
Multi-metal-dependent nucleic acid enzymes
Wenhu
Zhou
*a and
Juewen
Liu
 *b
*b
aXiangya School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410013, China. E-mail: zhouwenhuyaoji@163.com
bDepartment of Chemistry, Water Institute, and Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada. E-mail: liujw@uwaterloo.ca
First published on 24th October 2017
Abstract
Nucleic acid enzymes (NAEs) are catalytically active RNA and DNA molecules. NAEs with RNA-cleaving activity are most extensively studied for applications in analytical chemistry, gene therapy and nanotechnology. Most NAEs require metal ions for activity. From a biochemical standpoint, these NAEs are reminiscent of metalloprotein enzymes with metal binding sites. While most NAEs require a single metal for the reaction, more and more recent examples have emerged that use two or even three metals for the reaction. The metal binding profile is sharper for these NAEs if they use the same metal ion due to cooperativity. Detailed studies have indicated examples of lanthanide and Ca2+ binding DNAzymes, where the metals interact with the non-bridging oxygen atoms in the scissile phosphate, and these DNAzymes often have a very strong thio effect that cannot be rescued by adding thiophilic metals. Another type uses multiple different metals, where one metal interacts with the scissile phosphate and the other binds to the catalytic loop for allosteric interactions. Such allosteric NAEs can also be obtained via rational design or intentional selection based on existing NAEs. These multi-metal NAEs might be useful as logic gates with metal ions as inputs. In this article, we review different types of NAEs based on their use of metal ions. The NAEs reviewed include ribozymes, DNAzymes and rationally designed aptazymes. Finally, their emerging applications are discussed, and some future research opportunities are proposed.
 Wenhu Zhou | Dr Wenhu Zhou received his BS (2010) and PhD (2016) degrees in Xiangya School of Pharmaceutical Science from Central South University (CSU), China. From 2014 to 2016, he worked at Prof. Juewen Liu's lab in the University of Waterloo as a visiting PhD student. He is currently a Professor at CSU and his research activities are focused on functional DNA for biomedical and pharmaceutical applications, and nanomaterial-based drug delivery. |
 Juewen Liu | Prof. Juewen Liu received his BS degree from the University of Science & Technology of China in 2000, and PhD degree from the University of Illinois at Urbana-Champaign in 2005. He is currently a Full Professor of Chemistry at the University of Waterloo, and holds a University Research Chair position. He received the Fred Beamish Award from the Canadian Society for Chemistry in 2014 for his contribution in bioanalytical chemistry. He is an Associate Editor for Analytical Methods and is on the editorial advisory board of Langmuir. He is an expert on functional nucleic acids chemistry, in vitro selection, bioanalytical chemistry, and biointerface chemistry. He has published over 180 papers, receiving over 14 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations.
000 citations.1. Introduction
It has been long thought that all enzymes are proteins. To exert catalytic functions, over 40% of enzymes recruit metal ions for catalysis, and these are called metalloenzymes. To elucidate the reaction mechanism, efforts have been devoted to studying metal binding sites and their role in catalysis.1 Interestingly, some metalloenzymes require multiple metal ions, of either the same or different metals. For example, both Cu2+ and Zn2+ are contained in superoxide dismutase (SOD).2 The iron–sulfur clusters found in many redox enzymes contain multiple iron ions.3The scope of enzymes has significantly broadened since the discovery of ribozymes (RNA-based enzymes) in the early 1980s.4 Later, in 1994, the first DNA-based enzyme (deoxyribozyme or DNAzyme) was also isolated by Breaker and Joyce through a combinatorial process called in vitro selection.5 This standard process has been used by many labs to isolate new ribozymes and DNAzymes, which are collectively termed nucleic acid enzymes (NAEs). So far, numerous NAEs have been discovered to catalyze a wide range of chemical reactions.6–8 Among them, RNA-cleaving NAEs have been the most extensively studied.
An interesting property of NAEs is their metal-dependent activity. By properly setting the selection conditions, DNAzymes specific for many metal ions have been obtained for biosensor applications, such as Pb2+,5 Zn2+,9,10 Cu2+,11 Cd2+,12 UO22+,13 Hg2+,14 Ca2+,15 Na+,16,17 Ag+,18 and lanthanides.19–21 In addition, these enzymes have been used as metal-dependent switches and actuators.22–25
The role of metal ions in NAEs has been extensively studied.26–29 While many NAEs require only one metal ion for catalysis, in the past few years, quite a few DNAzymes have been reported to require multiple metal ions. The multiple metals can work cooperatively, independently, or allosterically, showing the versatility of nucleic acids in utilization of metals. Compared to single-metal-binding NAEs, multi-metal-binding ones may have sharper binding curves and are useful as switches or as logic gates with metal ions as inputs. These NAEs also provide interesting scaffolds for understanding metal binding by nucleic acids. In this article, we review NAEs that require multiple metal ions for catalysis, present known mechanistic studies, and discuss their potential applications. Finally, some future research opportunities are speculated. This topic has never been reviewed previously, although scattered examples exist in papers describing DNAzymes in general.
2. Metal binding sites in nucleic acids
Before presenting examples of multi-metal binding NAEs, we first briefly introduce metal binding sites in nucleic acids. Nucleic acids are a polymer of nucleotides, which contain a (deoxy)ribose, a nucleobase and a phosphate (Fig. 1A). Each phosphate carries a negative charge (pKa below 2), while all the bases are neutral. The five types of natural nucleobases are shown in Fig. 1B. The interactions between nucleic acids and metal ions have been extensively studied.30–33 At the simplest level, nucleic acids require metal ions for charge screening to form and stabilize their active structures.34 Group 1A and 2B metals (e.g. Na+ and Mg2+) are often used for this purpose, and these metals are often treated as diffusive charges around the phosphate backbone (Fig. 1C). Metals can also interact with DNA through their bridging water to form outer-sphere binding (Fig. 1D), or through direct coordination for strong inner-sphere interactions (Fig. 1E).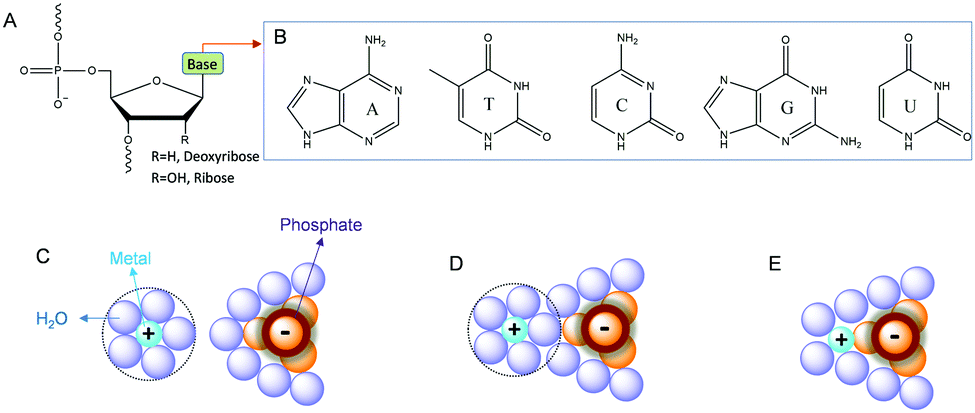 | ||
| Fig. 1 (A) Chemical structure of a nucleotide in nucleic acids. (B) Structures of the five types of natural nucleobases. Interactions of metal ions with phosphate through (C) electrostatic attraction, (D) outer-sphere binding and (E) inner-sphere coordination.28 | ||
The properties of metal ions, such as size, charge, coordination geometries, pKa of metal bound water, ligand exchange rate and thiophilicity, can strongly influence their binding to nucleic acids. For example, some metals with a high charge density and low polarizability (e.g. hard metals like Na+, K+, Mg2+, and Ca2+) prefer to interact with the negatively charged phosphate backbone via non-specific diffusive binding.33 In a few cases, specific interactions were also reported such as G-quadruplexes for K+ binding,35 and recently a Na+-specific aptamer.36,37 Soft metals such as Hg2+ and Ag+ mainly bind to nucleobases, while some other metals such as trivalent lanthanides and Pb2+ can bind both phosphate and nucleobases. A few metal ions are able to mediate DNA base pair formation, such as T–Hg2+–T,38 and C–Ag+–C,39 while Pt2+ can strongly bridge two guanine bases.40 For RNA, the 2′-hydroxyl group (2′-OH) is also an important site for metal binding. It can act as both a general acid and a general base (after deprotonation), which may explain RNA's versatility in metal recognition.41,42 In several NAEs, the 2′-OH at the cleavage site directly participates in metal binding (vide infra).
3. Roles of metals in NAEs
Metal binding to DNA in general stabilizes the DNA duplex, although a high concentration of transition metals can also destabilize DNA, since they bind to DNA bases.43 To better understand the role of metal ions in NAEs, we briefly describe the mechanism of the RNA-cleavage reaction, which is the most studied reaction for NAEs. A typical cleavage junction is presented with a chimeric structure of 5′-rAG (a ribo-adenine and a deoxyribo-guanine) (Fig. 2A). Compared to DNA, RNA has an extra 2′-OH group, which makes it ∼106-fold less stable than DNA.44 The 2′-OH acts as an internal nucleophile to attack the scissile phosphate, forming a highly negatively charged penta-coordinated transition state. Then, the 5′-OH leaves the intermediate to complete the cleavage reaction, and a 2′,3′-cyclic phosphate is formed as the other product.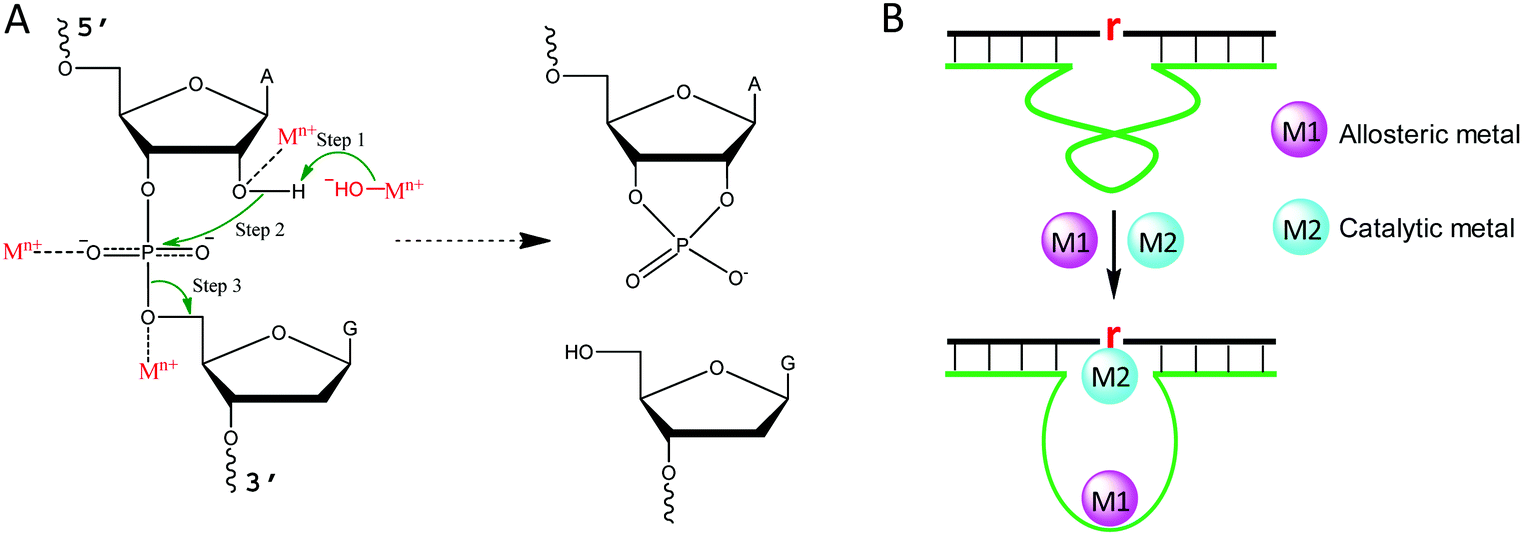 | ||
| Fig. 2 (A) The general mechanism of the RNA-cleavage reaction, and the possible roles of metal ions in catalysis. The 2′-OH group acts as an internal nucleophile to attack the phosphorus center. To facilitate this reaction, a metal hydroxide may function as a general base or a metal cation may act as a Lewis acid to facilitate deprotonation of the 2′-OH. In addition, the penta-coordinated intermediate can be stabilized by metals by binding to the non-bridging oxygens or 5′-leaving oxygen. Finally, the 5′-OH departs from the transition state to finish the reaction, forming a 2′,3′-cyclic phosphate, and metal species may also assist this step. For different NAEs, metal ions may take one or more of the above roles. (B) An example of a NAE with multiple metal binding sites, where the first metal (M1) plays an allosteric role, while the second metal (M2) interacts with the scissile phosphate as described in (A). The lower case “r” denotes the scissile RNA linkage. | ||
For each step described above, metal ions can participate in and accelerate the reaction. For example, a metal bound hydroxyl can act as a general base to deprotonate the 2′-OH, making it a stronger nucleophile.45 For this role, a water bound metal with a lower pKa tends to have better activity, which was reported for the 8–17 and 10–23 DNAzymes.46,47 In addition, a metal can be a Lewis acid to bind the 2′-OH through inner-sphere coordination, thereby lowering its pKa and accelerating its deprotonation (Fig. 2A).48 A crystallographic study on leadzyme provided direct evidence of a lead-bound hydroxide ion [Pb(OH)+].49 It needs to be noted though, that most ribozymes use a guanine to activate the 2′-OH.27
Another important role of metal ions is to stabilize the scissile phosphorane transition state (Fig. 2A). For example, the activity of the hammerhead ribozyme is proportional to the phosphate binding affinity of the corresponding metal.50 We recently isolated a series of lanthanide-dependent DNAzymes, and metals at a concentration of a few μM are enough to achieve saturated activity.12,19,21,51 This is because lanthanides can bind phosphate oxygen very tightly. We also reported an interesting DNAzyme called EtNa.15,52 It requires ∼1 M Na+ but only ∼1 mM Ca2+, which can be explained by the stronger Ca2+ binding affinity of the phosphate. Finally, metal ions may play allosteric roles in NAEs that indirectly regulate activity. In this case, NAEs undergo a conformational change upon metal binding to form the active structure (Fig. 2B).
