in 5% DMSO in PBS buffer, pH 7.4) afforded moderate to high ϕF values (0.67, 0.24 and 0.31 for L1, L2 and L3, respectively) (Table 1). The pH dependence (pH 2–12) of the fluorophores was also investigated, and the fluorescent intensities and emission maxima was unchanged in a wide range (pH 4–12, see ESI,† Fig. S2). However, a reduced intensity was observed at pH 2, likely due to protonation of the benzothia(dia)zole fragments.32 Despite the blue-shifted absorption and emission maxima for L1 we further investigate all three compounds (L1–L3) for their amyloid binding potential in solution and in AD brain tissue.
Table 1 Spectroscopic data for compounds L1–L3 measured in 5% DMSO in phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4)
| Compound |
λ
abs (nm) |
λ
em (nm) |
Stoke's shift (nm) |
ε [M−1 cm−1] |
ϕ
F
|
|
Determined relative to quinine sulfate in aqueous H2SO4 (0.1 M).
|
|
L1
|
313 |
391 |
78 |
9000 |
0.67 |
|
L2
|
353 |
494 |
141 |
11000 |
0.24 |
|
L3
|
387 |
531 |
144 |
8000 |
0.31 |
2.2.2. Photophysical characterization of the benzothiazole-triazole (L1) and benzothiadiazole-triazole derivatives (L2 and L3) towards recombinant Aβ1–42 amyloid fibrils.
In order to evaluate potential interactions with protein aggregates, excitation and emission spectra of the ligands (L1–L3) were recorded in the absence and the presence of amyloid-like fibrils, generated from recombinant Aβ1–42 (Fig. 3). Initially, the emission spectra of L1–L3 in the presence of 10 μM Aβ-fibrils were collected using different concentrations of the ligands (0.3, 0.6, 0.9, 1.2, 10 and 100 μM) (spectra not shown). However, a ligand concentration of 50 μM was chosen for further analysis due to the optimal linear-response between absorbance and integrated fluorescence intensity (ESI,† Fig. S4). Interestingly, all three ligands show reduced fluorescence (intensity area), approx. 20% (reduced excitation intensity: L1 = 16%, L2 = 17%, L3 = 23%; reduced emission intensity: L1 = 17%, L2 = 21%, L3 = 17%), when mixed with the fibrils in solution, quite the contrary to ThT, which shows an increase in emission intensity. Interestingly, the absorbance-integrated luminescence intensity curves for L1–L3 (ESI,† Fig. S4) show fluorescence quenching (L3 > L1 > L2) at high concentrations (>100 μM) in the absence of aggregates, and we thus hypothesize that the decreased intensity in the fibril experiment could originate from proximity-induced self-quenching of the ligands upon interaction with the Aβ-fibrils.33–37 Even if the ligands have a different photophysical behavior in comparison to ThT, the approx. 20% reduced intensity provides a useful and pronounced evidence of ligand–fibril interaction without approaching the limit of detection.
