
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for widespread, severe brain copper deficiency in Alzheimer's dementia†
Jingshu
Xu
 abc,
Stephanie J.
Church
bd,
Stefano
Patassini
abc,
Paul
Begley
bd,
Henry J.
Waldvogel
c,
Maurice A.
Curtis
c,
Richard L. M.
Faull
c,
Richard D.
Unwin
bd and
Garth J. S.
Cooper
*abcd
abc,
Stephanie J.
Church
bd,
Stefano
Patassini
abc,
Paul
Begley
bd,
Henry J.
Waldvogel
c,
Maurice A.
Curtis
c,
Richard L. M.
Faull
c,
Richard D.
Unwin
bd and
Garth J. S.
Cooper
*abcd
aSchool of Biological Sciences, Faculty of Science, and the Maurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: g.cooper@auckland.ac.nz; garth.cooper@manchester.ac.uk; Tel: +64 (9) 923 7394
bCentre for Advanced Discovery and Experimental Therapeutics, Central Manchester University Hospitals NHS Foundation Trust (CMFT), Manchester M13 9WL, UK
cCentre for Brain Research, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
dDivision of Cardiovascular Sciences, School of Medical Sciences, Faculty of Biology, Medicine and Health, The University of Manchester, and Manchester Academic Health Science Centre, Manchester M13 9NT, UK
First published on 16th June 2017
Abstract
Datasets comprising simultaneous measurements of many essential metals in Alzheimer's disease (AD) brain are sparse, and available studies are not entirely in agreement. To further elucidate this matter, we employed inductively-coupled-plasma mass spectrometry to measure post-mortem levels of 8 essential metals and selenium, in 7 brain regions from 9 cases with AD (neuropathological severity Braak IV–VI), and 13 controls who had normal ante-mortem mental function and no evidence of brain disease. Of the regions studied, three undergo severe neuronal damage in AD (hippocampus, entorhinal cortex and middle-temporal gyrus); three are less-severely affected (sensory cortex, motor cortex and cingulate gyrus); and one (cerebellum) is relatively spared. Metal concentrations in the controls differed among brain regions, and AD-associated perturbations in most metals occurred in only a few: regions more severely affected by neurodegeneration generally showed alterations in more metals, and cerebellum displayed a distinctive pattern. By contrast, copper levels were substantively decreased in all AD-brain regions, to 52.8–70.2% of corresponding control values, consistent with pan-cerebral copper deficiency. This copper deficiency could be pathogenic in AD, since levels are lowered to values approximating those in Menkes’ disease, an X-linked recessive disorder where brain-copper deficiency is the accepted cause of severe brain damage. Our study reinforces others reporting deficient brain copper in AD, and indicates that interventions aimed at safely and effectively elevating brain copper could provide a new experimental-therapeutic approach.
Significance to metallomicsThe work reported here shows that severe copper deficiency is widespread in the brains of patients with Alzheimer's dementia, whereas by contrast perturbations of other physiological metals are regionally localised and less marked. Severe copper deficiency occurred in all seven brain regions studied, despite them undergoing different degrees of structural-functional damage. The low brain-copper levels in Alzheimer's approximate those causing neurodegeneration and death in Menkes’ disease, a genetic disorder of copper transport. Brain-copper deficiency probably contributes to neurodegeneration in Alzheimer's and is an emergent target for therapeutic intervention to restore copper regulation and delay or prevent the onset/progression of dementia. |
Introduction
AD is the predominant cause of ageing-related dementia.1 Some 35 million people world-wide were living with dementia in 2010 and the estimated global prevalence will reach ∼115 million affected individuals by 2050.2 Consequently, AD represents the largest unmet medical need in neurology.3AD is characterized by the presence of extracellular amyloid-β (Aβ) plaques and intra-neuronal neurofibrillary tangles formed from tau protein in affected regions of the brain. These aggregates have been implicated in the causation of neuronal loss and neurodegeneration, which ultimately lead to the characteristic global impairment of cognitive function, but their role in pathogenesis remains unresolved and they may represent phenomena down-stream from the aetiological insult(s) that cause the disease.4 Moreover, therapeutic interventions aimed at these protein-level phenomena have not yielded an effective disease-modifying therapy.5
Perturbed brain-metal homeostasis has been associated with cognitive decline and neurodegeneration and hence may play a role in AD pathogenesis.6–9 However, the contribution of defective metal regulation to the pathogenesis of AD is unresolved and no available therapy targeting metal metabolism has an accepted place in the pharmacotherapy of AD.10 The brain is a highly oxidative organ, and contains abundant antioxidant mechanisms that may counteract the detrimental effects of reactive oxygen species (ROS) produced during the reactions of redox-active transition metals with molecular oxygen, superoxide and other oxygen-containing species.11 Metals may not only affect rates of synthesis, degradation and clearance of Aβ, but are also thought to play potential roles in the aggregation of Aβ and tau.7,12 Furthermore, as an integral component of many metalloenzymes, transition metals can modulate the function of numerous important pathways and, therefore, the metabolic processes they support.13 Results of therapeutic interventions aimed at Aβ or tau have been uniformly disappointing to date, so new ideas concerning the pathogenesis of AD leading to identification of more tractable targets and interventions, are required to meet the continuing unmet clinical need.5,14
Previous studies investigating changes in metal concentrations in human AD-brain have typically reported tissue content of one or a few metals (often iron (Fe), zinc (Zn), and/or copper (Cu)) in seriously affected regions such as the hippocampus. Furthermore, results of studies comparing levels of metals in the brains of AD patients and controls reported in the literature are somewhat heterogeneous,15 possibly due to experimental variability arising from factors such as differences in tissue processing: (e.g. fresh or formalin-fixed), handling, numbers of samples, and intrinsic variation in the distribution of metals in the brain tissue, resulting perhaps from both compartmentalization and inter-regional differences.7,16,17 Therefore, further mapping of the regional distribution of metals in the brain is required to substantiate and extend our understanding of the content of metals in various brain regions, and to associate these with region-specific variation in AD pathology. As one example, Cu, Fe and Zn are enriched in amyloid plaques from AD brain:18,19 by contrast, measurement of levels of these metals in whole-brain tissue from affected regions has yielded somewhat inconsistent values.20,21
To provide further evidence concerning the changes in concentration and regional distribution of metals in brain from both non-demented aged and cases with AD, we report a study that we performed using short-delay, fresh-frozen post-mortem human brain tissue by applying a validated inductively-coupled-plasma mass spectrometry (ICP-MS) method.22 We made parallel measurements of eight metals and selenium (Se), and compared and contrasted results across seven functionally distinct brain regions from cases with AD and matched controls without clinical dementia. Since some brain regions are more heavily affected by AD than others,23 we further aimed to contrast representative brain regions that are considered to be severely-affected: hippocampus (HP), entorhinal cortex (ENT), and middle-temporal gyrus (MTG);23,24 moderately-affected: sensory cortex (SCX), motor cortex (MCX), and cingulate gyrus (CG); along with one control region, cerebellum (CB), which is relatively spared in the disease process.25,26
Materials and methods
Acquisition and sampling of human brain
Whole brains from cases and matched controls were obtained from the New Zealand Neurological Foundation Douglas Human Brain Bank, in the Centre for Brain Research, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand.27 All procedures in this study were approved by the University of Auckland Human Participants Ethics Committee with informed consent from all families, and its performance was consistent with international best practice for such studies.After receipt into the Brain Bank, each brain was dissected under the supervision of neuroanatomists, to ensure accurate identification of each brain region to be studied.27,28 Two sets of tissue samples (each of 50 ± 5 mg) were dissected from each region and stored at −80 °C until analysis.
