
Primary microglia maintain their capacity to function despite internalisation and intracellular loading with carbon nanotubes†
Cyrill
Bussy
 *a,
Alberto
Bianco
b,
Maurizio
Prato
cde and
Kostas
Kostarelos
*a
*a,
Alberto
Bianco
b,
Maurizio
Prato
cde and
Kostas
Kostarelos
*a
aNanomedicine Lab, Faculty of Biology, Medicine and Health & National Graphene Institute, University of Manchester, AV Hill Building, Manchester M13 9PT, UK. E-mail: cyrill.bussy@manchester.ac.uk; kostas.kostarelos@manchester.ac.uk
bCNRS, Institut de Biologie Moléculaire et Cellulaire, UPR 3572, Immunopathologie et Chimie Thérapeutique, Strasbourg, France
cCenter of Excellence for Nanostructured Materials, Department of Chemical and Pharmaceutical Sciences, University of Trieste, Trieste, Italy
dCarbon Nanobiotechnology Laboratory, CIC biomaGUNE, Paseo de Miramón 182, 20009 Donostia-San Sebastian, Spain
eBasque Science Foundation, Ikerbasque, Bilbao 48013, Spain
First published on 15th June 2017
Abstract
Biomedical applications of functionalised carbon nanotubes (f-CNTs) for imaging as well as drug or gene delivery in the brain have recently gained interest. Several studies have demonstrated the potential of f-CNTs to offer treatment options for neurological conditions with success. However, there is also evidence that f-CNTs accumulate preferentially within microglial cells when introduced in the brain. Considering the key immunological role of these cells in the brain and the limited knowledge regarding the interaction of CNTs with microglial cells, it is imperative to understand whether accumulation of CNTs in microglial cells can alter their physiological functions or trigger pro-inflammatory signalling. The aim of the present study was to investigate the basic physiological functions of isolated primary microglial cells over time, following their exposure to multi-walled carbon nanotubes functionalized via different surface chemistries. We rationalized that some chemical strategies may be more deleterious for microglial cell functions than others. We used rat primary microglial cells that can be maintained in cell culture for a long period of time without undergoing cell division. Cell viability, phagocytosis, migration, and pro-inflammatory factor release were studied over one month, following a single exposure to a non-cytotoxic concentration of three different f-CNTs (carboxylated, aminated and both carboxylated and aminated). We show that f-CNTs do not induce inflammation and do not affect the basic functions of microglial cells under these conditions. Importantly, this was the case from day 1 to day 28 post-exposure, even though microglial cells had internalized f-CNTs in subcellular compartments and retained the nanotube load in their cytoplasm over time.
Conceptual insightsResident immune cells of the brain exposed to a single dose of functionalised carbon nanotubes (f-CNTs) maintain their physiological functions up to one month after exposure despite the massive internalisation and persistence of materials in endocytic vesicles. Not only did the microglial cells survive the exposure and operate, but they also showed no sign of pro-inflammatory activation (division and release of pro-inflammatory factors) over the course of the study. Interestingly, we found that the more oxidised the f-CNTs were the more they stimulated the migration of the f-CNT-laden cells. In comparison, amino-functionalised CNTs did not stimulate the migration of f-CNT-laden cells. This work emphasizes that the response of cells is greatly influenced by the surface functionalisation of the f-CNTs they have internalised. It also highlights the amino-functionalised CNTs as the most inert materials in terms of cell response, which is an important advantage toward their development as gene delivery platforms. Even though CNTs have poor biodegradation kinetics which may be considered as a barrier for clinical translation, functionalised CNTs are still valuable tools for nanomedicine as they do not activate or injure the main immune cells (resident macrophages) that they will likely encounter upon administration into any body part. |
Introduction
In recent years, numerous nanomaterials have been utilised in an attempt to overcome the great challenge of drug delivery to the central nervous system.1–3 Among them, carbon nanotubes (CNTs) have lately shown great promise4 due to their intrinsic properties, but also their unique ability to cross cellular barriers.5 For instance, acetylcholine adsorbed onto single-walled CNTs (SWNTs) was efficiently delivered to cells in a kainic acid-induced Alzheimer's Disease (AD) mouse model.6 Amine functionalized multi-walled CNTs (MWNTs) have also been used successfully to deliver Caspase 3 siRNA into neurons with the aim of reducing ischemic injuries in the motor cortex of an endothelin-1 induced stroke model.7 Similarly, amine functionalized SWNTs were injected into mouse brain ventricles in order to decrease the effects of ischemia and inflammation in an artery occlusion model of stroke.8 The SWNT treated animals showed better histopathological and behavioural profiles after injury, indicating that aminated SWNTs could achieve a neuroprotective effect even without carrying any therapeutic agent. More recently, PEG-SWCNTs have also been proposed as solutions to alleviate glutamate excitotoxicity of neurons by increasing astrocytic glutamate uptake.9Once localised in the brain parenchyma, knowledge of the fate and biological effects of these functionalised nanomaterials remains however limited. This raises the need for a better understanding of the interactions of carbon nanomaterials with different cell subpopulations in the central nervous system (CNS). While neurons might be a preferred target for therapeutic purposes, nanomaterials in the brain parenchyma are likely to interact with other cell types including the resident macrophages of the brain, namely the microglial cells. Even in the case of nanoscale vectors bearing neuron-targeting moieties, microglial cells have indeed been shown to be the cell type in which a significant fraction of the nanomaterials are accumulated.10 Considering that microglial cells are the main cellular components of the immune system in the brain, any nanomaterial that is intended for intracerebral applications should therefore have a controlled and well-understood interaction with these cells.
In terms of our current understanding of CNT–microglia interactions, microglial cells have been reported to play a major role in internalising functionalised CNTs (f-CNTs) in comparison to other cell types following stereotactic administration of f-CNTs in the cortex of mice.7,11,12 Similar results were obtained in vitro in mixed cell cultures of primary glial cells, whereby microglial cells displayed a higher uptake of functionalised MWNTs (f-MWNTs) compared to astrocytes.13 Interestingly, following internalisation in microglial cells, we have observed the partial degradation of f-MWNTs over time, both in vivo over 14 days12 and in vitro in a primary cell culture model up to 90 days.14,15 Alteration within 24 h of the graphitic structure of f-MWNTs in the immortalised N9 microglial cell line offered independent validation.16
These degradation results suggested that despite the internalisation of f-CNTs, microglial cells were able to maintain one of their primary functions over time: their ability to degrade foreign materials.12,14,17 In contrast, MWNTs dispersed in horse serum were shown in vitro to alter the microglial functions such as migration and phagocytosis, and also cell division in a BV2 immortalised microglial cell model after their internalisation.18 The main hypothesis to explain these alterations was the direct interaction of MWNTs with the cytoskeleton of BV2 cells leading to an impairment of all cytoskeleton related functions including migration and phagocytosis. The present study was therefore designed to interrogate the impact of internalisation and retention of f-CNTs in microglial cells on their capacity to perform normal cellular and macrophage-specific functions, other than their degradation ability that was previously demonstrated.14,15
In addition, using different chemical derivatives of f-CNTs, surface properties have been revealed as essential determinants that could modulate both CNT–microglia interactions and the bioreactivity of CNTs towards microglial cells. Firstly, the type of surface functionalisation was demonstrated to influence the uptake.19 Phosphatidyl-serine coated SWNTs were engulfed by primary rat microglia while phosphatidyl-choline coated and non-coated SWNTs were not. Similarly, microglial cells were shown to be very efficient at internalising oxidised MWNTs but were not fully able to break apart the agglomerates of pristine MWNTs and to internalise them.16 Secondly, the type of functionalisation was shown to modulate the response of microglial cells. While both pristine and oxidised MWNTs did not alter cell viability, pro-inflammatory cytokine release or nitric oxide (NO) production, a significant increase of reactive oxygen species production was measured after exposure to pristine MWNTs, suggesting a possible impact on cell viability in the long term.16 Accordingly, using an in vivo cortical stereotactic injection model, brain cells and microglial ones in particular were reported to differently respond to f-CNT exposure depending on the surface functionalisation of the materials injected.11 Functionalisation not only impacted the localisation and distribution pattern of the injected materials within the brain, but also modulated the inflammatory profile of different f-CNTs. Oxidised MWNTs were shown to induce and sustain a microglial cell mediated inflammatory response while aminated MWNTs only transiently induced inflammation.
Taken together, these studies demonstrate the major importance of material surface properties and in particular the type of surface functionalisation in the biological outcomes of the material–cell interaction. Moreover, the ability of CNTs to induce inflammation and impact on microglial cell physiological functions are two key endpoints that should be systematically investigated and correlated with the surface chemistry of the materials in order to determine the best biocompatibility profile for putative CNT-based brain delivery systems.
The objectives of the present study were therefore to investigate whether microglial cell functions can be affected by the internalisation of f-CNTs, whether varying the surface chemistry of f-CNTs can modulate the cell response, and whether the impact of these different f-CNTs on microglial cell functions may evolve over time. Oxidation of CNTs has been shown to increase the bioreactivity of these materials for macrophages;11,20 we therefore hypothesised that the degree of oxidation/carboxylation is determinant to modulate the biological responses and can affect microglial cell functions while not inducing cell death. To assess this hypothesis we selected three MWNTs with different carboxylation degrees (purely carboxylated, purely aminated and one type bearing both carboxyl and amine groups). We additionally developed a primary rat microglial cell model that demonstrated a long lifespan in vitro without undergoing division to monitor over time the evolution of microglial cell functions after internalisation of the materials. Cytotoxicity, phagocytosis, migration and pro-inflammatory mediator release were studied at different time points and up to 28 days following single exposure to a non-cytotoxic concentration of the three selected f-CNTs. We report that f-MWNTs, irrespective of their chemical functionalisation, did not affect microglial cell viability under our conditions and did not induce the release of pro-inflammatory mediators at any time points. Moreover, despite an important perinuclear internalisation of materials, microglial cells maintained their abilities to phagocytose and to migrate. Interestingly, oxidation of MWNTs stimulated the migration ability of microglial cells that have engulfed those CNTs in comparison to aminated MWNT laden cells.