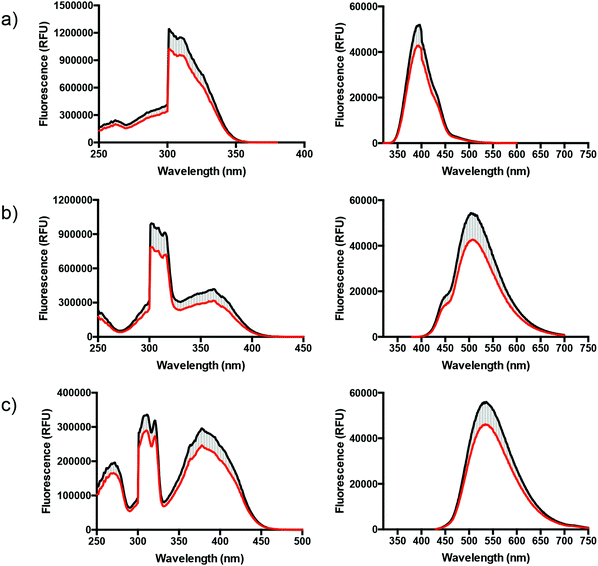 |
| | Fig. 3 Excitation (left) and emission (right) spectra of L1 (a), L2 (b), and L3 (c) in the presence (red line) or absence (black line) of recombinant Aβ1–42 fibrils in 5% DMSO in phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4). The reduced emission intensities are represented by the light grey striped area. The measurements were performed in a 5:1 ratio, ligand to fibrils, using the concentrations 50 μM and 10 μM, respectively. | |
2.3. Staining of Aβ-plaques in brain tissue sections from transgenic mice
Aβ plaques in the APP/PS1 mouse model of AD pathology38 were stained simultaneously with the ligands L1, L2 or L3 and with an anti-Aβ antibody (6E10) and then imaged via channels selective for one or the other stain (excitation at 405 and 680 nm, respectively). The resulting images (Fig. 4) clearly demonstrate that all three ligands bind to Aβ plaques with specificity and accuracy with L3 affording the best signal to background contrast. All three ligands display preferential binding to the plaque cores analogous to what has been described previously for this mouse model when stained with ThT or CN–PIB, which is expected due to the overall similarity in chemical structure between the ligands (L1–L3) and the well-established counterparts.39 This in turn holds promise that these new ligands may play a significant role as scaffolds for further development of amyloid-binding ligands with specific designs for various purposes using click chemistry for rapid multipurpose modification to improve binding affinities and pharmacokinetics.
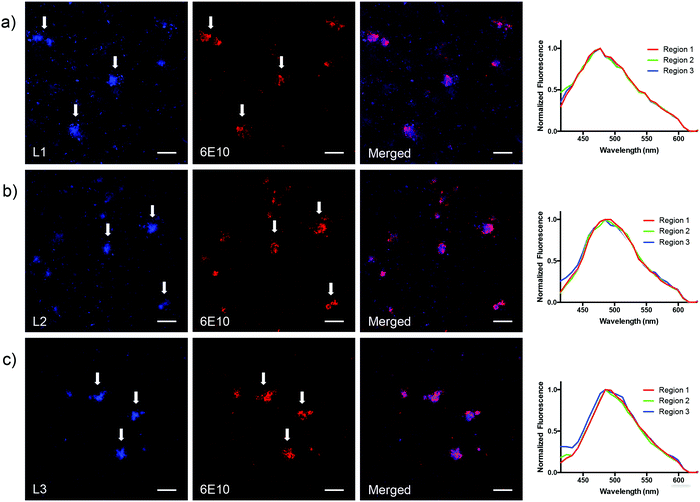 |
| | Fig. 4 Fluorescence microscopy images of Aβ deposits in brain sections from APP/PS1 mice stained with L1 (a) L2 (b) and L3 (c), and with an anti-Aβ antibody (6E10). White arrows point towards the localization of three representative amyloid deposits in each image stained with ligand (panels to the left) or anti-Aβ antibody (6E10) (panels in the middle). Panels to the right show both channels in order to clarify the co-localization between ligand (seen in blue) and the anti-Aβ antibody (seen in red). The emission spectra (excitation at 405 nm) of each ligand binding to the core of three different Aβ plaques (labeled as region 1, 2 and 3) are shown to the far right. The scale bar in the lower right corner of each image represents 50 μm. | |
Notably, as depicted in Fig. 4a (to the far right), the emission profile for L1 is more redshifted (λem ∼ 470 nm) in the fluorescent microscopy experiment (using histology samples) in comparison with the photophysical characterization measurements in 5% DMSO in PBS buffer (λem = 394 nm) (Table 1). We hypothesized that this result was not an optical effect that originates from binding to the Aβ deposits in brain tissue, and instead a result from using a longer excitation wavelength (i.e. 405 nm instead of 310 nm) in the tissue experiment. Furthermore, the redshift was verified by measuring the emission spectra of L1 in 5% DMSO in phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4) when exciting at 405 nm, which afforded an emission maxima at 472 nm (see ESI,† Fig. S5a). However, even though it is possible to excite L1 at a lower energy and generate a red-shifted emission profile, we clearly observed that the benzothiadiazole derivatives L2 and L3 are much brighter, affording higher signal-to-noise ratios in the fluorescent microscopy experiments using brain tissue sections. These derivatives are therefore superior ligands for further development.