Diagnosis and severity
All cases had clinical dementia, whereas age- and sex-matched controls did not. Study-group characteristics are shown in Table 1, and each patient's details, including cause(s) of death as certified by post-mortem examination, are summarized in Table 2. A consultant neuropathologist diagnosed or excluded AD by applying the Consortium to Establish a Registry for AD Disease (CERAD) criteria,29 and also determined the neuropathological severity by assigning the Braak stage25 to each brain (Tables 1 and 2).| Variable | Control | AD |
|---|---|---|
| Values are: age, post-mortem delay and brain-wt, median (range); wet-wt/dry-wt ratio, mean (±95% CI) averaged across all samples: *P = 0.005, **P < 0.0001 compared with control; all other differences are non-significant. | ||
| Number | 13 | 9 |
| Age | 73 (61–78) | 72 (60–80) |
| Male sex, n (%) | 7 (53.8) | 5 (55.6) |
| Post-mortem delay (h) | 12 (5.5–15.0) | 7 (4.0–12.0)* |
| Brain-wt (g) | 1260 (1094–1461) | 1062 (831–1355)* |
| Wet-wt/dry-wt | 5.7 (5.6–5.9) | 5.5 (5.4–5.6) |
| Plaques, n (%) | 1 (7.7) | 9 (100)** |
| Tangles, n (%) | 1 (7.7) | 9 (100)** |
| Code | Group | Age/sex | Ante-mortem assessment of brain disease/mental state | Cause of death | Braak stage | PMD (h) | Brain wt (g) |
|---|---|---|---|---|---|---|---|
| Abbreviations: GI, gastrointestinal; MI, myocardial infarction; PMD, post-mortem delay; wt, weight. Cause of death was determined by post-mortem examination, and brain pathology and Braak stage were assigned by specialist neuropathological examination. Causes of death were the primary causes listed on the death certificate. Patient H241 was found to have post-mortem signs consistent with AD and was therefore diagnosed with prodromal disease: data corresponding to this patient have been retained in the main analysis presented in this manuscript. | |||||||
| H155 | Control | 61/M | No brain disease or dementia | Ischaemic heart disease | 0 | 7.0 | 1258 |
| H121 | Control | 64/F | No brain disease or dementia | Pulmonary embolism | 0 | 5.5 | 1260 |
| H132 | Control | 63/F | No brain disease or dementia | Ruptured aorta | 0 | 12.0 | 1280 |
| H122 | Control | 72/F | No brain disease or dementia | Emphysema | 0 | 9.0 | 1230 |
| H204 | Control | 66/M | No brain disease or dementia | Ischaemic heart disease | 0 | 9.0 | 1461 |
| H241 | Control | 76/F | No brain disease or dementia | Metastatic carcinoma | II | 12.0 | 1094 |
| H164 | Control | 73/M | No brain disease or dementia | Ischaemic heart disease | 0 | 13.0 | 1315 |
| H123 | Control | 78/M | No brain disease or dementia | Ruptured aortic aneurysm | 0 | 7.5 | 1260 |
| H150 | Control | 78/M | No brain disease or dementia | Ruptured MI | 0 | 12.0 | 1416 |
| H168 | Control | 63/M | No brain disease or dementia | Ischaemic heart disease | 0 | 9.0 | 1432 |
| H137 | Control | 77/F | No brain disease or dementia | Coronary atherosclerosis | 0 | 12.0 | 1227 |
| H131 | Control | 73/F | No brain disease or dementia | Ischaemic heart disease | 0 | 13.0 | 1210 |
| H157 | Control | 66/M | No brain disease or dementia | Ischaemic heart disease | 0 | 15.0 | 1360 |
| AZ42 | AD | 60/M | Alzheimer's dementia | Alzheimer's disease | VI | 7.0 | 1020 |
| AZ71 | AD | 62/F | Alzheimer's dementia | Alzheimer's disease | VI | 6.0 | 831 |
| AZ48 | AD | 63/F | Alzheimer's dementia | Bronchopneumonia | VI | 7.0 | 1080 |
| AZ72 | AD | 70/F | Alzheimer's dementia | Lung cancer | V | 7.0 | 1044 |
| AZ90 | AD | 73/M | Alzheimer's dementia | GI haemorrhage | IV | 4.0 | 1287 |
| AZ96 | AD | 74/F | Alzheimer's dementia | Metastatic cancer | V | 8.5 | 1062 |
| AZ39 | AD | 74/M | Alzheimer's dementia | Pseudomonas bacteraemia | VI | 12.0 | 1355 |
| AZ80 | AD | 77/M | Alzheimer's dementia | Myocardial infarction | VI | 4.5 | 1180 |
| AZ38 | AD | 80/M | Alzheimer's dementia | Bronchopneumonia/pulmonary oedema | V | 5.5 | 1039 |
Brain extracts
We determined metal concentrations normalized on a per-dry-weight basis.30–33 Brain samples of 50 ± 5 mg wet-weight were first dried to constant weight in a centrifugal concentrator (Savant Speedvac; Thermo-Fisher, Waltham, MA) and dry-weight was measured using a Mettler Toledo New Classic ML balance (ML204/01): the coefficient of variation (CV) across 25 × 10 mg measurements was 0.68%. Wet-weight/dry-weight ratios are recorded in Table 1. Samples were digested in 2 mL microcentrifuge tubes (eppendorf) as described below.Digestion
Tissue was digested using concentrated nitric acid (A509 Trace Metal Grade; Fisher, Loughborough, UK) with added 5% (v/v) Agilent Internal Standard mixture (5183-4681; Agilent Technologies, Cheadle, UK). An internal standard solution was added to the digestion solution to account for evaporation-associated volume loss during the digestion protocol. This internally standardized acid was also used at appropriate dilutions to provide rinse and calibration solutions, at 2% (v/v) final nitric acid concentration. Calibration solutions were produced by appropriate dilutions of Environmental Calibration Standard (Agilent 5183-4688).Acid digestion was carried out using an ‘open-vessel’ method. Tissue aliquots were briefly centrifuged to ensure that the tissue sat at the bottom of the tube. The tube lids were punctured to prevent pressure build-up, and 0.2 mL standard-containing nitric acid added. Tubes were then inserted into a “Dri-block” heater, which was initially at room temperature. Plasma (SRM 1950) from the National Institute of Standards and Technology (NIST), Gaithersburg, USA was processed in parallel with experimental samples as standard reference material (Table S1, ESI†). Tube s with standard-containing acid but no sample were also processed in each batch to provide “digestion” blanks (Table S2, ESI†). Temperature was then set to 60 °C and the block switched on. After 30 min, the set temperature was increased to 100 °C, and digestion continued for a further 210 min. After digestion, the tubes were allowed to cool overnight.
Aliquots of 100 μL were taken from each digestion and added to 15 mL Falcon tubes (Greiner) containing 5 mL LC-MS grade water, to produce solutions for analysis at a final nitric acid concentration of 2% (v/v).
ICP-MS
Metal concentrations were measured using an Agilent 7700x ICP-MS spectrometer equipped with a MicroMist nebulizer (Glass Expansion, Melbourne, Australia) and a Scott double-pass spray chamber. Nickel sample and skimmer cones were used. Sample introduction was by using an Agilent Integrated autosampler (I-AS) with helium as the collision gas. A multi-element method including all those present in the calibration solution was applied as previously reported.22 Calibration solutions were produced by appropriate dilutions of Environmental Calibration Standard (Agilent 5183-4688). Scandium was used as the internal standard for all elements except Zn and Se, where germanium was used, and Mo, where indium was used. Two collision cell gas modes were applied, and all elements were analysed in helium mode (5.0 mL min−1 helium), except for Se which was analysed in high-energy helium mode (10 mL min−1 helium). Germanium and indium internal standards were analysed in both modes. Mode selection followed Agilent's recommendations to minimize interference for measured elements by e.g. isobaric cluster ions. Integration times were 0.1 s for sodium (Na), magnesium (Mg), potassium (K) and calcium (Ca), 0.3 s for manganese (Mn), Cu, Zn and molybdenum (Mo), 0.01 s for Fe, and 3 s for Se. For each analytical batch, multi-element calibration was performed using serial dilutions of the calibration standard (Fig. S1, ESI†). An intermediate concentration from this calibration series was used as a periodic quality-control (QC) sample throughout each analytical batch. Instrument and digestion blanks were also interspersed through each set of randomized samples. Detection limit, limit of quantitation and background equivalent concentration for each physiological metal measured in this study (Table S3, ESI†) were automatically calculated by the software employed (Mass Hunter, Agilent).ICP-MS generates measurements of metals in their elemental states, from samples that have been rendered into plasmas: therefore, where the result of an ICP-MS-based measurement is referred to in this manuscript, the elemental symbol has been used (e.g. K for potassium). On the other hand, most metals are present in the body as cations: therefore, when the physiological role(s) of metals and metal-related processes are discussed, the symbol for the physiological cation has been employed (e.g. K+ for the potassium ion, or Cu(II) for the physiological divalent-Cu cation).
Data analysis
Datasets were exported to Microsoft Excel worksheets and individual values of each sample normalized by the corresponding sample dry-weight. Weight-adjusted datasets were then log-transformed for statistical analysis. Means (±95% CI) of the log-transformed data were calculated and the significance of between-group differences was examined by unpaired Welch's t-tests to allow for unequal variances and sample sizes. Means (±95% CI) were back-transformed to reflect the actual elemental concentrations of elements. Statistical calculations were performed using GraphPad v6.04 (Prism; La Jolla, CA). P-values <0.05 have been considered significant, and those of 0.05 ≤ P < 0.10 are also tabulated.Results
We measured concentrations of eight essential metals, Na, Mg, K, Ca, Mn, Fe, Cu, and Zn, and the metalloid Se, in seven regions of human post-mortem brains from nine AD cases and 13 controls matched for age and sex (Tables 1 and 2). Median (range) brain weight was 1062 g (831–1355) in the patient group and 1260 g (1094–1461; P = 0.005) in the controls (Tables 1 and 2): the ∼16% decrease in mean brain weight in cases is consistent with the histological severity.34 The post-mortem delay in the AD group, 7 h (4.0–12.0), was significantly shorter than in the control group, 12 h (5.5–15.0; P = 0.005; Tables 1 and 2). The AD group comprised sporadic cases only, as determined by clinical, CERAD and Braak criteria. One control case also had neuropathological findings consistent with early-stage AD (Braak II; Table 2) and was therefore diagnosed with premanifest disease: this finding is consistent with the known frequency of asymptomatic AD in similarly-aged groups in the study population.35 Wet-wt/dry-wt ratios did not differ significantly between cases or controls (Table 1).In AD, measured Na levels were higher in severely affected regions (HP, ENT and MTG, where values ranged from 0.55 to 0.59 mol per kg dry-wt) compared with corresponding regions in control brains (whose values ranged from 0.30 to 0.41 mol per kg dry-wt; Tables 3–5). Mean Na levels were the highest in CB of normal brain (Fig. 1a and Tables 3–9).
