
Photoinduced charge separation in single-walled carbon nanotube/protein integrated systems†
Lenore
Kubie
,
Amanda R.
Amori
,
Saikat
Chakraborty
,
Kara L.
Bren
* and
Todd D.
Krauss
 *
*
Department of Chemistry, University of Rochester, Rochester, NY 14627, USA. E-mail: krauss@chem.rochester.edu; bren@chem.rochester.edu
First published on 31st January 2017
Abstract
Zinc-substituted cytochrome c (Zn-cyt c) is noncovalently bound to single-walled carbon nanotubes (SWNTs), causing the Zn-cyt c fluorescence to be quenched by up to 95%, primarily due to photoinduced charge transfer. Deposition of Zn-cyt c/SWNT films onto conductive oxides allows for harvesting of photoexcited electrons with an internal quantum efficiency of over 5%.
Conceptual insightsWe report a new and facile strategy to attach a semisynthetic chromophore to single walled carbon nanotubes (SWNTs) by making use of a modified protein, zinc-substituted cytochrome c (Zn-cyt c). Together the Zn-cyt c–SWNT complex is quite unique as the integrated nano-bio system forms an efficient and robust donor–acceptor complex for photoinduced charge-transfer with excellent aqueous solubility. In particular, we found electrons transferred from the photoactive zinc protoporphyrin IX in the protein to the SWNT quenched SWNT fluorescence with up to a 95% efficiency. Our approach to modifying nanotubes with proteins that contain chromophores directly addresses several challenges in the field, being highly advantageous because it avoids the use of complicated synthetic methods to produce a water-soluble chromophore and avoids covalent modification of the SWNTs. The biologically inspired method of SWNT modification with photochemical reductant molecules presents excellent opportunities for the rational design of highly engineered bio-nano assemblies for solar energy conversion. |
The superior charge transport properties of carbon nanotubes (NTs) make them attractive components in systems requiring efficient photoinduced charge separation.1–5 To use NTs in solar energy conversion applications, it is advantageous to closely associate a photosensitizer with a NT for efficient injection of photoelectrons.1,6–10 However, attaching a photosensitizer to a NT is nontrivial. If covalent modification is used,1,7,11 lattice defects are introduced whereby sp2 hybridized orbitals become sp3 hybridized,12–14 disrupting the extended π-electron system of the SWNTs and hindering electrical conduction. Noncovalent attachment of chromophores to NTs through direct pi–pi stacking or engineering electrostatic interactions with bridging molecules is an alternative approach, but is often limited to use in organic solvents and can involve synthetic complexity.1,5,8,9,15,16
Biopolymers can associate with NTs noncovalently and are compatible with aqueous solutions, suggesting an attractive alternative approach. For example, NTs can be wrapped with polyanionic DNA, which suspends them in water and serves to electrostatically bind cationic chromophores to form donor–acceptor complexes.8 Herein, we report a new strategy to noncovalently attach a semisynthetic chromophore to NTs by making use of a modified protein, zinc-substituted cytochrome c (Zn-cyt c). Cytochromes c (cyts c) are attractive biomolecules for this purpose because they contain an iron protoporphyrin IX (heme) covalently bound to the polypeptide via two thioether bonds (Fig. 1). Substitution of the cyt c heme iron with zinc creates a photoactive Zn(II) protoporphyrin-IX (ZnP) center with an excited state reduction potential ∼−1.3 V vs. SCE.17,18 Because the ZnP is covalently bound to the polypeptide, multiple sites for interaction with NTs are provided, which yields a robust donor–acceptor complex and enhanced aqueous solubility.19–23 The preparation of Zn-cyt c–NT donor–acceptor systems is relatively simple, based on solution-based processing. Further, using Zn-cyt c or a related system to tether ZnP photosensitizers to NTs has the additional advantages that complex chromophore synthesis is avoided, and the protein–NT interaction may be tuned by engineering the porphyrin-bearing polypeptide moiety using molecular biology techniques.24–26 This study demonstrates the preparation of aqueous suspensions of SWNTs modified with Zn-cyt c (SWNT:Zn-cyt c) for photoinduced charge injection into the SWNTs.