3. Conclusions
We have shown that the benzothiazole-triazole and the benzothiadiazole-triazole scaffolds constitute new promising classes of ligands for amyloid-β detection. The ligands show a significant reduction of the emission (approx. 20%) in the presence of recombinant Aβ1–42 amyloid fibrils in solution, an effect that likely originates from self-quenching of the ligands upon interaction with the protein fibrils. Histology staining of Aβ-plaques in mouse brain sections demonstrates specific binding to amyloid deposits for all three ligands, which were verified by co-labeling with an anti-Aβ antibody (6E10). Even though the present ligands are not superior in comparison to ThT for sensitive fluorescence detection of protein aggregates, we foresee that the synthetic pathway (azide–alkyne cycloaddition) described herein, will offer an avenue to design a variety of structurally diverse ligands that exhibit specific binding to different protein aggregates. Suggestively, the approach also enables the introduction of substituents that includes radioactive isotopes (e.g. [18F]) for PET or SPECT imaging. Our intention is to further investigate a library of benzothiadiazole ligands, formed via click chemistry, to generate amyloid binding agents and that can distinguish between different protein aggregates in solution and in amyloid containing brain tissue.
4. Experimental
4.1. Chemistry
All reagents and solvents were of analysis or synthesis grade and were used without further purification unless otherwise indicated. 1H and 13C NMR were recorded on a Bruker AVANCE III 400 MHz NMR (at 400 and 100 MHz, respectively) or on a Varian 300 instrument (Varian Inc., Santa Clara, CA, USA) (at 300 and 75 MHz, respectively) in CDCl3 or DMSO-d6. Chemical shift is reported in ppm with the solvent residual peak as reference; CDCl3 (δH 7.26, δC 77.0), DMSO-d6 (δH 2.50, δC 39.5) and CD3OD (δH 4.87, δC 49.0). The reactions were monitored by thin-layer chromatography (TLC), on silica plated (Silica gel 60 F254, E. Merck) aluminium sheets, detecting spots by UV (254 and 365 nm). Flash chromatography was performed using Merck Silica Gel 60 (40–63 μm). Mass spectra were obtained with the positive ion mode electrospray ionization [ESI(+)] source on an Agilent 6210 time of-flight liquid chromatography/mass spectrometry (MS) system. High-resolution mass data (ES-ToF) were obtained from the Division of Mass spectrometry, Department of Chemistry, Imperial College London.
4.1.1. 2-Iodo-5-methoxybenzo[d]thiazole (2)24.
The reaction was carried out under nitrogen atmosphere in order to have an evacuation line for the N2 gas that is formed during the reaction. A solution of NaNO2 (0.153 g, 2.22 mmol) and KI (0.461 g, 2.77 mmol) in H2O (0.7 mL) was added dropwise to an ice-cooled suspension of 2-amino-6-methoxy-benzothiazole (1) (0.2 g, 1.11 mmol) and p-toluenesulfonic acid monohydrate (0.633 g, 3.33 mmol) in acetonitrile (5.5 mL). The reaction was allowed to reach room temperature and was stirred overnight. The reaction mixture was quenched with H2O (20 mL), basified with 2 M NaHCO3(aq.) until pH reached 8–9, follow by the addition of Na2S2O3(aq.) (2.5 mL) to give a yellow/brown precipitate. The precipitate was filtered of and dried in vacuo to give compound 2 as a brown/gold power (0.245 g, 76%). 1H NMR (400 MHz, CDCl3) δ 3.86 (s, 3H), 7.03 (dd, J = 2.6, 9.0 Hz, 1H), 7.28 (d, J = 2.6 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 55.8, 101.5, 103.0, 115.6, 123.0, 149.4, 149.0, 158.0. ESI(+)-MS (m/z): [M + H]+ calcd for C8H7INOS, 291.9288. Found, 291.9318.
4.1.2. 6-Methoxy-2-((trimethylsilyl)ethynyl)benzo[d]thiazole (3)25,26.