| Element | Units | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 13) and AD (n = 9) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.41 (0.36–0.46) | 0.59 (0.53–0.66) | <0.0001 |
| Mg | (mol per kg dry-wt) | 24Mg | 0.028 (0.025–0.031) | 0.028 (0.025–0.031) | ns |
| K | (mol per kg dry-wt) | 39K | 0.37 (0.31–0.43) | 0.31 (0.27–0.37) | ns |
| Ca | (mmol per kg dry-wt) | 44Ca | 8.87 (7.31–10.79) | 10.84 (8.79–13.34) | ns |
| Mn | (μmol per kg dry-wt) | 55Mn | 28 (26–30) | 35 (30–42) | 0.010 |
| Fe | (mmol per kg dry-wt) | 56Fe | 3.71 (3.25–4.23) | 5.62 (4.40–7.18) | 0.0047 |
| Cu | (μmol per kg dry-wt) | 63Cu | 330 (260–419) | 183 (128–261) | 0.0066 |
| Zn | (μmol per kg dry-wt) | 66Zn | 1274 (1059–1531) | 1400 (1148–1702) | ns |
| Se | (μmol per kg dry-wt) | 78Se | 11 (9–14) | 11 (9–12) | ns |
| Element | Concentration unit | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 13) and AD (n = 9) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.32 (0.26–0.39) | 0.57 (0.46–0.70) | 0.0003 |
| Mg | (mol per kg dry-wt) | 24Mg | 0.025 (0.023–0.028) | 0.031 (0.029–0.034) | 0.0017 |
| K | (mol per kg dry-wt) | 39K | 0.33 (0.29–0.38) | 0.36 (0.3–0.43) | ns |
| Ca | (mmol per kg dry-wt) | 44Ca | 6.71 (4.65–9.68) | 10.57 (8.15–13.71) | 0.037 |
| Mn | (μmol per kg dry-wt) | 55Mn | 27 (18–41) | 38 (31–46) | ns |
| Fe | (mmol per kg dry-wt) | 56Fe | 4.61 (3.98–5.35) | 5.88 (5.15–6.71) | 0.013 |
| Cu | (μmol per kg dry-wt) | 63Cu | 309 (281–340) | 202 (146–281) | 0.018 |
| Zn | (μmol per kg dry-wt) | 66Zn | 968 (804–1167) | 1422 (1117–1807) | 0.011 |
| Se | (μmol per kg dry-wt) | 78Se | 12 (10–15) | 13 (10–16) | ns |
| Element | Concentration Unit | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 13) and AD (n = 9) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.3 (0.25–0.37) | 0.55 (0.44–0.68) | 0.0002 |
| Mg | (mol per kg dry-wt) | 24Mg | 0.027 (0.025–0.029) | 0.030 (0.028–0.032) | 0.0212 |
| K | (mol per kg dry-wt) | 39K | 0.39 (0.35–0.44) | 0.37 (0.33–0.41) | ns |
| Ca | (mmol per kg dry-wt) | 44Ca | 10.09 (5.12–19.95) | 13.12 (6.38–27.04) | ns |
| Mn | (μmol per kg dry-wt) | 55Mn | 23 (19–28) | 26 (23–30) | ns |
| Fe | (mmol per kg dry-wt) | 56Fe | 5.22 (4.70–5.81) | 6.67 (5.89–7.57) | 0.0035 |
| Cu | (μmol per kg dry-wt) | 63Cu | 356 (300–422) | 239 (195–293) | 0.0035 |
| Zn | (μmol per kg dry-wt) | 66Zn | 1042 (923–1175) | 1413 (1233–1618) | 0.0013 |
| Se | (μmol per kg dry-wt) | 78Se | 14 (11–18) | 14 (11–18) | ns |
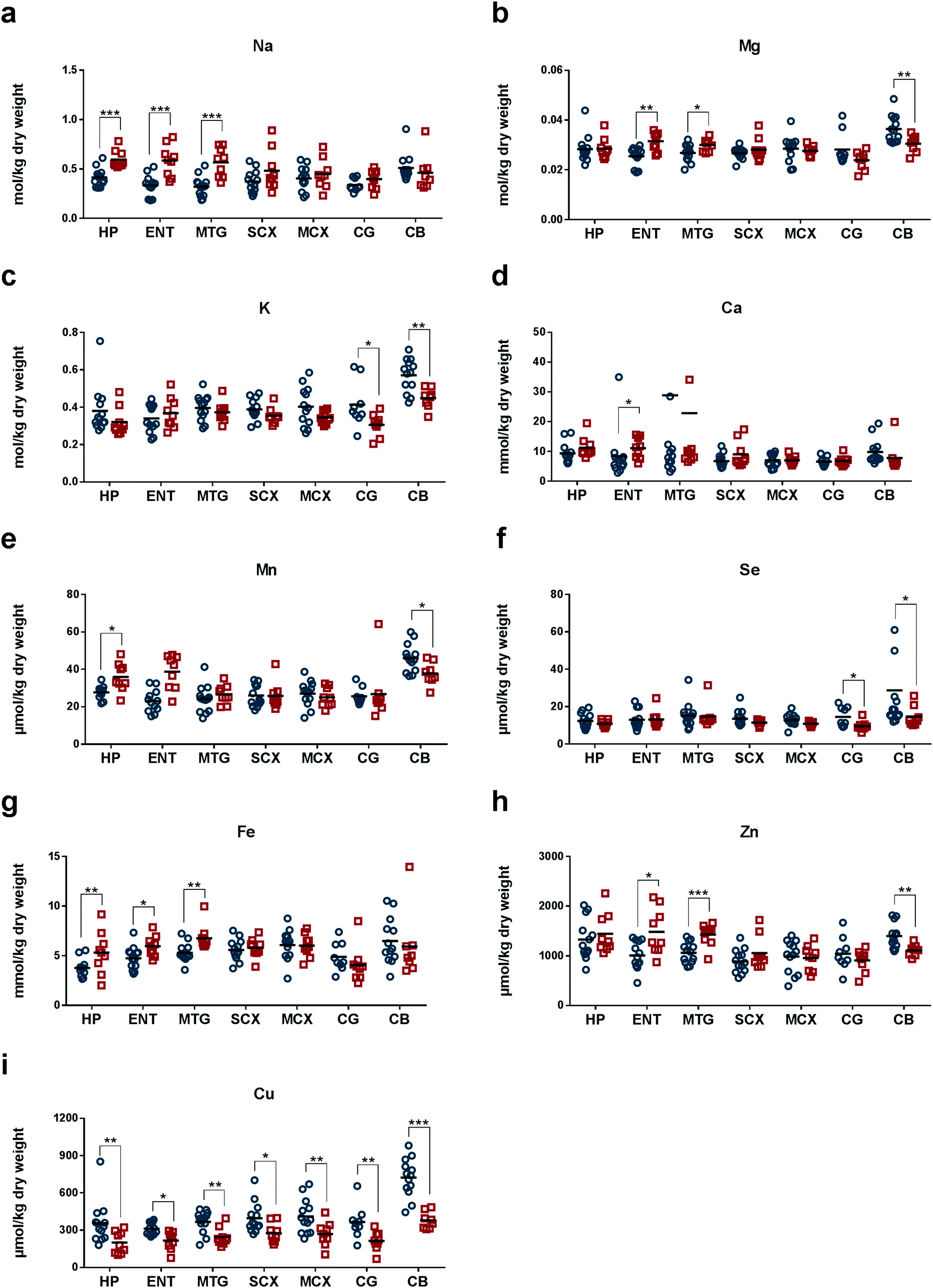 | ||
| Fig. 1 Concentrations of nine physiologically essential elements (a–i) in seven brain regions compared between AD cases and matched controls. Shown are elemental concentrations in each region of human post-mortem brain tissue from control (blue) and AD (red) subjects. Mean and between-group significance of calcium levels shown in (d), MTG, were determined from all measurements; two outlying values, one from each group, were excluded from the plot for clarity and all other data sets are complete as shown. Abbreviations: hippocampus, HP; entorhinal cortex, ENT; middle-temporal gyrus, MTG; sensory cortex, SCX; motor cortex, MCX; cingulate gyrus, CG; and cerebellum, CB: *, <0.05; **, ≤0.01; ***, ≤0.001 cases vs. controls. | ||
| Element | Concentration unit | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 13) and AD (n = 9) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.36 (0.3–0.43) | 0.45 (0.33–0.61) | ns |
| Mg | (mol per kg dry-wt) | 24Mg | 0.027 (0.025–0.028) | 0.028 (0.025–0.031) | ns |
| K | (mol per kg dry-wt) | 39K | 0.38 (0.35–0.42) | 0.35 (0.32–0.38) | ns |
| Ca | (mmol per kg dry-wt) | 44Ca | 6.41 (5.31–7.73) | 8.24 (5.91–11.48) | ns |
| Mn | (μmol per kg dry-wt) | 55Mn | 25 (22–29) | 25 (21–30) | ns |
| Fe | (mmol per kg dry-wt) | 56Fe | 5.48 (4.88–6.18) | 5.73 (4.99–6.58) | ns |
| Cu | (μmol per kg dry-wt) | 63Cu | 382 (323–451) | 268 (218–330) | 0.0079 |
| Zn | (μmol per kg dry-wt) | 66Zn | 861 (735–1012) | 1021 (826–1259) | ns |
| Se | (μmol per kg dry-wt) | 78Se | 13 (11–15) | 11 (10–13) | ns |
| Element | Concentration unit | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 13) and AD (n = 9) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.39 (0.32–0.47) | 0.43 (0.33–0.55) | ns |
| Mg | (mol per kg dry-wt) | 24Mg | 0.028 (0.025–0.031) | 0.028 (0.026–0.029) | ns |
| K | (mol per kg dry-wt) | 39K | 0.39 (0.33–0.46) | 0.34 (0.32–0.37) | ns |
| Ca | (mmol per kg dry-wt) | 44Ca | 6.73 (5.55–8.15) | 6.86 (5.92–7.96) | ns |
| Mn | (μmol per kg dry-wt) | 55Mn | 26 (22–31) | 25 (21–29) | ns |
| Fe | (mmol per kg dry-wt) | 56Fe | 5.88 (4.93–7.00) | 5.90 (5.06–6.90) | ns |
| Cu | (μmol per kg dry-wt) | 63Cu | 390 (319–478) | 252 (184–347) | 0.019 |
| Zn | (μmol per kg dry-wt) | 66Zn | 925 (728–1175) | 925 (743–1151) | ns |
| Se | (μmol per kg dry-wt) | 78Se | 12 (11–15) | 11 (10–12) | 0.097 |
| Element | Concentration unit | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 9) and AD (n = 9) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.33 (0.29–0.38) | 0.39 (0.32–0.47) | ns |
| Mg | (mol per kg dry-wt) | 24Mg | 0.028 (0.024–0.032) | 0.024 (0.021–0.027) | 0.093 |
| K | (mol per kg dry-wt) | 39K | 0.40 (0.32–0.5) | 0.30 (0.26–0.35) | 0.028 |
| Ca | (mmol per kg dry-wt) | 44Ca | 6.41 (5.42–7.59) | 6.64 (5.63–7.84) | ns |
| Mn | (μmol per kg dry-wt) | 55Mn | 25 (22–29) | 25 (18–33) | ns |
| Fe | (mmol per kg dry-wt) | 56Fe | 4.71 (3.77–5.88) | 3.79 (2.84–5.07) | ns |
| Cu | (μmol per kg dry-wt) | 63Cu | 346 (264–454) | 198 (141–280) | 0.010 |
| Zn | (μmol per kg dry-wt) | 66Zn | 998 (777–1282) | 880 (708–1094) | ns |
| Se | (μmol per kg dry-wt) | 78Se | 14 (10–18) | 9.3 (7.7–11) | 0.019 |
| Element | Concentration unit | Reference isotope | Control | AD | P-value |
|---|---|---|---|---|---|
| Data are means (±95% CI); P-values for significance of between-group differences were calculated by Welch's t-tests based on measurements from control (n = 13) and AD (n = 8) brains. | |||||
| Na | (mol per kg dry-wt) | 23Na | 0.50 (0.44–0.57) | 0.44 (0.33–0.58) | ns |
| Mg | (mol per kg dry-wt) | 24Mg | 0.036 (0.033–0.039) | 0.030 (0.028–0.033) | 0.0073 |
| K | (mol per kg dry-wt) | 39K | 0.56 (0.51–0.62) | 0.45 (0.4–0.5) | 0.0016 |
| Ca | (mmol per kg dry-wt) | 44Ca | 9.16 (7.35–11.43) | 6.98 (4.82–10.09) | ns |
| Mn | (μmol per kg dry-wt) | 55Mn | 45 (41–50) | 37 (33–43) | 0.018 |
| Fe | (mmol per kg dry-wt) | 56Fe | 6.11 (4.90–7.62) | 5.38 (3.75–7.71) | ns |
| Cu | (μmol per kg dry-wt) | 63Cu | 708 (617–811) | 374 (323–434) | <0.0001 |
| Zn | (μmol per kg dry-wt) | 66Zn | 1380 (1242–1535) | 1104 (1012–1205) | 0.0018 |
| Se | (μmol per kg dry-wt) | 78Se | 23 (15–34) | 14 (10–18) | 0.034 |
Mg levels were significantly higher in ENT and MTG, but lower in CB of AD-brain (Fig. 1b and Tables 4, 5, 9). In these brain regions, Mg concentrations were all around 0.03 mol per kg dry-wt in AD-brain. However, in normal brain, Mg levels varied among different brain regions. In the control brains, mean Mg concentrations were highest in CB (0.036 mol per kg dry-wt; Table 9), and much lower in ENT and MTG (0.025–0.027 mol per kg dry-wt; Tables 4 and 5). Similar to Na and Mg, K showed the highest mean concentrations in CB (0.56 mol per kg dry-wt) in normal brain (Fig. 1c and Table 9). The mean concentration of K was the second highest in CG in the normal brain (0.40 mol per kg dry-wt; Fig. 1c and Table 8). Compared to normal brain tissue, levels of K were significantly lowered in CG and CB of AD (Fig. 1c and Tables 8, 9).