Results and discussion
Chemical structure and characterisation of the MWNT suspensions
The selection of MWCNTs over other CNT types such as single walled- or double walled-CNTs was motivated by our previous studies on the interactions of f-MWNTs with brain cells7,11–13,21 and microglial cells in particular.14,15 We have also demonstrated that these functionalised nanomaterials can be used as delivery platforms for drugs,22 genes7,23 or contrast agents.24 The type of chemical functionalisation was previously identified by some of us as a major determinant of the in vivo ability of f-MWNTs to induce inflammation in the brain.11 Comparing two types of f-MWNTs, one that has been pre-oxidised and then aminated with one that has been aminated only, oxidation/carboxylation was revealed to cause inflammation, even though the effects of MWNTs bearing only carboxyl groups (i.e. negative charges) were not directly investigated. Here, three chemically functionalised MWNTs with different levels of oxidation were synthesised from the same starting materials in order to limit batch-to-batch variations. Their chemical structure is presented in Fig. 1 and they include: (i) carboxylated MWNTs (ox-MWNTs) prepared by treatment of the starting materials under strong acid conditions;25 (ii) amino-functionalized oxidised MWNTs (ox-MWNT–NH3+) prepared by a 1,3 dipolar-cycloaddition reaction after an initial oxidation as described previously;26 and (iii) amino-functionalized MWNTs (MWNT–NH3+) prepared following the 1,3 dipolar-cycloaddition reaction on the starting materials as described previously.27,28 The length of MWNT–NH3+ remained similar to the that of the starting materials (between 0.5–2 μm) while it was reduced for materials undergoing oxidation (shortened to 300–400 nm for ox-MWNTs and ox-MWNT–NH3+). Suspensions of MWNTs were prepared by hydrating a dry powder with a 5% dextrose water solution, which was then sonicated until the MWNTs reached good dispersion (about 40 min). It was essential to achieve a dispersion of similar quality for the three materials to prevent non-intrinsic differences amongst them. Difference in dispersion quality is indeed known to induce differences in biological responses, especially inflammation,29,30 and could therefore lead to biases in the experimental outcomes and misinterpretation with regard to the role of chemical functionalisations. To assess the stability of the 3 MWNT dispersions in 5% dextrose water solution, analysis of the absorbance (used as an index of dispersion, well dispersed materials absorbing more than agglomerated ones) was performed at 0 h and 24 h (Fig. S1, first column, ESI†). Despite the intrinsic differences between the 3 materials (highest absorbance: ox-MWNT–NH3+ > MWNT–NH3+ > ox-MWNT), no difference between the absorbances at 0 h and 24 h could be observed for any material. All 3 materials were stable over 24 h in 5% dextrose. Similar stability results over 24 h were observed when the materials initially suspended in 5% dextrose were diluted in cell culture medium supplemented with 12% FBS (Fig. S1, second column, ESI†). However, when materials initially suspended in 5% dextrose were then diluted in cell culture medium without FBS, the UV-Vis spectrometric analysis evidenced a clear sedimentation for all 3 materials, regardless of their surface chemistries (Fig. S1, third column, ESI†). These results suggested that MWNT suspensions were not stable in cell culture medium without the addition of FBS, and would sediment at the bottom of the cell culture wells under these conditions, increasing the potential of the materials to interact with the monolayer of cells. In contrast the addition of FBS would not be favourable to interaction with the cells as most materials would stay in suspension, above the cell monolayer.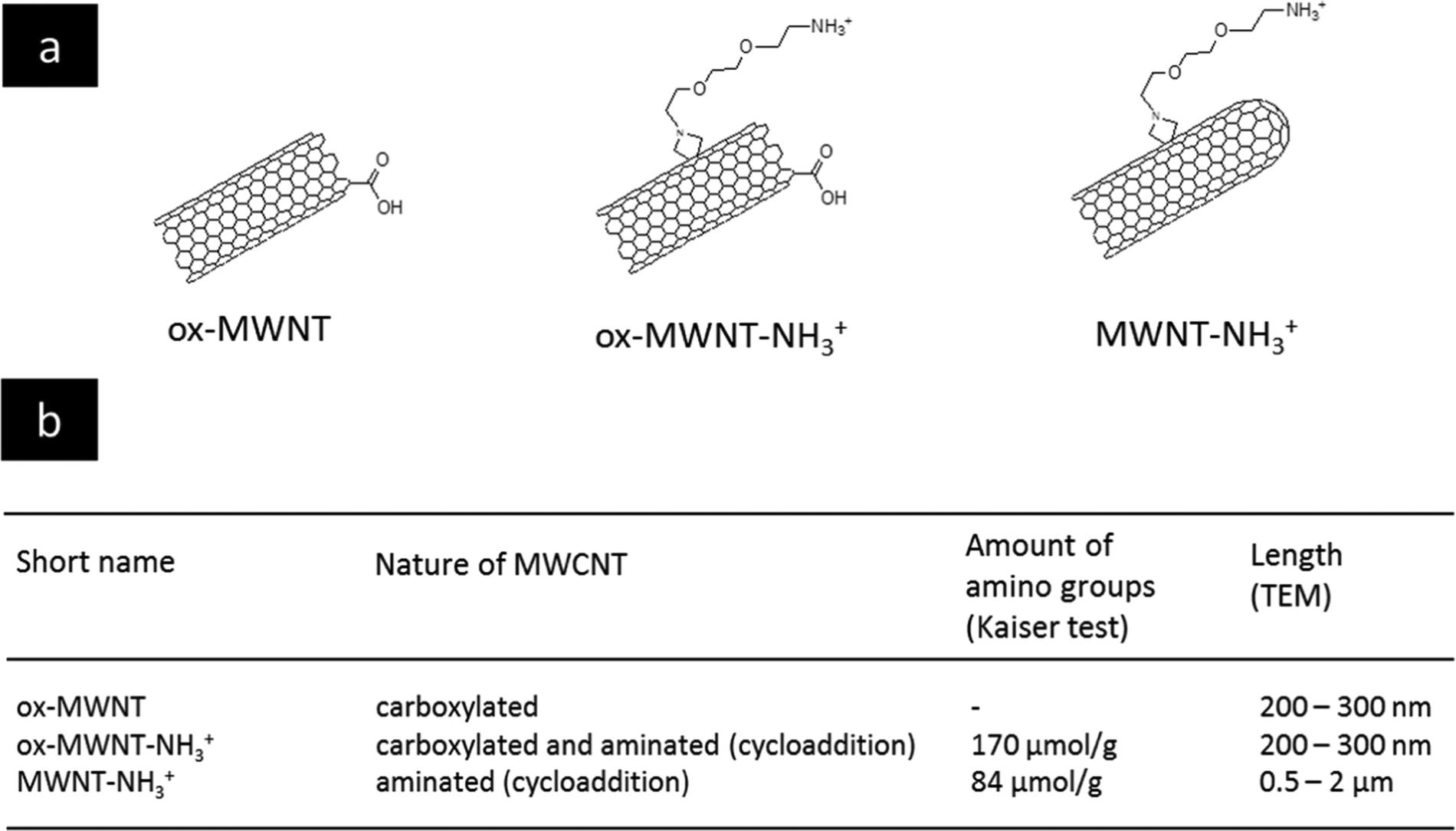 | ||
| Fig. 1 Characteristics of the different types of functionalised MWNTs. (a) Chemical structures of the different chemically functionalised MWNTs. (b) Table providing the abbreviations used in the manuscript, and the length and the amount of amino groups for the different MWNTs. | ||
Functionalised nanotubes had no effect on microglial cell viability
Microglial cells were revealed as the main cell population in which f-MWNTs are accumulated when infused in the brain parenchyma,7,11,12 or when exposing mixed glial cell cultures (containing mainly astrocytes and microglial cells).13,14 However, the impact of this preferential accumulation remains elusive and contradictory in the literature. We therefore first questioned whether the exposure to different f-MWNTs using a non-overload concentration will affect the viability of microglial cells over time. To answer this question, a model of primary microglial cell cultures that can be maintained over a long period of time without cell division was used. It was rationalised that once in the brain parenchyma, f-MWNTs will diffuse throughout the tissue as observed for other nanomaterials31 and therefore each microglial cell will likely be exposed to a limited amount of nanotubes. To ensure that microglial cells interacted with the MWNTs, we favoured the CNT sedimentation by exposing the cells to MWNTs in a serum free environment for 2 h, following which the cell culture medium was supplemented with 12% FBS. Following this single exposure to a unique dose of 10 μg mL−1, we used a modified LDH assay developed in our lab32 to assess the impact of f-MWNTs on microglial cell viability at 1, 7, 14 and 28 days after the initial exposure (Fig. 2). No effect on the viability of exposed microglial cells compared to untreated cells was observed irrespective of the f-MWNTs used or time after exposure. However, a statistically significant difference was observed at day 1 for ox-MWNT–NH3+ treated cells when compared to their respective control (untreated cells). This could be ascribed to the primary nature of the cell cultures used here (more dying cells in untreated cells than in ox-MWNT–NH3+ treated cells)14 or to a stimulation of viability as primary microglial cells preserve the ability to divide under stimulatory conditions.33 Further investigations will be needed to address this question. Overall, these results suggested that microglial cells are tolerant to f-MWNT exposure under the conditions used here, and that this tolerance is not dependent on the type of chemical functionalisation for this concentration. These results were in agreement with previous observations of a dose dependent cytotoxicity appearing after 24 h above 10 μg mL−1.13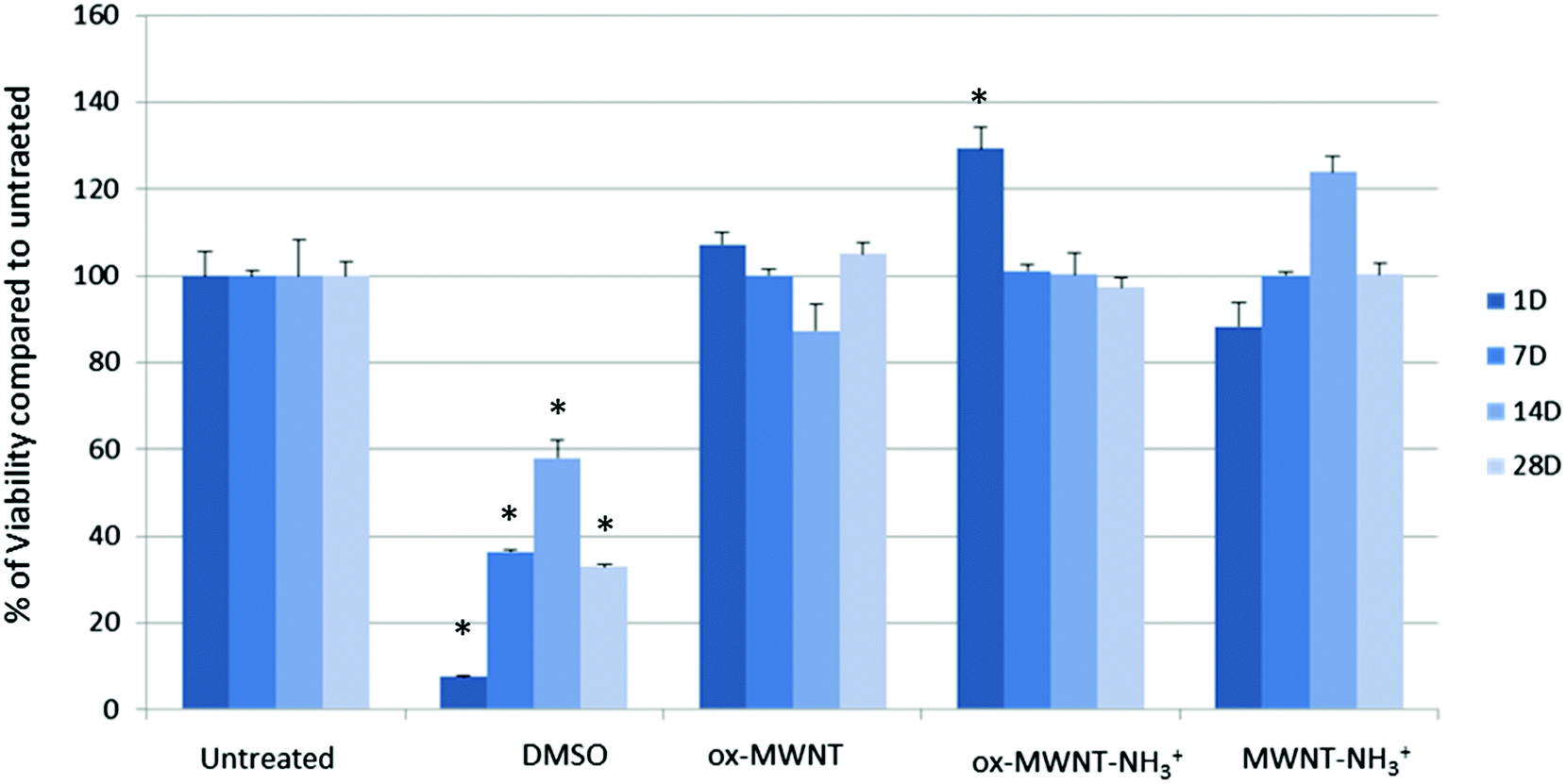 | ||