The reaction was carried out under inert conditions (N2). Ethynyltrimethylsilane (0.49 mL, 3.44 mmol) was added to a suspension of compound 2 (0.5 g, 1.72 mmol), PdCl2(PPh3)2 (0.024 g, 0.03 mmol), and CuI (6.5 mg, 0.03 mmol) in dry dichloromethane (2.5 mL). The reaction was refluxed for 3 hours and was then allowed to reach room temperature. The mixture was filtered through a layer of Celite and concentrated under reduced pressure. The crude product was purified by column chromatography (10% EtOAc in hexanes) to give compound 3 as a light yellow powder (0.312 g, 69%). The product was used in the next reaction step immediately or stored in the freezer for a limited time due to stability issues. 1H NMR (400 MHz, CDCl3) δ 0.30 (s, 9H), 3.88 (s, 3H), 7.11 (dd, J = 2.5, 9.1 Hz, 1H), 7.28 (d, J = 2.5 Hz, 1H), 7.92 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 0.5, 55.8, 96.9, 102.2, 103.3, 116.5, 124.2, 136.7, 145.6, 147.2, 158.8. ESI(+)-MS m/z: [M + H]+ calcd for C13H16NOSSi, 262.0716. Found 262.0738.
4.1.3. 3-(4-(5-Methoxybenzo[d]thiazol-2-yl)-1H-1,2,3-triazol-1-yl)propan-1-ol (L1)25,26.
TBAF (1 M in THF, 4.56 mL) was added dropwise to a stirred solution of 3 (0.298 g, 1.14 mmol) in THF (10 mL). Subsequently, 3-azidopropan-1-ol (0.231 g, 2.28 mmol) was added and then a solution of CuSO4·H2O (0.114 g, 0.456 mmol) and L-sodium ascorbate (0.090 g, 0.456 mmol) in H2O (10 mL). The reaction mixture was stirred at room temperature for 3 days and then Chelex resin (approx. a small spoon) was added to remove the Cu catalyst. The solution was filtered after 30 min, to remove the Chelex resin, and was then concentrated under reduced pressure. The crude residue was purified by column chromatography (100% EtOAc) to give the product (L1) as an off white powder (0.182 g, 55%). Mp 161–162 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.00–2.09, m, 2H), 3.44 (dd, J = 6.0, 11.1 Hz, 2H), 3.85 (s, 3H), 4.53 (t, J = 7.1 Hz, 2H), 4.71 (t, J = 5.0 Hz, 1H), 7.13 (dd, J = 2.5, 8.9 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.90 (d, J = 8.9 Hz, 1H), 8.85 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 32.6, 47.3, 55.7, 57.4, 104.9, 115.9, 123.1, 223.4, 135.3, 141.9, 147.6, 156.5, 157.5. HRMS (ES-ToF) [M + H]+ calcd for C13H15N4O2S, 291.0916. Found, 291.0924.
4.1.4. 3-(4-(Benzo[c][1,2,5]thiadiazol-4-yl)-1H-1,2,3-triazol-1-yl)propan-1-ol (L2)30.