Compared to controls, mean concentrations of Ca in AD brain trended higher in all brain regions except for CB (although values reached statistical significance only in ENT; Fig. 1d and Tables 3–9).
In normal brain, mean Mn concentration was the highest in CB (45 μmol per kg dry-wt) and similar in all other regions (23–28 μmol per kg dry-wt; Fig. 1e and Tables 3–9). In AD, Mn levels were significantly higher in HP (Table 3) but lower in CB (Table 9). In control brain, Se concentrations were highest in CB (23 μmol per kg dry-wt), and second highest in CG and MTG (14 μmol per kg dry-wt), whereas the rest of the brain regions fell within the range of 11–13 μmol per kg dry-wt (Fig. 1f and Tables 3–9). In AD tissue, Se levels were significantly lower in CG (9.3 μmol per kg dry-wt) and CB (14 μmol per kg dry-wt) when compared to controls (Tables 8 and 9).
In normal brain, Fe levels were highest in CB (6.1 mmol per kg dry-wt) and lowest in HP (3.7 mmol per kg dry-wt; Fig. 1g and Tables 3–9). AD tissue showed significantly higher levels of Fe in HP, ENT, and MTG (Tables 3–5), whereas no significant differences were observed in other brain regions. Interestingly, mean Fe levels were lower in CG and CB in AD compared to controls (Tables 8 and 9). Similarly, Zn concentrations were higher in heavily affected regions, ENT and MTG (Tables 4 and 5) but lower in CB (Table 9) in AD compared to control. Mean Zn concentrations were highest in CB (1.9 mmol per kg dry-wt) and HP (1.3 mmol per kg dry-wt; Fig. 1h and Tables 1–9).
Mean Cu concentrations in controls were highest in CB (710 μmol per kg dry-wt) and lowest in ENT (310 μmol per kg dry-wt). Remarkably, Cu levels were significantly decreased in all seven regions of AD-brain compared with corresponding controls (Fig. 1i and Tables 3–9), consistent with the presence of pan-cerebral Cu deficiency. This pattern distinguished Cu from all other elements measured.
Brain-metal concentrations in one control case (H241; Table 2), who had signs of premanifest AD (Braak stage II), are presented in Table 10: these values do not enable clear differentiation from either experimental group.
| Element | Concentration unit | Reference isotope | Brain regions | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HP | ENT | MTG | SCX | MCX | CG | CB | |||
| Abbreviations: hippocampus, HP; entorhinal cortex, ENT; middle-temporal gyrus, MTG; sensory cortex, SCX; motor cortex, MCX; cingulate gyrus, CG; and cerebellum, CB. | |||||||||
| Na | (mol per kg dry-wt) | 23Na | 0.43 | 0.35 | 0.25 | 0.46 | 0.37 | 0.30 | 0.49 |
| Mg | (mol per kg dry-wt) | 24Mg | 0.027 | 0.027 | 0.027 | 0.028 | 0.031 | 0.024 | 0.031 |
| K | (mol per kg dry-wt) | 39K | 0.34 | 0.41 | 0.37 | 0.43 | 0.47 | 0.42 | 0.46 |
| Ca | (mmol per kg dry-wt) | 44Ca | 16.26 | 7.03 | 12.30 | 10.06 | 5.60 | 6.05 | 7.52 |
| Mn | (μmol per kg dry-wt) | 55Mn | 30 | 23 | 24 | 32 | 29 | 25 | 60 |
| Fe | (mmol per kg dry-wt) | 56Fe | 2.96 | 5.77 | 6.76 | 5.36 | 5.87 | 4.80 | 8.68 |
| Cu | (μmol per kg dry-wt) | 63Cu | 231 | 316 | 231 | 360 | 384 | 325 | 709 |
| Zn | (μmol per kg dry-wt) | 66Zn | 1512 | 1152 | 943 | 1106 | 1089 | 1080 | 1268 |
| Se | (μmol per kg dry-wt) | 78Se | 15 | 20 | 20 | 17 | 11 | 19 | 23 |
Discussion
Here we report measurements of physiologically-essential metal concentrations in seven regions of post-mortem brain in cases with AD and controls matched for age and male-to-female ratio, whose ante-mortem brain structure and function had been assessed as normal. Although post-mortem tissue metal values may be affected by ion gradients between cells and extracellular compartments, they are neither metabolized nor exchanged with other tissues after death, due to cessation of blood circulation. There is no pronounced correlation between post-mortem delay and tissue metal content: hence, it has been suggested that total brain-tissue metal content does not change significantly after death.36 Therefore, the relatively small differences in post-mortem delay between case and control groups in this study are unlikely to have affected the accuracy of our measurements and hence, the validity of comparisons between AD and control groups.The maintenance of physiological intracellular Na+ and K+ levels is critical for controlling the homoeostasis of cell volume and prevention of apoptosis.37 The marked increase of Na levels in the most-affected regions of AD-brain (HP, ENT, and MTG) observed here is consistent with another report of increased Na concentrations in AD in the medial temporal lobes,38 and frontal and parietal cortex.36 We also observed, in moderately affected regions (MCX, SCX, and CG), that there were non-significant trends towards increased Na levels. Na levels in CB did not differ between groups, consistent with another report that cerebellar Na levels did not differ between post-mortem human AD and control brain.36 Increased Na content has also been inversely correlated with brain volume in the HP.38 Therefore, given that heavily-affected brain regions reportedly undergo volume losses at greater rates during disease progression,39 our observation is consistent with the idea that Na+ levels increase in regions of severe neurodegeneration/volume loss in AD, perhaps due to diminished energy supply required for aspects of cell-Na+ homeostasis.
Shrinkage of grey matter is commonly regarded as a consequence of neuronal cell death.40 Cell death and cell shrinkage are distinguishable processes that are intrinsically linked, in that cell shrinkage is a universal feature of apoptosis that is conserved among species. Intracellular Na+ reportedly increases transiently to initiate apoptotic signalling prior to cell-volume loss, and a significant decrease in intracellular K+ and Na+ is known to occur during apoptotic cell-shrinkage.37,41 Whether the elevation in Na levels observed here is associated with neuronal apoptosis is undetermined. While a previous study has reported decreased brain K levels in AD,42 here we found no changes of K levels in the severely affected regions of AD-brain. Instead, we identified significant lowering of K in CG and CB, at possible variance with a reported increase in K concentration in CB of AD-brain.36 It was also suggested that K+ efflux mediates cell death by apoptosis.37,43 Consistently, our observations may represent a result of K+ efflux associated with apoptosis in these regions prior to death in AD patients, possibly due to insufficient energy supply to maintain K+ homeostasis. CSF levels of Na and K did not differ between AD cases and controls in another report,36 consistent with the altered concentrations of both metals observed in our current study, which may reflect changes in the concentrations of both in the intracellular compartment.