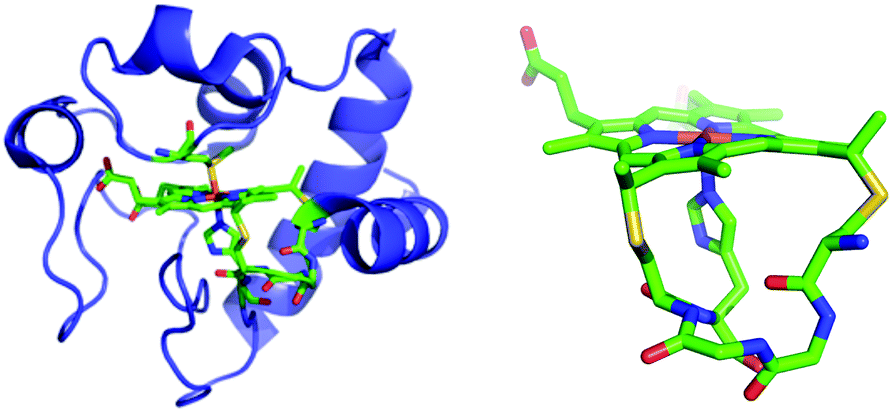 | ||
| Fig. 1 Left: Horse heart cyt c. Right: Cytochrome c heme attachment motif showing covalent bonds from Cys residues to heme and the axial His. From structure of horse heart cyt c (PDB: 1HRC).27 | ||
To prepare aqueous suspensions of SWNT:Zn-cyt c, suspensions of CoMoCAT SWNTs were first prepared via sonication with sodium cholate (SC) in 50 mM HEPES buffer, pH 7.5.28 Known volumes of Zn-cyt c stock solutions were combined with SC-suspended SWNTs to spontaneously form SWNT:Zn-cyt c. Aggregates form within minutes when Zn-cyt c is sonicated with SWNTs, thus SC is required to prepare a stable NT suspension. Control experiments were performed to verify a direct interaction between the SWNT and Zn-cyt c. In the absence of SC, the SWNTs with attached protein form an insoluble pellet upon centrifugation with no detectable protein in the supernatant (monitored by absorption spectroscopy). In the absence of SWNTs, if the protein is sonicated and centrifuged under the same conditions, the protein remains in solution (Fig. S1, ESI†). Because the Zn-cyt c is removed from the solution upon centrifugation only when SWNTs are present, we conclude that there is a direct interaction between the SWNT and Zn-cyt c. Additional information regarding sample preparation can be found in the ESI.†
Absorption spectra of SWNTs with and without Zn-cyt c are shown in Fig. S2 (ESI†). SWNTs have excitonic optical transitions associated with their Van Hove singularities; the E11 transitions occur in the IR region around 800 to 1200 nm, the E22 transitions occur in the visible region around 500 to 700 nm, and the E33 transition occurs in the UV.29–31 Zn-cyt c in 1 mg mL−1 SC, 50 mM HEPES, pH 7.5 has a Soret band absorption maximum of 422 nm and Q-band absorption maxima at 547 nm and 581 nm. These maxima are comparable to those recorded for Zn-cyt c in aqueous solution (423 nm, 549 nm and 585 nm respectively).32 Upon forming SWNT:Zn-cyt c, the Zn-cyt c Soret band does not shift (i.e. less than 0.5 nm), whereas previous studies of ZnP–SWNT interactions reported typical shifts of around 4 nm, although more subtle shifts are sometimes observed.5,33,34 One possible explanation for the lack of spectral shifts in the current system is that there is little direct electronic interactions between the ZnP and SWNT. Thus, it seems that the Zn-cyt c polypeptide, and not the ZnP itself, is interacting directly with the SWNT.15,34,35 The SWNT absorption spectrum also remains unchanged upon addition of the protein, as expected if there is little change in the dielectric environment surrounding the NT upon protein binding.36
Fluorescence spectroscopy measurements demonstrate significant influence of the SWNTs on ZnP fluorescence efficiency in SWNT:Zn-cyt c. Upon addition of Zn-cyt c to a solution of suspended SWNTs in 1 mg mL−1 SC, 50 mM HEPES, pH 7.5, ZnP fluorescence is strongly quenched (Fig. 2a). Zn-cyt c samples without SWNTs, but in 1 mg mL−1 SC, 50 mM HEPES, pH 7.5, were compared to SWNT:Zn-cyt c samples (with SC) to calculate the percent quenching. The concentration of Zn-cyt c was identical for each sample, and the optical density at the (6,5) SWNT E11 peak was also adjusted to be identical across all SWNT-containing samples. The total fluorescence intensity of each sample was determined by fitting the Zn-cyt c fluorescence spectrum with two Gaussian functions centered at the 580 nm and