4. Representative NAEs binding one metal
Although this article is mainly about NAEs binding multiple metals, we start with some classic single-metal-binding ones to facilitate our discussion (Fig. 3). Each trans-cleaving NAE contains a substrate and an enzyme strand. The substrate hybridizes to the enzyme via two base paired regions. The substrate can be a RNA/DNA chimera containing a single RNA linkage, or it can be a full RNA.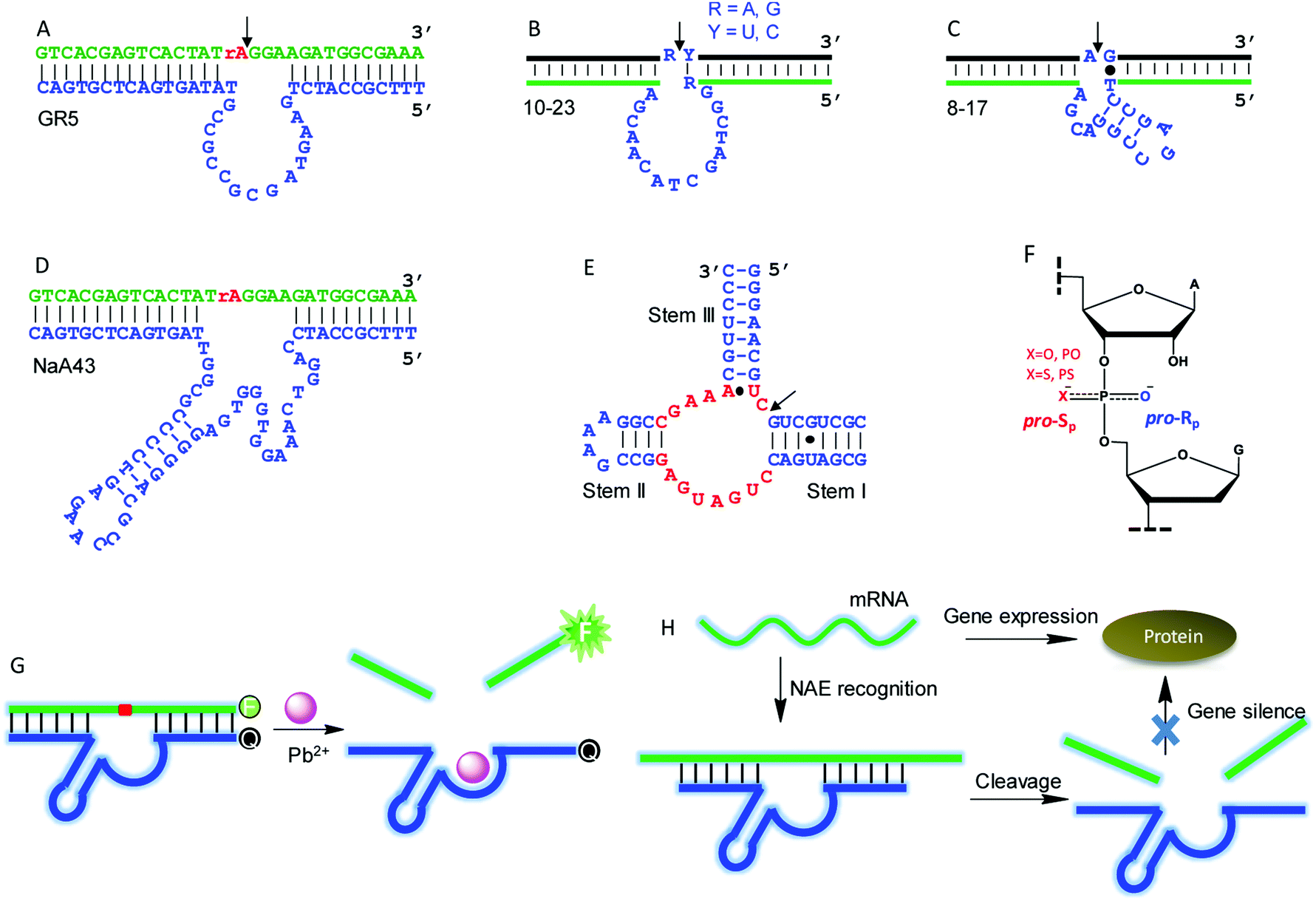 | ||
| Fig. 3 (A–E) The secondary structures of several representative single-metal binding NAEs: (A) GR5; (B) 10–23; (C) 8–17; (D) NaA43; (E) HHRz. The conserved nucleotides in (E) are highlighted in red. (F) The structure of a normal phosphodiester (PO) and a phosphorothioate (PS) linkage. The pro-Rp and pro-Sp positions are shown. (G) A typical NAE-based catalytic beacon for metal detection using the 8–17 DNAzyme as an example. (H) A scheme showing mRNA recognition and cleavage by NAEs for gene silence. | ||
GR5 was the first reported DNAzyme (Fig. 3A), and it requires Pb2+ for activity. It cleaves only chimeric substrates (not full RNA) with a high activity.5 With 10 μM Pb2+, GR5 can reach a rate >10 min−1.53 In 1997, the 10–23 (Fig. 3B) and 8–17 (Fig. 3C) DNAzymes were isolated to cleave full-RNA substrates.54 The 10–23 DNAzyme has a wider substrate range (cleaves all purine–pyrimidine junctions), and thus it has been studied more for intracellular RNA cleavage.55–58 The 8–17 DNAzyme was found to be highly active with Pb2+, and it has been used as a model system to develop biosensors for Pb2+.59–61 In 2015, Lu and coworkers isolated the NaA43 DNAzyme (Fig. 3D), which is highly specific for Na+, reaching a rate of 0.1 min−1 in the presence of 400 mM Na+ alone.17 The hammerhead ribozyme (HHRz) (Fig. 3E) is a class II ribozyme that contains 15 highly conserved nucleotides flanked by three duplex stems (I to III).62,63 HHRz in general requires divalent metal ions, although >1 M monovalent cations can also activate it.64
A commonly used technique to probe metal binding is to replace one of the non-bridging oxygens in the scissile phosphate with sulfur (i.e. phosphorothioate or PS modification, Fig. 3F). If a PS shifts the metal preference from hard to soft, metal binding to the scissile phosphate might be important. An interesting example is the lanthanide-dependent Ce13d DNAzyme. After PS modification, Ce13d works better with Cd2+ and Pb2+.61 Each PS modification produces two diastereomers, named Rp and Sp (Fig. 3F). In general, the Sp-PS modified DNAzyme is still quite active (within 5-fold), while the Rp-PS modified DNAzyme is strongly inhibited (∼100-fold or more). Therefore, the pro-Rp oxygen of the scissile phosphate is responsible for metal binding in most NAEs such as the 10–23 DNAzyme,65 HHRz,66 HDV ribozyme,67 and RNase P.68 Studying metal binding with nucleobases is a more difficult task, and a recent review has summarized the current biochemical and biophysical methods to explore such interactions.29 To explore important nucleobases that may directly participate in the chemical reaction or metal binding, mutation studies are a good starting point. They are performed by changing each nucleotide in the NAE core to the other three, and the activity of each mutant is measured. A nucleotide is regarded to be highly important if such mutations cannot be made. This technique has been widely used to study different DNAzymes.52,69–71
NAEs have many important applications, and the most straightforward one is metal sensing. This was first demonstrated by the Lu group, who converted the 8–17 DNAzyme into a catalytic beacon for Pb2+ detection (Fig. 3G).59 They labeled a fluorophore at one end of the substrate, and a quencher at the corresponding end of the enzyme with initially quenched fluorescence. Pb2+ activates the enzyme for substrate cleavage to produce a signal, and the rate of fluorescence increase was used for Pb2+ quantification. Later, various design strategies have been developed to improve the beacon performance.72–75 NAEs have also been used as gene therapy tools for intracellular mRNA cleavage. They can be designed to recognize and cleave a specific mRNA sequence, thus silencing gene expression (Fig. 3H). So far, NAE-based anti-cancer, anti-viral, anti-bacterial and anti-asthma studies have been performed,55,76,77 although concerns have been expressed regarding the lack of a sufficient metal concentration in cells leading to low NAE activity.78
5. Ribozymes using multiple metal ions
After introducing NAEs that bind one metal, we continue our discussion on multiple-metal-dependent ones and start with examples from natural ribozymes. Group I introns are large ribozymes that catalyze self-splicing through two sequential phosphoryl-transfer reactions. First, the 5′-oxygen of an exogenous guanosine nucleotide acts as a nucleophile. The guanosine attaches to the intron, also creating a 3′-OH on the 5′-exon. Then the newly formed terminal 3′-oxygen of the exon attacks the 3′-intron-exon junction to finish the splicing.79–81The group I introns require Mg2+ or other divalent metals for catalysis. In 2005, Strobel and co-worker reported a 3.4 Å crystal structure of a catalytically active group I intron splicing intermediate, in which two Mg2+ ions are positioned 3.4 Å apart and directly involved in the exon ligation reaction (Fig. 4A).82 Specifically, M1 directly coordinates to the 3′-oxygen nucleophile of U-1, and the pro-Rp oxygen of the scissile phosphate, while M2 makes inner-sphere contacts with the pro-Rp oxygen, 2′-OH and 3′-oxygen leaving group of the ωG. This crystal structure study offers a vivid picture of a two-metal binding model in a group I intron.
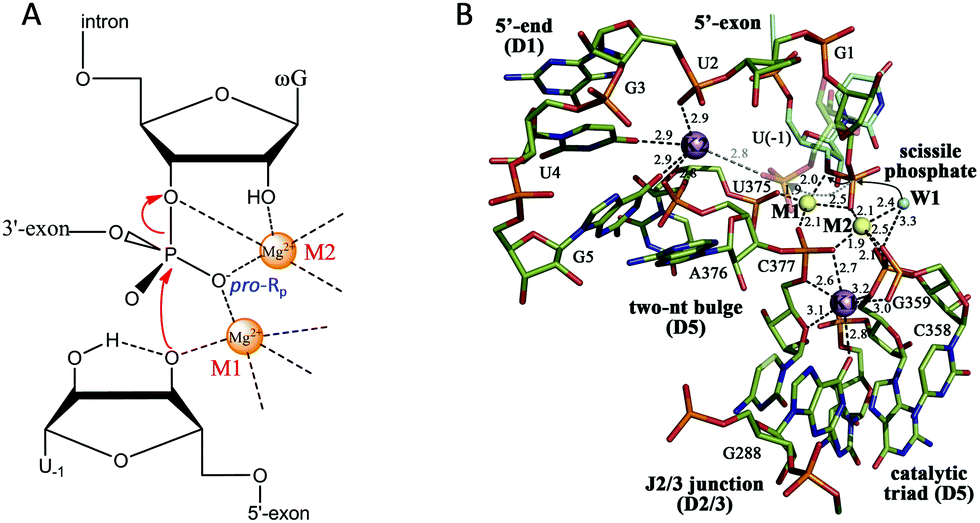 | ||
| Fig. 4 (A) The proposed two-metal mechanism in the active site of the group I intron during the second step of splicing. Note that the Mg2+ ions also coordinate with phosphate at other sites (not shown). (B) The structure responsible for the splicing step of a group II intron. Two Mg2+ ions (M1 and M2, yellow spheres) and two K+ ions (purple spheres) coordinate the nucleotides involved in catalysis. The water molecule (W1, cyan sphere) is in an ideal position for nucleophilic attack on the scissile phosphate (black arrows). Upon hydrolysis, the scissile phosphate leaves the active site (semi-transparent sticks and gray dotted arrow) to interact with K2 electrostatically (gray dotted line). Adapted from ref. 91 with permission. Copyright 2014 Elsevier Science. | ||
A study on the first step of the cleavage showed that substitution of the 3′-oxygen of the leaving group with sulphur resulted in a significant activity decrease in the presence of Mg2+, and this can be rescued by more thiophilic metals such as Mn2+ and Zn2+.83 This result indicates the interaction between the metals and the 3′-oxygen in the leaving group. Based on microscopic reversibility, the 3′-oxygen nucleophile is also a metal binding site during the following splicing step. Cech and coworker used 3′-(thioinosylyl)-(3′ → 5′)-uridine, IspU, as a substrate to emulate the exon ligation reaction, which provided evidence that the leaving group binds the metal as well for structure stabilization, supporting the model of a two-metal binding mechanism for the second step reaction.84
Group II introns also self-catalyze two consecutive phosphoryl-transfer reactions for splicing. Instead of using an exogenous guanosine, group II introns initiate the first step reaction using either an endogenous 2′-OH group or a water molecule as a nucleophile to attack the 5′-exon junction, yielding a lariat or linear intermediate, respectively. The second step proceeds the same as that for group I introns, using the 3′-OH from the previous step to attack the 3′-exon junction. For both steps, divalent metals such as Mg2+ or Mn2+ are essential cofactors.85 From PS substitution experiments, the importance of metal coordination to the 3′-leaving group was demonstrated for stabilization of the charge accumulation.86–88 Interestingly, the loss of activity after the Rp-PS modification could not be rescued by thiophilic metals, suggesting a special coordination mode.89 In addition to biochemical evidence, X-ray crystallography also indicated that group II introns employ at least two metals for the second step of the splicing reaction, in which the metal binding sites and the binding patterns are quite similar to that in group I introns.90
Pyle and co-workers performed a comprehensive crystallographic study on a group II intron through a set of 14 crystal structures at different stages of the catalysis.91 In this work, they identified a heteronuclear four-metal catalytic center, including two essential Mg2+ ions and two K+ ions (Fig. 4B). Both Mg2+ ions coordinate to the pro-Rp oxygen atom at the scissile phosphate. In addition, one of the Mg2+ ions also coordinates to the attacking aqua ligand, whereas the other binds the 3′-OH for stabilization. The two K+ ions impart additional electrostatic modulation and structural stabilization at the active site. The first K+ might modulate the conformational evolution between the first and the second splicing steps, while the second one plays a dual-role ensuring a correct arrangement of the 5′ splice junction and stabilizing the state after the first step of splicing.