The reaction was performed under inert conditions (N2). Ethynyltrimethylsilane (0.28 mL, 1.95 mmol) was added to a stirred solution of 4 (0.3 g, 1.39 mmol), PPh3 (0.038 g, 0.14 mmol), CuI (0.037 g, 0.195 mmol) and PdCl2(PPh3)2 (6.0 mg, 0.008 mmol) in NEt3 (3 mL). The mixture was heated at 90 °C for 18 hours and was then allowed to reach room temperature. The reaction mixture was diluted with dichloromethane (10 mL) and was then quickly filtered through a small silica column. The filtrate was concentrated under reduced pressure to give a crude solid that was used immediately in the next reaction step due to stability issues. TBAF (1 M in THF, 2.79 mL) was added dropwise to a stirred solution of the crude material in THF (10 mL). Subsequently, 3-azidopropan-1-ol (0.282 g, 2.79 mmol) was added followed by the addition of a solution of CuSO4·H2O (0.139 g, 0.56 mmol) and L-sodium ascorbate (0.111 g, 0.56 mmol) in H2O (10 mL). The reaction mixture was stirred at room temperature for 3 days and then Chelex resin (approx. a small spoon) was added to remove the Cu catalyst. The solution was stirred for 30 min and was then filtered in order remove the Chelex resin. The filtrate was concentrated under reduced pressure and the crude residue was purified by column chromatography (100% EtOAc) to give L2 as yellow powder (0.110 g, 30%). Mp 114–115 °C 1H NMR (400 MHz, CD3OD) δ 2.21 (ddd, J = 6.1, 7.0, 13.1 Hz, 2H), 3.65 (t, J = 6.1 Hz, 2H), 4.66 (t, J = 7.0 Hz, 2H), 7.76 (dd, J = 7.1, 8.8 Hz, 1H), 8.00 (dd, J = 1.0, 8.8 Hz, 1H), 8.38 (dd, J = 1.0, 7.1 Hz, 1H), 8,89 (s, 1H). The OH-signal was not observed in the spectrum, due to solvent exchange. 13C NMR (100 MHz, CD3OD) δ 34.1, 48.5, 59.3, 121.9, 124.6, 126.2, 126.3, 131.0, 143.9, 153.0, 156.6. HRMS (ES-ToF) [M + H]+ calcd for C11H12N5OS, 262.0763. Found, 262.0772.
4.1.5. 3,3′-(Benzo[c][1,2,5]thiadiazole-4,7-diylbis(1H-1,2,3-triazole-4,1-diyl))bis(propan-1-ol) (L3)30.
The reaction was performed under inert conditions (N2). Ethynyltrimethylsilane (0.4 mL, 2.86 mmol) was added to a stirred solution of 5 (0.3 g, 1.02 mmol), PPh3 (0.027 g, 0.1 mmol), CuI (0.027 g, 0.143 mmol) and PdCl2(PPh3)2 (4.3 mg, 0.006 mmol) in NEt3 (5 mL). The mixture was heated at 90 °C for 4 hours and was then allowed to reach room temperature. The reaction mixture was diluted with dichloromethane (10 mL) and was then quickly filtered through a small silica column. The filtrate was concentrated under reduced pressure to give a crude solid that was used immediately in the next reaction step due to stability issues. TBAF (1 M in THF, 4.08 mL) was added dropwise to a stirred solution of the crude material in THF (10 mL). Subsequently, 3-azidopropan-1-ol (0.248 g, 2.45 mmol) was added followed by the addition of a solution of CuSO4·H2O (0.051 g, 0.20 mmol) and L-sodium ascorbate (0.040 g, 0.2 mmol) in H2O (10 mL). The reaction mixture was stirred at room temperature for 3 days and then Chelex resin (approx. a small spoon) was added to remove the Cu catalyst. The solution was stirred for 30 min and was then filtered in order remove the Chelex resin. The filtrate was concentrated under reduced pressure and the crude residue was purified by column chromatography (100% EtOAc → 10% EtOH in EtOAc) to give the L3 as yellow powder (0.274 g, 69%). Mp 170–171 °C; 1H NMR (400 MHz, CD3OD) δ 2.18 (m, 4H), 3.66 (t, J = 6.1 Hz, 4H), 4.67 (t, J = 7.0 Hz, 4H), 8.49 (s, 2H), 9.01 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 33.0, 46.9, 57.4, 122.3, 124.7, 125.3, 141.7, 151.5. HRMS (ES-ToF) [M + H]+ calcd for C16H19N8O2S, 387.1352. Found, 387.1338.