the 690 nm peaks and adding the integrated areas under the peaks. As the Zn-cyt c to SWNT ratio is decreased (by decreasing the Zn-cyt c concentration) the relative quenching increases, reaching a maximum value of 95% at a ratio of ∼100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 2b). Above a ratio of ∼200:1, the ZnP fluorescence quenching efficiency decreases linearly (inset b), suggesting that SWNTs become effectively saturated with Zn-cyt c under these conditions. The amount of Zn-cyt c per SWNT to reach full quenching depends on several poorly defined factors, such as the conformational state of the protein, the average length of the SWNTs after sonication, and the exciton mobility. We hypothesize this decreased quenching efficiency is a result of free Zn-cyt c in solution. For ratios much greater than 100:1, we suggest some unbound ZnP fluorescence likely will be quenched (although less efficiently) due to collisions with SWNTs, and thus the dependence of percent quenching on Zn-cyt c to SWNT ratio is sub-linear.
:1 (Fig. 2b). Above a ratio of ∼200:1, the ZnP fluorescence quenching efficiency decreases linearly (inset b), suggesting that SWNTs become effectively saturated with Zn-cyt c under these conditions. The amount of Zn-cyt c per SWNT to reach full quenching depends on several poorly defined factors, such as the conformational state of the protein, the average length of the SWNTs after sonication, and the exciton mobility. We hypothesize this decreased quenching efficiency is a result of free Zn-cyt c in solution. For ratios much greater than 100:1, we suggest some unbound ZnP fluorescence likely will be quenched (although less efficiently) due to collisions with SWNTs, and thus the dependence of percent quenching on Zn-cyt c to SWNT ratio is sub-linear.
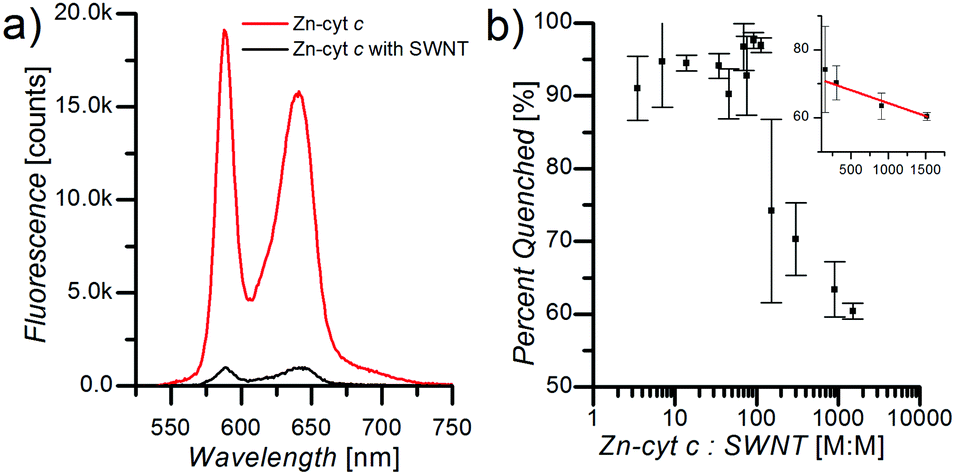 | ||
| Fig. 2 (a) Q-band fluorescence from Zn-cyt c (red curve). Fluorescence is quenched (96.7 ± 3.2)% when SWNTs are present at a ratio of 220:1 Zn-cyt c:SWNT (black curve). (b) Concentration dependence of Zn-cyt c fluorescence quenching. As the Zn-cyt c:SWNT ratio decreases, the quenching increases, reaching a maximum of ∼95%. The inset shows a linear relationship of the ZnP:SWNT ratio to the percent quenching once the quenching begins to decrease. | ||
Two possible mechanisms for the observed fluorescence quenching are energy transfer and electron transfer. Energy transfer from the ZnP to the SWNTs can be detected in a photoluminescence excitation (PLE) map.37 The PLE map in Fig. S3 (ESI†) shows primarily (6,5), (8,3) and (7,5) SWNT emission, which is typical of CoMoCAT SWNTs.29 If energy transfer were the cause of the Zn-cyt c fluorescence quenching, SWNT fluorescence would be seen at the E11 energies of the respective SWNT structure (i.e. 965, 985 and 1035 nm for (8,3), (6,5) and (7,5) SWNTs respectively) when the Zn-cyt c Soret band is photoexcited (at 422 nm). Data collected with Q-band excitation does not definitively prove or disprove energy transfer processes because excitation at these wavelengths overlaps with common SWNT absorption peaks in the E22 region. However, as seen in Fig. S3 (ESI†), the absence of SWNT emission peaks at the Zn-cyt c Soret excitation wavelength indicates negligible energy transfer in the ZnP–SWNT conjugates. Thus, PLE data are consistent with fluorescence quenching being due to a photoinduced electron transfer event from the ZnP to the SWNT.