Leadzyme is a small ribozyme that was initially selected from a library of yeast tRNAs with 9–10 randomized positions using Pb2+ as a cofactor.92 Different from many other ribozymes, it is quite specific for Pb2+.93 The crystal structure shows the role of Pb2+ for scissile 2′-OH binding to activate the enzyme.49 Later, Sugimoto and Ohmichi discovered that its activity can be enhanced by combining Pb2+ with a lanthanide (Ln3+) such as Nd3+, suggesting multiple metal binding sites in the enzyme.94 The Ln3+ is believed to bind the phosphate oxygen, while Pb2+ is believed to deprotonate the 2′-OH.95
6. NAEs using multiple metals from in vitro selection
While ribozymes exist in nature, all of the DNAzymes reported so far are from in vitro selection. This is not surprising since most DNAs in biological systems are double-stranded and thus cannot carry out the required catalytic functions. A representative cycle of selecting RNA-cleaving DNAzymes is shown in Fig. 5A. The library contains a single RNA linkage (ribo-adenosine, rA) serving as the potential cleavage site, and a 50-nucleotide random region is also included. The rest of the sequences are mainly for folding the library into a designed secondary structure and for PCR primer binding. If some of the sequences can be cleaved at the rA site by the added metal (step 1), the cleaved shorter products are then separated using gel electrophoresis (step 2, PAGE) followed by two rounds of PCR amplification (steps 3 and 4) to regenerate the full-length library. This selection cycle is continued until the activity of the library reaches a plateau, and then the library is sequenced. Some selections intentionally used multiple different metal ions,96–98 while others did not. Some of the selected NAEs can bind multiple metals. | ||
| Fig. 5 (A) A scheme showing the typical steps of selecting RNA-cleaving DNAzymes: 1, metal induced cleavage; 2, PAGE-based separation; 3, polymerase chain reaction 1 (PCR1) to produce a full-length library; 4, PCR2 to introduce an rA cleavage site and FAM fluorophore label. F denotes a fluorophore, and rA is the RNA linkage as the cleavage site. (B) A scheme of allosteric ribozyme selection with four main steps: 1, library generation by in vitro transcription; 2, negative selection; 3, positive selection; 4, reverse transcription (RT)-PCR to amplify the active allosteric HHRz sequences. | ||
Another way of selecting multiple-metal binding NAEs is by appending a randomized region to an existing NAE, so that the binding of the target metal allosterically activates the enzyme.99,100 Using this method, the obtained NAEs have an allosteric metal binding domain (for the metal effector) and a catalytic domain (for the metal cofactor). Using the HHRz as an example, the library was constructed by replacing stem II of HHRz with a domain of 40 nt random sequence (Fig. 5B, step 1). Then negative selections with Mg2+ only (step 2), and positive selections with also a target metal (step 3) were alternated. Sequences cleaved in the negative selections were discarded, while those cleaved in the positive selections were harvested and amplified. Using this method, Breaker and coworkers selected a series of allosteric HHRzs with transition metals as effectors, such as Cd2+, Co2+, Mn2+, Ni2+ and Zn2+.100 Interestingly, some of these allosteric HHRzs even bind two metals in the allosteric domain, making the total number of metals needed for the reaction three.
6.1 Multiple metal ions binding to the scissile phosphate
As introduced above regarding the reaction mechanism, the scissile phosphate is one of the major metal binding sites for RNA-cleaving NAEs. Metal binding can stabilize the phosphorane transition state, thus facilitating the subsequent cleavage reaction. Even without the enzyme, some metals can still non-specifically hydrolyze RNA, due to their high affinity towards phosphate. For example, low mM Ln3+ such as Tm3+, Yb3+ and Lu3+ fully hydrolyzes RNA under physiological conditions within a few minutes.101 It was proposed that two metals were involved in the pentacoordinated transition state to alleviate the negative charge density at the phosphorus center. Inspired by this, the Liu lab performed lanthanide-dependent selections for new RNA-cleaving DNAzymes. The DNAzymes work optimally with low μM Ln3+.19–21,51,102Tm7 is a representative lanthanide-dependent DNAzyme (Fig. 6A), and it achieves a cleavage rate of 1.6 min−1 with 10 μM Er3+ at pH 7.8.20 Further study on the cleavage rate as a function of Er3+ concentration showed a sigmoidal curve suggesting multiple metal binding (Fig. 6B). The double log plot of the cleavage rate versus Er3+ concentration (insert of Fig. 6B) had an initial slope of 2.7, indicating that three Er3+ ions were involved in the reaction. After a PS modification at the cleavage junction, the Tm7 dropped its activity to the background level for both the Rp and Sp isomers, indicating the importance of metal binding to the scissile phosphate for catalysis. In addition, this activity loss cannot be rescued by thiophilic Cd2+, or a mixture of Er3+ and Cd2+, suggesting that three Er3+ ions synergistically instead of independently bind to both non-bridging oxygen atoms. Based on all these observations, we proposed a three-metal binding mechanism for Tm7 catalysis (Fig. 6C). In this model, two of the Er3+ ions directly coordinate to both non-bridging oxygen atoms to stabilize the transition state, and the third one through its metal-bound water interacts with the 2′-OH for enzyme activation.
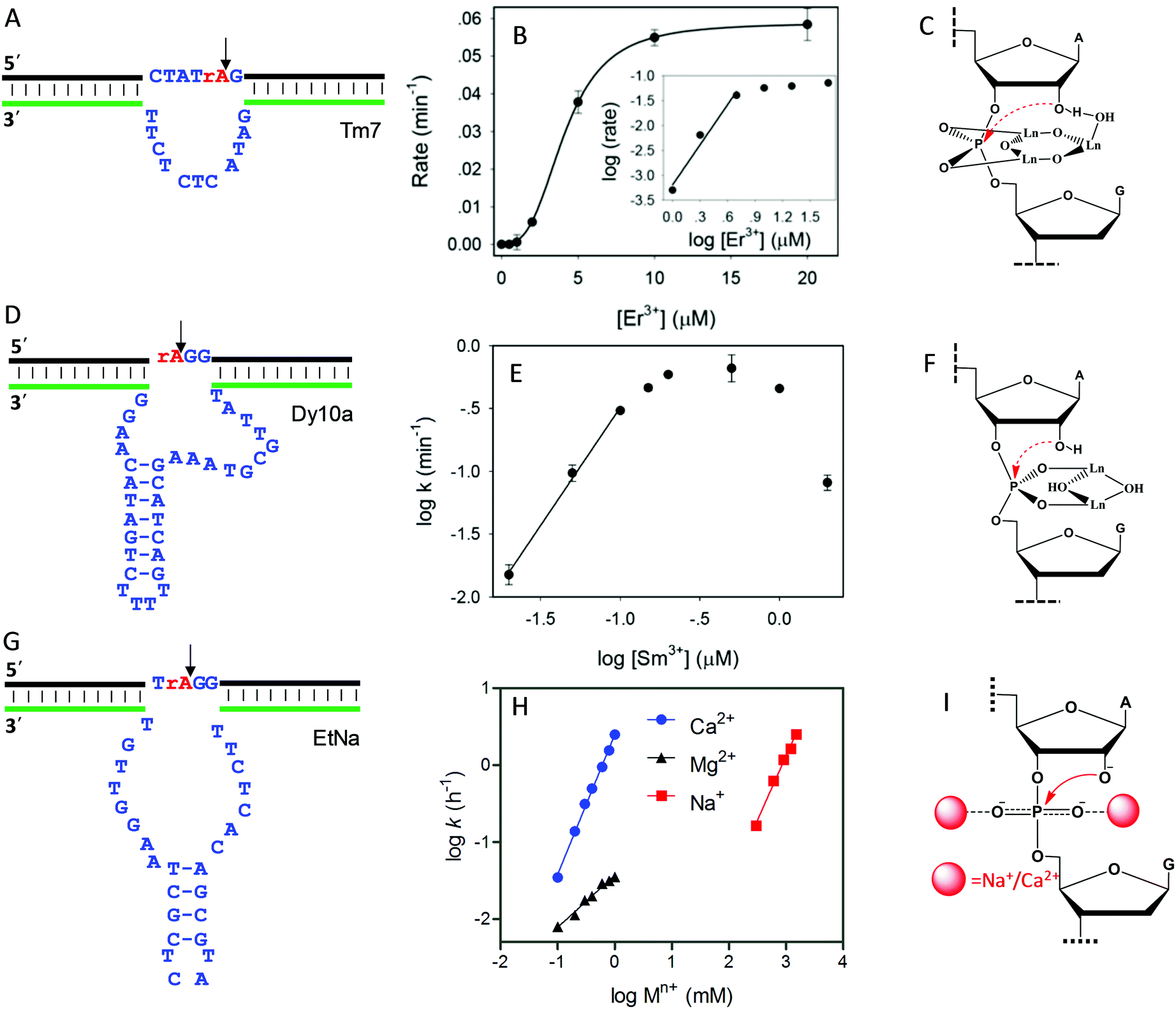 | ||
| Fig. 6 The secondary structures, the double log plots of cleavage rate against metal concentration, and the proposed metal binding mechanisms of the (A–C) Tm7, (D–F) Dy10a, and (G–I) EtNa DNAzymes. Panel (B) was from ref. 20, and (H) was re-drawn from ref. 15. (E) was adapted from ref. 21. With permission. Copyright 2016 American Chemical Society. | ||
Dy10a is another lanthanide-dependent DNAzyme selected using Dy3+ as a cofactor (Fig. 6D).21 It is among the most efficient lanthanide-dependent enzymes reported so far, achieving a rate of 0.6 min−1 at pH 6 with just 200 nM Sm3+. In addition, it is also one of the tightest metal binding DNAzymes, with an apparent dissociation constant (Kd) of 100 nM Sm3+. We also plotted the double logarithm data of initial rate against Sm3+ concentration, and a slope of 1.8 was obtained (Fig. 6E), suggesting two-metal binding. Both PS isomers decreased the rate by >1000-fold compared to the normal phosphate oxygen (PO) substrate. Therefore, both non-bridging oxygens contribute equally to metal binding. This strong thio effect was not rescued by the addition of Cd2+ either. Based on this, a two-metal binding mechanism of Dy10a was proposed in Fig. 6F, inspired from the RNA cleavage reaction of free Ln3+.101 Aside from these two, the other selected Ln3+-dependent DNAzymes only use one metal for catalysis.19,51,102
Besides lanthanide-dependent DNAzymes, we identified another one called EtNa (Fig. 6G) that requires the cooperative binding of two Na+ or two Ca2+ ions to the scissile phosphate for function.15,16,52 EtNa was initially isolated during the ethanol/isopropanol precipitation step of the selection process, which showed a large rate acceleration effect by several organic solvents.16 In aqueous solution, EtNa needs molar concentrations of Na+ to achieve substantial cleavage, while in 54% ethanol, it reached a rate of 2.0 h−1 in the presence of 120 mM Na+ alone, with a Kd of 21 mM Na+. Its Na+ binding mechanism was then carefully studied and compared with other Na+-dependent DNAzymes.52 While it is difficult to achieve saturated binding with Na+ in water, EtNa showed cooperative binding with two Na+ ions during catalysis, as demonstrated by its [Na+]-dependent rate profile (Fig. 6H, red trace, with a slope of 1.7 for the double logarithm plotting). PS modification probes revealed that both non-bridging oxygen atoms are involved in Na+ binding, and it is probable that each oxygen atom is responsible for one Na+ ion (Fig. 6I).
While the affinity of EtNa is quite low for Na+ in water, EtNa can be activated by divalent Ca2+ with a much higher affinity (low mM Ca2+), which is likely due to the higher binding affinity of Ca2+ with phosphate.15 Using Ca2+ as a metal cofactor, EtNa shares quite similar biochemical properties to those obtained in the presence of Na+, such as two metal binding (Fig. 6H, blue trace, the plot of the logarithm of the rate against the logarithm of the Ca2+ concentration gives a slope of 1.8), and an unrescuable thio effect. Therefore, we reason that Na+ and Ca2+ share the same binding mechanism (Fig. 6I).
Compared to typical one-metal binding DNAzymes, the metal-dependent response curves of these DNAzymes are sharper, and thus they might be useful as switches in DNA nanotechnology. An interesting observation is that the EtNa DNAzyme has excellent selectivity for Ca2+ over Mg2+ (around 90-fold). A careful study showed that EtNa binds only one Mg2+ (Fig. 6H, black trace, the double logarithm plot displaying a slope of 0.7), and thus Ca2+ and Mg2+ use completely different mechanisms for activating the DNAzyme. The best previously reported Ca2+-dependent DNAzyme was a mutant of the 8–17 DNAzyme,103 but it had only around 10-fold selectivity. We rationalized this by only one Ca2+ or Mg2+ being used by that DNAzyme. Therefore, the difference between metal ions might be amplified by using multiple of them. We need to emphasize that although the phosphate interaction is believed to be the major factor, other nucleotides must also be involved so that high metal specificity can be explained. These multi-metal binding DNAzymes indicate different ways of metal ions interacting with nucleic acids.
6.2 Metal ions binding to both the phosphate and catalytic loop
The DNAzymes in Section 6.1 use the same metal for cooperative binding to the scissile phosphate. We now describe examples that require different metal ions working at distinct sites. For example, one metal binds to the scissile phosphate to facilitate the cleavage reaction, and the other interacts with nucleobases in the catalytic core. These are similar to aptazymes, although they might not be intentionally selected for this purpose.An interesting example is the Ce13d DNAzyme (Fig. 7A). Ce13d was originally isolated using a lanthanide as the metal cofactor, and it is highly active with all of the Ln3+ and Y3+ ions, reaching a cleavage rate of 0.25 min−1 with 10 μM Ce3+ at pH 6.19 It was converted to a DNAzyme beacon for Ln3+ detection, showing a detection limit down to 1.7 nM Ce3+. By introducing a PS modification at the scissile phosphate, Ce13d shifts its activity from being dependent on Ln3+ to soft thiophilic metals such as Cu2+, Cd2+, Hg2+ and Pb2+ (Fig. 7B), suggesting a metal binding site at the phosphate group.61 Based on this observation, a systematic study of Ce13d was further performed by using a PO substrate (with Ce3+) and a PS substrate (with Cd2+), respectively.70 For both systems, Ce13d showed quite similar biochemical properties, such as metal-dependent activity, pH-rate profile and mutation studies, suggesting that the main role of the metal is to neutralize the negative charge on the phosphate.