| Fig. 2 Cytotoxic effects of different MWNTs on primary microglial cell cultures evaluated using the modified LDH assay. Microglial cell cultures were exposed for 24 h to a unique dose of different MWNTs (10 μg mL−1). Following different periods of time after the exposure, the LDH assay was performed. No significant effect was observed compared to untreated cells or the positive control (DMSO 10%). A one way ANOVA, Kruskal-Wallis test with Dunn's multiple comparison test was used to analyse the differences in the viability assay. p < 0.05 (*) was considered as significantly different. | ||
Primary microglial cells internalised and retained nanotubes over time
CNTs found primarily free in the cytoplasm were reported to reduce proliferation and induce apoptosis in BV2 cells, an immortalised model of microglial cells.18 In order to explain the difference between this study and the viability results of the present study (Fig. 2), optical microscopy was used to investigate the internalisation pattern and evolution of CNT loading over time (Fig. 3a, Fig. S2 and S3, ESI†). For all three f-MWNTs and irrespective of the time point considered, the distribution of nanotubes observed as black materials in bright-field mode (Fig. S2 and S3, ESI†) was characteristic, with an accumulation of materials around the nucleus (perinuclear region), the absence of black materials in the nucleus, and no apparent accumulation in the cytoplasm periphery close to the cell boundaries (Fig. S3, ESI†). No obvious difference in terms of CNT loading from 1 day to 28 days was observed for all three f-MWNTs, suggesting that exocytosis was limited. In addition, using immunocytochemistry (anti-CD68), a singular pattern of CD68 immunolabelling was observed, with black materials appearing as entrapped in vesicles positive for CD68 immunoreactivity (Fig. S4, ESI†). The vesicular pattern was enhanced and contrasted by the black materials contained within the CD68 positive vesicles. CD68 is predominantly an intracellular glycosylated transmembrane protein with minimal surface expression in unactivated macrophages.34 The perinuclear position of CNT accumulation, the absence of diffuse CNT-related black signal across the cytoplasm, and the vesicular pattern of CD68 immunoreactivity suggested a vesicular accumulation of f-MWNTs. | ||
| Fig. 3 Imaging of primary microglial cells exposed to different functionalised MWNTs via optical microscopy and confirmation of CNTs via Raman spectroscopy. Primary microglial cell cultures were exposed for 24 h to a unique dose of different MWNTs (10 μg mL−1) and then left for different periods of time before imaging. (a) Live imaging (phase contrast) of primary microglial cells at day 1 to 28 for different exposures. LPS activation (200 ng mL−1) was used as a control that induced a change in cell shape (cells flattened and spread). No significant difference in terms of the shape or number of cells was observed between the non-treated cells and the cells exposed to MWNTs at any time of recovery after the initial exposure. (b) At the end of each desired time of recovery, primary microglial cells were fixed with −20 °C pre-cooled methanol and then air dried for at least one hour before further analysis. The Raman analysis of regions randomly selected within the cell boundaries was performed (as presented in Fig. S5c, ESI†). Intensities were normalised to the intensity of the G band (IG = 1). For all MWNTs and for all time points considered, the typical Raman signature of MWNTs with D, G and D′ bands was obtained in regions which appeared black under bright-field microscopy. No Raman signature of CNTs was detected in white regions (nucleus and cell periphery). All scale bars shown are 100 μm. | ||
In order to confirm that the punctuated black halo around the nucleus (Fig. S5b, ESI†) was indeed due to localised accumulation of carbon nanotubes, Raman spectroscopy analysis was performed at a single cell level and at different places in the considered cell (Fig. 3b). The characteristic peaks at 1360 cm−1 (D band), 1585 cm−1 (G band) and 1620 cm−1 (D′ band) were used to confirm the CNT nature of the accumulation.35,36 Following exposure to f-CNTs, all three microglial cell cultures exhibited the specific Raman signature of MWNTs (i.e. D, G, and D′ bands) in the perinuclear region (Fig. S5b, ESI†). For each f-CNT, the Raman profile found in the cells (Fig. 3b) was similar to the Raman profile of the starting materials used to treat the cells (Fig. S5a, ESI†), confirming the adequacy and specificity of Raman fingerprints to characterise carbon nanomaterials. In addition, the Raman signature of MWNTs was observed at each time points tested (i.e. 1, 7, 14 and 28 days), confirming the persisting perinuclear presence of f-CNTs in each case (Fig. 3b). Taken together, these results show that the type of chemical functionalisation had no influence on the internalisation of f-MWNTs, their distribution within the cells, and their retention over time in microglial cells. However, both endocytosis and exocytosis of f-MWNTs in primary microglial cells will need further investigations to fully describe the mechanisms and kinetics of the processes at stake. In particular, further studies investigating the amount of materials internalised and maintained in cells over time for each f-MWNT considered will be important, as over time degradation in microglial cells in a surface chemistry dependent manner has been recently confirmed using two different methodological approaches.14,15 Importantly, as mentioned above, the persistent retention of f-MWNTs over 28 days around the nucleus of microglial cells had no effect on viability, which demonstrated the absence of correlation between intracellular accumulation of materials and cytotoxicity under the considered conditions.