Thin SWNT:Zn-cyt c films on indium tin oxide (ITO) fabricated using a layer-by-layer technique were used to test the hypothesis that photoinduced electron transfer occurs from ZnP to SWNTs.1,38 Zn-cyt c is positively charged at a neutral pH, while the SC-suspended SWNTs have an overall negative surface charge. Thus, thin films are easily assembled using electrostatic interactions. Photoexcitation of the films resulted in a measureable photocurrent above background noise current levels (Fig. 3). Approximately 5.6 nA of current was generated upon illumination, and the current dropped back to baseline when the light source was blocked (Fig. 3 inset).
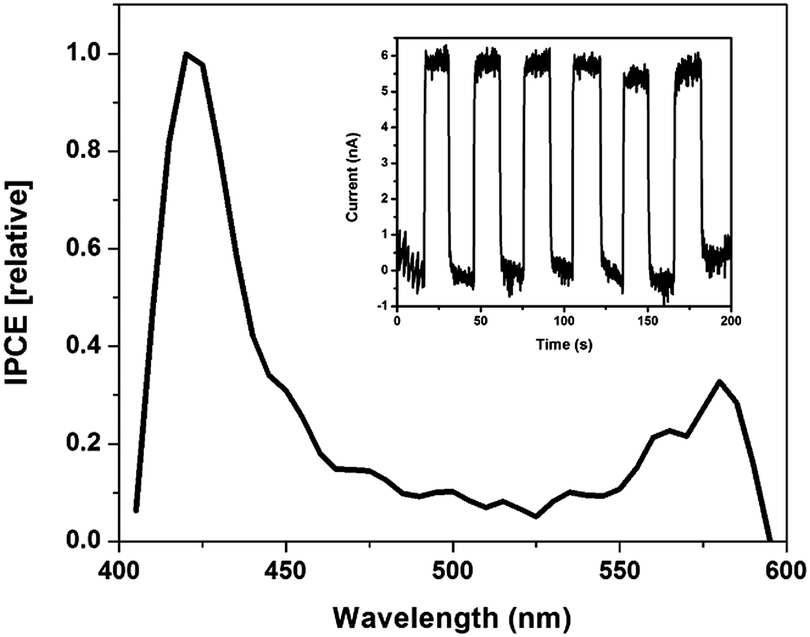 | ||
| Fig. 3 Photoaction spectrum of SWNT:Zn-cyt c films in 50 mM HEPES buffer, 100 mM NaCl at pH = 7.5 and 10% triethanolamine (TEOA) with a 0 mV bias. The inset shows repeatable current generation with 421 nm monochromatic light by manually blocking and unblocking the light source every 100 seconds. | ||
A photoaction spectrum taken between 400–600 nm confirms that the photocurrent was generated by excited state electron transfer from Zn-cyt c to the SWNT. As seen in Fig. 3, photocurrent is generated upon illumination of both the Soret band and Q-band regions of the system, but not at other wavelengths. Two control films were made using the same method, one of alternating layers of poly-L-lysine and SWNTs and the other of alternating layers of poly(acrylic acid) and Zn-cyt c (Fig. S4, ESI†). Neither control film produced any measureable photocurrent. The maximum internal quantum efficiency (IQE) of the 20-layer Zn-cyt c/SWNT system was calculated to be 7.65%, with an average IQE across tested assemblies of 5.33%. This result, accompanied by the excellent fluorescence quenching of the Zn-cyt c/SWNT system, suggests facile intrinsic charge transport. We also note that a 10-layer Zn-cyt c/SWNT film on ITO was also made, yielding an IQE of 0.46% (Fig. S5, ESI†). The poorer performance of this film is attributed to the higher resistivity that is seen per the increased rise time to reach the on-state.