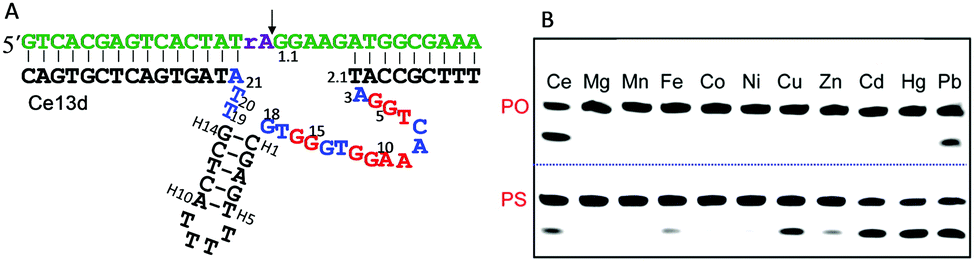 | ||
| Fig. 7 (A) The secondary structure of the Ce13d DNAzyme. The nucleotides in red in the catalytic core are highly conserved for activity.70 (B) A gel image showing the cleavage of Ce13d in the presence of 10 μM different metal ions with either the PO or the PS substrate after 30 min of reaction. Figure adapted from ref. 61 with permission. Copyright 2014 American Chemical Society. | ||
Later, it was discovered that the sequences of Ce13d and the Na+-specific NaA43 DNAzyme have a high degree of similarity with a continuous stretch of 16 nucleotides being identical (Fig. 3C).17 Therefore, Ce13d might contain a Na+ aptamer.104,105 To unveil their relationship, we performed a comprehensive biochemical study, and found that Ce13d was active only in the presence of both Ce3+ and Na+.105 Its cleavage rate depends linearly on the concentration of each metal, and the DNAzyme requires one of each metal.
To understand the reason for Na+ specificity, we first used Tb3+-sensitized luminescence to study the global folding of Ce13d.36,105 Free Tb3+ ions have poor luminescence due to a very small absorption cross-section, while after binding to DNA (especially to guanine), its emission significantly enhanced because of energy transfer from DNA to Tb3+. Compared with other monovalent metal ions, Na+ can efficiently displace Tb3+ from Ce13d (Fig. 8A), thus decreasing the luminescence (Fig. 8B). This study suggests specific Na+ binding by the DNAzyme.
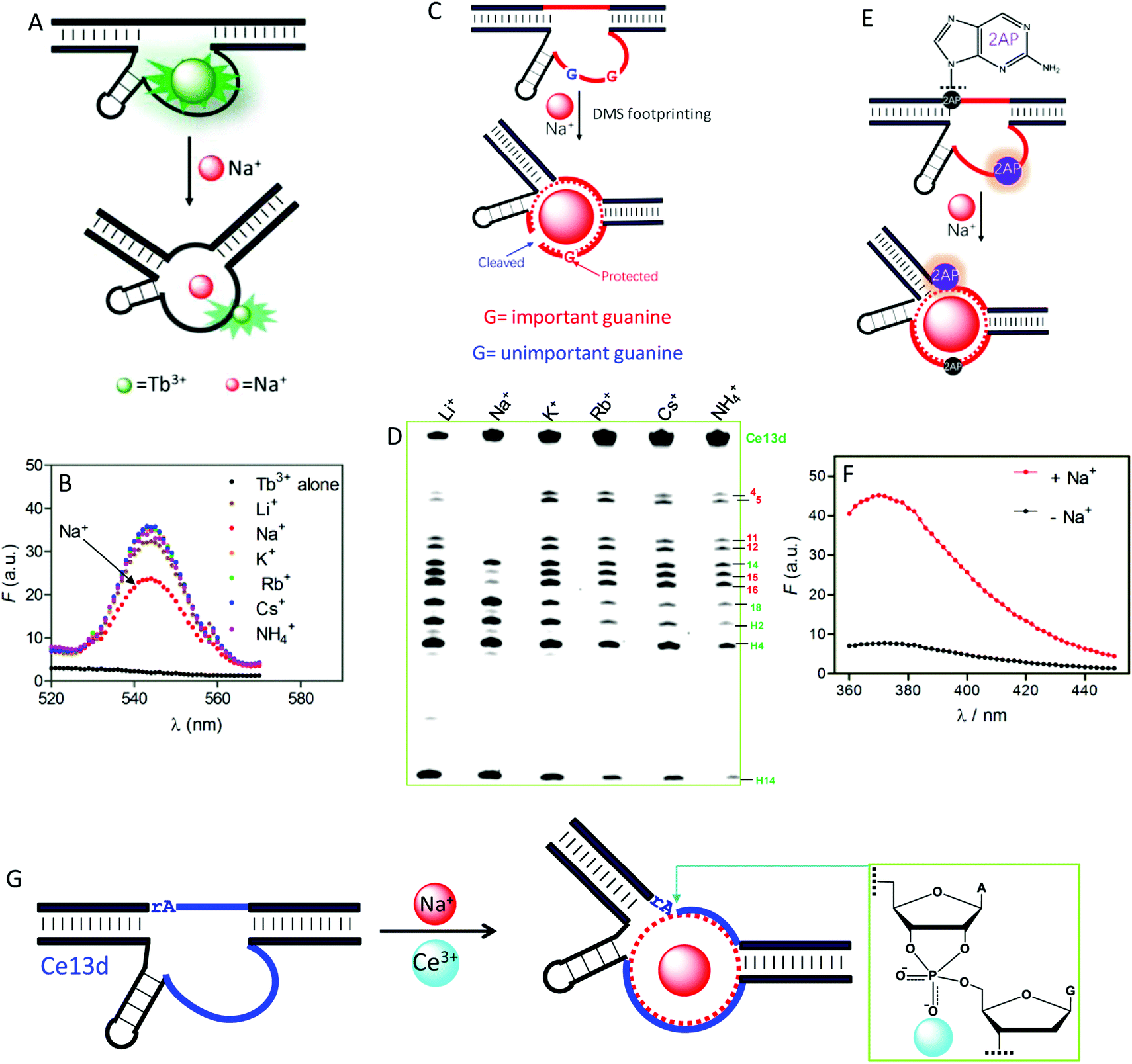 | ||
| Fig. 8 (A) A scheme showing Na+ displacement of Tb3+ from Ce13d, resulting in luminescence decrease. (B) Sensitized Tb3+ luminescence spectra (5 μM) and after mixing with 5 μM Ce13d in the presence of a 25 mM concentration of various monovalent metal ions.105 (C) A carton showing Na+-induced folding protecting a conserved guanine (in red) from cleavage under DMS footprinting conditions, while the unimportant guanine (in blue) is cleaved. (D) A DMS footprinting gel of Ce13d with a 600 mM concentration of various monovalent metal ions. The numbering of each band corresponds to the guanines in Fig. 7A.105 (E) A scheme showing Na+-induced Ce13d folding and 2AP fluorescence change. Na+ binding enhances the 2AP fluorescence at the A1.1 position (relaxed base stacking) and quenches the 2AP emission at the A8 position (enhanced base stacking). (F) Fluorescence spectra of an optimized Ce13d mutant with a 2AP-modified substrate (at A1.1 site) before and after adding 100 mM Na+.37 (G) Proposed metal binding mechanism of Ce13d. Ce13d binds Na+ through its aptamer motif to fold the enzyme into an active configuration, and Ce3+ interacts with the scissile phosphate to stabilize the phosphorane transition state for catalysis. Reprinted with permission from ref. 52. Copyright 2017 John Wiley & Sons, Inc. | ||
Dimethyl sulfate (DMS) footprinting was utilized to probe the important guanines in the enzyme loop of Ce13d for Na+ binding.105 DMS methylates guanines at the N7 site, leading to subsequent cleavage by piperidine. The relative cleavage yield at each guanosine site reflects its accessibility to the DMS probe. Na+ binding indeed protects conserved guanines in the enzyme loop from cleavage under footprinting conditions (Fig. 8C), resulting in reduced band intensity in the PAGE gel (Fig. 8D). Based on this result, the guanines that participate in Na+ binding were proposed.
We further exploited 2-aminopurine (2AP), a fluorescent adenine analog, to study the Ce13d local folding in response to Na+.37 Its fluorescence is sensitive to the local base stacking environment, and thus is useful for probing the DNA local folding. By introducing a 2AP at the substrate cleavage site, we observed a strong fluorescence increase upon addition of Na+ (Fig. 8F). However, when an unessential adenine in the enzyme strand was replaced by 2AP, Na+-dependent folding quenched the fluorescence. These results provide clear evidence of the Na+ binding information in Ce13d (Fig. 8E). Finally, the Na+-dependent folding was studied by using fluorescence resonance energy transfer (FRET).106
All of the above characterizations revealed a well-defined Na+ aptamer in Ce13d with a binding affinity of 20–50 mM Na+, based on the probing technique and mutation. Therefore, Ce13d is an aptazyme:Na+ binding is fulfilled by the aptamer motif to achieve allosteric regulation, while Ce3+ binds to the scissile phosphate for enzyme activation (Fig. 8G). Through systematic characterization of Ce13d, we have established a set of methods for studying metal binding in such DNAzymes, which might be useful for obtaining insights into other NAEs.
Recently, we isolated another DNAzyme called Ag10c using Ag+ as a metal cofactor (Fig. 9A).18 It is indeed highly specific for Ag+, displaying a cleavage rate of 0.41 min−1 with 10 μM Ag+ at pH 7.5 with 200 mM NaNO3. It was converted into a catalytic beacon for Ag+ detection, with a detection limit of 24.9 nM. Detailed characterization was performed on Ag10c by using the above techniques to understand its metal binding.107 The effect of Ag+ concentration on enzyme activity was found to follow a sigmoidal curve, indicating cooperative binding of multiple Ag+ ions.107 The double-log plot showed a slope of 2, suggesting that the DNAzyme binds two Ag+ ions (Fig. 9B).107 Since Ag+ is a highly thiophilic metal with a low affinity towards phosphate, we suspect that Ag+ is unlikely to directly interact with the scissile phosphate.
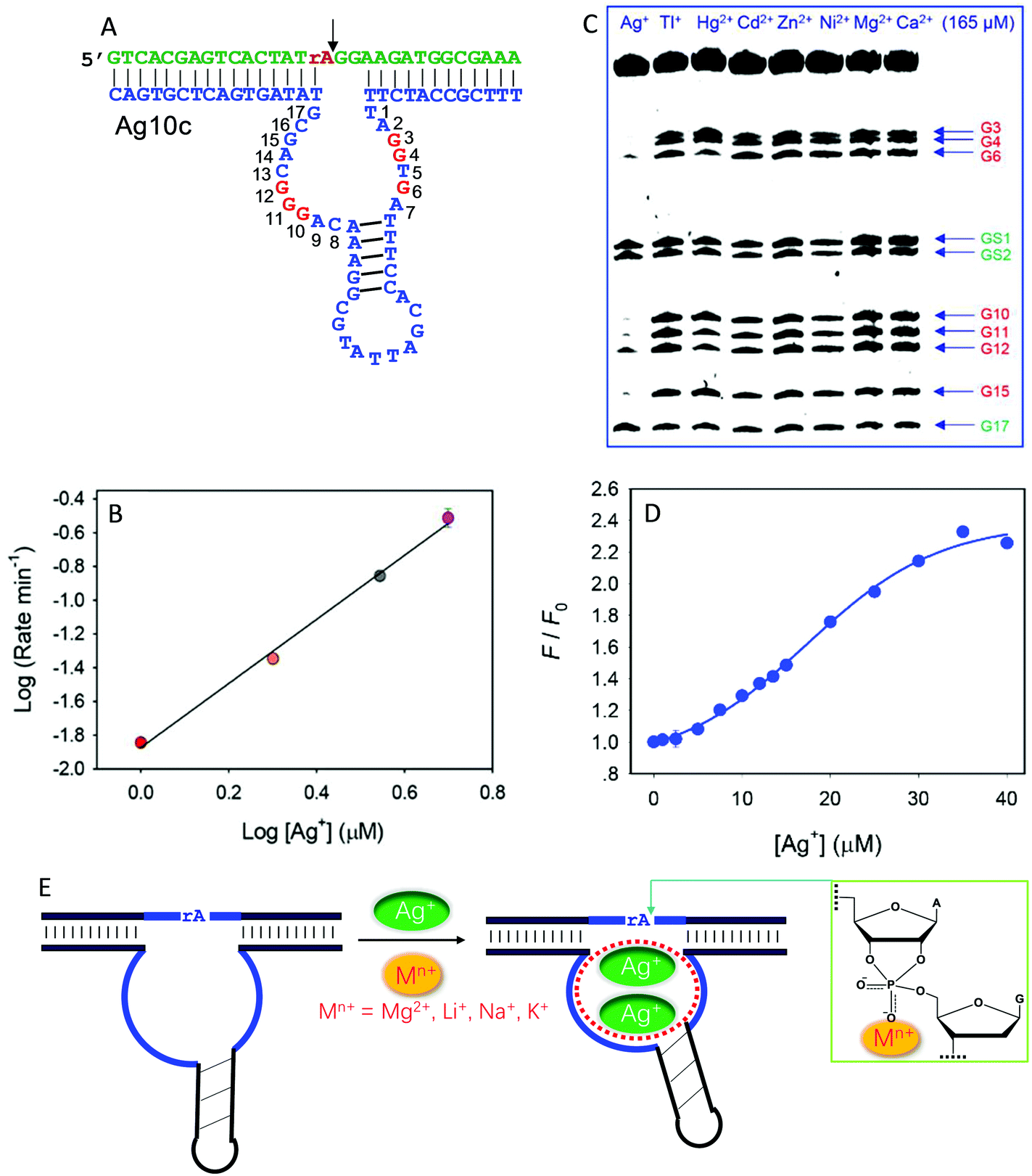 | ||
| Fig. 9 (A) The secondary structure of the Ag10c DNAzyme. The protected guanines under the DMS footprinting conditions are highlighted in red. (B) The double-log plot of cleavage rate versus Ag+ concentration. (C) A DMS footprinting gel image of Ag10c in the presence of 165 μM of different metal ions. The numbering of each guanine is in (A). (B and C) are adapted from ref. 107. With permission. Copyright 2017 American Chemical Society. (D) The 2AP fluorescence increase as a function of Ag+ concentration with 2AP incorporating at the A9 site.108 (E) A proposed mechanism of metal binding by Ag10c. Two Ag+ ions bind to the loop aptamer structure, and another metal ion (e.g., Na+, Li+, Na+, or Mg2+) binds to the scissile phosphate to directly facilitate catalysis. | ||
DMS footprinting indeed suggests a highly specific Ag+ aptamer in Ag10c, since only Ag+ could protect certain guanine nucleotides in the enzyme loops, while the guanines in the hairpin were not protected (Fig. 9C).107 Specific Ag+ binding was also supported by 2AP probing.108 For one labeling site, the fluorescence enhancement as a function of [Ag+] also showed sigmoidal binding with a Hill coefficient of 2.17 (Fig. 9D), consistent with two Ag+ ions binding, as obtained from the previous cleavage activity based assays. Therefore, there is some similarity between Ag10c and Ce13d, each containing a metal aptamer.