Neuronal Mg2+ not only participates in energy production (e.g. as a component of purine nucleoside complexes), intracellular signalling and synaptic neurotransmission, but also modulates synaptic density and plasticity.44 Mg2+ displays a non-homogeneous distribution among different regions of normal brain, and significant decreases have been reported in AD.45 Here we found that brain Mg showed a heterogeneous distribution, with CB having the highest levels in normal brain. In AD however, Mg concentrations were increased in ENT and MTG but relatively decreased in CB, resulting in a more homogenous distribution of Mg across different brain regions as compared with normal brain. Prolonged exposure to elevated Mg2+ is said to exert an overall inhibitory effect on neuronal transmission resulting in decreased neuronal survival,44 which could be consistent with increased Mg levels in the heavily affected regions of ENT and MTG. On the contrary, whether the markedly decreased Mg concentration in the CB is associated with improved neuronal survival remains uncertain.
Calcium homeostasis is critical for maintenance of normal brain function since Ca2+ ions mediate neuronal signal transduction and modulate processes including synaptic plasticity and apoptosis.46–48 Characteristic lesions of AD have been associated with increased intracellular Ca2+, including Aβ accumulation, tau hyperphosphorylation, and neuronal death.48 Excessive Ca2+ is implicated as a cause of acute neuronal injury, possibly through neurotoxicity induced by excitatory amino acids.47,49 Here, Ca levels were significantly increased only in ENT, possibly reflecting a Ca2+-associated neurotoxicity localized in this brain region, which is known to be an initial site of neurodegeneration in AD. Our findings also suggest alteration in total Ca concentrations in the affected regions of AD tissue following progressive neurodegeneration, although they do not exclude the possibility that disturbed Ca2+ homeostasis occurs before neurodegeneration.
As an important antioxidant, Se plays a protective role against oxidative stress, and its concentration is well-maintained by the brain, even during dietary Se deficiency.50 Se deficiency has been correlated with altered cognitive function51,52 and decreased serum and plasma Se is reported in AD.53–55 However, systematic examination of Se levels in AD-brain has not been reported to our knowledge. Here we found that Se concentrations were not altered in the heavily or moderately affected brain regions. Rather, significant decreases were observed in CG and CB of AD-brain. This finding may be consistent with previously observed co-localization of the Se-transport protein, selenoprotein P, with Aβ plaques and neurofibrillary tangles,56 possibly serving as a compensatory mechanism to maintain Se levels in affected locations in AD brain.
Oxidative damage is an important aspect of AD pathology. Cu, Zn and Mn participate in oxidative defence mechanisms as necessary components of the superoxide dismutase (SOD) enzymes, including Mn-SOD (SOD2), present mainly in mitochondria,57 and two Cu/Zn-SODs (SOD1, SOD3), present respectively in the cytosol and mitochondria, and in the extracellular space.58
Mitochondrial oxidative damage and impaired energy production are considered to be early pathological events that lead to neurodegeneration. Mice with brain-specific Mn-SOD deficiency exhibit spongiform neurodegeneration of the brain accompanied by increased lipid peroxidation59 and Mn-SOD may play a protective role against apoptosis and neuronal degeneration.60 Furthermore, Mn-deficient animals exhibit depressed Mn-SOD, with associated increase in lipid peroxidation.61 Additionally, Mn excess confers neurotoxicity in the central nervous system, through both oxidative damage and involvement of Mn in the activation of the enzyme glutamine synthetase and therefore, in glutaminergic and GABAergic neurotransmission.62,63 Previous studies reported decreased serum Mn levels in AD patients.55 However, data concerning altered brain levels of Mn remain inconclusive in the current literature; independent studies have reported either increased Mn levels64 or no significant alteration65 in Mn levels in AD post mortem brain. This is not surprising considering the inter-regional difference in Mn levels in the brain as observed in this study. Our results indicate that Mn homeostasis is disturbed in both HP and CB, but evidently in opposite directions, since Mn levels were increased in HP and decreased in CB of AD-brain. Also, acting through mitochondrial electron transport chain (mETC) complex II, Mn2+ reportedly elicits mitochondrial hydrogen peroxide production.66,67 The increase in Mn concentration observed here in the HP could be associated with severe neurodegeneration through increased oxidative stress via this mechanism.
The metal hypothesis of AD pathogenesis was originally built upon a triad of altered homeostasis of the transition elements: Fe, Zn and Cu. In AD brain, dysregulation of metal homeostasis is postulated to engender amyloidogenic and oxidative stress as a consequence, and, in reverse, AD-related proteins (for example, APP and tau) may play significant roles in brain-metal regulation.8,16 Previous studies reported elevated levels of Fe68 and Zn69 in extracts of AD brain and increased levels of labile Fe in the serum of AD patients.55 Furthermore, Fe levels were reported to be markedly increased in degenerating regions of AD brain.70 Here we also found significant increases in Fe and Zn levels that were localized in heavily affected regions. This effect could possibly be associated with overall load of plaques and tangles in these regions, considering the reported enrichment of Fe and Zn in and around amyloid plaques in AD.19 Decreased transferrin in cortex may impair brain-Fe utilization and subsequently drive oxidative damage and neuronal degeneration in affected regions.71
In the brain, Zn2+ is co-localized with glutamate and modulates both excitatory and inhibitory neurotransmission.72 Zn2+ not only elicits potential roles as a (putative) neurotransmitter and modulator of synaptic plasticity, but is also thought to play a potential role in AD pathogenesis related to its interactions with APP and Aβ, reportedly participating in APP processing, Aβ aggregation and Aβ clearance. Enrichment of Zn in the core of Aβ plaques might disrupt Zn homeostasis in brain regions important for memory and vulnerable to AD pathogenesis.73,74 Our observations of increased Zn in ENT and MTG mirrors previous reports of its elevation in AD brain,69 which may accord with the (proposed) increase of Zn2+-sequestration in heavily affected brain regions, potentially leading to functional unavailability of Zn2+ required for physiological processes, regardless of any increase in total tissue-Zn. Whether or how the previously reported decrease in serum Zn levels in AD55 is associated with altered brain-Zn regulation is unclear. A previous study has suggested that HP may contain the highest Zn content among those brain-regions examined:17 consistently, we found Zn levels to be higher in HP than other regions, although they approximated those in CB.
Cu is a necessary component of certain metalloenzymes that play key roles in essential metabolic processes such as energy metabolism, antioxidant-defence mechanisms, Fe metabolism, and neurotransmitter synthesis.75 It is still controversial as to whether it is Cu excess or Cu deficiency that might contribute to the pathogenesis of AD.76 Here we found Cu levels to be significantly depressed in all seven regions of AD-brain examined (Fig. 1). There are several reports that tissue-Cu concentration is decreased in several regions of post-mortem brain in AD.68,69 These findings, taken together, are consistent with reported depression of cytochrome C oxidase (COX) activity in mitochondria isolated from AD-brain.77 COX is a metalloenzyme that acts at the terminus of the mitochondrial respiratory chain (in complex IV); its catalytic core is formed by three subunits encoded by the mitochondrial genome: two of these, COI (cytochrome C oxidase subunit I) and COII, which cooperate to catalyse the reduction of molecular oxygen to form water, are linked Cu enzymes that together contain three bound Cu atoms. Marked decreases in tissue-Cu content will not only have a significant impact on cellular energy production, but are also likely to facilitate increased oxidative damage in the AD-brain by impairing Cu-mediated antioxidant defences, for example that catalysed by SOD1. Cu also plays roles in regulation of other transition metals: therefore, the possibility that altered Cu homeostasis in AD could play roles in observed alterations in levels of some of the other transition metals merits consideration.
Increased concentrations of caeruloplasmin (the major plasma ferroxidase and Cu-containing protein in plasma) in HP, ENT and frontal cortex of AD-brain, have been suggested to represent a localized acute phase-type response and/or a compensatory increase to counter oxidative stress.78 In AD patients, caeruloplasmin-copper dysregulation has been associated with elevated labile-Cu levels in the blood.55,76,79 In light of this observation, global decreases in brain Cu as observed in this study, combined with previously-described Cu enrichment in and around amyloid plaques in AD-brain,19 have been interpreted as consistent with marked depletion of Cu-availability for normal cellular functions (such as energy production and anti-oxidant defence) in all brain regions examined in the current study.
Another interesting pattern is evident in our data. There were markedly higher levels of Cu in the normal CB, compared to all other brain regions examined. It is noteworthy that the metal concentrations in normal CB measured in this study (on a wet-weight basis) are consistent with CB metal concentrations previously reported (for Cu, Fe, Zn, Mn, and Se only).80 The intrinsically higher levels of several metals measured in CB, including Ca, Mg, Zn, Fe and Cu may be attributable to higher mitochondrial density in this region. Consistently, AD-brain reportedly exhibits significant reduction in mitochondrial membrane fluidity in cerebral regions that may result from lipid peroxidation: the exception to this observation was noted to be the CB.81
What might be the significance of lowered brain Cu in AD? Cu plays central roles in antioxidant defence via its structural and functional actions in SOD1, SOD3, and COI and COII. We have previously shown that, in the context of diabetic cardiomyopathy (DCM, another chronic disease linked to altered Cu regulation), deficient cardiac Cu impairs antioxidant defences that are restored by drug treatment, thereby establishing the pathogenic action of lowered tissue Cu.58,82 In DCM, lowering of myocardial Cu may be caused by the action of divalent Cu bound to N-ε-carboxymethyllysine (CML) groups in modified collagen, to suppress CTR1-mediated Cu(II) uptake, leading to deficient Cu(II) insertion into metalloenzymes.82–84 The depression in brain-Cu levels in AD is similar to that in the diabetic heart,82 and may well be caused by a similar mechanism; for example, extracellular CML is also elevated in AD,85 where it could well serve as a marker for this Cu-related pathogenic process.84 Therefore, these low brain-Cu levels are consistent with the causation of neurodegeneration and may well be pathogenic in AD.