Uptake of f-MWNTs did not induce pro-inflammatory reaction in microglial cells
Since internalisation of CNTs had no effect on microglial cell viability over time, it was then questioned whether the CNT uptake could induce the release of pro-inflammatory factors. The Griess assay was therefore used to measure the amount of NO released by MWNT exposed microglial cells at 1, 7, 14 and 28 days after the initial exposure (Fig. 4). NO is one of the main markers for microglial cell activation and pro-inflammation induction, and is also a toxic agent for neurons.17,37 A statistically significant release of NO compared to untreated cells was observed 24 h after the initial exposure to CNTs for all three f-MWNTs. However, this positive release of pro-inflammatory markers was largely limited in comparison to LPS stimulated microglial cells. At later time points, the release of NO became insignificant compared to untreated cells and disappeared at 28 days. These data showed that f-MWNTs, irrespective of their surface functionalisation, have limited ability to induce inflammation in vitro under the conditions used here, and that their internalisation and persistence in microglial cells over time did not lead to a chronic pro-inflammatory reaction, as would have been expected if microglial cells were activated and inducing an immune response, as previously observed with other MWNTs.16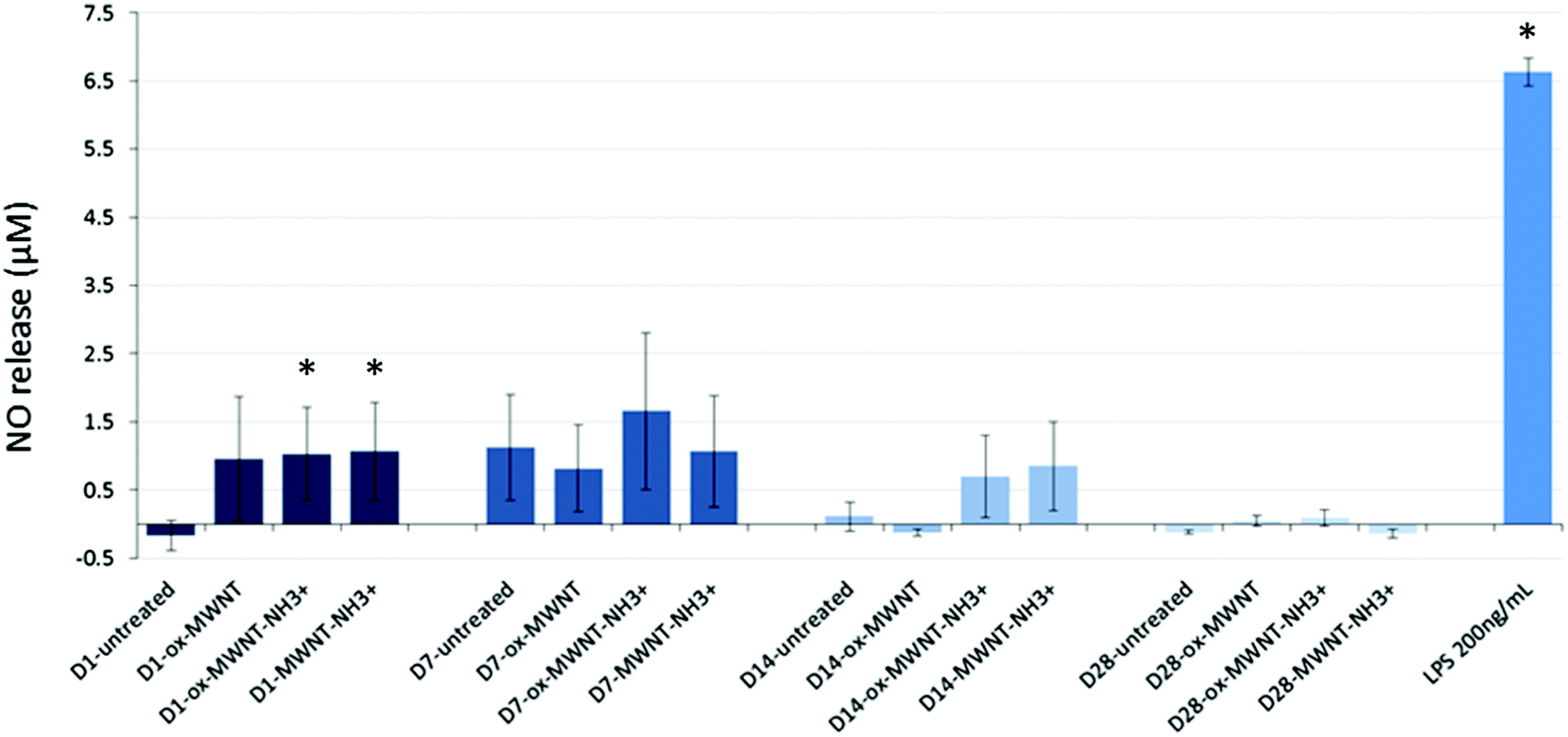 | ||
| Fig. 4 Inflammation reaction assessed by a measurement of NO release using the Griess reagent assay. Microglial cell cultures were exposed for 24 h to a unique dose of different MWNTs (10 μg mL−1). Following different periods of time after the exposure, the Griess reagent system assay was performed. No significant effect was observed compared to untreated cells or the positive control (LPS 200 ng mL−1). Only at day 1 after the initial CNT exposure, the NO release was higher than the untreated cells but minimal compared to the NO release induced by LPS stimulation. | ||
Uptake of f-MWNTs did not alter internalisation or migration abilities
The presence of f-MWNTs inside microglial cells for 28 days did not seem to affect their viability or to induce an inflammatory reaction. The next question was then to determine whether other basic physiological functions characteristic of resident macrophages would stay unimpaired by f-MWNT exposure and persistence over time in the cells.Firstly, the internalisation ability of CNT-laden microglial cells was tested, using 0.5 μm fluorescently labelled polystyrene beads. It was rationalised that if the internalisation ability was maintained, the uptake of polystyrene beads for each microglial cell considered would be limited only by the amount of materials (i.e. CNTs) already inside these MWNT-preloaded cells. The internalisation of polystyrene beads inside microglia previously exposed to MWNTs was assessed over 8 h and compared to naïve microglial cells (i.e. unexposed to MWNTs) and activated microglial cells (i.e. unexposed to MWNTs but stimulated by LPS (200 ng mL−1)) (Fig. 5 and Fig. S6, ESI†). The internalisation assay was performed only at day 7 after exposure to CNTs because it was observed that engulfment of polystyrene beads was leading to acute cytotoxicity at later time points (14 and 28 days) for all cells, including naïve microglial cells. This was attributed to the age of the primary cells, which have been isolated from brain tissue about 1.5 months before the day 14 time point. Similarly, the test was not performed at day 1 after exposure to MWNTs because the cells were exposed to MWNTs the day before and some free nanotubes might still be in the supernatant and would be in direct competition with the polystyrene beads for internalisation. At the earliest time point (1 h after the bead treatment), there was no major difference between groups, including untreated cells or LPS stimulated cells (Fig. 5c and Fig. S6, ESI†). While non-statistically significant, the amount of polystyrene beads (i.e. fluorescence) in microglial cells previously exposed to oxidised MWNTs (i.e. both ox-MWNTs and ox-MWNT–NH3+) was however lower compared to untreated cells (Fig. 5c and Fig. S6, ESI†). After 2 h, the LPS stimulation greatly enhanced the polystyrene bead internalisation compared to the other groups (Fig. 5a and c). In addition, the internalisation of polystyrene beads was slightly higher for microglia exposed to aminated MWNTs (i.e. both ox-MWNT–NH3+ and MWNT–NH3+) compared to naïve microglia or microglia exposed to ox-MWNTs. After 8 h, there was no significant difference between the various groups suggesting that all groups of cells had reached their highest loading of polystyrene beads possible (Fig. 5b and c). It also suggested that despite their initial CNT loading, CNT-laden microglial cells were able to internalise polystyrene beads to the same extent as cells not pre-loaded with CNTs. These results showed that the internalisation ability of microglial cells was not perturbed by the uptake of f-MWNTs, irrespective of the type of chemical functionalisation tested. These results also underlined the relative tolerance of cells to f-MWNT exposure, especially when considering non-overload non-toxic doses (i.e. 10 μg mL−1).