The activity of Ag10c was found to be enhanced by salt concentration in the buffer (such as Na+, Li+, K+ and Mg2+). This was not seen in other DNAzymes, where salt often screens the interaction between the scissile phosphate and metal ions. We rationalized this by such group 1A and 2A metals interacting with the scissile phosphate. Since their phosphate binding affinity is weak, they require much higher metal concentrations for better activity. Further PS modification indicated that these buffer salts bind to the pro-Rp oxygen.107 Taken together, the metal binding mechanism of Ag10c is summarized in Fig. 9E. Two Ag+ ions bind to the catalytic loop to exert its allosteric function, while a hard metal (e.g., Na+, Li+, Na+, or Mg2+) interacts with the scissile phosphate, leading to a total of three metals binding.
6.3 Examples with unidentified mechanisms.
In addition to the above examples, a few other NAEs are also known to require multiple metals, but their mechanism of metal binding remains to be fully explored. Bipartite II is one such example discovered by the Sen Lab.109 They initially performed an in vitro selection using Mg2+ as a cofactor, and isolated a DNAzyme called Bipartite I that is active with many divalent ions such as Mg2+, Ca2+, Mn2+, Zn2+ and Co2+. A re-selection was then carried out by partially randomizing the Bipartite I structure, through which Bipartite II was selected. Bipartite II can cleave all RNA substrates with a catalytic efficiency comparable to naturally occurring ribozymes.109 Its multiple-metal binding properties were revealed by a metal concentration-dependent study, which gave a Hill coefficient for Mg2+ of 2, suggesting the binding of two Mg2+ ions.110 In addition, this DNAzyme also displays a distinct bell-shaped pH-rate profile with two deprotonation steps,110 which may be related to its two metal-binding property. The detailed mechanism, such as whether the Mg2+ ions are involved in binding to the scissile phosphate, is yet to be fully elucidated.The pistol ribozyme, a small ribozyme discovered through a bioinformatics strategy by Breaker and coworkers, was also found to perform self-cleaving reaction via a two-metal-mechanism.111 The pistol ribozyme catalyzes the strand scission reaction with Mg2+.112 At low Mg2+ concentrations, the double logarithm plot between Mg2+ concentration and Kobs showed an initial slope higher than 1, indicating that at least two Mg2+ ions were involved in the function of the ribozyme.
6.4 Rationally designed multiple-metal-dependent NAEs
The above reviewed multi-metal NAEs were all from in vitro selection. Here, we review some examples based on rational design, where a metal binding motif is incorporated into existing DNAzymes, and the resulting products are termed aptazymes. For example, Liu and Lu introduced T–T mismatches into the hairpin part of a UO22+-specific DNAzyme to construct an aptazyme that requires Hg2+ and UO22+ for function (Fig. 10).113 The catalytic core of the enzyme contains an eight-nucleotide bulge, which is highly conserved for activity. In addition, it contained a hairpin structure that can be replaced by other hairpins. After introducing the T–T mismatches in the hairpin, in the absence of Hg2+, the T–T mismatches were not paired, leading to inactivation of the enzyme. The addition of Hg2+ mediated the formation of T–Hg2+–T mismatches, folding the DNAzyme into an active structure for substrate cleavage with the aid of UO22+. In general, the more T–T mismatches in the stem, the higher the activation effect by Hg2+. Therefore, this system used up to six Hg2+ ions and one UO22+. This aptazyme was used for Hg2+ detection with a detection limit of 2.4 nM Hg2+ and excellent selectivity.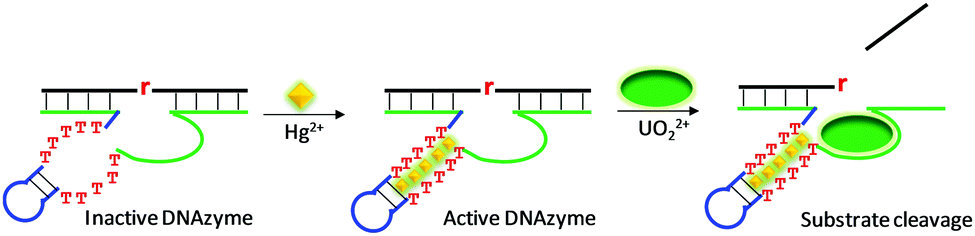 | ||
| Fig. 10 A schematic presentation of rational aptazyme design by introducing several T–T mismatches into the stem region of a UO22+-dependent DNAzyme. Re-drawn from ref. 113. | ||
Willner and coworkers replaced the stem loop region in a Mg2+-dependent DNAzyme with a Hg2+ or Ag+ aptamer (T–Hg2+–T or C–Ag+–C mismatch structure), and the specific metal binding by the aptamers was used to activate the DNAzyme.114 To achieve a reversible switch, chelating ligands were added to remove Hg2+/Ag+ in the system to deactivate the enzyme, and the system could be reactivated by adding more metals. Another general way of rationally designing aptazymes is to incorporate an aptamer into the DNAzyme at the substrate binding stem.115 It is well-known that the substrate binding stems of DNAzymes are usually interchangeable as long as base pairing interactions are maintained. Their sequences have little effect on DNAzyme activity.54,116 This strategy has been used to design aptazymes for non-metal targets.117,118
7. Multiple-metal-dependent NAEs for other chemical reactions
While we have been focusing mainly on RNA-cleavage for multi-metal NAEs, there are also such examples catalyzing other reactions. As mentioned above, NAEs can catalyze a wide range of chemical reactions comparable to the range catalyzed by protein enzymes. We summarize all of the NAEs using multiple metals in Table 1, and in this section, we highlight a few examples of such enzymes catalyzing other types of reactions.| Reaction | Type of NAEs | Metal requirement (name of the NAE, if available) | Ref. |
|---|---|---|---|
| Self-splicing | Group I introns | Two Mg2+ | 82 |
| Group II introns | Two Mg2+ + two K+ | 91 | |
| RNA cleavage | Ribozyme | Pb2+ + Ln3+ (leadzyme) | 94 and 95 |
| Ribozyme | Two Mg2+ (pistol ribozyme) | 111 and 112 | |
| DNAzyme | Three Ln3+ (Tm7) | 20 | |
| DNAzyme | Two Ln3+ (Dy10a) | 21 | |
| DNAzyme | Two Na+ or two Ca2+ (EtNa) | 16 and 52 | |
| DNAzyme | Ce3+ + Na+ (Ce13d) | 105 | |
| DNAzyme | Two Ag+ + Na+/Li+/K+/Mg2+ (Ag10c) | 107 and 108 | |
| DNAzyme | Two Mg2+ (Bipartite II) | 110 | |
| Aptazyme | Multiple Hg2+ + UO22+ | 113 | |
| DNA cleavage | DNAzyme | Zn2+ + Mn2+ (10MD5) | 119 |
| DNAzyme | Zn2+ + Mn2+ + Mg2+ | 120 | |
| DNAzyme | Ln3+ + Zn2+ | 96 | |
| DNA adenylation | DNAzyme | Mg2+ + Cu2+ + Na+ | 121 |
| RNA labeling | DNAzyme | Mg2+ + Tb3+ | 122 |
| Peptide cleavage | DNAzyme | Zn2+ + Mn2+ | 119 |
| Nucleopeptide linkage formation | DNAzyme | Zn2+ + Mn2+ + Mg2+ | 123 |
| Conjugation of DNA to tyrosine | DNAzyme | Mn2+ + Mg2+ | 124 |
| Tyrosine phosphorylation | DNAzyme | Zn2+ + Mg2+ | 125 |
| Phosphoserine lyase activity | DNAzyme | Zn2+ + Mn2+ + Mg2+ | 126 |
| Diels–Alder cycloaddition | Ribozyme | Eight Mg2+ | 127 |
7.1 DNA cleavage
Unlike RNA with an internal 2′-OH nucleophile, DNA with a deoxyribose can only rely on external nucleophiles or oxidative mechanisms for cleavage. With the assistance of a high concentration of Ce(IV), free DNA can be hydrolyzed under physiological conditions.101 It was proposed that two Ce(IV) ions cooperatively coordinate to a phosphate residue to activate the phosphodiester linkage for nucleophilic attack by the hydroxide group on one of the Ce(IV) ions, forming the pentacoordinated intermediate (Fig. 11A). Then, a metal bound water on the other Ce(IV) ion acts as an acid catalysis to facilitate the removal of the 5′-OH from the phosphorus atom, resulting in 5′-phosphate and 3′-hydroxyl products. | ||
| Fig. 11 (A) A proposed mechanism of Ce(IV)-induced DNA hydrolysis. (B) The attack of the α-phosphorus center of ATP by DNA through its 5′-phosphate oxygen to form a 5′,5′-pyrophosphate cap. (C) Labeling of RNA by the attack of an RNA 2′-OH on the α-phosphorus center of a ribose-modified guanosine triphosphate. | ||
DNAzyme-catalyzed DNA cleavage was also reported by the Silverman lab. They performed in vitro selections in the presence of metal mixtures and isolated a series of such DNAzymes that strictly require multiple metal ions.96,119,120 In an initial study, they reported an efficient DNAzyme to hydrolyze DNA in the presence of Zn2+ and Mn2+.119 Re-selection was then carried out by partially randomizing the DNAzyme sequence and they identified a variant that uses Zn2+ alone.128 Therefore, for these DNAzymes, Zn2+ is likely involved in the catalytic mechanism directly, while Mn2+ might only play a structural role. Since these DNAzymes can only cleave the substrate junction of ATG^T, they then performed new selections aiming to expand the substrate generality. The resulting DNAzyme also requires multiple metals of Zn2+, Mn2+ and Mg2+, but it was able to cleave any N^G junction.120 In a follow-up work, they further included Ln3+ as a cofactor during selection, and isolated a few DNAzymes that strictly require Ln3+ and Zn2+ for activity.96 Compared to RNA cleavage, the mechanism of DNA cleavage is more complex and the role of metals has yet to be fully studied in these DNAzymes.
Breaker and coworkers reported a very small Zn2+-dependent DNA-cleaving DNAzyme named I-R3 that can hydrolytically cleave DNA with a rate of ∼1 min−1, rivalling the kinetics of RNA-cleaving DNAzymes. Although the paper did not explicitly study the number of Zn2+ ions involved in the reaction, from the jump in activity between 0.5 and 1 mM Zn2+, we can deduce that it has a strong dependency on metal cooperativity and must require multiple Zn2+ ions.129 Under the neutral conditions used by this DNAzyme, Zn2+ is easily hydrolyzed to form hydroxide or oxide species,130 and these complexes might contribute to the multiple Zn2+ requirement. It is interesting to note that Ce(IV), Ln3+ and Zn2+ are all easily hydrolyzed and condensed at neutral pH, and these water, hydroxyl, or oxo-bridged multinuclear complexes might be important for achieving DNA cleavage.