4.2. Photophysical measurements
4.2.1. Photophysical characterization of compounds L1–L3.
All photophysical measurements were performed in 5% DMSO in 0.1 M PBS buffer (pH 7.4) using 1 cm path length quartz cuvettes. Absorption spectra were collected using a Varian CARY 5000 UV-visible-near IR spectrophotometer or a Hitachi U-1900 spectrophotometer. The emission data were collected using a Photon Technology International 814 photomultiplier detection system or a Horiba Jobin-Yvon Fluoromax-4 instrument, and the signals were collected at 90° to the incident excitation beam. The final concentrations of ligands were approximately 100 μM and 100 nM for the absorption and emission measurements, respectively (ESI,† Fig. S1). Extinction coefficients were determined from samples of known concentrations. Quantum yields (QY) were measured relative to quinine sulfate (QY = 0.577)40 in aqueous H2SO4 (0.1 M). The refraction index for the solvent mixture (5% DMSO/H2O) was included in the calculations (η = 1.3390).41 The concentration-absorbance relationship and the absorbance-integrated fluorescence intensity relationship were measured at known concentrations (20, 40, 60, 80, 100 and 250 μM) using a Tecan Saphire 2 micro plate reader (Tecan Group Ltd, Männerdorf, Switzerland). All the measurements were repeated in triplicates with an excitation wavelength of 310, 350 and 370 nm for L1, L2 and L3, respectively.
4.2.2. Photophysical characterization of the benzothiazole-triazole (L1) and benzothiadiazole-triazole derivatives (L2 and L3) towards recombinant Aβ1–42 amyloid fibrils.
Recombinant Aβ1–42 amyloid fibrils were prepared as previously reported in the literature; recombinant Aβ1–42 peptide lyophilized in hydroxyfluoroisopropanol (rPeptide, Athens, GA, USA) was dissolved in aqueous NaOH (2 mM) to a stock concentration of 1 mg mL−1. The alkaline solution of Aβ1–42 was further diluted with phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4) (Medicago, Sweden) to a final concentration of 10 μM and added to the wells of a microtiter plate (Corning). The microtiter plate was incubated at 37 °C in quiescent mode for 48 hours and the presence of recombinant Aβ1–42 amyloid fibrils was confirmed by thioflavin T (ThT) staining. Stock solutions of L1, L2 and L3 (1 mM in DMSO) was added to phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4) or to the 10 μM solution of Aβ1–42 fibrils in phosphate buffered saline (PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4), respectively, in a 96-well plate to give a final probe concentration of 50 μM (5% DMSO in phosphate buffered saline, PBS, 10 mM phosphate, 140 mM NaCl, 2.7 mM KCl, pH 7.4). Emission spectra were collected using a Tecan Saphire 2 micro plate reader (Tecan Group Ltd, Männerdorf, Switzerland). All measurements were repeated in triplicates and the excitation wavelengths used for L1, L2 and L3 were 310, 365 and 400 nm, respectively.
4.3. Staining of Aβ plaques in mouse brain sections
This study did not directly involve experiments or handling of live animals. The brain tissue sections with AD pathology were provided by Prof. Frank Heppner, Department of Neuropathology, Charité – Universitätsmedizin Berlin, Germany. Animal handling to provide the histology samples used herein was carried out in accordance with relevant ethical guidelines of breeding and keeping transgenic animals as well as culling and collection of tissue for ex vivo experiments (approved by the Swiss cantonary veterinary office or the regional offices for health and social services in Berlin). 10 μm cryosections of a 531 days old APP/PS1 mouse38 were placed on superfrost ultraplus glass slides. The sections were fixed in 99% ethanol for 10 minutes and rehydrated with increasing proportion of dH2O followed by equilibration in PBS pH 7.4 (Medicago, Sweden) for 10 minutes. Monoclonal anti-Aβ antibody 6E10 was diluted 1:200 in PBS, administered to the sections and left at 4 °C overnight followed by extensive washing with PBS. Goat-antimouse secondary antibody with fluorescence detection maximum at 647 nm (GAM650, Dako, Denmark) was diluted 1:500 and incubated on the sections for 30 minutes followed by washing with PBS. The ligands were diluted from 5 mM stocks in DMSO to 10 μM working solution in PBS immediately prior to use. The ligand working solutions were applied to the sections and left at room temperature for 30 minutes followed by destaining by dipping in PBS for 10 minutes. The glass slides were left to dry in ambient air and finally mounted with Dako fluorescence mounting medium and a cover slip. Images were collected using an inverted Zeiss LSM 780 confocal microscope (Carl Zeiss, Oberkochen, Germany) with excitation wavelength at 405 nm (L1–L3) and 680 nm (anti-Aβ antibody 6E10).