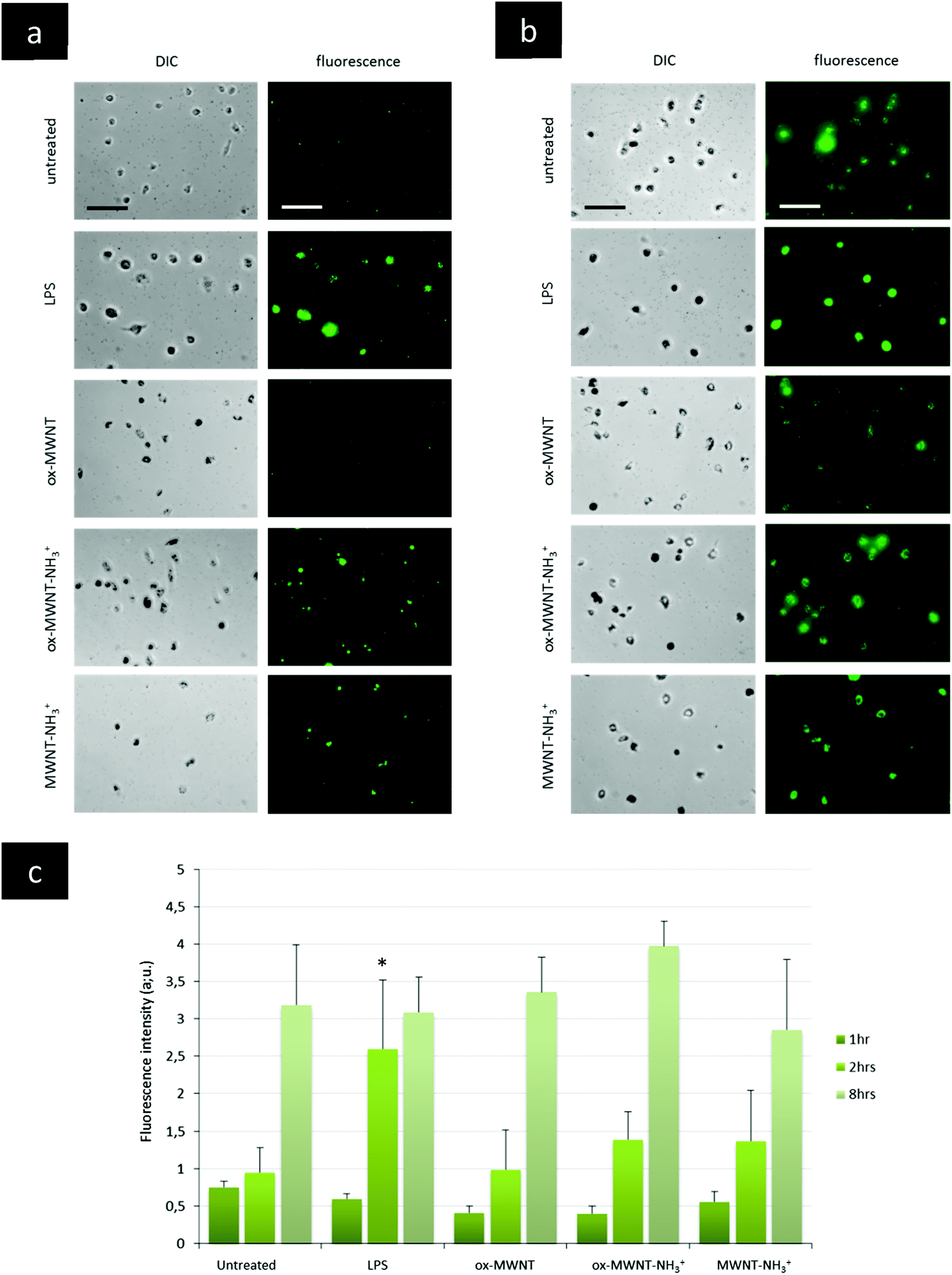 | ||
| Fig. 5 Internalisation ability assessed using a fluorescently labelled microsphere assay. Microglial cell cultures were exposed for 24 h to a unique dose of different MWNTs (10 μg mL−1). On day 7 after this initial exposure, microglial cells were treated with fluorescently labelled microspheres (0.5 μm). The internalisation of the fluorescent microspheres was recorded at 2 h (a) and 8 h (b) after the treatment. For the LPS control, untreated cells were exposed to LPS (200 ng mL−1) 24 h before the microsphere treatment in order to separate LPS stimulation and microsphere exposure, as for the MWNT exposure. A quantitative measurement of the fluorescence obtained in each picture was analysed via ImageJ and is presented in (c). For 8 h, the results presented here are divided by a factor of 10 for all conditions. After 1 and 8 h, no significant internalisation was observed compared to untreated or LPS stimulated cells. At 2 h following microsphere exposure, LPS stimulated cells internalised more microspheres than untreated cells. All scale bars shown are 100 μm. | ||
The migration ability of CNT-laden microglial cells was then tested. Following the observation that an unintended scratch on the plastic cell culture surface during the renewal of cell culture medium induced the migration of microglial cells toward the scratch, a specific migration assay based on “scratch stimulated” migration was developed. The migration assay was performed at day 7, 14 and 28 after the initial exposure to one of the three f-MWNTs and compared to the results with naïve microglial cells and microglial cells stimulated with LPS (200 ng mL−1) (Fig. 6 and Fig. S7, ESI†). The assay was not performed on the day following the exposure to CNTs as free CNTs might still be present in the supernatant and perturb the assay. Unexpectedly, the migration of microglial cells stimulated by LPS was inhibited at all three time points tested compared to untreated cells (Fig. S7, ESI† and Fig. 6). LPS is indeed known to stimulate migration and should have acted as a positive control/stimulator of migration.38 It was previously reported that the amount of LPS used during an assay is a critical parameter that can influence the motility of primary microglial cells.39 While low (0.1 ng mL−1) or high (100 ng mL−1) concentrations were shown to have no effect on promoting migration, 10 ng mL−1 was reported as the adjusted dose which induces higher migration compared to control cells. In our case, the reduced motility of microglial cells under LPS conditions can possibly be explained by the deviation of normal macrophage functionalities toward an activation profile dedicated to pro-inflammatory factor secretion due to the overloaded LPS dose (i.e. 200 ng mL−1 was used for both the NO release and migration assay). This will require further investigations to fully understand the reason for this unusual behaviour of LPS stimulated microglial cells and confirm the optimal concentration of 10 ng mL−1 for promoting migration.39
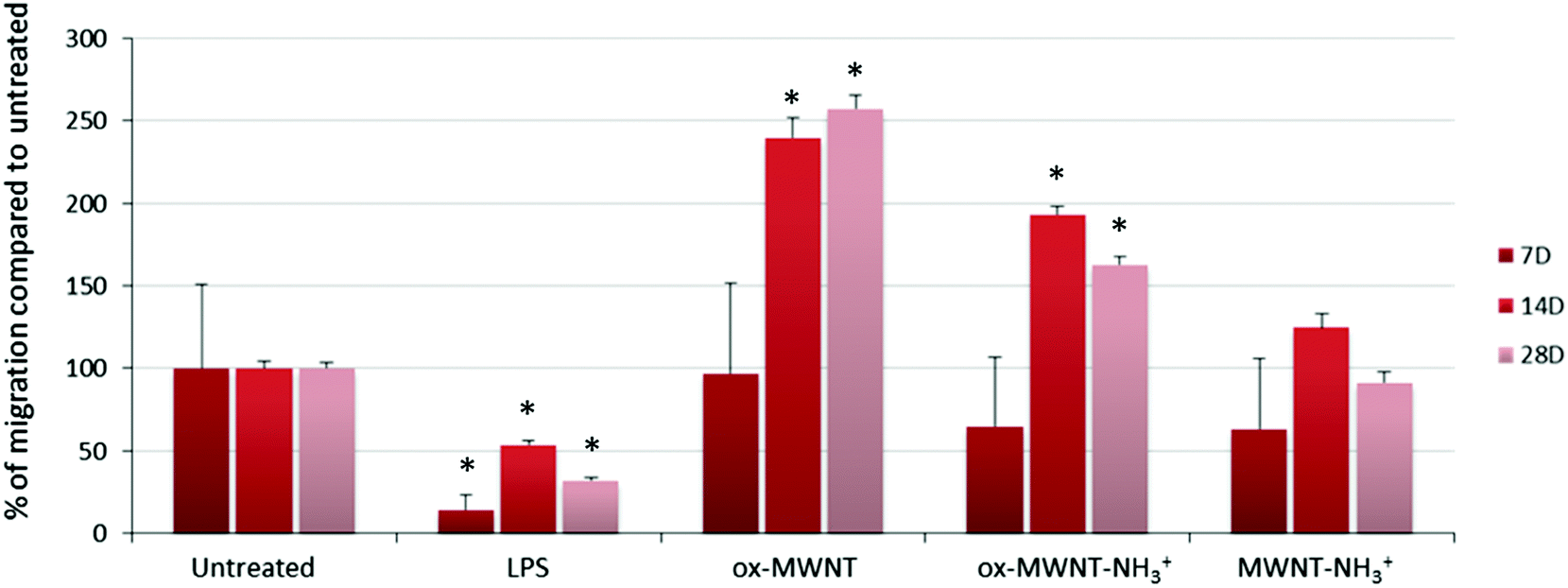 | ||
| Fig. 6 Migration ability assessed using a scratch assay. Microglial cell cultures were exposed for 24 h to a unique dose of different MWNTs (10 μg mL−1). On day 7, 14 and 28 after the initial exposure, the plastic surface of the microglial cell cultures was subjected to scratches. Seventy-two hours after the scratch, the amount of cells that have migrated toward the scratch was imaged and counted using ImageJ. For the LPS control, untreated cells were exposed to LPS (200 ng mL−1) 24 h before the formation of the scratch on the plastic surface. The migration is presented as the percentage compared to untreated cells. LPS treatment was found to inhibit the migration of microglial cells. The exposure to ox-MWNTs or ox-MWNT–NH3+ was found to significantly stimulate microglial cell migration at both 14 and 28 days, with ox-MWNTs having a higher stimulation than ox-MWNT–NH3+. In contrast, MWNT–NH3+ showed a limited effect on the microglial cell migration ability compared to untreated cells. | ||
As regards to f-MWNTs, a chemical functionalisation dependent modification of the microglial cell migration ability was observed (Fig. 6). At any time point, microglial cells exposed to ox-MWNTs had higher migration ability than the cells exposed to aminated MWNTs (i.e. both ox-MWNT–NH3+ and MWNT–NH3+) with MWNTs bearing both chemical functions (i.e. ox-MWNT–NH3+) in between ox-MWNTs and MWNT–NH3+. At day 7, microglial cells exposed to ox-MWNTs migrated to the same extent as untreated cells whereas microglial cells exposed to aminated MWNTs (i.e. both ox-MWNT–NH3+ and MWNT–NH3+) moved less than untreated cells. At day 14, surprisingly, microglial cells exposed to any type of MWNTs migrated more than the untreated cells. At day 28, only microglial cells exposed to oxidised MWNTs (i.e. both ox-MWNTs and ox-MWNT–NH3+) migrated more than the untreated cells or the cells exposed to MWNT–NH3+, which both show similar migration ability. These results showed that internalisation of f-MWNTs did not alter microglial cell migration ability and that exposure to (and uptake of) f-MWNTs can actually stimulate migration ability in a chemical functionalisation dependent fashion. In contrast to amination, oxidation of MWNTs seemed to stimulate the migration ability. Similar enhanced migration ability in comparison to the untreated cells has been recently observed when peritoneal macrophages were exposed to nanographene oxide flakes and to a higher extent when exposed to nanographene oxide functionalised with PEG groups.40 In this study, enhanced cell migration was attributed to the activation of the cells towards a pro-inflammatory profile upon exposure to graphene oxide flakes. A correlation between enhanced cell migration and activation was not identified in the present study (Fig. 4 and 6) and would therefore require further investigations to reveal the mechanism at the origin of the enhanced migration ability of cells exposed to oxidised MWNTs.