Lowered brain-Cu levels, measured here in AD cases, approximate to those reported in Menkes’ disease, an X-linked recessive disorder caused by mutation in ATP7A, which encodes a Cu-transporting ATPase, causing severe neurodegeneration via brain-Cu deficiency.86 Similar lowering of brain Cu with neurodegeneration also occurs in animals with spontaneous or induced mutations in Atp7a.87 Metabolic studies reveal that trafficking of ATP7A after NMDA (N-methyl-D-aspartate) receptor activation triggers rapid release of copper ions from hippocampal neurons; ATP7A is directly required for this copper efflux, because parallel studies in hippocampal neurons derived from mice lacking functional ATP7A do not release copper.88 Thus, ATP7A plays a critical role in the availability of an NMDA receptor-dependent, releasable pool of copper in hippocampal neurons and affords a unique mechanism linking copper homeostasis to neuronal activation in the central nervous system. In addition, NMDA receptor activity is modulated by cellular prion protein (PrPC) in a copper-dependent manner.89 Moreover, copper ions potently modulate NMDA-receptor kinetics via modulation of PrPC, providing a mechanism that potentially links defective copper regulation to neurodegeneration. Thus, disruption of the NMDA synaptic system could provide a mode of copper dysregulation in AD. Furthermore, brain-Cu deficiency also causes neurodegeneration in other contexts, for example, insufficient dietary-Cu uptake.90–93 Taken together, these observations suggest that a treatment that can safely and effectively restore brain Cu might exert beneficial effects in the treatment of AD.10 However, elevating oral Cu intake had no effect on cognition in patients with mild AD in a pilot phase 2 clinical trial94 although, in that study, restoration of brain-Cu levels was not demonstrated. An effective method for in vivo measurement of brain Cu would be helpful for monitoring future intervention trials that aim to restore brain Cu but, to our knowledge, such a method is not yet available. Also required is an effective means of therapeutically raising brain-Cu levels: in this regard, the divalent-Cu-selective chelator, triethylenetetramine demonstrably elevates tissue Cu in another metabolic context, namely localized myocardial Cu deficiency in diabetic cardiomyopathy.10,82,95 Recent data demonstrating severe diabetes-like metabolic perturbations in the brain in AD may also be relevant to this objective.9,96
In conclusion, we have presented a study of post-mortem levels of nine essential elements in seven regions of the human brain in cases of AD and matched controls. The inter-regional difference in metal concentrations in both diseased and healthy brains as observed in this study: (1) demonstrates the importance of spatial resolution when studying a complex organ such as brain, and (2) provides potential explanations for some of the apparently differing findings in the literature. AD-associated alterations in metal homeostasis were evident in all brain regions studied, but changes in metals other than Cu were present only in a subset. By contrast, these data provide compelling evidence for pan-cerebral Cu deficiency in AD. Widespread brain-Cu deficiency may well contribute to the pathogenesis of neurodegeneration and dementia, probably acting via Cu-deficiency-induced defects in energy utilization and anti-oxidant defences. We propose that new therapeutic interventions that can safely and effectively restore brain Cu levels, could have a place in the experimental therapeutics of Alzheimer's dementia.
Duality of interest statement
GJC is named as inventor in patents that disclose the use of the Cu-selective chelator, triethylenetetramine (TETA) for the treatment of diabetic organ damage: these have been assigned and he has no financial interest in them. All other authors declare no duality of interest.Acknowledgements
We thank Cynthia Tse and Michael Anderson for their assistance with the management of these studies. We acknowledge Professor Sir John Scott for his encouragement, moral support, and helpful discussions with us, and thank the families of patients who supported this research through their donation of brains to the New Zealand Neurological Foundation Douglas Human Brain Bank. This work was supported by the Endocore Research Trust [60147]; the Maurice and Phyllis Paykel Trust [3627036; and Travel funding for JX]; Lottery Health New Zealand [3626585; 3702766]; the Maurice Wilkins Centre for Molecular Biodiscovery [Tertiary Education Commission 9341-3622506; and Doctoral Scholarship for JX]; the Health Research Council of New Zealand [3338701; 3627373]; the University of Auckland [Doctoral Student PReSS funding JXU058]; the Oakley Mental Health Research Foundation [3456030; 3627092; 3701339; 3703253; 3702870]; the Ministry of Business, Innovation & Employment [UOAX0815]; the Neurological Foundation of New Zealand; the Medical Research Council [UK, MR/L010445/1 and MR/L011093/1]; Alzheimer's research UK (ARUK-PPG2014B-7); the University of Manchester, the CMFT, and the Northwest Regional Development Agency through a combined programme grant to CADET; and was facilitated by the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network.References
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer and M. Scazufca, Global prevalence of dementia: a Delphi consensus study, Lancet, 2005, 366, 2112–2117 CrossRef.
- M. Prince, R. Bryce, E. Albanese, A. Wimo, W. Ribeiro and C. P. Ferri, The global prevalence of dementia: a systematic review and metaanalysis, Alzheimer's Dementia, 2013, 9, 63–75 CrossRef PubMed.
- M. Citron, Alzheimer's disease: strategies for disease modification, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS PubMed.
- V. J. De-Paula, M. Radanovic, B. S. Diniz and O. V. Forlenza, Alzheimer's disease, Subcell. Biochem., 2012, 65, 329–352 CAS.
- J. L. Cummings, T. Morstorf and K. Zhong, Alzheimer's disease drug-development pipeline: few candidates, frequent failures, Alzheimer's Res. Ther., 2014, 6, 37 CrossRef PubMed.
- R. Squitti, Metals in Alzheimer's disease: a systemic perspective, Front. Biosci., 2012, 17, 451–472 CrossRef CAS.
- M. Sastre, C. W. Ritchie and N. Hajji, Metal Ions in Alzheimer's disease brain, JSM Alzheimer's Dis. Related Dementia, 2015, 2, 1014 Search PubMed.
- A. I. Bush, The metal theory of Alzheimer's disease, J. Alzheimer's Dis., 2013, 33, S277–281 Search PubMed.
- J. Xu, P. Begley, S. J. Church, S. Patassini, S. McHarg, N. Kureishy, K. A. Hollywood, H. J. Waldvogel, H. Liu, S. Zhang, W. Lin, K. Herholz, C. Turner, B. J. Synek, M. A. Curtis, J. Rivers-Auty, C. B. Lawrence, K. A. B. Kellett, N. M. Hooper, E. R. L. C. Vardy, D. Wu, R. D. Unwin, R. L. M. Faull, A. W. Dowsey and G. J. S. Cooper, Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer's disease: metabolic basis for dementia, Sci. Rep., 2016, 6, 27524 CrossRef CAS PubMed.
- G. J. S. Cooper, Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes and Alzheimer's disease, Drugs, 2011, 71, 1281–1320 CrossRef CAS PubMed.
- D. G. Smith, R. Cappai and K. J. Barnham, The redox chemistry of the Alzheimer's disease amyloid beta peptide, Biochim. Biophys. Acta, 2007, 1768, 1976–1990 CrossRef CAS PubMed.
- P. A. Adlard and A. I. Bush, Metals and Alzheimer's disease, J. Alzheimer's Dis., 2006, 10, 145–163 CrossRef PubMed.
- B. R. Roberts, T. M. Ryan, A. I. Bush, C. L. Masters and J. A. Duce, The role of metallobiology and amyloid-beta peptides in Alzheimer's disease, J. Neurochem., 2012, 120, 149–166 CrossRef CAS PubMed.
- J. L. Cummings, Alzheimer's disease, N. Engl. J. Med., 2004, 351, 56–67 CrossRef CAS PubMed.
- M. Schrag, C. Mueller, U. Oyoyo, M. A. Smith and W. M. Kirsch, Iron, zinc and copper in the Alzheimer's disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion, Prog. Neurobiol., 2011, 94, 296–306 CrossRef CAS PubMed.
- S. Ayton, P. Lei and A. I. Bush, Metallostasis in Alzheimer's disease, Free Radical Biol. Med., 2013, 62, 76–89 CrossRef CAS PubMed.
- P. Ramos, A. Santos, N. R. Pinto, R. Mendes, T. Magalhaes and A. Almeida, Anatomical region differences and age-related changes in copper, zinc, and manganese levels in the human brain, Biol. Trace Elem. Res., 2014, 161, 190–201 CrossRef CAS PubMed.
- A. S. Pithadia and M. H. Lim, Metal-associated amyloid-beta species in Alzheimer's disease, Curr. Opin. Chem. Biol., 2012, 16, 67–73 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- H. Akatsu, A. Hori, T. Yamamoto, M. Yoshida, M. Mimuro, Y. Hashizume, I. Tooyama and E. M. Yezdimer, Transition metal abnormalities in progressive dementias, Biometals, 2012, 25, 337–350 CrossRef CAS PubMed.
- A. Rembach, D. J. Hare, M. Lind, C. J. Fowler, R. A. Cherny, C. McLean, A. I. Bush, C. L. Masters and B. R. Roberts, Decreased copper in Alzheimer's disease brain is predominantly in the soluble extractable fraction, Int. J. Alzheimer's Dis., 2013, 623241 Search PubMed.
- S. J. Church, P. Begley, N. Kureishy, S. McHarg, P. N. Bishop, D. A. Bechtold, R. D. Unwin and G. J. Cooper, Deficient copper concentrations in dried-defatted hepatic tissue from ob/ob mice: A potential model for study of defective copper regulation in metabolic liver disease, Biochem. Biophys. Res. Commun., 2015, 460, 549–554 CrossRef CAS PubMed.