Acknowledgements
This work was supported by an International postdoctoral grant from the Swedish Research Council, Dnr: 350-2012-239 (to C. D.), The Swedish Alzheimer's Foundation (to S. N.), Ciência sem Fronteiras (Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil, Proc. No. 245405/2012-7) and Fundação CAPES (Bolsista CAPES, Projeto PVE 118/2013) (to R. P. V.), and a Michael Smith Foundation for Health Research career Investigator Award (to T. S.). The authors also thank Alexander Sandberg and Leif B. G. Johansson for providing the amyloid-like fibrils, generated from recombinant Aβ1–42. The brain tissue sections with AD like pathology was provided by Prof. Frank Heppner, Department of Neuropathology, Charité – Universitätsmedizin Berlin, Germany.
Notes and references
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741 CAS.
- V. H. Finder, J. Alzheimer's Dis., 2010, 22, 5 Search PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205 CrossRef CAS PubMed.
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608 RSC.
- M. Xu, W. Ren, X. Tang, Y.-H. Hu and H. Zhang, Acta Pharmacol. Sin., 2016, 37, 719 CrossRef CAS PubMed.
- M. Shimojo, M. Higuchi, T. Suhara and N. Sahara, Front. Neurosci., 2015, 9, 482 Search PubMed.
- K. P. Nilsson, FEBS Lett., 2009, 583, 2593 CrossRef CAS PubMed.
- P. A. Adlard, B. A. Tran, D. I. Finkelstein, P. M. Desmond, L. A. Johnston, A. I. Bush and G. F. Egan, Front. Neurosci., 2014, 8, 327 Search PubMed.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244 CrossRef CAS PubMed.
- H. LeVine, 3rd, Protein Sci., 1993, 2, 404 CrossRef PubMed.
- E. S. Voropai, M. P. Samtsov, K. N. Kaplevskii, A. A. Maskevich, V. I. Stepuro, O. I. Povarova, I. M. Kuznetsova, K. K. Turoverov, A. L. Fink and V. N. Uverskii, J. Appl. Spectrosc., 2003, 70, 868 CrossRef CAS.
- A. A. Reinke, G. A. Abulwerdi and J. E. Gestwicki, ChemBioChem, 2010, 11, 1889 CrossRef CAS PubMed.
- M. R. Nilsson, Methods, 2004, 34, 151 CrossRef CAS PubMed.
- H. Bennhold, Muench. Med. Wochenschr., 1922, 69, 1537 Search PubMed.
- P. Divry, J. Neurol. Psychiatry, 1927, 27, 643 Search PubMed.
- P. Frid, S. V. Anisimov and N. Popovic, Brain Res. Rev., 2007, 53, 135 CrossRef CAS PubMed.
- G. T. Westermark, K. H. Johnson and P. Westermark, Methods Enzymol., 1999, 309, 3 CAS.
- L. Zhu, K. Ploessl and H. F. Kung, Chem. Soc. Rev., 2014, 43, 6683 RSC.
- W. E. Klunk, H. Engler, A. Nordberg, Y. Wang, G. Blomqvist, D. P. Holt, M. Bergström, I. Savitcheva, G.-F. Huang, S. Estrada, B. Ausén, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Långström, Ann. Neurol., 2004, 55, 306 CrossRef CAS PubMed.
- C. M. Clark, J. A. Schneider, B. J. Bedell, T. G. Beach, W. B. Bilker, M. A. Mintun, M. J. Pontecorvo, F. Hefti, A. P. Carpenter, M. L. Flitter, M. J. Krautkramer, H. F. Kung, R. E. Coleman, P. M. Doraiswamy, A. S. Fleisher, M. N. Sabbagh, C. H. Sadowsky, E. P. Reiman, S. P. Zehntner and D. M. Skovronsky, JAMA, 2011, 305, 275 CrossRef CAS PubMed.
- S. R. Choi, J. A. Schneider, D. A. Bennett, T. G. Beach, B. J. Bedell, S. P. Zehntner, M. J. Krautkramer, H. F. Kung, D.