In the present in vitro study, no activation of pro-inflammation reaction following the exposure of primary microglial cells to different types of f-MWNTs was observed, despite the persistence over time of the nanomaterials in the cytoplasm, and in vesicles (Fig. S4 and inset of Fig. S5, ESI†), and regardless of the type of surface functionalisation. These results are in agreement with other in vitro studies performed using the BV2 microglial cell line exposed to non-functionalised MWNTs, in which neither cytotoxicity nor induction of expression of major inflammation markers were observed despite CNT internalisation at 48 h.41 Similarly, no cytotoxicity or pro-inflammatory cytokine release or NO production were observed in vitro in the N9 microglial cell line exposed to oxidised MWNTs despite internalisation.16 In contrast, using similar MWNTs and similar surface functionalisation (ox-MWNT–NH3+ and MWNT–NH3+), in vivo cortical injection of f-MWNTs was demonstrated to induce a neuroinflammatory response that was surface chemical functionalisation dependent.11 One month after injection, the degree of oxidation of the f-MWNTs was suggested to be a major factor driving the immune tissue response to explain the higher amount of CD11b positive (microglial cell marker) and GFAP positive (astrocyte marker) cells around the site of injection for animals injected with f-MWNTs compared to vehicle injected animals.
In contrast to previous reports, no effects due to the interaction of f-MWNTs with the cytoskeleton were observed here. It was indeed previously speculated that if CNTs are free in the cytoplasm and interacting with the cytoskeleton, eventually forming bundles with microtubules and microfilaments, they could possibly rigidify cells and consequently reduce both the migration and phagocytosis/internalisation abilities of the cells.18 However, both internalisation and migration abilities were here found to be normal in f-MWNT exposed microglial cells, despite a high internalisation of the materials illustrated by a black halo around the nucleus region and confirmed to be made of CNTs using Raman spectroscopy. This suggested that f-MWNTs were likely not free in the cytoplasm but in vesicular compartments, which is in agreement with the microscopic results presented in Fig. 3a, Fig. S2–S5 (ESI†). Secondly, this also meant that although their cytoplasms were relatively congested with CNT loading, the cells were still elastic enough to perform basic cytoskeleton-mediated functions and especially cell migration, one of the most important cell functions in vivo for clearance of foreign materials.17,37 This led us to believe that in vivo the tested materials would probably undergo brain tissue emigration through microglial cell-mediated drainage finishing in the lymphatic system without harming microglial cells. However, further studies are needed to address this specific hypothesis and to assess whether free f-MWNTs (not engulfed in any brain cells) would be able to use the brain solute elimination system (i.e. glymphatic clearance system).42,43
Theoretically, CNT–cytoskeleton interactions can also hamper the cytoskeleton dynamics required during cell division, and therefore modify the cell cycle, which would eventually lead to apoptosis. Using the dividing BV2 microglial cell model, MWNTs were indeed shown to induce apoptosis at the relatively low concentration levels of 0.6 and 6 μg mL−1 (35% and 66%, respectively).18 Induction of the apoptotic pathway was also explained by cell cycle arrest due to CNT interactions with cytoskeleton molecules.18,44 In contrast to these studies, no cell death at 10 μg mL−1 regardless of the time point considered was observed here. Seclusion of f-MWNT loading in vesicular compartments over time is likely the best explanation for the difference between the present results and previous reports. This will require complementary investigations on the subcellular fate of f-MWNTs, notably to confirm the nature of the vesicular compartmentalisation of the CNT content and its potential dynamics (endocytosis/exocytosis) over time. Moreover, a complete dose–response relationship correlated with the physicochemical features of the materials should be performed in order to identify the full extent of microglial tolerance to f-MWNTs and its limitations.
Degradation and clearance of cell debris and foreign materials are also a fundamental role of microglial cells for maintaining brain homeostasis.17 After stereotactic administration of aminated MWNTs in the cortex of mice, resident microglial cells were reported to internalise and then alter the structural integrity of the injected materials.12 Analysis of tissue sections via Raman spectroscopy and electron microscopy gave evidence of the initiation of a degradative process within two days following the injection. Similarly, it was shown that after 24 h, the N9 microglial cells were able to alter the graphitic structure of oxidised MWNTs.16 And finally, but more importantly using the same model as described in the present study (same primary cells, same f-MWNTs, and same 10 μg mL−1 dose), primary microglial cells were shown to degrade f-MWNTs over time, irrespective of the surface functionalisation of MWNTs.14,15 These results demonstrate that in addition to conserving their internalisation and migration abilities as reported here, microglial cells had preserved their degradation functions despite their CNT loading. Taken together, these results confirm that microglial cells exposed to 10 μg mL−1 of the f-MWNTs used here are still fully functional cells.
The agglomeration and colloidal properties of the three CNT suspensions were not found to be different (Fig. S1, ESI†). Similarly, variations in the surface chemical functionalisation were not found to be deterministic in the impact of the three CNTs apart from the migration ability that seemed to be stimulated by the two oxidised CNTs, in an oxidation degree dependent manner (ox-MWNTs ≫ ox-MWNT–NH3+; Fig. 6). But alongside surface chemical functionalisation and agglomeration, dimensions (both length and diameter) are important parameters for CNT biological impact.45–48 In this respect, the type of CNTs (multi-walled rather than single-walled or double-walled) could also be important as dimensions together with agglomeration will highly differ between CNT types. Therefore, the present results obtained using MWNTs are likely to be different if SWNTs are used.
Conclusion
In summary, primary microglial cells when exposed to a low concentration of f-CNTs maintained their basic resident macrophage functions (phagocytosis/internalisation, migration, and degradation) and did not initiate an inflammatory reaction despite a large internalisation of materials in cytoplasmic vesicles. Considering the key role of microglial cells within the brain, the demonstration of the relative absence of the bioreactivity of f-CNTs upon interaction with microglial cells is an important step towards their development as nanovectors for drug delivery purposes in the central nervous system.Experimental
Materials, MWNTs and preparation of MWNT suspensions for cell exposure
Chemicals and solvents were obtained from Merck/Sigma-Aldrich (UK). Cell culture reagents (PBS, trypsin, foetal bovine serum, DMEM:F12) were purchased from Gibco (ThermoScientific, UK). Multi-walled carbon nanotubes (MWNTs) were purchased from Nanostructured and Amorphous Materials Inc. (Houston, TX, USA; Lot # 1240XH, 95%). The different chemical functionalisations of the MWNTs were performed as previously reported.25,27,28,32 The amount of COOH (on both ox-MWNTs and ox-MWNT–NH3+) after the oxidation reaction has been previously calculated using thermogravimetric analysis, corresponding to 1.7 μmol g−1,49 while the amount of amino groups measured using Kaiser's test was 170 μmol g−1 for ox-MWNT–NH3+ and 84 μmol g−1 for MWNT–NH3+. Preparation of f-MWNT dispersions for cell exposure was performed as previously reported.14Stability of f-MWNT suspension in different media assessed by UV-Vis spectrophotometry
A suspension of the three f-MWNTs originally prepared in 5% dextrose water solution (1 mg mL−1) was diluted to 10 μg mL−1 in 5% dextrose water solution, cell culture medium (DMEM:F12) supplemented with 12% FBS or cell culture medium (DMEM:F12) without FBS. Absorbance spectra were acquired at 0 h and 24 h after preparing different 10 μg mL−1 suspensions using a Cary 50 Bio UV/Vis spectrophotometer (Varian Inc., Agilent Technologies, UK). For measurements, the prepared suspensions were transferred from a microtube (0.5 mL) to a 1 mL quartz cuvette with 1 cm path length. Measurements were performed at room temperature, using a dual beam mode with baseline correction based on 5% dextrose water solution, cell culture medium with 12% FBS or cell culture medium without FBS, as blank controls to create the baseline.Primary microglial cell cultures and exposure to functionalised CNTs
The preparation of primary microglial cell cultures was performed as previously reported14 and according to a previously described method, based on mild trypsinisation of mixed glial cell cultures50 prepared from rat embryonic stratial extracts. Microglial cell cultures were then exposed for 2 h to a non-cytotoxic 10 μg mL−1 CNT suspension prepared in serum free cell culture medium (DMEM:F12 medium). After 2 h of incubation at 37 °C without serum, the cell culture medium was completed with heat inactivated foetal bovine serum (12%). After 24 h of incubation at 37 °C, the supernatant containing CNTs was removed, the cells were washed twice with pre-warmed PBS and then incubated with CNT free complete medium (DMEM:F12 with 12% serum) for 1, 7, 14 and 28 days (recovery periods). The CNT-free complete medium was changed every 3 days.Assessing cell viability using a modified LDH assay
Cell viability was assessed using a modified LDH assay as previously described.32 The Promega Cytotox 96 © Non-radioactive cytotoxicity assay (Promega, UK) was modified to avoid interference between CNTs and LDH. 10% DMSO was used as a positive control of the assay. Microglial cell cultures were generated in 24 well plates. At the desired time points (1, 7, 14 and 28 days), the cells were lysed with 10 μL of lysis buffer (0.9% triton X100 in distilled water) per 100 μL of serum free media and left for 45 min at 37 °C. After centrifuging (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 5 min), 50 μL of cell lysate supernatant was mixed with 50 μL of the substrate mixture in a new microtiter plate and incubated for 6 min at room temperature. LDH content was determined by reading the absorbance at 490 nm using a multiwell plate reader (Fluostar Omega, BMG Labtech Ltd). The amount of LDH detected after cell lysis indicated the number of cells that have survived CNT exposure. The percentage cell survival is expressed as [LDH released from tested cells − Blank (media alone)/LDH released control cells − Blank (media alone)] × 100. Each assay was performed at least 3 times, using 3 wells per condition at each considered time.