- W. Wu and S. A. Small, Imaging the earliest stages of Alzheimer's disease, Curr. Alzheimer Res., 2006, 3, 529–539 CrossRef CAS PubMed.
- K. A. Jobst, A. D. Smith, M. Szatmari, M. M. Esiri, A. Jaskowski, N. Hindley, B. McDonald and A. J. Molyneux, Rapidly progressing atrophy of medial temporal lobe in Alzheimer's disease, Lancet, 1994, 343, 829–830 CrossRef CAS.
- H. Braak and E. Braak, Neuropathological stageing of Alzheimer-related changes, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS PubMed.
- K. M. Bradley, V. T. O'Sullivan, N. D. Soper, Z. Nagy, E. M. King, A. D. Smith and B. J. Shepstone, Cerebral perfusion SPET correlated with Braak pathological stage in Alzheimer's disease, Brain, 2002, 125, 1772–1781 CrossRef CAS PubMed.
- H. J. Waldvogel, J. Y. Bullock, B. J. Synek, M. A. Curtis, W. M. van Roon-Mom and R. L. Faull, The collection and processing of human brain tissue for research, Cell Tissue Banking, 2008, 9, 169–179 CrossRef CAS PubMed.
- H. J. Waldvogel, M. A. Curtis, K. Baer, M. I. Rees and R. L. Faull, Immunohistochemical staining of post-mortem adult human brain sections, Nat. Protoc., 2006, 1, 2719–2732 CrossRef CAS PubMed.
- S. S. Mirra, A. Heyman, D. McKeel, S. M. Sumi, B. J. Crain, L. M. Brownlee, F. S. Vogel, J. P. Hughes, G. van Belle and L. Berg, The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease, Neurology, 1991, 41, 479–486 CrossRef CAS PubMed.
- G. S. Fell, H. Smith and R. A. Howie, Neutron activation analysis for copper in biological material applied to Wilson's disease, J. Clin. Pathol., 1968, 21, 8–11 CrossRef CAS PubMed.
- W. W. Harrison, M. G. Netsky and M. D. Brown, Trace elements in human brain: copper, zinc, iron, and magnesium, Clin. Chim. Acta, 1968, 21, 55–60 CrossRef CAS.
- K. V. Kowdley and M. M. K. Kaplan, Iron chelation therapy with oral deferiprone – toxicity or lack of efficacy?, N. Engl. J. Med., 1998, 339, 468–469 CrossRef CAS PubMed.
- J. N. Cumings, The copper and iron content of brain and liver in the normal and in hepatolenticular degeneration, Brain, 1948, 71(Pt 4), 410–415 CrossRef CAS PubMed.
- H. Braak and E. Braak, Neuropathological stageing of Alzheimer-related changes, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS PubMed.
- I. Skoog, Detection of preclinical Alzheimer's disease, N. Engl. J. Med., 2000, 343, 502–503 CrossRef CAS PubMed.
- V. M. Vitvitsky, S. K. Garg, R. F. Keep, R. L. Albin and R. Banerjee, Na+ and K+ ion imbalances in Alzheimer's disease, Biochim. Biophys. Acta, 2012, 1822, 1671–1681 CrossRef CAS PubMed.
- C. D. Bortner and J. A. Cidlowski, Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death, J. Biol. Chem., 2003, 278, 39176–39184 CrossRef CAS PubMed.
- E. A. Mellon, D. T. Pilkinton, C. M. Clark, M. A. Elliott, W. R. Witschey, 2nd, A. Borthakur and R. Reddy, Sodium MR imaging detection of mild Alzheimer disease: preliminary study, Am. J. Neuroradiol., 2009, 30, 978–984 CrossRef CAS PubMed.
- R. I. Scahill, J. M. Schott, J. M. Stevens, M. N. Rossor and N. C. Fox, Mapping the evolution of regional atrophy in Alzheimer's disease: unbiased analysis of fluid-registered serial MRI, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4703–4707 CrossRef CAS PubMed.
- B. H. Anderton, Ageing of the brain, Mech. Ageing Dev., 2002, 123, 811–817 CrossRef CAS PubMed.
- C. D. Bortner, M. Gomez-Angelats and J. A. Cidlowski, Plasma membrane depolarization without repolarization is an early molecular event in anti-Fas-induced apoptosis, J. Biol. Chem., 2001, 276, 4304–4314 CrossRef CAS PubMed.
- B. R. Roberts, J. D. Doecke, A. Rembach, L. F. Yevenes, C. J. Fowler, C. A. McLean, M. Lind, I. Volitakis, C. L. Masters, A. I. Bush and D. J. Hare, Rubidium and potassium levels are altered in Alzheimer's disease brain and blood but not in cerebrospinal fluid, Acta Neuropathol. Commun., 2016, 4, 119 CrossRef PubMed.
- S. P. Yu, C. H. Yeh, S. L. Sensi, B. J. Gwag, L. M. Canzoniero, Z. S. Farhangrazi, H. S. Ying, M. Tian, L. L. Dugan and D. W. Choi, Mediation of neuronal apoptosis by enhancement of outward potassium current, Science, 1997, 278, 114–117 CrossRef CAS PubMed.
- W. H. Dribben, L. N. Eisenman and S. Mennerick, Magnesium induces neuronal apoptosis by suppressing excitability, Cell Death Dis., 2010, 1, e63 CrossRef CAS PubMed.
- E. Andrasi, S. Igaz, Z. Molnar and S. Mako, Disturbances of magnesium concentrations in various brain areas in Alzheimer's disease, Magnesium Res., 2000, 13, 189–196 CAS.
- M. J. Berridge, Neuronal calcium signaling, Neuron, 1998, 21, 13–26 CrossRef CAS PubMed.
- K. Hyrc, S. D. Handran, S. M. Rothman and M. P. Goldberg, Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators, J. Neurosci., 1997, 17, 6669–6677 CAS.
- F. M. LaFerla, Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease, Nat. Rev. Neurosci., 2002, 3, 862–872 CrossRef CAS PubMed.
- A. Frandsen and A. Schousboe, Excitatory amino acid-mediated cytotoxicity and calcium homeostasis in cultured neurons, J. Neurochem., 1993, 60, 1202–1211 CrossRef CAS PubMed.
- J. Chen and M. J. Berry, Selenium and selenoproteins in the brain and brain diseases, J. Neurochem., 2003, 86, 1–12 CrossRef CAS PubMed.
- T. N. Akbaraly, I. Hininger-Favier, I. Carriere, J. Arnaud, V. Gourlet, A. M. Roussel and C. Berr, Plasma selenium over time and cognitive decline in the elderly, Epidemiology, 2007, 18, 52–58 CrossRef PubMed.
- S. Gao, Y. Jin, F. W. Unverzagt, C. Liang, K. S. Hall, J. Cao, F. Ma, J. R. Murrell, Y. Cheng, P. Li, J. Bian and H. C. Hendrie, Selenium level and depressive symptoms in a rural elderly Chinese cohort, BMC Psychiatry, 2012, 12, 72 CrossRef PubMed.
- B. R. Cardoso, T. P. Ong, W. Jacob-Filho, O. Jaluul, M. I. Freitas and S. M. Cozzolino, Nutritional status of selenium in Alzheimer's disease patients, Br. J. Nutr., 2010, 103, 803–806 CrossRef CAS PubMed.
- H. Vural, H. Demirin, Y. Kara, I. Eren and N. Delibas, Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer's disease, J. Trace Elem. Med. Biol., 2010, 24, 169–173 CAS.
- R. Gonzalez-Dominguez, T. Garcia-Barrera and J. L. Gomez-Ariza, Homeostasis of metals in the progression of Alzheimer's disease, Biometals, 2014, 27, 539–549 CrossRef CAS PubMed.
- F. P. Bellinger, Q. P. He, M. T. Bellinger, Y. Lin, A. V. Raman, L. R. White and M. J. Berry, Association of selenoprotein p with Alzheimer's pathology in human cortex, J. Alzheimer's Dis., 2008, 15, 465–472 CrossRef CAS PubMed.
- R. A. Weisiger and I. Fridovich, Superoxide dismutase. Organelle specificity, J. Biol. Chem., 1973, 248, 3582–3592 CAS.
- G. J. S. Cooper, Selective divalent copper chelation for the treatment of diabetes mellitus, Curr. Med. Chem., 2012, 19, 2828–2860 CrossRef CAS PubMed.
- N. Izuo, H. Nojiri, S. Uchiyama, Y. Noda, S. Kawakami, S. Kojima, T. Sasaki, T. Shirasawa and T. Shimizu, Brain-Specific Superoxide Dismutase 2 Deficiency Causes Perinatal Death with Spongiform Encephalopathy in Mice, Oxid. Med. Cell. Longevity, 2015, 2015, 238914 Search PubMed.
- J. N. Keller, M. S. Kindy, F. W. Holtsberg, D. K. St Clair, H. C. Yen, A. Germeyer, S. M. Steiner, A. J. Bruce-Keller, J. B. Hutchins and M. P. Mattson, Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction, J. Neurosci., 1998, 18, 687–697 CAS.
- S. Zidenberg-Cherr, C. L. Keen, B. Lonnerdal and L. S. Hurley, Superoxide dismutase activity and lipid peroxidation in the rat: developmental correlations affected by manganese deficiency, J. Nutr., 1983, 113, 2498–2504 CAS.
- N. C. Burton and T. R. Guilarte, Manganese neurotoxicity: lessons learned from longitudinal studies in nonhuman primates, Environ. Health Perspect., 2009, 117, 325–332 CrossRef CAS PubMed.
- R. B. Hernandez, M. Farina, B. P. Esposito, N. C. Souza-Pinto, F. Barbosa, Jr. and C. Sunol, Mechanisms of manganese-induced neurotoxicity in primary neuronal cultures: the role of manganese speciation and cell type, Toxicol. Sci., 2011, 124, 414–423 CrossRef CAS PubMed.