M. Skovronsky, F. Hefti and C. M. Clark, Alzheimer Dis. Assoc. Disord., 2012, 26, 8 CrossRef CAS PubMed.
- M. R. Jones, E. L. Service, J. R. Thompson, M. C. P. Wang, I. J. Kimsey, A. S. DeToma, A. Ramamoorthy, M. H. Lim and T. Storr, Metallomics, 2012, 4, 910 RSC.
- M. R. Jones, C. Dyrager, M. Hoarau, K. J. Korshavn, M. H. Lim, A. Ramamoorthy and T. Storr, J. Inorg. Biochem., 2016, 158, 131 CrossRef CAS PubMed.
- E. A. Krasnokutskaya, N. I. Semenischeva, V. D. Filimonov and P. Knochel, Synthesis, 2007, 81 CrossRef CAS.
- J. Qi, M. S. Han, Y. C. Chang and C. H. Tung, Bioconjugate Chem., 2011, 22, 1758 CrossRef CAS PubMed.
- J. Qi and C. H. Tung, Bioorg. Med. Chem. Lett., 2011, 21, 320 CrossRef CAS PubMed.
- B. Wang, S. W. Tsang, W. Zhang, Y. Tao and M. S. Wong, Chem. Commun., 2011, 47, 9471 RSC.
- B. A. Coombs, B. D. Lindner, R. M. Edkins, F. Rominger, A. Beeby, H. Uwe and F. Bunz, New J. Chem., 2012, 36, 550 RSC.
- R. C. Lirag, H. T. Le and O. S. Miljanic, Chem. Commun., 2013, 49, 4304 RSC.
- A. V. Moro, P. C. Ferreira, P. Migowski, F. S. Rodembusch, J. Dupont and D. S. Ludtke, Tetrahedron, 2013, 69, 201 CrossRef CAS.
- B. A. Neto, P. H. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625 CrossRef CAS PubMed.
- X. Zhuang, T. Ha, H. D. Kim, T. Centner, S. Labeit and S. Chu, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14241 CrossRef CAS PubMed.
-
D. C. Harris, Quantitative Chemical Analysis, W. H. Freeman, 2010 Search PubMed.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy. Springer, US, 2007 Search PubMed.
-
S. M. Auerbach, K. A. Carrado and P. K. Dutta, Handbook of Layered Materials, CRC Press, 2004 Search PubMed.
- J. Zhang, Y. Fu and J. R. Lakowicz, J. Phys. Chem. C, 2007, 111, 1955 CAS.
- R. Radde, T. Bolmont, S. A. Kaeser, J. Coomaraswamy, D. Lindau, L. Stoltze, M. E. Calhoun, F. Jaggi, H. Wolburg, S. Gengler, C. Haass, B. Ghetti, C. Czech, C. Holscher, P. M. Mathews and M. Jucker, EMBO Rep., 2006, 7, 940 CrossRef CAS PubMed.
- S. Nystrom, K. M. Psonka-Antonczyk, P. G. Ellingsen, L. B. Johansson, N. Reitan, S. Handrick, S. Prokop, F. L. Heppner, B. M. Wegenast-Braun, M. Jucker, M. Lindgren, B. T. Stokke, P. Hammarstrom and K. P. Nilsson, ACS Chem. Biol., 2013, 8, 1128 CrossRef PubMed.
- J. W. Eastman, Photochem. Photobiol., 1967, 6, 55 CrossRef CAS.
- A. S. L. Gouveia, L. C. Tomé and I. M. Marrucho, J. Chem. Eng. Data, 2016, 61, 83 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nj01703g |
| ‡ Division of Organic Chemistry, Department of Physics, Chemistry and Biology, Linköping University, SE-58183, Linköping, Sweden. E-mail: http://christine.dyrager@liu.se; Tel: +46 13281311. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Rafael Pinto
Vieira
acd,
Sofie
Nyström
b,
K. Peter R.
Nilsson
b and
Tim
Storr
a
*ab,
Rafael Pinto
Vieira
acd,
Sofie
Nyström
b,
K. Peter R.
Nilsson
b and
Tim
Storr
a