000 rpm, 5 min), 50 μL of cell lysate supernatant was mixed with 50 μL of the substrate mixture in a new microtiter plate and incubated for 6 min at room temperature. LDH content was determined by reading the absorbance at 490 nm using a multiwell plate reader (Fluostar Omega, BMG Labtech Ltd). The amount of LDH detected after cell lysis indicated the number of cells that have survived CNT exposure. The percentage cell survival is expressed as [LDH released from tested cells − Blank (media alone)/LDH released control cells − Blank (media alone)] × 100. Each assay was performed at least 3 times, using 3 wells per condition at each considered time.
Raman spectroscopy
A Thermo Scientific DXR Raman microscope, equipped with an Olympus microscope (100× objective lens) and a 633 nm laser (1 mW of laser power) was used to perform Raman spectroscopy. Collection of Raman spectra was performed as previously described.14Immunocytochemistry of microglial cells
Microglial cell cultures were prepared on poly-L-lysine coated plastic tissue-culture treated dishes (35 mm dia.). At the end of exposure, the cells were fixed with pre-cooled methanol (−20 °C) for 10 min, air-dried and then washed twice with PBS containing 0.1% triton (TPBS, pH 7.4) before staining. Staining with primary antibody was performed after 1 h of incubation (i.e. blocking) with 5% normal goat serum in TPBS. The primary antibodies used were: mouse monoclonal anti-CD11b/c (1:200) (Ox42, Abcam) or mouse monoclonal anti-CD68 (ED1, Abcam, UK), both diluted in 2% BSA in TPBS and incubated for 2 h at room temperature. The secondary antibody (goat anti-mouse Cy3 labelled) was used at the dilution of 1:250 in 2% BSA in TPBS for 1 h at room temperature. After washing, the cell preparations were mounted in appropriate medium containing an anti-fading agent and DAPI (Vectashield, Vector laboratories LTD). Immunoreactivity was assessed on an inverted epifluorescence microscope (Carl Zeiss Axio Observer A1).
Assessing inflammation markers (NO release) using the Griess reagent assay
Inflammation reaction was based on the release of NO, a well-known biomarker of inflammation for microglial cells. As for the cytotoxicity assay, microglial cell cultures were generated in 24 well plates. The cells were then exposed to 10 μg mL−1 of the three different CNT suspensions or to CNT free medium. LPS (E. coli, O26:B6, 200 ng mL−1) was used as a positive control for inflammation and was applied 24 h before performing the measurement on the cell supernatants. At day 1, 7, 14 and 28 after CNT exposure, the amount of nitrite (stable breakdown products of NO) was measured using the Griess reagent system assay (Promega, UK) following the manufacturer's instructions. In brief, 50 μL of each experimental sample was mixed with 50 μL of the sulfanilamide solution. Following 8 min of incubation, 50 μL of the NED solution was added to all wells for 8 min of incubation before reading the absorbance at 530 nm. The amount of nitrite was determined by comparison to a nitrite standard reference curve, prepared and measured in parallel to each sample measurement.Assessing phagocytosis/internalisation ability using a fluorescent polystyrene bead assay
To assess the internalisation ability of CNT-laden microglial cells, we used 0.50 μm Fluoresbrite® YellowGreen Microspheres (Polysciences, Inc.) and reduced the cell density of the microglial cell cultures (1/2 of the usual cell density when preparing mixed glia cell cultures). No assay was performed at day 1 after the CNT exposure as some CNTs might still be present in the cell culture medium and compete with the beads for internalisation, despite carefully washing. The assays were therefore performed at day 7 of the recovery period. At this time point, 2 μL of the initial bead suspension was diluted in 2 mL of complete cell culture medium. This bead-diluted suspension was used to treat CNT-laden microglial cells prepared and cultured in 35 mm cell culture dishes. The phagocytosis of the fluorescently labelled beads was observed using an inverted epifluorescence microscope tuned on the FITC channel (Carl Zeiss Axio Observer A1) and recorded at 1, 2 and 8 h after bead treatment. Green fluorescence present in each picture was measured using ImageJ image processing and analysis software (NIH, USA).Assessing migration ability using a scratch assay
To assess the migration ability of CNT-laden microglial cells, we developed a migration assay based on the well-known wound-healing assay and using the formation of scratches at the bottom of the plastic cell culture support. Using a reduced cell density of microglial cell cultures (1/2 of usual mixed glia cell density), we first exposed cells to 10 μg mL−1 of the different f-MWNT suspension for 24 h for loading purposes. As for the internalisation assay, no migration assay was performed at day 1 after CNT exposure because it was considered too soon after exposure and the presence of free CNTs on the cell culture surface might bias the migration assessment of CNT-laden microglial cells. On day 7, 14 and 28 of the recovery period, we generated straight scratches on the plastic surface of the 35 mm cell culture dishes by gently drawing straight lines using a sterile plastic pipette tip. To allow a maximum number of cells to move towards the scratches, the migration ability was assessed 72 h after generation of the scratches. Pictures were centred on the scratch and a ROI was drawn around the scratch to count the cells present in the ROI of each picture (ImageJ image processing and analysis software (NIH, USA)) as illustrated in the ESI,† Fig. S7.Statistics
The results are expressed as mean ± SD. Statistical differences were analysed using the Student's t test, and p < 0.05 (*) was considered as significantly different. Each assay was performed independently at least 3 times. A one way ANOVA, Kruskal-Wallis test with Dunn's multiple comparison test was used to analyse the differences in the viability assay and p < 0.05 (*) was considered as significantly different.Acknowledgements
This work was supported by the European Commission, under the FP-7 Marie Curie actions (Career Development Intra-European Fellowship, PIEF-GA-2010-276051, project NANONEUROHOP). AB wishes to thank the CNRS financial support from PICS (Project for International Scientific Cooperation). MP, as the recipient of the AXA Chair, is grateful to the AXA Research Fund for financial support. MP was also supported by the Spanish Ministry of Economy and Competitiveness MINECO (project CTQ2016-76721-R), by the University of Trieste and by Diputación Foral de Gipuzkoa program Red (101/16).References
- P. Blasi, S. Giovagnoli, A. Schoubben, M. Ricci and C. Rossi, Adv. Drug Delivery Rev., 2007, 59, 454–477 CrossRef CAS PubMed.
- T. Patel, J. Zhou, J. M. Piepmeier and W. M. Saltzman, Adv. Drug Delivery Rev., 2012, 64, 701–705 CrossRef CAS PubMed.
- S. B. Pehlivan, Pharm. Res., 2013, 30, 2499–2511 CrossRef PubMed.
- J. T. Wang and K. T. Al-Jamal, Nanomedicine, 2015, 10, 2639–2642 CrossRef CAS PubMed.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J. P. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- Z. Yang, Y. Zhang, Y. Yang, L. Sun, D. Han, H. Li and C. Wang, Nanomedicine, 2010, 6, 427–441 CrossRef CAS PubMed.
- K. T. Al-Jamal, L. Gherardini, G. Bardi, A. Nunes, C. Guo, C. Bussy, M. A. Herrero, A. Bianco, M. Prato, K. Kostarelos and T. Pizzorusso, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10952–10957 CrossRef CAS PubMed.
- H. J. Lee, J. Park, O. J. Yoon, H. W. Kim, D. Y. Lee, D. H. Kim, W. B. Lee, N. E. Lee, J. V. Bonventre and S. S. Kim, Nat. Nanotechnol., 2011, 6, 120–124 Search PubMed.
- M. K. Gottipati, E. Bekyarova, R. C. Haddon and V. Parpura, Amino Acids, 2015, 47, 1379–1388 CrossRef CAS PubMed.
- G. Tosi, A. Vilella, R. Chhabra, M. J. Schmeisser, T. M. Boeckers, B. Ruozi, M. A. Vandelli, F. Forni, M. Zoli and A. M. Grabrucker, J. Controlled Release, 2014, 177, 96–107 CrossRef CAS PubMed.