- R. A. Srivastava and J. C. Jain, Scavenger receptor class B type I expression and elemental analysis in cerebellum and parietal cortex regions of the Alzheimer's disease brain, J. Neurol. Sci., 2002, 196, 45–52 CrossRef CAS PubMed.
- D. Religa, D. Strozyk, R. A. Cherny, I. Volitakis, V. Haroutunian, B. Winblad, J. Naslund and A. I. Bush, Elevated cortical zinc in Alzheimer disease, Neurology, 2006, 67, 69–75 CrossRef CAS PubMed.
- E. Bonke, K. Zwicker and S. Drose, Manganese ions induce H2O2 generation at the ubiquinone binding site of mitochondrial complex II, Arch. Biochem. Biophys., 2015, 580, 75–83 CrossRef CAS PubMed.
- Y. Liu, D. S. Barber, P. Zhang and B. Liu, Complex II of the mitochondrial respiratory chain is the key mediator of divalent manganese-induced hydrogen peroxide production in microglia, Toxicol. Sci., 2013, 132, 298–306 CrossRef CAS PubMed.
- S. F. Graham, M. B. Nasaruddin, M. Carey, C. Holscher, B. McGuinness, P. G. Kehoe, S. Love, P. Passmore, C. T. Elliott, A. A. Meharg and B. D. Green, Age-associated changes of brain copper, iron, and zinc in Alzheimer's disease and dementia with Lewy bodies, J. Alzheimer's Dis., 2014, 42, 1407–1413 CAS.
- M. Schrag, A. Crofton, M. Zabel, A. Jiffry, D. Kirsch, A. Dickson, X. W. Mao, H. V. Vinters, D. W. Domaille, C. J. Chang and W. Kirsch, Effect of cerebral amyloid angiopathy on brain iron, copper, and zinc in Alzheimer's disease, J. Alzheimer's Dis., 2011, 24, 137–149 CAS.
- D. J. Hare, E. P. Raven, B. R. Roberts, M. Bogeski, S. D. Portbury, C. A. McLean, C. L. Masters, J. R. Connor, A. I. Bush, P. J. Crouch and P. A. Doble, Laser ablation-inductively coupled plasma-mass spectrometry imaging of white and gray matter iron distribution in Alzheimer's disease frontal cortex, NeuroImage, 2016, 137, 124–131 CrossRef CAS PubMed.
- J. R. Connor, B. S. Snyder, J. L. Beard, R. E. Fine and E. J. Mufson, Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer's disease, J. Neurosci. Res., 1992, 31, 327–335 CrossRef CAS PubMed.
- T. G. Smart, A. M. Hosie and P. S. Miller, Zn2+ ions: modulators of excitatory and inhibitory synaptic activity, Neuroscientist, 2004, 10, 432–442 CrossRef CAS PubMed.
- T. J. Craddock, J. A. Tuszynski, D. Chopra, N. Casey, L. E. Goldstein, S. R. Hameroff and R. E. Tanzi, The zinc dyshomeostasis hypothesis of Alzheimer's disease, PLoS One, 2012, 7, e33552 CAS.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer's disease, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS PubMed.
- I. F. Scheiber, J. F. Mercer and R. Dringen, Metabolism and functions of copper in brain, Prog. Neurobiol., 2014, 116, 33–57 CrossRef CAS PubMed.
- R. Squitti and C. Salustri, Agents complexing copper as a therapeutic strategy for the treatment of Alzheimer's disease, Curr. Alzheimer Res., 2009, 6, 476–487 CrossRef CAS PubMed.
- W. D. Parker, Jr., J. Parks, C. M. Filley and B. K. Kleinschmidt-DeMasters, Electron transport chain defects in Alzheimer's disease brain, Neurology, 1994, 44, 1090–1096 CrossRef.
- D. A. Loeffler, P. A. LeWitt, P. L. Juneau, A. A. Sima, H. U. Nguyen, A. J. DeMaggio, C. M. Brickman, G. J. Brewer, R. D. Dick, M. D. Troyer and L. Kanaley, Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders, Brain Res., 1996, 738, 265–274 CrossRef CAS PubMed.
- R. Squitti, C. C. Quattrocchi, G. D. Forno, P. Antuono, D. R. Wekstein, C. R. Capo, C. Salustri and P. M. Rossini, Ceruloplasmin (2-D PAGE) pattern and copper content in serum and brain of Alzheimer disease patients, Biomarker Insights, 2007, 1, 205–213 Search PubMed.
- K. M. Davies, D. J. Hare, V. Cottam, N. Chen, L. Hilgers, G. Halliday, J. F. Mercer and K. L. Double, Localization of copper and copper transporters in the human brain, Metallomics, 2013, 5, 43–51 RSC.
- P. Mecocci, M. F. Beal, R. Cecchetti, M. C. Polidori, A. Cherubini, F. Chionne, L. Avellini, G. Romano and U. Senin, Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain, Mol. Chem. Neuropathol., 1997, 31, 53–64 CrossRef CAS PubMed.
- S. Zhang, H. Liu, G. V. Amarsingh, C. C. H. Cheung, S. Hogl, U. Narayanan, L. Zhang, S. McHarg, J. Xu, D. Gong, J. Kennedy, B. Barry, Y. S. Choong, A. R. J. Phillips and G. J. S. Cooper, Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation, Cardiovasc. Diabetol., 2014, 13, 100 CrossRef PubMed.
- G. J. S. Cooper, A. R. J. Phillips, S. Y. Choong, B. L. Leonard, D. J. Crossman, D. H. Brunton, L. Saafi, A. M. Dissanayake, B. R. Cowan, A. A. Young, C. J. Occleshaw, Y. K. Chan, F. E. Leahy, G. F. Keogh, G. D. Gamble, G. R. Allen, A. J. Pope, P. D. Boyd, S. D. Poppitt, T. K. Borg, R. N. Doughty and J. R. Baker, Regeneration of the heart in diabetes by selective copper chelation, Diabetes, 2004, 53, 2501–2508 CrossRef CAS PubMed.
- M. Kamalov, P. W. R. Harris, C. G. Hartinger, G. M. Miskelly, G. J. S. Cooper and M. A. Brimble, Physicochemical studies on the copper(II) binding by glycated collagen telopeptides, Org. Biomol. Chem., 2015, 13, 3058–3063 CAS.
- N. Sasaki, R. Fukatsu, K. Tsuzuki, Y. Hayashi, T. Yoshida, N. Fujii, T. Koike, I. Wakayama, R. Yanagihara, R. Garruto, N. Amano and Z. Makita, Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases, Am. J. Pathol., 1998, 153, 1149–1155 CrossRef CAS PubMed.
- S. G. Kaler, C. S. Holmes, D. S. Goldstein, J. Tang, S. C. Godwin, A. Donsante, C. J. Liew, S. Sato and N. Patronas, Neonatal diagnosis and treatment of Menkes disease, N. Engl. J. Med., 2008, 358, 605–614 CrossRef CAS PubMed.
- J. F. Mercer and R. M. Llanos, Molecular and cellular aspects of copper transport in developing mammals, J. Nutr., 2003, 133, 1481S–1484S CAS.
- M. L. Schlief, A. M. Craig and J. D. Gitlin, NMDA receptor activation mediates copper homeostasis in hippocampal neurons, J. Neurosci., 2005, 25, 239–246 CrossRef CAS PubMed.
- P. K. Stys, H. You and G. W. Zamponi, Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders, J. Physiol., 2012, 590, 1357–1368 CrossRef CAS PubMed.
- M. Mandelbrote, M. W. Stanier, R. H. S. Thompson and M. N. Thruston, Studies on copper metabolism in demyelinating diseases of the central nervous system, Brain, 1948, 71, 212–228 CrossRef PubMed.
- J. M. Howell and A. N. Davison, The copper content and cytochrome oxidase activity of tissues from normal and swayback lambs, Biochem. J., 1959, 72, 365–368 CrossRef CAS PubMed.
- N. Kumar, Copper deficiency myelopathy (human swayback), Mayo Clin. Proc., 2006, 81, 1371–1384 CrossRef CAS PubMed.
- D. P. Griffith, D. A. Liff, T. R. Ziegler, G. J. Esper and E. F. Winton, Acquired copper deficiency: a potentially serious and preventable complication following gastric bypass surgery, Obesity, 2009, 17, 827–831 CrossRef CAS PubMed.
- H. Kessler, T. A. Bayer, D. Bach, T. Schneider-Axmann, T. Supprian, W. Herrmann, M. Haber, G. Multhaup, P. Falkai and F.-G. Pajonk, Intake of copper has no effect on cognition in patients with mild Alzheimer's disease: a pilot phase 2 clinical trial, J. Neural Transm., 2008, 115, 1181–1187 CrossRef CAS PubMed.
- G. J. S. Cooper, A. A. Young, G. D. Gamble, C. J. Occleshaw, A. M. Dissanayake, B. R. Cowan, D. H. Brunton, J. R. Baker, A. R. Phillips, C. M. Frampton, S. D. Poppitt and R. N. Doughty, A copper(II)-selective chelator ameliorates left-ventricular hypertrophy in type 2 diabetic patients: a randomised placebo-controlled study, Diabetologia, 2009, 52, 715–722 CrossRef CAS PubMed.
- J. Xu, P. Begley, S. J. Church, S. Patassini, K. A. Hollywood, M. Jüllig, M. A. Curtis, H. J. Waldvogel, R. L. M. Faull, R. D. Unwin and G. J. S. Cooper, Graded perturbations of metabolism in multiple regions of human brain in Alzheimer's disease: Snapshot of a pervasive metabolic disorder, Biochim. Biophys. Acta, 2016, 1862, 1084–1092 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mt00074j |
| This journal is © The Royal Society of Chemistry 2017 |