Conclusions
Using Zn-cyt c to donate electrons to NTs has several benefits over synthetic chromophores. First, Zn-cyt c is readily prepared by simple metal substitution. Additionally, Zn-cyt c contains a ZnP covalently bound to the polypeptide, which allows for solution processing without fear of losing the cofactor. Furthermore, peptide modifications, such as point mutations, chemical modification, or incorporation of unnatural amino acids can be readily made to alter the peptide/SWNT interaction, the immediate ZnP environment, or incorporate an antenna moiety to increase the spectral range of productive photocurrent generation via energy transfer to the ZnP center.39In conclusion, we report a simple method for noncovalently attaching a photoactive electron donor to SWNTs in water. This approach is advantageous because it avoids the use of complicated synthetic methods to produce a water-soluble chromophore and avoids covalent modification of the SWNTs. We observe photoinduced charge transfer from Zn-cyt c to SWNTs, a process that, according to fluorescence quenching data, may be up to 95% efficient. Photocurrent measurements on films containing Zn-cyt c–SWNTs confirm that photoinduced charge transfer occurs from the ZnP to the SWNT, and additional work will address developing thin films with better charge transport characteristics. The straightforward and biologically inspired method of SWNT modification with photochemical reductant molecules reported here may present novel opportunities for the implementation of SWNTs in novel bio-nano assemblies for energy conversion.
Acknowledgements
This work is supported by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy, Grant No. DE-FG02-09ER16121.References
- D. M. Guldi, G. M. A. Rahman, M. Prato, N. Jux, S. Qin and W. Ford, Angew. Chem., Int. Ed., 2005, 44, 2015–2018 CrossRef CAS PubMed.
- T. Hasobe, Phys. Chem. Chem. Phys., 2010, 12, 44–57 RSC.
- E. Kymakis and G. A. J. Amaratunga, Appl. Phys. Lett., 2002, 80, 112–114 CrossRef CAS.
- J. Geng and T. Zeng, J. Am. Chem. Soc., 2006, 128, 16827–16833 CrossRef CAS PubMed.
- F. D'Souza, A. S. D. Sandanayaka and O. Ito, J. Phys. Chem. Lett., 2010, 1, 2586–2593 CrossRef.
- M. Alvaro, P. Atienzar, L. C. P. De, J. L. Delgado, V. Troiani, H. Garcia, F. Langa, A. Palkar and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6626–6635 CrossRef CAS PubMed.
- D. Baskaran, J. W. Mays, X. P. Zhang and M. S. Bratcher, J. Am. Chem. Soc., 2005, 127, 6916–6917 CrossRef CAS PubMed.
- F. D'Souza, S. K. Das, M. E. Zandler, A. S. D. Sandanayaka and O. Ito, J. Am. Chem. Soc., 2011, 133, 19922–19930 CrossRef PubMed.
- C. Ehli, G. M. A. Rahman, N. Jux, D. Balbinot, D. M. Guldi, F. Paolucci, M. Marcaccio, D. Paolucci, M. Melle-Franco, F. Zerbetto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222–11231 CrossRef CAS PubMed.
- D. M. Guldi, G. M. A. Rahman, S. Qin, M. Tchoul, W. T. Ford, M. Marcaccio, D. Paolucci, F. Paolucci, S. Campidelli and M. Prato, Chem. – Eur. J., 2006, 12, 2152–2161 CrossRef CAS PubMed.
- C. Oelsner, M. A. Herrero, C. Ehli, M. Prato and D. M. Guldi, J. Am. Chem. Soc., 2011, 133, 18696–18706 CrossRef CAS PubMed.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853–1859 CrossRef CAS PubMed.
- J. L. Bahr and J. M. Tour, J. Mater. Chem., 2002, 12, 1952–1958 RSC.