- G. Bardi, A. Nunes, L. Gherardini, K. Bates, K. T. Al-Jamal, C. Gaillard, M. Prato, A. Bianco, T. Pizzorusso and K. Kostarelos, PLoS One, 2013, 8, e80964 Search PubMed.
- A. Nunes, C. Bussy, L. Gherardini, M. Meneghetti, M. A. Herrero, A. Bianco, M. Prato, T. Pizzorusso, K. T. Al-Jamal and K. Kostarelos, Nanomedicine, 2012, 7, 1485–1494 CrossRef CAS PubMed.
- C. Bussy, K. T. Al-Jamal, J. Boczkowski, S. Lanone, M. Prato, A. Bianco and K. Kostarelos, ACS Nano, 2015, 9, 7815–7830 CrossRef CAS PubMed.
- C. Bussy, C. Hadad, M. Prato, A. Bianco and K. Kostarelos, Nanoscale, 2016, 8, 590–601 RSC.
- J. Russier, L. Oudjedi, M. Piponnier, C. Bussy, M. Prato, K. Kostarelos, B. Lounis, A. Bianco and L. Cognet, Nanoscale, 2017, 9, 4642–4645 RSC.
- A. E. Goode, D. A. Gonzalez Carter, M. Motskin, I. S. Pienaar, S. Chen, S. Hu, P. Ruenraroengsak, M. P. Ryan, M. S. Shaffer, D. T. Dexter and A. E. Porter, Biomaterials, 2015, 70, 57–70 CrossRef CAS PubMed.
- L. C. Davies, S. J. Jenkins, J. E. Allen and P. R. Taylor, Nat. Immunol., 2013, 14, 986–995 CrossRef CAS PubMed.
- J. C. Villegas, L. Alvarez-Montes, L. Rodriguez-Fernandez, J. Gonzalez, R. Valiente and M. L. Fanarraga, Adv. Healthcare Mater., 2014, 3, 424–432 CrossRef CAS PubMed.
- N. V. Konduru, Y. Y. Tyurina, W. Feng, L. V. Basova, N. A. Belikova, H. Bayir, K. Clark, M. Rubin, D. Stolz, H. Vallhov, A. Scheynius, E. Witasp, B. Fadeel, P. D. Kichambare, A. Star, E. R. Kisin, A. R. Murray, A. A. Shvedova and V. E. Kagan, PLoS One, 2009, 4, e4398 Search PubMed.
- C. Bussy, M. Pinault, J. Cambedouzou, M. J. Landry, P. Jegou, M. Mayne-L'hermite, P. Launois, J. Boczkowski and S. Lanone, Part. Fibre Toxicol., 2012, 9, 46 CrossRef CAS PubMed.
- J. T. Wang, N. Rubio, H. Kafa, E. Venturelli, C. Fabbro, C. Menard-Moyon, T. Da Ros, J. K. Sosabowski, A. D. Lawson, M. K. Robinson, M. Prato, A. Bianco, F. Festy, J. E. Preston, K. Kostarelos and K. T. Al-Jamal, J. Controlled Release, 2016, 224, 22–32 CrossRef CAS PubMed.
- H. Ali-Boucetta, K. T. Al-Jamal, D. McCarthy, M. Prato, A. Bianco and K. Kostarelos, Chem. Commun., 2008, 459–461 RSC.
- R. Singh, D. Pantarotto, D. McCarthy, O. Chaloin, J. Hoebeke, C. D. Partidos, J. P. Briand, M. Prato, A. Bianco and K. Kostarelos, J. Am. Chem. Soc., 2005, 127, 4388–4396 CrossRef CAS PubMed.
- A. Servant, I. Jacobs, C. Bussy, C. Fabbro, T. da Ros, E. Pach, B. Ballesteros, M. Prato, K. Nicolay and K. Kostarelos, Carbon, 2016, 97, 126–133 CrossRef CAS.
- S. P. Li, W. Wu, S. Campidelli, V. Sarnatskaia, M. Prato, A. Tridon, A. Nikolaev, V. Nikolaev, A. Bianco and E. Snezhkova, Carbon, 2008, 46, 1091–1095 CrossRef CAS.
- C. Gaillard, G. Cellot, S. P. Li, F. M. Toma, H. Dumortier, G. Spalluto, B. Cacciari, M. Prato, L. Ballerini and A. Bianco, Adv. Mater., 2009, 21, 2903–2908 CrossRef CAS.
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760–761 CrossRef CAS PubMed.
- V. Georgakilas, N. Tagmatarchis, D. Pantarotto, A. Bianco, J. P. Briand and M. Prato, Chem. Commun., 2002, 3050–3051 RSC.
- P. Wick, P. Manser, L. K. Limbach, U. Dettlaff-Weglikowska, F. Krumeich, S. Roth, W. J. Stark and A. Bruinink, Toxicol. Lett., 2007, 168, 121–131 CrossRef CAS PubMed.
- L. Dong, C. M. Witkowski, M. M. Craig, M. M. Greenwade and K. L. Joseph, Nanoscale Res. Lett., 2009, 4, 1517–1523 CrossRef CAS PubMed.
- E. A. Salegio, H. Streeter, N. Dube, P. Hadaczek, L. Samaranch, A. P. Kells, W. San Sebastian, Y. Zhai, J. Bringas, T. Xu, J. Forsayeth and K. S. Bankiewicz, Front. Neuroanat., 2014, 8, 9 Search PubMed.
- H. Ali-Boucetta, K. T. Al-Jamal, K. H. Muller, S. Li, A. E. Porter, A. Eddaoudi, M. Prato, A. Bianco and K. Kostarelos, Small, 2011, 7, 3230–3238 CrossRef CAS PubMed.
- S. Ganter, H. Northoff, D. Mannel and P. J. Gebickeharter, J. Neurosci. Res., 1992, 33, 218–230 CrossRef CAS PubMed.
- M. P. Ramprasad, V. Terpstra, N. Kondratenko, O. Quehenberger and D. Steinberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14833–14838 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
- K. Saijo and C. K. Glass, Nat. Rev. Immunol., 2011, 11, 775–787 CrossRef CAS PubMed.
- T. Tajima, T. Murata, K. Aritake, Y. Urade, H. Hirai, M. Nakamura, H. Ozaki and M. Hori, J. Pharmacol. Exp. Ther., 2008, 326, 493–501 CrossRef CAS PubMed.
- H. Scheiblich, F. Roloff, V. Singh, M. Stangel, M. Stern and G. Bicker, Brain Res., 2014, 1564, 9–21 CrossRef CAS PubMed.
- N. Luo, J. K. Weber, S. Wang, B. Luan, H. Yue, X. Xi, J. Du, Z. Yang, W. Wei, R. Zhou and G. Ma, Nat. Commun., 2017, 8, 14537 CrossRef CAS PubMed.
- B. Kateb, M. Van Handel, L. Zhang, M. J. Bronikowski, H. Manohara and B. Badie, NeuroImage, 2007, 37(Suppl 1), S9S–17 CrossRef PubMed.
- L. Xie, H. Kang, Q. Xu, M. J. Chen, Y. Liao, M. Thiyagarajan, J. O'Donnell, D. J. Christensen, C. Nicholson, J. J. Iliff, T. Takano, R. Deane and M. Nedergaard, Science, 2013, 342, 373–377 CrossRef CAS PubMed.
- J. J. Iliff, M. Wang, Y. Liao, B. A. Plogg, W. Peng, G. A. Gundersen, H. Benveniste, G. E. Vates, R. Deane, S. A. Goldman, E. A. Nagelhus and M. Nedergaard, Sci. Transl. Med., 2012, 4, 147ra111 Search PubMed.
- L. Rodriguez-Fernandez, R. Valiente, J. Gonzalez, J. C. Villegas and M. L. Fanarraga, ACS Nano, 2012, 6, 6614–6625 CrossRef CAS PubMed.
- S. Lanone, P. Andujar, A. Kermanizadeh and J. Boczkowski, Adv. Drug Delivery Rev., 2013, 65, 2063–2069 CrossRef CAS PubMed.
- H. Nagai, Y. Okazaki, S. H. Chew, N. Misawa, Y. Yamashita, S. Akatsuka, T. Ishihara, K. Yamashita, Y. Yoshikawa, H. Yasui, L. Jiang, H. Ohara, T. Takahashi, G. Ichihara, K. Kostarelos, Y. Miyata, H. Shinohara and S. Toyokuni, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1330–1338 CrossRef PubMed.
- C. A. Poland, R. Duffin, I. Kinloch, A. Maynard, W. A. Wallace, A. Seaton, V. Stone, S. Brown, W. Macnee and K. Donaldson, Nat. Nanotechnol., 2008, 3, 423–428 CrossRef CAS PubMed.
- C. Muhlfeld, C. A. Poland, R. Duffin, C. Brandenberger, F. A. Murphy, B. Rothen-Rutishauser, P. Gehr and K. Donaldson, Nanotoxicology, 2012, 6, 867–879 CrossRef PubMed.
- J. Russier, C. Menard-Moyon, E. Venturelli, E. Gravel, G. Marcolongo, M. Meneghetti, E. Doris and A. Bianco, Nanoscale, 2011, 3, 893–896 RSC.
- J. Saura, J. M. Tusell and J. Serratosa, Glia, 2003, 44, 183–189 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nh00055c |
| This journal is © The Royal Society of Chemistry 2017 |