7.2 DNA adenylation (capping)
Using multiple metal ions, Breaker isolated a series of DNAzymes that promote an ATP-dependent self-capping reaction, which catalyze the transfer of the AMP moiety of an ATP to the 5′ phosphate group of DNA, forming a 5′,5′-pyrophosphate linkage (Fig. 11B).121 This reaction replicates the first step of DNA ligation catalyzed by T4 DNA ligase. A representative DNAzyme requires both Mg2+ and Cu2+ for function, displaying a catalytic efficiency of 104 M−1 min−1. While Mg2+ can be replaced by Ca2+ without activity loss, Cu2+ is an essential cofactor with an apparent Kd of ∼3 μM. However, when the Cu2+ concentration was higher than 20 μM, the reaction was strongly inhibited. Interestingly, this DNAzyme also requires Na+ to facilitate the catalysis, with an optimal concentration of 250 mM Na+, and it cannot be replaced by other monovalent ions. Therefore, Na+ may also be involved in the catalysis, and this is reminiscent of our Ce13d DNAzyme requiring both Ln3+ and Na+.1057.3 RNA labeling
Site-specific labeling of RNA with functional molecules is of great interest in the field of RNA chemical biology. By harnessing an RNA ligating DNAzyme that catalyzes the synthesis of 2′,5′-branched RNA,131 Hoebartner and coworkers established a mild and efficient labeling method for RNA using ribose-modified guanosine triphosphates as a labeling reagent (Fig. 11C).122 The DNAzyme requires over 20 mM Mg2+ for catalysis, while the efficiency is strongly improved by adding 100 μM Tb3+, showing Mg2+-dependent and Tb3+-assisted catalysis.7.4 Reactions with substrates beyond RNA/DNA
Efforts have also been made to expand the chemical repertoire of NAEs for catalysis involving substrates other than RNA/DNA. For example, the Silverman group selected several bimetallic and trimetallic DNAzymes that were able to catalyze a wide range of chemical reactions, such as peptide cleavage,119 nucleopeptide linkage formation,123 conjugation of DNA to tyrosine,124 tyrosine phosphorylation,125 and phosphoserine lyase activity.126 These studies have added new examples of multiple-metal-dependent DNAzymes, although the fundamental aspect of metal binding was less explored. Besides DNAzymes, ribozymes were also selected to use multiple metal ions for different chemical reactions. For example, the Diels–Alder ribozyme, a 49 nt RNA structure obtained through in vitro selection with Diels–Alder cycloaddition activity,132 was demonstrated to contain eight Mg2+ ions in its crystal structure.127 Among the eight Mg2+ ions, six of them are involved in structural stabilization, while the remaining two are believed to mediate the contact between RNA molecules.8. Methods to study multi-metal binding by NAEs
Thus far, we have extensively reviewed examples of NAEs that require multiple metal ions for activity. Their metal binding mechanisms have been studied in detail for some NAEs as shown above, while the mechanism is still quite elusive for the rest. In this section, we summarize some useful methods, which may facilitate future work in this research front.8.1 Activity assays
Since enzymes are involved, the most direct way is to measure their catalytic rate as a function of metal concentration. When multiple metal ions (the same type of metal) act cooperatively, we expect a sigmoidal binding curve, and the number of metal ions involved can be fitted from the double log plot (e.g.Fig. 6B, E and H) based on the mass action law. If metal binding can induce changes in other types of measurable properties, those signals can also be used for fitting, such as FRET, Tb3+-sensitized luminescence, and 2-aminopurine fluorescence. It needs to be noted that when more than one type of metal is involved, such as the case of Ce13d (one Na+ and one Ce3+) and Ag10c (one Na+ and two Ag+), each metal needs to be probed individually. This method can only provide information on the number of metal ions involved in each reaction, while the chemical details of the metal binding sites need to be further studied by other methods.8.2 PS modification and mutation
PS modification is a quite simple and cost-effective way to probe metal binding to the scissile phosphate, mainly for RNA-cleaving DNAzymes. If the DNA backbone phosphate is involved in metal binding, PS can also be used for that purpose. In addition, RNA ligation reactions can also be probed by this method.133 For most of the single-metal-binding RNA-cleaving enzymes, when a PS is made at the scissile phosphate, the activity of the Rp isomer becomes strongly inhibited by ∼100-fold, while the Sp isomer is affected only marginally (typically within 5-fold). This is because the metal binds to the pro-Rp oxygen and replacing this oxygen with sulfur decreases the metal binding affinity. This general conclusion does not hold true for enzymes that bind multiple metals. For the Dy10a DNAzyme that binds two lanthanide ions, either isomer was suppressed by ∼1000 fold, suggesting that both non-bridging oxygen atoms in the scissile phosphate are involved in metal binding.21 The same was observed also for the Ca2+-dependent EtNa DNAzyme.15 The Tm7 DNAzyme binds three lanthanides, and this enzyme was completely inactivated with the PS-modified substrate.20 Therefore, the data from the PS probing and activity assay in Section 8.1 can be analysed side-by-side.In addition to PS modification, base mutation is another way to study metal binding sites in general. It is quite easy synthetically to make such mutations. In addition to the four standard bases, further mutation to non-natural nucleobases such as 2-aminopurine and inosine can also be performed to probe metal binding at the atomic level. For example, we have investigated Ag10c binding to Ag+ by systematically mutating the aptamer loop,107 and concluded that Ag+ binding is not based on the commonly known C–Ag+–C mechanism.134
8.3 DNA footprinting
To probe the nucleobase binding, footprinting techniques are powerful tools. Among them, DMS footprinting specifically probes the guanine bases, and we have shown two such examples in this article, namely Ce13d binding Na+ and Ag10c binding Ag+ (Fig. 8D and 9C). Nuclease S1 footprinting is a complementary method to map the whole nucleobase information for ligand binding.135,136 Nuclease S1 selectively cleaves exposed ssDNA, and metal induced NAE folding could protect the structure from nuclease S1 recognition, resulting in less degradation. In-line probing is another way to study the metal binding in RNA structures.137 Phosphodiester linkages in RNA are susceptible to spontaneous cleavage in the presence of a high concentration of Mg2+ under basic conditions. The relative cleavage rate depends on the local structural characteristics of each RNA linkage. When the linkage position is folded into a metal binding structure, it is less available for “in-line” nucleophilic attack by the 2′-oxygen, resulting in reduced cleavage. The analysis of relative cleavage provides detailed information about structural characteristics of RNA. Therefore, this method is suitable for studying metal binding in ribozymes.8.4 Other methods
The above techniques are quite commonly used since they can be carried out in most labs. Other techniques that might also be used include a suite of biophysical methods such as isothermal titration calorimetry (ITC), X-ray crystallography and NMR. ITC measures the heat of binding reactions and can provide binding stoichiometry. In theory, ITC is ideal for metal binding. For DNA, however, because of its strong negative charge, heat from non-specific binding needs to be carefully subtracted. While ITC has been used to measure aptamer binding,138 we are not aware of any examples of ITC being used to measure NAE binding.X-ray crystallography is extremely powerful in providing an atomic scale mechanistic understanding of metal ions in NAEs. It has been very successful in ribozymes.27 Crystallization of DNAzymes, however, has been more challenging and the first active RNA-ligating DNAzyme crystal was solved only last year.133 In that example, interestingly though, the required Mg2+ ions were not identified in the active site. NMR is another powerful method complementary to X-ray crystallography providing information in the solution phase. Again, no NMR structures on DNAzyme were reported. The lack of structural information has limited deeper insights into DNAzymes, while at the same time, this fact also suggests their structural floppiness and metal ions may not need to be tightly bound to exert their function.
9. Applications related to multi-metal binding NAEs
For most NAEs, metal ions are required cofactors for activity. This property has been widely utilized to design NAE-based metal sensors, especially for RNA-cleaving DNAzymes. The detection limit of these sensors can often reach low nM or even pM levels, rivaling those of traditional analytical instruments.13,59 Compared with single-metal-dependent DNAzymes, multi-metal-binding ones can have sharper switch-like binding curves. In addition, cooperative binding may allow better metal specificity. For example, the EtNa DNAzyme cooperatively binds two Ca2+ ions but only one Mg2+ for activation, and it is 90-fold more active in Ca2+ than in Mg2+ (about 10-fold better than previous single-metal-binding DNAzymes).15 Given this property, EtNa was engineered for Ca2+ sensing, showing a detection limit of 17 μM Ca2+ with exquisite selectivity. Aside from these typical analytical applications, we highlight a few emerging applications that are unique for multiple-metal-binding NAEs.9.1 Folding-based metal sensing
While DNAzyme-cleavage based sensors are quite robust and sensitive for metal detection, they have several limitations. Since an enzymatic reaction is involved in signal production, the sensing process is irreversible, disabling its application for the continuous monitoring of the fluctuation of metal concentration. Aptamers, on the other hand, only rely on binding and thus can monitor time-dependent signal changes. Several DNAzymes introduced above, such as Ce13d and Ag10c, contain an aptamer motif for metal binding, which may be useful for constructing folding-based sensors. In this case, the RNA linkage at the cleavage site is replaced by its DNA analog to avoid cleavage reactions. Alternatively, the catalytic metal (e.g., for scissile phosphate binding) should be removed from the system, so that the DNAzyme is inactive and intact during detection. It needs to be noted that metals can always induce folding of nucleic acids, both non-specifically and specifically. For example, the 17E DNAzyme can be folded by Mg2+, Zn2+ and Pb2+ when probed using FRET.139,140 With appropriate probe design, such embedded aptamers allow highly specific detection. To contain an aptamer and still retain catalytic activity, these enzymes by default use multiple metals (except for the very special case of NaA43 using Na+ alone for catalysis).To produce a folding-based signal, a fluorophore is usually incorporated into DNA. A good choice is fluorescent base analogs, since they can be incorporated with minimal perturbation of DNAzyme structures.141,142 Among them, 2-aminopurine (2AP) is the most popular one. After optimization, Na+-induced more than 600% fluorescence increase in the Ce13d DNAzyme, suggesting a substantial conformation change upon Na+ binding. At the same time, other monovalent metals did not result in a signal increase, indicating that non-specific signaling was inhibited. This turn-on fluorescence sensor achieved a detection limit of 0.4 mM Na+ with a saturated signal produced in just 10 s. Since 2AP emits in the UV region, future work on other base analogs (e.g. pyrrolo-cytosine emitting at 473 nm in the blue region) to extend the emission to the visible region is needed for cellular analysis.143,144
9.2 Identification of new metal aptamers from multiple-metal-dependent NAEs
Metal binding aptamers have long been sought after. A few nucleotides can specifically coordinate certain metal ions, such as thymine for Hg2+,38 and cytosine for Ag+,39 and thus rationally designed aptamers have been achieved for these metals. However, for most other metals, their aptamers have to be selected through a combinatorial process called systematic evolution of ligands by exponential amplification (SELEX).145,146 Typical aptamer selection requires immobilization of the target, in this case metal ions. Due to technical difficulties, limited success was reported using this direct selection strategy.41,147 Although this issue can be solved by immobilizing a DNA library,148,149 or by compartmentalization,150 the number of high quality metal aptamers is still quite low.149–152The study of the Ce13d and Ag10c DNAzymes has clearly indicated an aptamer motif in each DNAzyme. Therefore, aptamers might be isolated from the multiple-metal-dependent NAEs. We reason that this method can be applied for identification of other metal aptamers. In fact, the Li lab has selected many RNA-cleaving fluorescent DNAzymes (RFDs) targeting the crude extracellular mixture (CEM) produced by certain bacterial pathogens,153 and cancer cells.154 Although their targets there were mainly proteins, the selection of such aptazymes can certainly be applied to metal ions.
9.3 DNA nanotechnology
Due to its excellent programmability, DNA has found wide applications in nanotechnology. DNAzymes provide an excellent research tool for DNA nanotechnology, since it can cleave and ligate DNA for more complex functions beyond simple hybridization.155,156 We reason that multi-metal-dependent NAEs allow for the construction of logic gates and computing circuits using metal ions as inputs.22 Willner and coworkers designed an AND logic gate by simultaneously using the UO22+-dependent and Mg2+-dependent DNAzymes (Fig. 12A).157 This system consists of a long substrate strand with two cleavage sites, and two enzyme strands are respectively hybridized. The substrate also contains a G-rich domain X, which encodes the sequence of the horseradish peroxidase (HRP)-mimicking G-quadruplex DNAzyme. Only when both UO22+ and Mg2+ are added (i.e., AND gate), both substrate fragments are cleaved, releasing the G-rich sequence to form the active G-quadruplex/hemin DNAzyme for the production of a colorimetric signal.In the above work, two different DNAzymes were used. We reason that such an AND logic gate can also be fabricated by using a single DNAzyme, if the enzyme requires multiple metal ions for activation. For example, although not explicitly described as a logic gate, the Ce13d DNAzyme catalytic beacon is indeed such a device. It requires both Na+ and Ce3+ for signal production (Fig. 12B).19,105 Similarly, the rationally designed Hg2+ sensor based on the UO22+ DNAzyme is also such an example, where both metals are needed for signaling.158 Compared with the above two-DNAzyme design, the use of a multiple-metal-dependent DNAzyme makes the system simpler.
 | ||
| Fig. 12 (A) An “AND” logic gate design by using two DNAzymes linked to a common substrate with UO22+ and Mg2+ as inputs. A G-quadruplex based peroxidase mimicking DNAzyme was used for oxidation of ABTS to produce a colorimetric signal. (B) A schematic presentation of an “AND” logic gate consisting of a multiple-metal-dependent DNAzyme using fluorescence signaling. | ||
9.4 Metallomics
Metal ions are extremely important to maintain the normal biological functions and physiology of living systems. To describe the integrated bio-metal science, the term “metallomics” was coined in 2004.159 The detection and speciation of metal ions with high temporal and spatial resolution is an important aspect of metallomics. However, sensing metal ions in biological system is a long standing challenge in bioanalytical chemistry, chemical biology and bioinorganic chemistry.160–163 While standard instrumental methods such ICP-AES and ICP-MS have excellent sensitivity, in situ metal monitoring in biological systems mainly relies on small molecule probes and fluorescent proteins.164–167 In the past few years, DNAzyme-based sensors have emerged for such a purpose.17,168–171For intracellular metal sensing, a major challenge is DNAzyme delivery into cells. This has been achieved by using cationic lipids/polypeptides/polymers, gold nanoparticles and DNA nanostructures.168,169,172,173 In addition, to achieve temporal resolution of metal sensing, caged DNAzymes have been developed.17,173,174 Caged DNAzymes have a covalently attached photolabile group that blocks their activity. Upon exposure to a particular wavelength of light, the capping group falls off to recover the DNAzyme activity. Therefore, this technique is quite useful for in vivo monitoring as light can be conveniently applied to cells and thin tissues with precisely controlled intensity, location and timing. DNAzyme-based metal measurement has also been applied to other biological samples such as blood serum. Compared with intracellular sensing, serum does not require a delivery step. However, serum scatters light strongly and serum proteins chelate metal ions tightly, presenting two major challenges for such applications. Due to these limitations, detection of metal in serum by DNAzymes has only been reported for some abundant metals such as Mg2+ and Ca2+ using gel-based and electrochemical methods.175,176
A unique aspect of multi-metal-dependent DNAzymes is their potential for multiplex metal sensing, which may provide more detailed biological mechanisms. In addition, cooperative metal binding may allow more sensitive and specific metal detection, which could be useful if the metal range falls in the quick rising stage of the DNAzyme. To fully realize such goals, new DNAzymes with higher metal binding affinities are needed, especially when the targets are transition metals. Transition metals are often sequestered by proteins and the free metal concentrations are very low. Therefore, modified DNA with tighter metal binding ligands might be required to achieve this goal.9,14,28,177,178 For realistic profiling of metals in biological samples, it is unlikely that any single DNAzyme can be used. More realistically, a DNAzyme sensor array with multiple DNAzymes each detecting one or two metals will be the platform for detection.28,179
10. Conclusions and future directions
In summary, we have reviewed RNA-cleaving NAEs that require multiple metal ions, of both the same and different metals. Such NAEs were found in both natural ribozymes and also in many in vitro selected or rationally designed ribozymes/DNAzymes. Fundamental biochemical studies on their metal binding mechanisms allow us to classify them into a few types. One type uses all the metal ions to interact with the scissile phosphate group for catalysis, and these enzymes exhibit a sharp metal binding curve and an often unrescuable thio effect. The other type binds different metals at different sites, fulfilling the catalytic and allosteric roles, respectively. This type generally contains a metal binding aptamer. Most of their applications have focused on metal sensing and nanotechnology. Given this progress, we postulate some future directions on this topic that may provide new research opportunities.(1) Selection of new DNAzymes and characterization. Most previously reported RNA-cleaving DNAzymes are single-metal-dependent. By tuning the selection conditions, more multiple-metal-dependent DNAzymes might be obtained. Exciting advancements have been made by the discovery of Ce13d and Ag10c. We believe that there is still a lot of room for selecting new DNAzymes. The choice of metal ion is important. For example, we obtained a few DNAzymes using multiple lanthanide ions for catalysis, and this might be related to the tendency of lanthanides to hydrolyze forming multinuclear complexes. The selection conditions might also be important. For example, we obtained EtNa in isopropanol. So far, this is still a trial-and-error process. A combination of hard metals (e.g. Na+, Mg2+ and Ca2+) and soft metals (e.g. Hg2+ and Ag+) might promote the production of aptazymes for the soft metals. A good example is the Ag10c example. Whether this strategy can be applied to other metals is yet to be tested.