- S. Niyogi, M. A. Hamon, H. Hu, B. Zhao, P. Bhowmik, R. Sen, M. E. Itkis and R. C. Haddon, Acc. Chem. Res., 2002, 35, 1105–1113 CrossRef CAS PubMed.
- D. M. Guldi, G. M. A. Rahman, N. Jux, N. Tagmatarchis and M. Prato, Angew. Chem., Int. Ed., 2004, 43, 5526–5530 CrossRef CAS PubMed.
- E. Maligaspe, A. S. D. Sandanayaka, T. Hasobe, O. Ito and F. D'Souza, J. Am. Chem. Soc., 2010, 132, 8158–8164 CrossRef CAS PubMed.
- E. Magner and G. McLendon, J. Phys. Chem., 1989, 93, 7130–7134 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- N. M. B. Cogan, C. J. Bowerman, L. J. Nogaj, B. L. Nilsson and T. D. Krauss, J. Phys. Chem. C, 2014, 118, 5935–5944 CAS.
- G. R. Dieckmann, A. B. Dalton, P. A. Johnson, J. Razal, J. Chen, G. M. Giordano, E. Munoz, I. H. Musselman, R. H. Baughman and R. K. Draper, J. Am. Chem. Soc., 2003, 125, 1770–1777 CrossRef CAS PubMed.
- K. Matsuura, T. Saito, T. Okazaki, S. Ohshima, M. Yumura and S. Iijima, Chem. Phys. Lett., 2006, 429, 497–502 CrossRef CAS.
- T. J. McDonald, D. Svedruzic, Y.-H. Kim, J. L. Blackburn, S. B. Zhang, P. W. King and M. J. Heben, Nano Lett., 2007, 7, 3528–3534 CrossRef CAS PubMed.
- V. Zorbas, A. L. Smith, H. Xie, A. Ortiz-Acevedo, A. B. Dalton, G. R. Dieckmann, R. K. Draper, R. H. Baughman and I. H. Musselman, J. Am. Chem. Soc., 2005, 127, 12323–12328 CrossRef CAS PubMed.
- W. B. Asher and K. L. Bren, Protein Sci., 2010, 19, 1830–1839 CrossRef CAS PubMed.
- W. B. Asher and K. L. Bren, Chem. Commun., 2012, 48, 8344–8346 RSC.
- J. G. Kleingardner and K. L. Bren, Metallomics, 2011, 3, 396–403 RSC.
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585–595 CrossRef CAS PubMed.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593–596 CrossRef PubMed.
- S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361–2366 CrossRef CAS PubMed.
- R. Saito, G. Dresselhaus and M. S. Dresselhaus, Physical Properties of Carbon Nanotubes, Imperial Press, 1998 Search PubMed.
- M. S. Dresselhaus, G. Dresselhaus and P. Avouris, Carbon Nanotubes: Synthesis, Structure, Properties, and Applications, Springer, 2001 Search PubMed.
- J. M. Vanderkooi, F. Adar and M. Erecinska, Eur. J. Biochem., 1976, 64, 381–387 CrossRef CAS PubMed.
- G. M. A. Rahman, D. M. Guldi, S. Campidelli and M. Prato, J. Mater. Chem., 2006, 16, 62–65 RSC.
- D. M. Guldi, G. N. A. Rahman, J. Ramey, M. Marcaccio, D. Paolucci, F. Paolucci, S. Qin, W. T. Ford, D. Balbinot, N. Jux, N. Tagmatarchis and M. Prato, Chem. Commun., 2004, 2034–2035 RSC.
- E. Stellwagen, Biochemistry, 1968, 7, 2893–2898 CrossRef CAS PubMed.
- V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge, R. E. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379–1382 CrossRef CAS.
- C. Roquelet, J.-S. Lauret, V. Alain-Rizzo, C. Voisin, R. Fleurier, M. Delarue, D. Garrot, A. Loiseau, P. Roussignol, J. A. Delaire and E. Deleporte, ChemPhysChem, 2010, 11, 1667–1672 CrossRef CAS PubMed.
- R. K. Iler, J. Colloid Interface Sci., 1966, 21, 569–594 CrossRef CAS.
- A. A. Ensign, I. Jo, I. Yildirim, T. D. Krauss and K. L. Bren, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10779–10784 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nh00172f |
| This journal is © The Royal Society of Chemistry 2017 |