After obtaining optimal DNAzymes, a subsequent step is to perform careful characterizations to understand their metal binding mechanisms, which would be beneficial for their following applications. Most current metal binding information was obtained by biochemical assays, which can give the number of metal ions involved and their approximate binding sites. However, to understand atomic scale mechanisms, structural biology data, such as crystal or NMR structures, are required. Recently, the first active DNAzyme has been crystallized,133 and this is an encouraging development and we expect more such examples to be reported in the near future.
(2) More sensitive and selective metal detection. RNA-cleaving DNAzymes are an excellent tool for metal sensing, and they can easily detect transition or lanthanide metals down to nM levels in clean buffers due to their strong DNA binding affinity. For biologically abundant metal ions such as Na+, Mg2+, K+ and Ca2+, DNAzymes might also be able to achieve the required sensitivity either directly,17 or via further signal amplification.171 To achieve better metal binding, modified nucleotides can be introduced during DNAzyme selection. For example, PS modification can enhance the soft metal binding,61 and modified nucleobases can improve the metal binding affinity for several particular metals.14 Metal sensing has already been performed by cells using riboswitches.180,181 In this regard, nucleic acids should have sufficient metal binding affinity for some metal ions at physiologically important concentration ranges.
We have not seen many examples of artificial sensors that can not only perform proof-of-concept studies but also provide useful biochemical and cellular insights. Given the programmability of DNA and its potential for signal amplification, using DNAzymes for metal sensing in cells is a promising direction and multi-metal-binding DNAzymes with sharp binding curves are potentially a useful tool for this goal.
(3) DNA nanotechnology applications. Aside from biosensing and metallomics related applications, multi-metal NAEs are also attractive for making stimuli-responsive DNA nanostructures. So far, multiple inputs including metal inputs have been achieved by using different DNAzymes. With the selection of a single DNAzyme that requires multiple different metals, more elegant structures can be envisioned.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for the work performed in the Liu lab was mainly from The Natural Sciences and Engineering Research Council of Canada (NSERC).References
- C. J. Wilson, D. Apiyo and P. Wittung-Stafshede, Q. Rev. Biophys., 2005, 37, 285–314 CrossRef PubMed.
- A. Battistoni, S. Folcarelli, L. Cervoni, F. Polizio, A. Desideri, A. Giartosio and G. Rotilio, J. Biol. Chem., 1998, 273, 5655–5661 CrossRef CAS PubMed.
- K. Brzoska, S. Meczynska and M. Kruszewski, Acta Biochim. Pol., 2006, 53, 685–691 CAS.
- K. Kruger, P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling and T. R. Cech, Cell, 1982, 31, 147–157 CrossRef CAS PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS PubMed.
- S. K. Silverman and T. P. Begley, Nucleic Acid Enzymes (Ribozymes and Deoxyribozymes): In Vitro Selection and Application, Wiley Encyclopedia of Chemical Biology, John Wiley & Sons, Inc., 2007 Search PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595–609 CrossRef CAS PubMed.
- S. W. Santoro, G. F. Joyce, K. Sakthivel, S. Gramatikova and C. F. Barbas, 3rd, J. Am. Chem. Soc., 2000, 122, 2433–2439 CrossRef CAS PubMed.
- J. Li, W. Zheng, A. H. Kwon and Y. Lu, Nucleic Acids Res., 2000, 28, 481–488 CrossRef CAS PubMed.
- P.-J. J. Huang and J. Liu, Anal. Chem., 2016, 88, 3341–3347 CrossRef CAS PubMed.
- P.-J. J. Huang and J. Liu, Nucleic Acids Res., 2015, 43, 6125–6133 CrossRef CAS PubMed.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS PubMed.
- M. Hollenstein, C. Hipolito, C. Lam, D. Dietrich and D. M. Perrin, Angew. Chem., Int. Ed., 2008, 47, 4346–4350 CrossRef CAS PubMed.
- W. H. Zhou, R. Saran, P. J. J. Huang, J. S. Ding and J. W. Liu, ChemBioChem, 2017, 18, 518–522 CrossRef CAS PubMed.
- W. Zhou, R. Saran, Q. Chen, J. Ding and J. Liu, ChemBioChem, 2016, 17, 159–163 CrossRef CAS PubMed.
- S.-F. Torabi, P. Wu, C. E. McGhee, L. Chen, K. Hwang, N. Zheng, J. Cheng and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5903–5908 CrossRef CAS PubMed.
- R. Saran and J. W. Liu, Anal. Chem., 2016, 88, 4014–4020 CrossRef CAS PubMed.
- P.-J. J. Huang, J. Lin, J. Cao, M. Vazin and J. Liu, Anal. Chem., 2014, 86, 1816–1821 CrossRef CAS PubMed.
- P. J. Huang, M. Vazin, Z. Matuszek and J. Liu, Nucleic Acids Res., 2015, 43, 461–469 CrossRef CAS PubMed.
- P. J. Huang, M. Vazin and J. Liu, Biochemistry, 2016, 55, 2518–2525 CrossRef CAS PubMed.
- R. Orbach, B. Willner and I. Willner, Chem. Commun., 2015, 51, 4144–4160 RSC.
- F. Wang, C.-H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881–2941 CrossRef CAS PubMed.
- O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528–2556 CrossRef CAS PubMed.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- R. K. Sigel and A. M. Pyle, Chem. Rev., 2007, 107, 97–113 CrossRef CAS PubMed.
- W. L. Ward, K. Plakos and V. J. DeRose, Chem. Rev., 2014, 114, 4318–4342 CrossRef CAS PubMed.
- W. Zhou, R. Saran and J. Liu, Chem. Rev., 2017, 117, 8272–8325 CrossRef CAS PubMed.
- K. Hwang, P. Hosseinzadeh and Y. Lu, Inorg. Chim. Acta, 2016, 452, 12–24 CrossRef CAS PubMed.
- M. Pechlaner and R. K. Sigel, Met. Ions Life Sci., 2012, 10, 1–42 CAS.
- H. Sigel, Chem. Soc. Rev., 1993, 22, 255–267 RSC.
- R. M. Izatt, J. J. Christensen and J. H. Rytting, Chem. Rev., 1971, 71, 439–481 CrossRef CAS PubMed.
- R. K. Sigel and H. Sigel, Acc. Chem. Res., 2010, 43, 974–984 CrossRef CAS PubMed.
- Z. J. Tan and S. J. Chen, Met. Ions Life Sci., 2011, 9, 101–124 CAS.
- H. Ueyama, M. Takagi and S. Takenaka, J. Am. Chem. Soc., 2002, 124, 14286–14287 CrossRef CAS PubMed.
- W. Zhou, J. Ding and J. Liu, ChemBioChem, 2016, 17, 1563–1570 CrossRef CAS PubMed.
- W. H. Zhou, J. S. Ding and J. W. Liu, Nucleic Acids Res., 2016, 44, 10377–10385 CrossRef CAS PubMed.
- A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300–4302 CrossRef CAS PubMed.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 4825–4827 RSC.
- S. Cai, X. Tian, L. Sun, H. Hu, S. Zheng, H. Jiang, L. Yu and S. Zeng, Anal. Chem., 2015, 87, 10542–10546 CrossRef CAS PubMed.
- J. Ciesiolka, J. Gorski and M. Yarus, RNA, 1995, 1, 538–550 CAS.
- H. P. Hofmann, S. Limmer, V. Hornung and M. Sprinzl, RNA, 1997, 3, 1289–1300 CAS.
- G. L. Eichhorn and Y. A. Shin, J. Am. Chem. Soc., 1968, 90, 7323–7328 CrossRef CAS PubMed.
- Y. Li and R. R. Breaker, J. Am. Chem. Soc., 1999, 121, 5364–5372 CrossRef CAS.
- S. C. Dahm, W. B. Derrick and O. C. Uhlenbeck, Biochemistry, 1993, 32, 13040–13045 CrossRef CAS PubMed.
- M. Bonaccio, A. Credali and A. Peracchi, Nucleic Acids Res., 2004, 32, 916–925 CrossRef CAS PubMed.
- S. W. Santoro and G. F. Joyce, Biochemistry, 1998, 37, 13330–13342 CrossRef CAS PubMed.
- Y. Takagi, M. Warashina, W. J. Stec, K. Yoshinari and K. Taira, Nucleic Acids Res., 2001, 29, 1815–1834 CrossRef CAS PubMed.
- J. E. Wedekind and D. B. McKay, Nat. Struct. Mol. Biol., 1999, 6, 261–268 CAS.
- J. Schnabl and R. K. Sigel, Curr. Opin. Chem. Biol., 2010, 14, 269–275 CrossRef CAS PubMed.
- P.-J. J. Huang, M. Vazin and J. Liu, Anal. Chem., 2014, 86, 9993–9999 CrossRef CAS PubMed.
- W. Zhou, R. Saran, J. Ding and J. Liu, ChemBioChem, 2017, 18, 1828–1835 CrossRef CAS PubMed.
- R. Saran and J. Liu, Inorg. Chem. Front., 2016, 3, 494–501 RSC.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS.
- A. A. Fokina, D. A. Stetsenko and J.-C. François, Expert Opin. Biol. Ther., 2015, 15, 689–711 CrossRef CAS PubMed.
- C. R. Dass, P. F. Choong and L. M. Khachigian, Mol. Cancer Ther., 2008, 7, 243–251 CrossRef CAS PubMed.
- H. K. Karnati, R. S. Yalagala, R. Undi, S. R. Pasupuleti and R. K. Gutti, Tumor Biol., 2014, 35, 9505–9521 CrossRef CAS PubMed.
- L. M. Khachigian, R. G. Fahmy, G. Zhang, Y. V. Bobryshev and A. Kaniaros, J. Biol. Chem., 2002, 277, 22985–22991 CrossRef CAS PubMed.
- J. Li and Y. Lu, J. Am. Chem. Soc., 2000, 122, 10466–10467 CrossRef CAS.
- T. Lan, K. Furuya and Y. Lu, Chem. Commun., 2010, 46, 3896–3898 RSC.
- P.-J. J. Huang and J. Liu, Anal. Chem., 2014, 86, 5999–6005 CrossRef CAS PubMed.
- M. de la Peña, I. García-Robles and A. Cervera, Molecules, 2017, 22, 78 CrossRef PubMed.
- K. F. Blount and O. C. Uhlenbeck, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 415–440 CrossRef CAS PubMed.
- J. L. Boots, M. D. Canny, E. Azimi and A. Pardi, RNA, 2008, 14, 2212–2222 CrossRef CAS PubMed.
- B. Nawrot, K. Widera, M. Sobczak, M. Wojcik and W. J. Stec, Curr. Org. Chem., 2008, 12, 1004–1009 CrossRef CAS.
- S. Wang, K. Karbstein, A. Peracchi, L. Beigelman and D. Herschlag, Biochemistry, 1999, 38, 14363–14378 CrossRef CAS PubMed.
- P. Thaplyal, A. Ganguly, B. L. Golden, S. Hammes-Schiffer and P. C. Bevilacqua, Biochemistry, 2013, 52, 6499–6514 CrossRef CAS PubMed.
- J. M. Warnecke, J. P. Furste, W. D. Hardt, V. A. Erdmann and R. K. Hartmann, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8924–8928 CrossRef CAS.
- K. Schlosser, J. Gu, L. Sule and Y. Li, Nucleic Acids Res., 2008, 36, 1472–1481 CrossRef CAS PubMed.
- M. Vazin, P.-J. J. Huang, Z. Matuszek and J. Liu, Biochemistry, 2015, 54, 6132–6138 CrossRef CAS PubMed.
- A. K. Brown, J. W. Liu, Y. He and Y. Lu, ChemBioChem, 2009, 10, 486–492 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Fluorescent DNAzyme Biosensors for Metal Ions Based on Catalytic Molecular Beacons, in Fluorescent Energy Transfer Nucleic Acid Probes: Designs and Protocols, ed. V. V. Didenko, Humana Press, Totowa, NJ, 2006, pp. 275–288 Search PubMed.
- J. Liu and Y. Lu, Anal. Chem., 2003, 75, 6666–6672 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Org. Biomol. Chem., 2006, 4, 3435–3441 CAS.
- X. B. Zhang, Z. Wang, H. Xing, Y. Xiang and Y. Lu, Anal. Chem., 2010, 82, 5005–5011 CrossRef CAS PubMed.
- N. Krug, J. M. Hohlfeld, A.-M. Kirsten, O. Kornmann, K. M. Beeh, D. Kappeler, S. Korn, S. Ignatenko, W. Timmer, C. Rogon, J. Zeitvogel, N. Zhang, J. Bille, U. Homburg, A. Turowska, C. Bachert, T. Werfel, R. Buhl, J. Renz, H. Garn and H. Renz, N. Engl. J. Med., 2015, 372, 1987–1995 CrossRef PubMed.
- D. E. Weng, P. A. Masci, S. F. Radka, T. E. Jackson, P. A. Weiss, R. Ganapathi, P. J. Elson, W. B. Capra, V. P. Parker and J. A. Lockridge, Mol. Cancer Ther., 2005, 4, 948–955 CrossRef CAS PubMed.
- D. D. Young, M. O. Lively and A. Deiters, J. Am. Chem. Soc., 2010, 132, 6183–6193 CrossRef CAS PubMed.
- T. Inoue, F. X. Sullivan and T. R. Cech, J. Mol. Biol., 1986, 189, 143–165 CrossRef CAS PubMed.
- F. Michel, M. Hanna, R. Green, D. P. Bartel and J. W. Szostak, Nature, 1989, 342, 391–395 CrossRef CAS PubMed.
- M. D. Been and A. T. Perrotta, Science, 1991, 252, 434–437 CAS.
- M. R. Stahley and S. A. Strobel, Science, 2005, 309, 1587–1590 CrossRef CAS PubMed.
- J. A. Piccirilli, J. S. Vyle, M. H. Caruthers and T. R. Cech, Nature, 1993, 361, 85–88 CrossRef CAS PubMed.
- L. B. Weinstein, B. C. Jones, R. Cosstick and T. R. Cech, Nature, 1997, 388, 805–808 CrossRef CAS PubMed.
- N. Toor, A. R. Robart, J. Christianson and S. Zimmerly, Nucleic Acids Res., 2006, 34, 6461–6471 CrossRef CAS PubMed.
- L. Hunsicker-Wang, M. Vogt and V. J. DeRose, EPR Methods to Study Specific Metal-Ion Binding Sites in RNA, in Methods In Enzymology, Vol. 468: Biophysical, Chemical, And Functional Probes Of RNA Structure, Interactions And Folding, Pt A, ed. D. Herschalag, Elsevier Academic Press Inc., San Diego, 2009, vol. 468, pp. 335–367 Search PubMed.
- M. Podar, P. S. Perlman and R. A. Padgett, Mol. Cell. Biol., 1995, 15, 4466–4478 CrossRef CAS PubMed.
- R. A. Padgett, M. Podar, S. C. Boulanger and P. S. Perlman, Science, 1994, 266, 1685–1688 CAS.
- P. M. Gordon, E. J. Sontheimer and J. A. Piccirilli, Biochemistry, 2000, 39, 12939–12952 CrossRef CAS PubMed.
- P. M. Gordon, R. Fong and J. A. Piccirilli, Chem. Biol., 2007, 14, 607–612 CrossRef CAS PubMed.
- M. Marcia and A. M. Pyle, Cell, 2012, 151, 497–507 CrossRef CAS PubMed.
- T. Pan and O. C. Uhlenbeck, Biochemistry, 1992, 31, 3887–3895 CrossRef CAS PubMed.
- T. Pan, B. Dichtl and O. C. Uhlenbeck, Biochemistry, 1994, 33, 9561–9565 CrossRef CAS PubMed.
- N. Sugimoto and T. Ohmichi, FEBS Lett., 1996, 393, 97–100 CrossRef CAS PubMed.
- T. Ohmichi and N. Sugimoto, Biochemistry, 1997, 36, 3514–3521 CrossRef CAS PubMed.
- V. Dokukin and S. K. Silverman, Chem. Sci., 2012, 3, 1707–1714 RSC.
- Z. Liu, S. H. Mei, J. D. Brennan and Y. Li, J. Am. Chem. Soc., 2003, 125, 7539–7545 CrossRef CAS PubMed.
- S. H. Mei, Z. Liu, J. D. Brennan and Y. Li, J. Am. Chem. Soc., 2003, 125, 412–420 CrossRef CAS PubMed.
- S. Seetharaman, M. Zivarts, N. Sudarsan and R. R. Breaker, Nat. Biotechnol., 2001, 19, 336–341 CrossRef CAS PubMed.
- M. Zivarts, Y. Liu and R. R. Breaker, Nucleic Acids Res., 2005, 33, 622–631 CrossRef CAS PubMed.
- M. Komiyama, N. Takeda and H. Shigekawa, Chem. Commun., 1999, 1443–1451 RSC.
- W. Zhou, J. Ding and J. Liu, ChemBioChem, 2016, 17, 890–894 CrossRef CAS PubMed.
- A. Peracchi, J. Biol. Chem., 2000, 275, 11693–11697 CrossRef CAS PubMed.
- S.-F. Torabi and Y. Lu, J. Mol. Evol., 2015, 81, 225–234 CrossRef CAS PubMed.
- W. Zhou, Y. Zhang, P. J. Huang, J. Ding and J. Liu, Nucleic Acids Res., 2016, 44, 354–363 CrossRef CAS PubMed.
- W. Zhou, J. Ding and J. Liu, Org. Biomol. Chem., 2017, 15, 6959–6966 CAS.
- R. Saran, K. Kleinke, W. Zhou, T. Yu and J. Liu, Biochemistry, 2017, 56, 1955–1962 CrossRef CAS PubMed.
- R. Saran, L. Yao, P. Hoang and J. Liu, Biochimie, 2017 DOI:10.1016/j.biochi.2017.07.001.
- A. R. Feldman and D. Sen, J. Mol. Biol., 2001, 313, 283–294 CrossRef CAS PubMed.
- A. R. Feldman, E. K. Leung, A. J. Bennet and D. Sen, ChemBioChem, 2006, 7, 98–105 CrossRef CAS PubMed.
- K. A. Harris, C. E. Lünse, S. Li, K. I. Brewer and R. R. Breaker, RNA, 2015, 21, 1852–1858 CrossRef CAS PubMed.
- Z. Weinberg, P. B. Kim, T. H. Chen, S. S. Li, K. A. Harris, C. E. Lunse and R. R. Breaker, Nat. Chem. Biol., 2015, 11, 606–610 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46, 7587–7590 CrossRef CAS PubMed.
- S. Shimron, J. Elbaz, A. Henning and I. Willner, Chem. Commun., 2010, 46, 3250–3252 RSC.
- J. C. Achenbach, R. Nutiu and Y. Li, Anal. Chim. Acta, 2005, 534, 41–51 CrossRef CAS.
- A. K. Brown, J. Li, C. M. Pavot and Y. Lu, Biochemistry, 2003, 42, 7152–7161 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Anal. Chem., 2004, 76, 1627–1632 CrossRef CAS PubMed.
- Z. J. Zhou, L. Xiao, Y. Xiang, J. Zhou and A. J. Tong, Anal. Chim. Acta, 2015, 889, 179–186 CrossRef CAS PubMed.
- M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720 CrossRef CAS PubMed.
- Y. Xiao, R. J. Wehrmann, N. A. Ibrahim and S. K. Silverman, Nucleic Acids Res., 2011, 40, 1778–1786 CrossRef PubMed.
- Y. F. Li, Y. Liu and R. R. Breaker, Biochemistry, 2000, 39, 3106–3114 CrossRef CAS PubMed.
- L. Buttner, F. Javadi-Zarnaghi and C. Hobartner, J. Am. Chem. Soc., 2014, 136, 8131–8137 CrossRef PubMed.
- T. E. Velez, J. Singh, Y. Xiao, E. C. Allen, O. Y. Wong, M. Chandra, S. C. Kwon and S. K. Silverman, ACS Comb. Sci., 2012, 14, 680–687 CrossRef CAS PubMed.
- O. Y. Wong, P. I. Pradeepkumar and S. K. Silverman, Biochemistry, 2011, 50, 4741–4749 CrossRef CAS PubMed.
- S. M. Walsh, A. Sachdeva and S. K. Silverman, J. Am. Chem. Soc., 2013, 135, 14928–14931 CrossRef CAS PubMed.
- J. Chandrasekar, A. C. Wylder and S. K. Silverman, J. Am. Chem. Soc., 2015, 137, 9575–9578 CrossRef CAS PubMed.
- A. Serganov, S. Keiper, L. Malinina, V. Tereshko, E. Skripkin, C. Hobartner, A. Polonskaia, A. T. Phan, R. Wombacher, R. Micura, Z. Dauter, A. Jaschke and D. J. Patel, Nat. Struct. Mol. Biol., 2005, 12, 218–224 CAS.
- Y. Xiao, E. C. Allen and S. K. Silverman, Chem. Commun., 2011, 47, 1749–1751 RSC.
- H. Gu, K. Furukawa, Z. Weinberg, D. F. Berenson and R. R. Breaker, J. Am. Chem. Soc., 2013, 135, 9121–9129 CrossRef CAS PubMed.
- L. Z. Ma, B. W. Liu, P. J. J. Huang, X. Zhang and J. W. Liu, Langmuir, 2016, 32, 5672–5680 CrossRef CAS PubMed.
- C. Hobartner and S. K. Silverman, Angew. Chem., Int. Ed., 2007, 46, 7420–7424 CrossRef CAS PubMed.
- B. Seelig and A. Jäschke, Chem. Biol., 1999, 6, 167–176 CrossRef CAS PubMed.
- A. Ponce-Salvatierra, K. Wawrzyniak-Turek, U. Steuerwald, C. Höbartner and V. Pena, Nature, 2016, 529, 231–234 CrossRef CAS PubMed.
- A. Ono, H. Torigoe, Y. Tanaka and I. Okamoto, Chem. Soc. Rev., 2011, 40, 5855–5866 RSC.
- Y. J. Lee, S. R. Han, N. Y. Kim, S. H. Lee, J. S. Jeong and S. W. Lee, Gastroenterology, 2012, 143, 155–165 CrossRef CAS PubMed.
- D. D. Vo, T. P. A. Tran, C. Staedel, R. Benhida, F. Darfeuille, A. Di Giorgio and M. Duca, Chem. – Eur. J., 2016, 22, 5350–5362 CrossRef CAS PubMed.
- E. E. Regulski and R. R. Breaker, In-line probing analysis of riboswitches, in Methods in Molecular Biology, ed. J. Wilusz, Humana Press Inc, 999 Riverview Dr, Ste 208, Totowa, NJ 07512-1165, USA, 2008, vol. 419, pp. 53–67 Search PubMed.
- H. Torigoe, A. Ono and T. Kozasa, Chem. – Eur. J., 2010, 16, 13218–13225 CrossRef CAS PubMed.
- H. K. Kim, J. Liu, J. Li, N. Nagraj, M. Li, C. M. Pavot and Y. Lu, J. Am. Chem. Soc., 2007, 129, 6896–6902 CrossRef CAS PubMed.
- H.-K. Kim, I. Rasnik, J. Liu, T. Ha and Y. Lu, Nat. Chem. Biol., 2007, 3, 763–768 CrossRef CAS PubMed.
- N. Dai and E. T. Kool, Chem. Soc. Rev., 2011, 40, 5756–5770 RSC.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS PubMed.
- P. Chen and C. He, J. Am. Chem. Soc., 2004, 126, 728–729 CrossRef CAS PubMed.
- P. Chen, B. Greenberg, S. Taghavi, C. Romano, D. van der Lelie and C. He, Angew. Chem., Int. Ed., 2005, 44, 2715–2719 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- J. Ciesiolka and M. Yarus, RNA, 1996, 2, 785–793 CAS.
- R. Nutiu and Y. F. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS PubMed.
- M. Rajendran and A. D. Ellington, Anal. Bioanal. Chem., 2008, 390, 1067–1075 CrossRef CAS PubMed.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS PubMed.
- H. Wang, H. Cheng, J. Wang, L. Xu, H. Chen and R. Pei, Talanta, 2016, 154, 498–503 CrossRef CAS PubMed.
- Y. Wu, S. Zhan, L. Wang and P. Zhou, Analyst, 2014, 139, 1550–1561 RSC.
- M. M. Ali, S. D. Aguirre, H. Lazim and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 3751–3754 CrossRef CAS PubMed.
- S. He, L. Qu, Z. Shen, Y. Tan, M. Zeng, F. Liu, Y. Jiang and Y. Li, Anal. Chem., 2014, 87, 569–577 CrossRef PubMed.
- Y. Chen and C. Mao, J. Am. Chem. Soc., 2004, 126, 8626–8627 CrossRef CAS PubMed.
- Y. Tian, Y. He, Y. Chen, P. Yin and C. Mao, Angew. Chem., Int. Ed., 2005, 44, 4355–4358 CrossRef CAS PubMed.
- M. Moshe, J. Elbaz and I. Willner, Nano Lett., 2009, 9, 1196–1200 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 119, 7731–7734 CrossRef.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5–14 RSC.
- R. McRae, P. Bagchi, S. Sumalekshmy and C. J. Fahrni, Chem. Rev., 2009, 109, 4780–4827 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- J. Yin, Y. Hu and J. Yoon, Chem. Soc. Rev., 2015, 44, 4619–4644 RSC.
- G. R. Hamilton, S. K. Sahoo, S. Kamila, N. Singh, N. Kaur, B. W. Hyland and J. F. Callan, Chem. Soc. Rev., 2015, 44, 4415–4432 RSC.
- A. E. Palmer and R. Y. Tsien, Nat. Protoc., 2006, 1, 1057–1065 CrossRef CAS PubMed.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- R. J. Radford and S. J. Lippard, Curr. Opin. Chem. Biol., 2013, 17, 129–136 CrossRef CAS PubMed.
- P. Wu, K. Hwang, T. Lan and Y. Lu, J. Am. Chem. Soc., 2013, 135, 5254–5257 CrossRef CAS PubMed.
- L. Li, J. Feng, Y. Fan and B. Tang, Anal. Chem., 2015, 87, 4829–4835 CrossRef CAS PubMed.
- K. Hwang, P. Wu, T. Kim, L. Lei, S. Tian, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 13798–13802 CrossRef CAS PubMed.
- Z. Wu, H. Fan, N. S. R. Satyavolu, W. Wang, R. Lake, J.-H. Jiang and Y. Lu, Angew. Chem., Int. Ed., 2017, 56, 8721–8725 CrossRef CAS PubMed.
- H. Meng, X. Zhang, Y. Lv, Z. Zhao, N. Wang, T. Fu, H. Fan, H. Liang, L. Qiu and G. Zhu, ACS Nano, 2014, 8, 6171–6181 CrossRef CAS PubMed.
- K. Hwang, P. Wu, T. Kim, L. Lei, S. Tian, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2014, 126, 14018–14022 CrossRef.
- X. Wang, M. Feng, L. Xiao, A. Tong and Y. Xiang, ACS Chem. Biol., 2015, 11, 444–451 CrossRef PubMed.
- W. Zhou, Y. Zhang, J. Ding and J. Liu, ACS Sens., 2016, 1, 600–606 CrossRef CAS.
- X. Gao, H. Huang, S. Niu, H. Ye, Z. Lin, B. Qiu and G. Chen, Anal. Methods, 2012, 4, 947–952 RSC.
- M. Hollenstein, C. J. Hipolito, C. H. Lam and D. M. Perrin, Nucleic Acids Res., 2009, 37, 1638–1649 CrossRef CAS PubMed.
- L. Lermer, Y. Roupioz, R. Ting and D. M. Perrin, J. Am. Chem. Soc., 2002, 124, 9960–9961 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838–9839 CrossRef CAS PubMed.
- K. Furukawa, A. Ramesh, Z. Zhou, Z. Weinberg, T. Vallery, W. C. Winkler and R. R. Breaker, Mol. Cell, 2015, 57, 1088–1098 CrossRef CAS PubMed.
- E. E. Regulski, R. H. Moy, Z. Weinberg, J. E. Barrick, Z. Yao, W. L. Ruzzo and R. R. Breaker, Mol. Microbiol., 2008, 68, 918–932 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |