
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper binding and redox chemistry of the Aβ16 peptide and its variants: insights into determinants of copper-dependent reactivity†
Nineveh
Yako
,
Tessa R.
Young
,
Jade M.
Cottam Jones
,
Craig A.
Hutton
,
Anthony G.
Wedd
and
Zhiguang
Xiao‡
 *
*
School of Chemistry and The Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: z.xiao@florey.edu.au; Fax: +61 3 9347 5180; Tel: +61 3 9035 6072
First published on 24th January 2017
Abstract
The metal-binding sites of Aβ peptides are dictated primarily by the coordination preferences of the metal ion. Consequently, Cu(I) is typically bound with two His ligands in a linear mode while Cu(II) forms a pseudo-square planar stereochemistry with the N-terminal amine nitrogen acting as an anchoring ligand. Several distinct combinations of other groups can act as co-ligands for Cu(II). A population of multiple binding modes is possible with the equilibrium position shifting sensitively with solution pH and the nature of the residues in the N-terminal region. This work examined the Cu(II) chemistry of the Aβ16 peptide and several variants that targeted these binding modes. The results are consistent with: (i) at pH < 7.8, the square planar site in CuII–Aβ16 consists primarily of a bidentate ligand provided by the carboxylate sidechain of Asp1 and the N-terminal amine supported by the imidazole sidechains of two His residues (designated here as component IA); it is in equilibrium with a less stable component IB in which the carboxylate ligand is substituted by the Asp1-Ala2 carbonyl oxygen. (ii) Both IA and IB convert to a common component II (apparent transition pKa ∼7.8 for IA and ∼6.5 for IB, respectively) featuring a tridentate ligand consisting of the N-terminal amine, the Asp1-Ala2 amide and the Ala2-Pro3 carbonyl; this stereochemistry is stabilized by two five-membered chelate rings. (iii) Component IA is stabilized for variant Aβ16-D1H, components I (both IA and IB) are imposed on Aβ16-A2P while the less stable IB is enforced on Aβ16-D1A (which is converted to component II at pH ∼6.5); (iv) components IA and IB share two His ligands with Cu(I) and are more reactive in redox catalysis than component II that features a highly covalent and less reactive amide N− ligand. The redox activity of IA is further enhanced for peptides with a His1 N-terminus that may act as a ligand for either Cu(I) or Cu(II) with lower re-organization energy required for redox-shuttling. This study provided insights into the determinants that regulate the reactivity of Cu–Aβ complexes.
Significance to metallomicsCopper binding to Aβ peptides has been subjected to recent intensive studies due to its possible relevance to the aetiology of Alzheimer's disease. Multiple metal binding ligands are present in the soluble and unstructured N-terminal section of Aβ(x–16), where x > 1 indicates truncation. The nature of the copper binding modes is dictated by the coordination preference of the copper oxidation state and by the medium pH, as is the reactivity of the Cu–Aβ complexes. This study clarifies current controversy regarding several important Cu(II) binding modes and provides an improved understanding of the complex copper chemistry of Aβ peptides. |
Introduction
Disruption of metal homeostasis has been linked to Alzheimer's disease and undesirable interactions between Aβ peptides and copper ions may play roles in the promotion of toxic Aβ aggregation and/or in the catalytic production of reactive oxygen species (ROS).1,2 Monomeric Aβ peptides are flexible and unstructured and the copper binding sites and binding modes change dynamically with the oxidation state and pH,3,4 as does the reactivity of the metal centres towards ligand substitution and redox catalysis.5,6Previous studies have demonstrated that Aβ peptides bind Cu(I) in dynamic linear two-coordinate modes utilizing the three different combinations of the available His-6, -13 and -14 sidechains (Fig. 1a).7,8 In contrast, they typically bind Cu(II) in a square planar-based stereochemistry. The N-terminal amine nitrogen acts as a primary anchoring ligand. Depending on the conditions, several distinct combinations of other groups can act as co-ligands. The speciation depends sensitively on the medium pH and on the nature of the available co-ligands including the His residues, the sidechain of Asp1 and the carbonyl oxygen or deprotonated amide nitrogen of the Asp1-Ala2 peptide link (Fig. 1b).3,4 Note the presence of stabilizing six- or five-membered chelate rings in each Cu(II) complex.
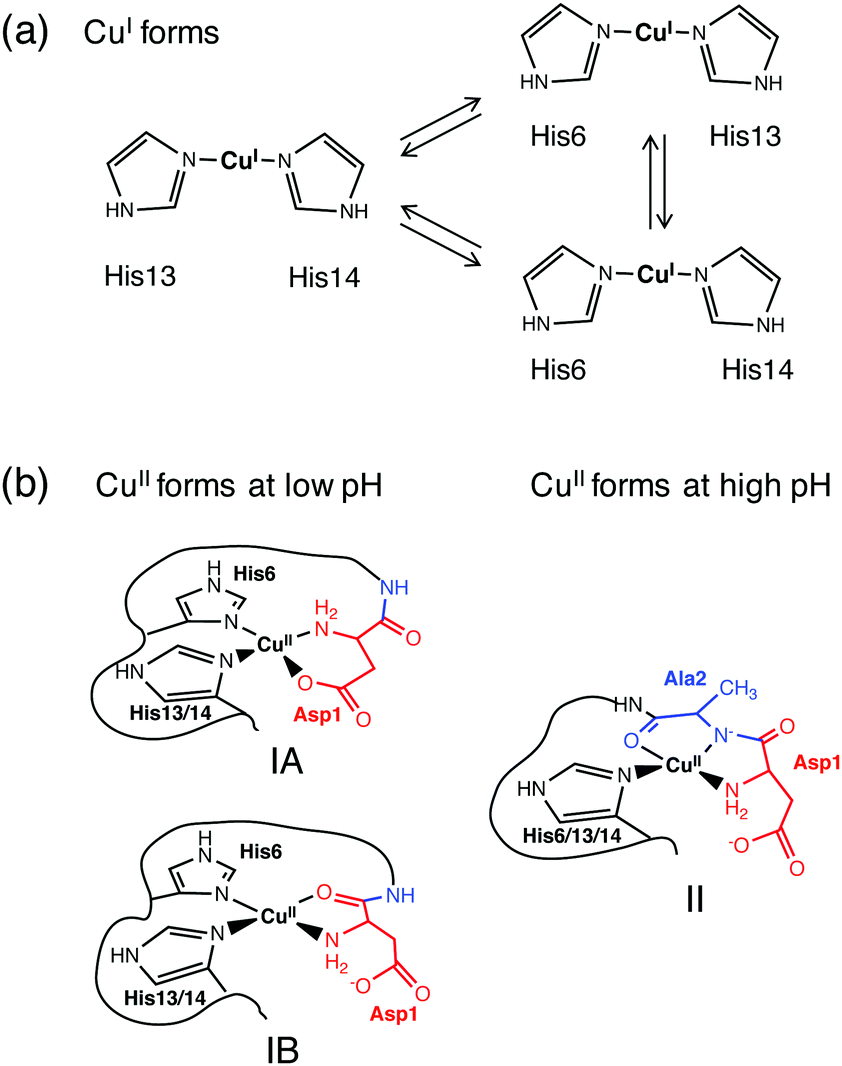 | ||
| Fig. 1 Proposed models of coordination of the Aβ peptide to Cu(I) (a) and Cu(II) (b). Note: only the equatorial ligands are indicated for the Cu(II) sites, and possible weakly binding axial ligands may include various side-chain carboxylates or water. | ||
The three distinct complexes in Fig. 1b are designated as components IA, IB and II in this work. Notably, they each can incorporate different combinations of His ligands as they appear to be conformationally available. Structure IA has frequently been proposed as a coordination mode at lower pH.9–11 However, recently IB has been preferred, based mainly on EPR studies.4,12,13
A transition from components IA/B to component II requires deprotonation of the peptide amide of Ala2 and is promoted by elevated pH. Any modification or truncation to the N-terminal residues, as occurs in native Aβ peptides,14,15 can alter the Cu(II) binding modes dramatically. For example, both the substitution of Asp1 with Asn and deletion of Asp1 to impose Ala2 as the N-terminal residue in the Aβ16 peptide lead to a decrease of over one pH unit in the pKa of transition I → II.16–19 Deletion of the first three residues, as detected in native Aβ peptides in high proportion,14,15 generates a classic N-terminal H2N-X-X-His3 motif known as the ATCUN (amino terminal copper and nickel) binding motif: the Cu(II) centre is stabilized by three chelate rings in a tetradentate ligand (Fig. 2b).20–23
 | ||
| Fig. 2 Proposed equatorial Cu(II) ligands in DP3 and DP4 (see Table 1 for sequences).21 Possible axial coordination ligand(s) include carboxylate(s) from Asp4 and/or Asp6 in DP3 and imidazole nitrogen ligand(s) from His1 and/or His4 in DP4. DNS = dansyl fluorophore. | ||
The different binding modes sensitively affect the reactivity of Cu–Aβ complexes towards ligand substitution and redox catalysis, especially for Cu(II).5,6,8,16,17 This study undertook a range of chemical and spectroscopic studies of the copper chemistry of the Aβ16 peptide to provide improved insight into the relationship between binding mode and reactivity. In particular, several variants that targeted the Cu(II) binding site specifically were examined. The work was facilitated by the recent development of a set of peptide-based fluorescent dansyl probes DP1–4 that are able to quantify Cu(II) binding stoichiometries and affinities from micromolar to femtomolar concentrations on a unified scale.8,21 The combined data demonstrated that (i) the Aβ16 peptide binds Cu(II) dominantly as component IA in equilibrium with less stable IB at low pH, while at higher pH, component II predominates (Fig. 1b); (ii) component I is more reactive than component II in redox catalysis.
Experimental section
Materials and general procedure
Materials purchased from Sigma included the ligands Ferene S (Fs), Ferrozine (Fz) and bicinchoninic anion (Bca) (as their sodium salts Na2Fs, NaHFz and Na2Bca), and the reductants NH2OH (as its H2SO4 salt) and ascorbic acid (Asc). They were all used as received. A standard Cu(II) solution was prepared by dissolving CuSO4 in Milli-Q water and the concentration calibrated via reaction with excess of the Cu(I)-binding ligand Bca in Mops buffer containing reductant NH2OH. Under such conditions, all copper ions are converted quantitatively to the well-defined chromophoric complex anion [CuI(Bca)2]3− with characteristic absorbance at 562 nm (ε = 7900 M−1 cm−1).24 The peptides and probes listed in Table 1 were purchased from GL Biochem (Shanghai) except for Aβ16-A2P and Aβ16-D1H/H14A which were synthesized on site by standard solid phase peptide techniques as detailed previously.25 The identity of each peptide was verified by electrospray ionization mass spectrometry (ESI-MS; Table 1) while the purity was confirmed to be >98% by HPLC. Peptide concentrations were estimated from absorbance maxima at ∼276 nm using εmax = 1410 M−1 cm−1 for Aβ16 peptides containing a single tyrosine residue. The concentration of dansyl peptide probes DP2–4 was estimated by absorbance maxima at ∼326 nm using the reported εmax = 4500 M−1 cm−1 for each probe.21 The concentrations obtained matched those estimated from fluorescence titrations with the copper standard assuming the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex.
:1 complex.
| Peptide or probe | Sequence | Molar mass (Da) | |
|---|---|---|---|
| Calcd | Found | ||
| a The residues changed are underlined in bold. | |||
| Aβ16 | DAEFRHDSGYEVHHQK | 1955.0 | 1954.5 |
| Ac-Aβ16 | Ac-DAEFRHDSGYEVHHQK | 1997.0 | 1997.5 |
| D1A |
![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) AEFRHDSGYEVHHQK AEFRHDSGYEVHHQK |
1911.0 | 1910.7 |
| D1E |
![[E with combining low line]](https://www.rsc.org/images/entities/b_char_0045_0332.gif) AEFRHDSGYEVHHQK AEFRHDSGYEVHHQK |
1969.0 | 1970.4 |
| D1N |
![[N with combining low line]](https://www.rsc.org/images/entities/b_char_004e_0332.gif) AEFRHDSGYEVHHQK AEFRHDSGYEVHHQK |
1954.0 | 1954.9 |
| D1H |
![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) AEFRHDSGYEVHHQK AEFRHDSGYEVHHQK |
1977.0 | 1978.0 |
| D1H/H14A |
AEFRHDSGYEVHQK |
1911.0 | 1911.4 |
| A2P | D![[P with combining low line]](https://www.rsc.org/images/entities/b_char_0050_0332.gif) EFRHDSGYEVHHQK EFRHDSGYEVHHQK |
1981.0 | 1981.5 |
| DP2 | DAE(KDNS)RHDH | 1240.3 | 1240.9 |
| DP3 | HP(KDNS)DHDH | 1118.2 | 1118.0 |
| DP4 | H(KDNS)HH | 790.8 | 790.4 |
All ligand stock solutions, reaction buffers, reductant solutions and protein solutions were prepared in rigorously deoxygenated Milli-Q water and stored in an anaerobic glove-box (<1 ppm O2). The working solution of ligand Fs was prepared freshly from aliquots of frozen stock solution stored in a freezer at temperature −80 °C.26 All reactions involving Cu(I) were performed anaerobically in the glove-box and transported outside in a capped cuvette for spectroscopic characterization.
Spectroscopic methods
UV-visible spectra were recorded on a Varian Cary 300 spectrophotometer in the dual beam mode with quartz cuvettes. All metal titrations were performed in appropriate buffers and corrected for baseline and dilution. Fluorescence emission spectra were recorded on a Varian Cary Eclipse spectrophotometer with a band pass of 20 nm for both excitation and emission. The excitation wavelength for the dansyl peptide (DP) probes was 330 nm and the emission spectra were recorded between 450 and 750 nm at a scan rate of 600 nm min−1. Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker Elexsys E 500 EPR spectrometer under the following instrument conditions: microwave frequency ν = 9.456–9.495 GHz but conversion to a single frequency of 9.475 GHz for reporting via relationship g = 714.4775 ν/F, where F is the magnetic field in the Gauss unit, the microwave power is ∼1 mW, the modulation amplitude is 4 G, the modulation frequency is 100 kHz, the sweep time is 40 s, the time constant is 20 ms, and the average number of scans is 10. All sample solutions (total 0.2 mL) were prepared in 50 mM buffer (Mes at pH 5.9–6.5; Mops at pH 6.6–8.0 or Ches at pH > 8) containing [Cu(II)]tot = 0.45 mM, [peptide or probe]tot = 0.5 mM and 5% glycerol. The solution pH of each sample was re-checked after recording each spectrum. The samples were snap-frozen in liquid nitrogen and the spectra recorded at 77 K in a liquid-nitrogen finger Dewar. The EPR parameters (g values and A values) were extracted directly from the recorded spectra without simulation.Quantification of Cu(II) binding
Cu(II) binding to Aβ peptides was quantified with a series of DP probes as reported recently.21 These probes feature a dansyl group and emit intense fluorescence at λmax ∼ 550 nm upon excitation at ∼330 nm. They bind one equiv. of Cu(II) with varying affinities and Cu(II) binding to the probes is detected sensitively via proportional fluorescence quenching. The quantification is based on the following relationships:| CuIIDP + Aβ ⇌ CuIIAβ + DP | (1) |
 | (2) |
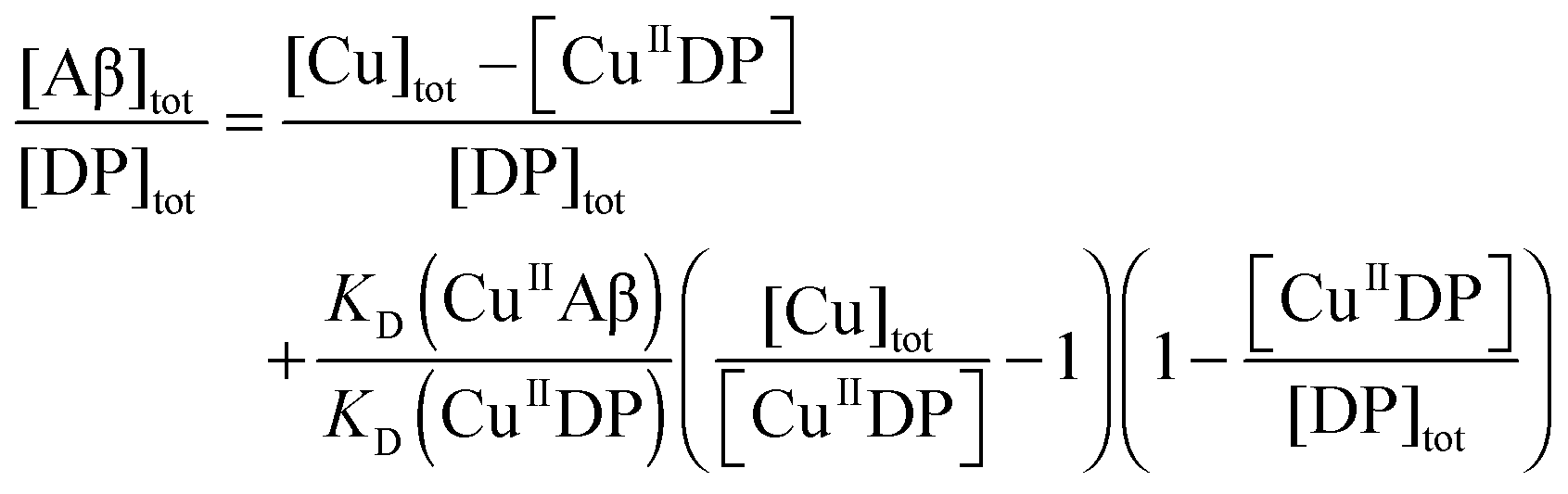 | (3) |
 | (4) |
The KD values for each peptide were determined similarly at a range of pH values. The pH-dependent affinities of the Cu(II) probes DP2–4 have been reported recently.21 If the Cu(II) binding site remains unchanged within a pH range and the ligand compositions are known, then the conditional KD within that pH range may be correlated with the absolute dissociation constant (KabsD) and the proton ionization constants (Kan) of each ligand function of the site according to eqn (5):
 | (5) |
The experiments were conducted in buffers (50 mM) Mes (pH 6.0–6.6), Mops (pH 7.0–7.4), Hepes (pH 7.8–8.2) and Ches (pH 8.5–9.2). On the other hand, the relationship between the determined KD and pH may also be employed to model the most likely binding site using eqn (5) to derive KabsD for the Cu(II) complex and Kan for the possible binding ligands.
Quantification of Cu(I) binding
Competition for Cu(I) with a suitable probe ligand L (L = Fs or Fz) was set up according to eqn (6) and (7):26| [CuIL2]3− + Aβ ⇌ CuIAβ + 2L2− | (6) |
 | (7) |
β2 = 13.7 for L = Fs and 15.1 for L = Fz).26 The experiments were performed via reported protocols.26
Catalytic aerobic oxidation of ascorbate
Experiments were conducted in air-saturated Mops buffers (50 mM; pH 6.6 and 7.4) and Ches buffer (50 mM, pH 8.7). To minimize the catalytic effect from unbound copper, a stock solution of each Cu–peptide complex was prepared with one-fold excess of the corresponding apo-peptide and the total copper content quantified accurately with the Cu(I) probe Bcs as a stable chromophoric complex [CuI(Bcs)2]3− (logβ2 = 19.9; ε483 = 13000 M−1 cm−1).27 The reactions were conducted using a quartz mixing cuvette where one half-cell was loaded with Asc (∼100 μM) and the other half-cell with the CuII–Aβ complex (varying in concentration between 2 and 40 μM, dependent on the catalytic activity). After recording the background reaction (if any), the catalytic reaction was started by quick mixing (note: the concentration of each component in the half-cells becomes half after the mixing) and followed in real time by the change in absorbance A265 of the reduced Asc. The oxidation rate was expressed as μM min−1 and calculated using the extinction coefficient ε265 = 14500 M−1 cm−1 for Asc.28 The catalytic activity of each Cu–Aβ complex was expressed as molar turn-over number per min (MTN/min) and was obtained from the slope of a linear plot of the Asc oxidation rate (μM min−1) versus the concentration (μM) of each Cu–Aβ complex. The background oxidation rate without added Cu has been removed from the determined oxidation rate.
Formal reduction potential
The formal reduction potential of the copper centres in each Cu–Ab16 complex was estimated from the relative affinities of the peptides for Cu(I) and Cu(II) under the same conditions via the Nernst relationship of eqn (8): | (8) |
Results and discussion
The N-terminal residues of Aβ peptides have a major impact on Cu(II) binding
Processing of the amyloid precursor protein (APP) by proteases β- and γ-secretase generates the Aβ peptides 1–(36–43) whose interaction with transition metal ions, and with copper, in particular, appears to be a source of toxicity in vivo.30 The possible effects include catalytic generation of reactive oxygen species (ROS) by toxic oligomers.2,31,32 All major Cu ligands are located within the first 16-residue water-soluble fragment and thus Aβ16 is a valid model for the study of the copper chemistry of full length Aβ.3 In particular, the N-terminal amine nitrogen and nearby residues play a critical role in Cu(II) coordination and in imposing both binding affinity and coordination modes.In this study, the two N-terminal residues Asp1 and Ala2 of Aβ16 were changed systematically via generation of the following variants (Table 1):
(i) D1A eliminates the sidechain of residue 1 as a potential ligand (cf. IA of Fig. 1b).
(ii) D1E, D1N, and D1H provide alternative residue 1 sidechains as potential ligands; D1H/H14A also removes His14 as a potential ligand.
(iii) A2P removes the amide of residue 2 as a potential anionic ligand (cf. II of Fig. 1b).
In addition, the N-terminus was acetylated (Ac-Aβ16) to perturb its metal binding properties. Detection of fine differences in properties is possible with a set of peptide-based dansyl fluorescent probes DP2–4 (Tables 1 and 2).21
KIID for Cu(II) of Aβ16 peptide and variants at different pH valuesa
| Aβ16 peptide or DP probe | logKIID |
K IID/KIID (wt) | Affinity standard | ||||
|---|---|---|---|---|---|---|---|
| pH 6.2 | pH 7.4 | pH 9.4 | pH 6.2 | pH 7.4 | pH 9.4 | ||
|
a The logKIID values were determined via ligand competition for Cu(II) between each peptide and the specified DP probe ligand in respective buffer (50 mM) of Mes (pH 6.2), Mops (pH 7.4) and Ches (pH 9.4). See the Experimental section for details.
b Too weak to be determined reliably.
c Following relationships demonstrated from a previous study21 were used to calculate the logKIID values given in the table: logKIID = 1.15–1.49 pH for DP2 and eqn (5) with KabsD and each pKa value given in Table 3 for DP3.
|
|||||||
| WT | −8.1 | −9.8 | −12.9 | 1.00 | 1.00 | 1.00 | DP2 |
| D1A | −7.2 | −9.1 | −12.9 | 7.94 | 5.01 | 1.00 | DP2 |
| D1E | −7.3 | −9.3 | −12.9 | 6.31 | 3.16 | 1.00 | DP2 |
| D1N | −7.9 | −9.5 | −12.7 | 1.58 | 2.00 | 1.58 | DP2 |
| A2P | −8.3 | −9.8 | −12.4 | 0.63 | 1.00 | 3.16 | DP2 |
| D1H | −9.7 | −11.7 | −13.3 | 0.03 | 0.01 | 0.40 | DP3 |
| D1H/H14A | −9.4 | −11.5 | −12.8 | 0.05 | 0.02 | 1.26 | DP3 |
| Ac-Aβ16 | ndb | −8.1 | −12.1 | ndb | 50 | 6.3 | DP2 |
| DP2c | −8.1 | −9.9 | −12.9 | Gly/His | |||
| DP3c | −9.9 | −12.2 | −13.5 | Gly/His | |||
A control titration of standardized CuSO4 solution with the DP2 probe alone generated a titration curve with an endpoint at [Cu(II)]tot/[DP2] = 1.0 (Fig. 3a and b, black trace), confirming that the probe binds a single Cu(II) ion of the highest affinity (logKD = −9.9).21 The equivalent titration into a 1:1 mixture of DP2 and Aβ16 (2.0 μM) in Mops buffer (50 mM, pH 7.4) generated a titration curve with an apparent endpoint at [Cu(II)]tot/[DP2] ∼ 2.0 (Fig. 3b, red trace). The essentially linear nature of the curve indicated that the peptide can bind one equiv. of Cu(II) with an affinity similar to that of DP2. Indeed, the Cu(II) binding sequence in the DP2 peptide was designed as a mimic of Aβ16 (Table 1).21
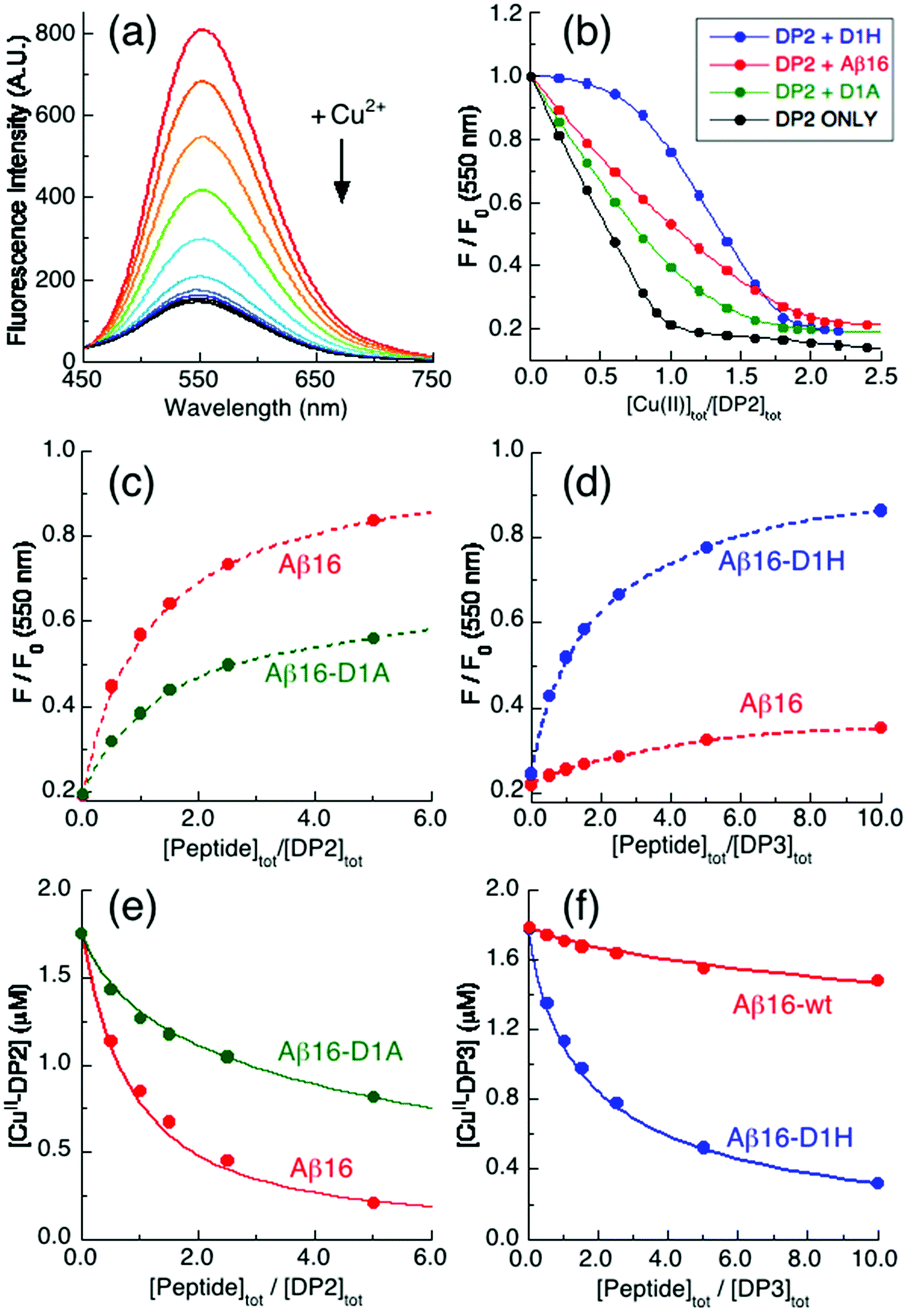 | ||
| Fig. 3 Quantification of Cu(II) binding to Aβ16 peptides in Mops buffer (50 mM, pH 7.4). (a) Illustrative quenching of the fluorescence spectrum of a DP2 probe (2.0 μM) upon titration with Cu2+ solution (λex = 330 nm; λem = 550 nm); (b) Cu2+ titration curves at 550 nm for 1:1 mixtures (2.0 μM) of DP2 and Aβ16 peptides plus control with DP2 only; (c and d) recovery of the Cu(II)-quenched fluorescence intensity of mixtures (c) CuII0.9–DP2 or (d) CuII0.9–DP3 (2.0 μM) upon titration with Aβ16 peptides; (e and f) correlation of (e) [CuII–DP2] versus [Aβ16 peptide]tot/[DP2]tot or (f) [CuII–DP3] versus [Aβ16 peptide]tot/[DP3]tot. The solid traces in (e and f) are the fits of eqn (3) to the experimental data, allowing estimation of the conditional logKIID for the Cu(II) complex of each Aβ16 peptide in the range of pH values listed in Table 1. | ||
Equivalent titrations for Aβ16-D1A and Aβ16-D1H also displayed apparent titration endpoints at [Cu(II)]tot/[DP2] ∼ 2.0 (Fig. 3b, green, blue), consistent with these peptides also binding a single Cu(II) ion. However, the different shapes of the titration curves are consistent with the affinity of Aβ16 being higher than that of Aβ16-D1A but lower than that of Aβ16-D1H. In fact, the probe does not compete effectively with the latter peptide until [Cu(II)]tot/[DP2] ∼ 1.
A more quantitative approach is possible with the fluorescence recovery analysis of titrations of apo-peptides into solutions of a copper–probe complex of appropriate affinity. Preliminary testing indicated that the appropriate experiments involved sub-stoichiometric Cu(II) in the systems CuII0.9–DPx (x = 2 for Aβ16-D1A and 3 for Aβ16-D1H and x = 2 or 3 for Aβ16). Analysis of the results suggested that, at pH 7.4, the Cu(II) binding affinity of Aβ16-D1A is 5-fold weaker than that of Aβ16 while the affinity of Aβ16-D1H is ∼100 times stronger (Fig. 3c–f and Table 2).
Equivalent experiments and analyses for the remaining peptides, with their respective optimal DP probes, lead to an increasing order of relative Cu(II) binding affinities at pH 7.4 as follows (Table 2):
| Ac-Aβ16 (50) < Aβ16-D1A (5.0) < Aβ16-D1E (3.2) |
| < Aβ16-D1N (2.0) < Aβ16 (1.0) ∼ Aβ16-A2P (1.0) |
| < Aβ16-D1H/H14A (0.02) < Aβ16-D1H (0.01) | (9) |
The numbers in the brackets refer to the dissociation constants normalized to that of Aβ16 and decrease in the order of increasing Cu(II) binding affinity. It is apparent that the residues in the N-terminal region have a major impact on the binding affinity and likely on the binding mode as well. A more complete analysis follows.
Component IA is favoured by the presence of a His1-based chelate ring
The two most significant changes in Cu(II) binding affinity are seen for Ac-Aβ16 and Aβ16-D1H (eqn (9)). The N-terminal amine nitrogen was blocked by acetylation in Ac-Aβ16, leading to a significant decrease in affinity under all conditions (Table 2), confirming the primary anchoring role of the N-terminal nitrogen in Cu(II) binding for each of the components IA, IB and II. Replacement of the carboxylate sidechain of Asp1 with the imidazole sidechain of His1 in Aβ16-D1H led to an increase in affinity of almost two orders of magnitude at pH 7.4 (Table 2). The presence of His1 allows the formation of a stabilizing bidentate ligand comprising the N-terminal amine and the His1 sidechain within a six-membered chelate ring. Other known examples of this binding mode include the DP3 probe employed in this work (Fig. 2a) and the bacterial copper binding proteins PcoC and CopC.24,33–35 The Cu(II) coordination sphere in CuII(Aβ16-D1H) may be depicted schematically by Fig. 4a(i), noting its similarity to the proposed IA site in Aβ16 (Fig. 1b). There are three other His residues (His-6, -13, -14) in the Aβ16-D1H sequence, but only two of them are required to constitute the proposed Cu(II) site. In fact, replacing one of them (His14) with Ala in Aβ16-D1H/H14A only marginally affects the Cu(II) affinity (Fig. 5a and Table 2). | ||
| Fig. 4 Proposed Cu(II) coordination modes of Aβ16 peptides and variants. (a) Component IA for CuII(Aβ16-D1H) and component IB for CuII(Aβ16-D1A). (b) Equilibria between compoments IA, IB and II for CuII(Aβ16). | ||
 | ||
| Fig. 5 Variation of conditional pKIID (= −logKIID) with solution pH: (a) Cu(II) complexes of various Aβ16 peptides (see Table S1 for the Experimental data, ESI†); (b) Cu(II) complexes of DP3 and DP4 probe peptides (data adapted from ref. 21). The dotted traces for peptides D1H, D1H/H14A and DP3 are the curve-fittings of the respective experimental data to eqn (5) generating KabsD and pKa for each ligand of the Cu(II) sites that are given in Table 3. Note: the data for D1H and D1H/H14A above pH 8.7 cannot be fitted to the model. | ||
The Cu(II) sites proposed above for CuII(Aβ16-D1H) and CuII(Aβ16-D1H/H14A) are supported by the fact that the pH-dependence of their affinities for Cu(II) (expressed as pKIID = –logKIID) can be modeled satisfactorily over the pH range 6.0–8.7, assuming that the dependence is driven by the basicity of the proposed ligands (Fig. 5a and eqn (5)). Their derived ligand pKa values are listed in Table 3. The I → II transition is not evident in these peptides and the pH dependence of the affinities is comparable to those of several documented examples including DP3 and several CopC proteins that adopt a DP3-type Cu(II) binding mode, shown in Fig. 2a.21,33
KD and pHa
| Peptide or protein | Cu(II) binding site | LogKabsD |
pKa for | Ref. | |
|---|---|---|---|---|---|
| NH2 | NIm | ||||
| a See Fig. 5 for experiments; parameters obtained by curve-fitting to eqn (5). | |||||
| Aβ16-D1H | NH2 (H1); 3 NIm (H1,6,13/14) | −12.7 | 8.3 | 6.4 | This work |
| Aβ16-D1H/H14A | NH2 (H1); 3 NIm (H1,6,13) | −12.3 | 8.1 | 6.4 | This work |
| DP3 probe | NH2 (H1), 3 NIm (H1,5,7) | −13.6 | 8.8 | 6.5 | 21 |
| Pf-CopC | NH2 (H1); 3 NIm (H1,3,85) | −17.2 | 8.8 | 6.6 | 33 |
| Ps-CopC | NH2 (H1); 2 NIm (H1,91); COO− (D89) | −14.4 | 8.2 | 6.1 | 33 |
| Ec-PcoC | NH2 (H1); 2 NIm (H1,92); COO− (D90) | −14.0 | 8.2 | 6.5 | 33 |
In contrast, the absence of His1 in the WT, D1A and A2P peptides leads to very different pH-dependencies that could not be modelled over the same pH range with any single set of ligands or a single binding mode (Fig. 5). The behaviour is attributed to the influence of the I → II transition, even for the A2P peptide at pH > 8.5 (see Fig. 6c). The Cu(II) site of component II features a deprotonated peptide amide ligand (Fig. 1b).
 | ||
| Fig. 6 Frozen solution EPR spectra of various Cu(II) complexes recorded at 77 K. The Cu(II) ligands are: (a) DP3 probe; (b) Aβ16-D1H; (c) Aβ16-A2P; (d) Aβ16; (e) Aβ16-D1A; (f) DP4 probe at various pH values as indicated (see the Experimental section for detail). Insets show enlargements of the spectra scaled up by a factor of 3. Component I is indicated by green dashed lines and component II by red dashed lines. | ||
pH-Dependent EPR spectra provide further support for the proposed binding models. The EPR signal of CuII(Aβ16-D1H) shows no change within the pH range 5.9–8.9 and essentially superimposes that of CuII–DP3 (Fig. 6a and b). It differs significantly from the component I EPR signals of CuII(Aβ16) or CuII(Aβ16-A2P) (cf., Fig. 6a–d and Table 4). This supports a Cu(II) site in CuII(Aβ16-D1H) of composition [NH2, 3Nim] that features the stabilizing bidentate ligand comprising the N-terminal amine and the His1 sidechain (Fig. 2a and 4a(i)).
| Ligand for the CuII complex | Component I | Component II | pKa for I ↔ II | ||||
|---|---|---|---|---|---|---|---|
| g ∥ | g ⊥ | A ∥ (G) | g ∥ | g ⊥ | A ∥ (G) | ||
| a Estimated directly from the respective spectrum, see Fig. 6, Fig. S1–S7 (ESI) for details. b Data adopted from ref. 21. c Data for CuII(Aβ16-D1H/H14A) were indistinguishable (see Fig. S5 and S6, ESI). d Data adopted from ref. 16. e Minor component at pH 7.4. | |||||||
| DP3 | 2.242b | 2.060b | 182b | — | — | — | — |
| Aβ16-D1Hc | 2.243 | 2.060 | 183 | — | — | — | — |
| Aβ16-A2P | 2.269 | 2.051 | 169 | — | — | — | ∼8.8 |
| Aβ16 | 2.268 | 2.050 | 170 | 2.202 | 2.042 | 160 | ∼7.8 |
| Aβ16-D1A | 2.269 | 2.051 | 169 | 2.204 | 2.041 | 161 | ∼6.5 |
| Aβ2-16 | 2.264d,e | — | 170d,e | 2.226d | — | 153d | — |
| DP4 | — | — | — | 2.178b | 2.050b | 200b | — |
| Aβ4-16 | — | — | — | 2.178d | 2.049d | 206d | — |
This particular His1 sidechain binding mode is suppressed in CuII–DP4. Although its four-residue peptide sequence (His-Lys-His-His) features a His1 residue, the presence of His3 provides the more favourable ATCUN binding mode that includes the N-terminal amine and two intervening deprotonated backbone amides (Fig. 2b).21 This conclusion is supported by the dramatic pH-dependence of its Cu(II) binding affinity and the unique EPR signal: both are readily distinguishable from those of components I and II (Fig. 5b and 6f).21 This same ATCUN binding mode is adopted by the N-truncated Aβ16 peptide, Aβ4-16 (sequence: FRHDSGYEVHHQK) that exhibits Cu(II) affinities and EPR spectra equivalent to those of DP4 (Table 4).16,21–23
Component IB is favoured by an Ala1 N-terminus at low pH but is less stable than IA
The variant Aβ16-D1A has a five-fold lower affinity for Cu(II) than Aβ16 at pH 7.4 (eqn (9); Fig. 5a; Table 2). The I → II transition in CuII(Aβ16) with pKa ∼ 7.8 is characteristic of deprotonation of the peptide amide of Ala2. The non-coordinating methyl sidechain of Ala1 eliminates the IA binding mode, enforcing a IB mode (Fig. 4a(ii)) that is converted to component II at pH ∼ 6.5, i.e., > 1 pH unit lower than that for the equivalent transition of CuII(Aβ16) (Fig. 6d and e). However, at higher pH values, the affinities of Aβ16 and Aβ16-D1A for Cu(II) become indistinguishable (Fig. 5a), consistent with a common tridentate chelate [–NH2, N−, CO] as part of the square planar array [–NH2, N−, CO, Nim], characteristic of component II (Fig. 1b). The binding affinity of the latter shows a different pH dependency and EPR signal to those of DP4, a Cu(II) ligand of the ATCUN binding mode (Fig. 2b, 5 and 6).The equivalent data for variants D1N and D1E provide further support for a role for Asp1 as an equatorial ligand: their Cu(II) binding properties are intermediate between those of Aβ16 and D1A (Table 2 and Fig. S1–S4, ESI†). The data are consistent with the weaker ligand properties of the sidechains of Asn1 (neutral carboxamide is a poor ligand for Cu(II)) and Glu1 (which would form a less favoured seven-membered chelate ring). In particular, substitution of Asp1 with Asn or Glu also leads to a decrease in Cu(II) binding affinity (Table 2) and in the pKa for the I → II transition (Fig. S1 and S4, ESI†).
Several previous studies provide support for this model. Deletion of Asp1 in Aβ16 generates a truncated peptide with an Ala N-terminus (sequence: AEFRHDSGYEVHHQK). Its qualitative Cu(II) binding properties are comparable to those of D1A: weaker binding affinity for Cu(II) and a lower I → II transition pKa.16 Similar effects were also observed for D1N,17–19 although the decreased pKa was attributed to the change of a putative hydrogen bond to the Cu(II) ligand.17
Component I is favoured in CuII(A2P)
The I → II transition relies on the availability of the Asp1-Ala2 peptide amide (Fig. 1b). Substitution of Ala2 with Pro eliminates this possibility and all properties observed for CuII(A2P) are consistent with the binding mode being locked in component I. Most obviously, KD is comparable to that of CuII(Aβ16) at lower pH values but becomes larger (i.e., lower affinity) at higher pH with the point of deviation occurring above pH 7.8, in line with the I → II transition for CuII(Aβ16) around this pH (Fig. 5a and 6d). The EPR signal confirmed that component I of CuII(Aβ16-A2P) was present up to at least pH 8.5 (Fig. 6c).At lower pH, component IA is preferred for Aβ peptides with an Asp1 N-terminus
The Cu(II) affinities of component I of CuII(Aβ16) and CuII(Aβ16-A2P) are similar. They are about one order of magnitude higher than that of component IB, which is favoured for CuII(Aβ16-D1A) at lower pH values such as pH 6.2 (Fig. 5a and Table S1, ESI†). This implies that component IA rather than IB is the preferred binding mode for both CuII(Aβ16) and CuII(Aβ16-A2P) at lower pH values (Fig. 1b). This is consistent with the negatively charged harder carboxylato ligand favouring the harder metal ion Cu(II) over a neutral peptide carbonyl oxygen for similar stereochemistries (a six-membered chelate ring in IA versus a five-membered chelate ring in IB; see Fig. 1b).However, the possibility for a IB binding mode for either CuII(Aβ16) or CuII(Aβ16-A2P) at low pH cannot be excluded. The EPR signals for both components IA and IB are indistinguishable (Fig. 6c–e and Table 4) and cannot be used to differentiate the binding modes at low pH. It is possible that components IA and IB are in fast exchange at lower pH for those peptides with an Asp1 N-terminus but with IA being the preferred binding mode. At higher pH, both components IA and IB are converted to component II: the KD values of CuII(Aβ16) and CuII(Aβ16-D1A) become indistinguishable at pH 9.4 (Fig. 5a). But the I → II conversion is restricted for CuII(Aβ16-A2P), weakening its Cu(II) binding affinity at high pH relative to other Aβ16 peptides (Fig. 5a).
It is unlikely that the differences in the properties of CuII(Aβ16), CuII(Aβ16-A2P) and CuII(Aβ16-D1A) at lower pH values are due to axial coordination by the Asp1 sidechain due to the following reasons:
(i) Given the possibility of axial coordination, mutation of Asp1 by Ala should lower the affinity across the whole pH range. However, there is no difference in the Cu(II) affinity for component II between WT and D1A.
(ii) A bidentate [NH2, COO−] binding mode for peptides with N-terminal aspartyl residues has been well-documented in many cases, including various Aβ peptides.11,36,37
(iii) The IA binding mode is confirmed for many peptides and proteins with a His1 N-terminus including Aβ16-D1H, DP3 probe, CopC and PcoC proteins, as surveyed above.
(iv) Contributions from axial coordination at Cu(II) centres are usually negligible due to the Jahn–Teller effect. There are many possible carboxylate sidechains (Asp1, Asp7, Glu3 or Glu11) which have been proven to be redundant. For example, an E3Q mutation showed no significant effect on the EPR spectrum.3
The IA coordination mode for Aβ16 has been proposed frequently as one of the two possible coordination modes of Asp1 at lower pH for component I (Fig. 1b).9–11 However, component IB has been preferred recently, based mainly on pulsed EPR studies.4,12,13 The chemical and spectroscopic study presented in this work, supported by previous Cu(II) binding studies for those peptides with N-terminal aspartyl residues,11,36 provides strong evidence that the preferred binding mode for component I in CuII(Aβ16) is IA, and not IB (Fig. 1b). It is recommended that the possibility of the IB binding mode can be re-investigated by pulsed EPR using CuII(Aβ16-D1H) and CuII(Aβ16-D1A) as controls.
Impact of N-terminal modification on Cu(I) binding
The first evidence of Cu(I) binding to Aβ peptides was implicated in an early study of Cu(II) potentiation of Aβ neurotoxicity.38 We now know that Aβ peptides bind Cu(I) mainly via a combination of two histidine residues within the first 16 residues.7,8 The Cu(I) binding stoichiometries and affinities of various Aβ16 peptides generated in this work were examined using Cu(I) probe ligands Ferene S (Fs) and Ferrozine (Fz) according to a recent protocol.26 Both probe ligands react with Cu(I) under reducing conditions to yield respective chromophoric Cu(I) complexes [CuIL2]3− with different formation constants (logβ2 = 13.7 for L = Fs and 15.1 for L = Fz).26 They were used to estimate the binding stoichiometries of Aβ16 peptides according to eqn (6) under weakly competing conditions and the binding affinities according to eqn (6) and (7) under strongly competing conditions. As demonstrated previously for Aβ16,8 most peptides listed in Table 1 bind a single equiv. of Cu(I) under Cu+ limiting conditions imposed by a slight excess of the weak probe ligand Fs (Fig. 7a). The exception was Aβ16-D1H that appeared to bind more than one equiv. Cu(I) under the same conditions (Fig. 7b).
 | ||
| Fig. 7 Change in [CuI(Fs)2]3− concentration in Mops buffer (50 mM, pH 7.4, 1.0 mM NH2OH) with increasing concentration of apo-peptide Aβ16 (a and c) or Aβ16-D1H (b and d) under weak competing conditions for (a and b) ([Cu]tot = 32.5 μM; [Fs]tot = 70 μM) or strong competing conditions for (c and d) ([Cu]tot = 38.0 μM; [Fs]tot = 140 μM). The dashed lines in (a and b) suggest Cu(I) binding stoichiometry under weak competing conditions; the solid traces in (c and d) are the curve-fitting of the experimental data to eqn (7), allowing estimation of the Cu(I) binding affinity for each peptide (Table 5). | ||
Increasing Fs ligand concentration or employing the stronger ligand Fz created more effective competing conditions for eqn (6) that allowed analysis and definition of Cu(I) binding affinity of each peptide with eqn (7) (Fig. 7c and d). There are only marginal differences in Cu(I) binding affinities for those peptide variants with substitution of a non-His residue in the sequence. But, the Cu(I) binding affinities of Aβ16-D1H and Aβ16-D1H/H14A are slightly higher than those of the other peptides (Table 5).
KID for Cu(I) of Aβ16 peptide variants at different pH valuesa
| Aβ16 peptide | logKID |
K ID/KID (wt) | Probe-affinity std | ||||
|---|---|---|---|---|---|---|---|
| pH 6.2 | pH 7.4 | pH 9.4 | pH 6.2 | pH 7.4 | pH 9.4 | ||
|
a The logKID values were determined via ligand competition for Cu(I) between each peptide and the specified probe ligands Fs and/or Fz in the respective buffers (50 mM) Mes (pH 6.2), Mops (pH 7.4) and Ches (pH 9.4). See the Experimental section for details.
|
|||||||
| WT | −9.6 | −10.6 | −11.0 | 1.00 | 1.00 | 1.00 | Fs |
| D1A | −9.3 | −10.6 | −11.0 | 2.00 | 1.00 | 1.00 | Fs |
| D1E | −9.5 | −10.4 | −10.9 | 1.26 | 1.58 | 1.26 | Fs |
| D1N | −9.5 | −10.4 | −10.8 | 1.26 | 1.58 | 1.58 | Fs |
| A2P | −9.9 | −10.7 | −11.0 | 0.50 | 0.79 | 1.00 | Fs |
| D1H | −10.2 | −11.1 | −11.5 | 0.25 | 0.32 | 0.32 | Fz |
| D1H/H14A | −10.1 | −10.9 | −11.3 | 0.32 | 0.50 | 0.50 | Fs/Fz |
| Ac-Aβ16 | −10.4 | 1.58 | Fs | ||||
The data are consistent with previous observations: (i) the non-His residues in the N-terminal region play little role in Cu(I) binding;8 (ii) two and only two His ligands are required for the flexible Aβ16 peptides to constitute a Cu(I) binding site (Fig. 1a);7,8,39–41 (iii) the Cu(I) bound by Aβ16 peptides is populated between different pairs of His ligands in a fast exchange mode that tends to increase the apparent binding affinities.7,8 There are three different combinations of His pairs for most peptides examined in this work (Fig. 1a) and little difference in their Cu(I) binding properties, except that for Aβ16-D1H/H14A is slightly higher (Table 5).
Aβ16-D1H contains an extra His1 residue to provide four possible His ligands. Its Cu(I) affinity is the highest of all the peptides. Theoretically, it may bind two equiv. of Cu(I) with similar affinities, but this does not seem to be the case (Fig. 7b). Binding of the first equiv. of Cu(I) appears to discourage the binding of a second equiv. of Cu(I) with the same affinity (Fig. 7b). Certainly, the first equiv. of Cu(I) bound in Aβ16-D1H may be populated among six different combinations of His pairs with considerable entropic gain, but binding of a further equiv. of Cu(I) is expected to restrict the binding sites to two His pairs only with considerable entropic loss.
Impact of the N-terminal modifications on the catalytic aerobic oxidation of ascorbate
A common feature in Alzheimer's disease is oxidative stress, a component of which may result from undesirable catalytic reduction of dioxygen by cellular reductants such as ascorbate (Asc) mediated by Cu complexes of Aβ peptides.2,31,32,38,46–48 The dioxygen reduction product H2O2 is liable to undergo further reduction via Fenton-type reactions to generate the hydroxyl radical that can cause damage not only to the direct copper ligands,49 but also to the surrounding residues.50 Indeed, Asc is abundant in the brain as an important neuromodulator and/or a neuroprotective agent and its concentration fluctuates dynamically in the range 200–400 μM in certain synaptic clefts.51,52 In reality, the oxidation rate of Asc by dioxygen is slow and must be catalysed by redox-active couples such as Cu(II)/Cu(I). It has been proposed that metal dyshomeostasis in the Alzheimer brain may result in binding of copper to Aβ peptides to initiate catalytic roles.53Aβ peptides can bind Cu(II) with the distinct binding modes of components I (including IA and IB) and II at physiological pH (Fig. 1b). Recent studies suggest that the different components available in Cu–Aβ16 systems (Fig. 1) display different reaction kinetics in both metal substitution and redox chemistry.5,6 This study has identified the critical role of the first two N-terminal residues in determining these Cu(II) binding modes and an integrated study of the thermodynamic and kinetic contributions of these binding modes in the generation of H2O2via catalytic aerobic oxidation of Asc was also undertaken. It should be noted that the reaction scheme in Fig. 8a is presented as an overall two-electron reaction process, but it may proceed via two consecutive one-electron steps with O2˙− as an immediate reduction intermediate that may be reduced further to H2O2 by a second CuI–Aβ complex.54
 | ||
| Fig. 8 (a) Catalytic aerobic oxidation of Asc monitored via A(265). Inset: Overall catalytic scheme. (b and c) Oxidation rate (μM min−1) catalyzed by various Cu–Aβ16 peptide complexes (1–20 μM) in the presence of one-fold excess of each respective apo-peptide in air-saturated buffers: (b) Mops buffer (50 mM, pH 6.6); (c) Mops buffer (50 mM, pH 7.4); (d) Ches buffer (50 mM, pH 8.7). The background oxidation rate (if any) that was detected in the presence of various concentrations of respective apo-peptides without added Cu has been removed from the determined rate. | ||
The experiments were performed in air-saturated buffers (50 mM) at three different pH values (Mops buffer at pH 6.6 and 7.4; Ches buffer at pH 8.7). The catalytic aerobic oxidation of Asc (50 μM) was undertaken with each Cu–Aβ16 complex (prepared with a one-fold excess of apo-peptide) at a range of concentrations (1.0–20 μM). The reaction was followed by the rate of decrease in absorbance at 265 nm for the reduced form of Asc (Fig. 8a). The catalytic activity of each Cu–Aβ16 peptide complex was determined from the slope of a linear correlation of the initial oxidation rates versus the catalyst concentrations and expressed as μM Asc oxidized/μM catalyst per minute, i.e., molar turnover number min−1 (see the Experimental section for details).
As observed previously,8,55 Asc is stable in metal-free air-saturated buffers within the experiment timescale (1–2 h) and all apo-peptides are inactive (at least after demetallation with EDTA). ‘Free’ Cu2+ and Cu+ have robust catalytic activities that are suppressed, but not silenced, by apo-peptides. The Cu–Aβ16 complexes are redox-active catalysts, but less active than ‘free’ copper. A one-fold excess of apo-peptide over each Cu–peptide was sufficient to minimize the catalytic contribution from ‘free’ Cu and to allow reliable detection of the catalytic activity of the Cu–complex. The results are summarized in Fig. 8, Fig. S8 (ESI†) and Table 6.
| Aβ16 peptide | Catalytic rate as molar velocity (min−1) at pH | Relative to that with Cu–D1H as catalyst at pH |
|
E 0′ (mV vs. SHE) reported at pHc | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6.6 | 7.4 | 8.7 | 6.6 | 7.4 | 8.7 | 6.2 | 7.4 | 9.4 | ∼6.5 | 7.4 | ∼8.5 | |
| a Conditions: [Asc]tot = 50 μM; [Cu–peptide]tot = 1–20 μM with a one-fold excess apo-peptide; buffers (50 mM) of Mops (pH 6.6 and 7.4) and Ches (pH 8.7). The background oxidation rate without the added Cu has been removed, see the Experimental section for details. b Calculated from eqn (8) using E0 = +153 mV for the redox couple Cu2+/Cu+ (note: the Cu(I) KD values at pH 6.6 and 8.7 were not determined and thus the calculations used data at pH 6.2 and 9.4 instead). c Determined by cyclic voltammetry, except a recent theoretical calculation predicted +280 mV for component I and –370 to –810 mV for component II.43 | ||||||||||||
| Aβ16 | 0.28 | 0.38 | 0.37 | 0.40 | 0.49 | 0.14 | +242 | +200 | +40.9 | +2555 | +9042 | −1535 |
| +28043 | +1807 | Lowc | ||||||||||
| +30044 | +27745 | |||||||||||
| D1A | 0.09 | 0.08 | 0.17 | 0.13 | 0.10 | 0.06 | +277 | +242 | +40.9 | |||
| D1E | 0.09 | 0.10 | 0.12 | 0.14 | 0.13 | 0.04 | +283 | +218 | +35.0 | |||
| D1N | 0.15 | 0.14 | 0.15 | 0.22 | 0.17 | 0.06 | +247 | +206 | +40.9 | |||
| A2P | 0.32 | 0.66 | 1.12 | 0.46 | 0.84 | 0.42 | +247 | +206 | +70.4 | |||
| D1H | 0.69 | 0.78 | 2.70 | 1.00 | 1.00 | 1.00 | +183 | +118 | +46.8 | |||
| D1H/H14A | 0.79 | 0.90 | 3.29 | 1.16 | 1.15 | 1.22 | +194 | +118 | +64.5 | |||
| Ac-Aβ16 | +277 | +2835 | −1305 | |||||||||
At pH 7.4, the molar catalytic activities (min−1) of the different Cu–Aβ16 complexes for Asc oxidation are given below in increasing order of their activity:
| D1A (0.08 min−1) ∼ D1E (0.10) ∼ D1N (0.14) < wt (0.38) |
| < A2P (0.66) < D1H (0.78) ∼ D1H/H14A (0.9) | (10) |
However, contradictions to these conclusions can be seen in Table 6. For example, while components IA and II dominate at pH 6.6 and 8.7, respectively, for CuII(Aβ16), they are both present in equilibrium at pH 7.4 (Fig. 6d and Fig. S1, ESI†), but the observed activities at pH 7.4 and 8.7 were essentially identical (Table 6). The complications arise from the fact that Asc is more stable towards oxidation at lower pH and less stable at higher pH values. Hence, there are two opposing effects acting on the Asc oxidation rates: (i) a transition from component I at lower pH to component II at higher pH decreases the catalytic rate; (ii) deprotonation of Asc that occurs at higher pH increases its oxidation rate. This latter effect is seen clearly for Cu(Aβ16-A2P), Cu(Aβ16-D1H) and Cu(Aβ16-D1H/H14A). Within the pH range 6–9, component IA dominates in the first complex and is locked in place in the latter two complexes (Fig. 6b and c). However, the catalytic oxidation rate increased with pH for each complex.
To separate these two opposing effects, the observed oxidation rates were normalized to that of Cu(Aβ16-D1H) under the same conditions (Table 6). The normalized data indicate: (i) the redox activities of Aβ16 and variants Aβ16-D1A, -D1E and -D1N decrease uniformly with increasing pH, consistent with the influence of the I → II transition (Fig. 6d, e and Fig. S1–S4, ESI†); (ii) the redox activity of Cu(Aβ16-A2P) is lowest at pH 8.7, consistent with a partial transition of the Cu(II) centre in this complex from component I to other unknown Cu(II) centre(s) (Fig. 6c) at high pH; (iii) the redox activities of the two Cu complexes with a His1 N-terminus are higher than all others and notably, the one with peptide Aβ16-D1H/H14A has the highest activity consistently at all pH values (Table 6). This high redox activity is attributed to a combined redox advantage of the Cu centres in both oxidation states and this is further analysed and discussed below.
Determinants that regulate the redox activity of Cu–Aβ peptides
In order to be an efficient redox catalytic centre for a particular process of oxidation and reduction, the formal reduction potential of the copper centre must be lower than that of the oxidant but higher than that of the reductant. At pH 7.4, the formal reduction potentials of ascorbate and dioxygen are ∼+50 mV56 and ∼+300 mV57 (vs. SHE), respectively.The formal reduction potentials of the Cu–Aβ complexes may be estimated via the Nernst relationship of eqn (8) from the respective Cu(I) and Cu(II) affinities of each Aβ16 peptide (Table 6). The reduction potentials are generally more negative for component II than for component I and decrease with increased pH. This is consistent with the more dramatic stabilization effect for Cu(II) coordination at higher pH, especially with the dominant coordination effect of the deprotonated amide nitrogen to the Cu(II) centre in component II.5,43 The calculated reduction potentials in this work from the respective dissociation constants generally match those determined experimentally by cyclic voltammetry or predicted from theoretical calculations (Table 6). However, it must be noted that the dissociation constants of the Cu–Aβ complexes are quite large (indicating relatively weak binding). In addition, the estimated values vary considerably with experimental conditions, as do the reduction potentials determined by cyclic voltammetry (Table 6). The redox processes are frequently not well-defined due to the different coordination environments for Cu(I) and Cu(II). Consequently, it is difficult to define the reduction potentials consistently and accurately as can be seen from the data in Table 6. Nevertheless, the statistical data seen there at pH 7.4 suggest that the reduction potentials of most Cu–Aβ complexes studied in this work fall within the potential window for catalytic oxidation of ascorbate by dioxygen (Table 6) and therefore should have no significant thermodynamic impact on their efficiency as catalysts for the aerial oxidation of ascorbate. In fact, there is no clear correlation between the calculated reduction potentials and the observed catalytic rates at the three pH values tested (Table 6).
It is apparent that the difference in the redox activity of the Cu centre in each Aβ16 peptide must be attributed primarily to differences in reaction kinetics that are closely related to the binding modes for both Cu(I) and Cu(II). As an efficient catalytic centre, the metal ion must be able to shuttle between different oxidation states with minimal reorganization energy,44 but this condition is often not met for Cu that prefers different ligands and binding modes for its two biologically accessible oxidation states. Blue copper proteins such as plastocyanin and azurin that accommodate robust redox active Cu centres feature conformational rigidity in the protein structures that impose an ‘entatic state' to the Cu centre for efficient electron transfer.58
Such ‘entatic states' for Aβ peptides, however, are problematic, given their flexibility and unstructured nature. The coordination spheres for Aβ peptides are decided primarily by the coordination preferences of each oxidation state of the Cu centre, as demonstrated above and in previous studies.3,8 Consequently, the Cu centres in Aβ peptides are generally not efficient redox catalysts since only a very small fraction of Cu centres in equilibrium are pre-organized for electron transfer44 and the catalytic activity of ‘free’ Cu is inhibited upon binding to Aβ peptides.8,55,59
At physiological pH, Aβ16 binds one equiv. of Cu(I) or Cu(II) with sub-nanomolar affinities (Fig. 1). Component I in the Cu(II) system is more reactive in the catalytic oxidation of Asc than component II for several reasons: (i) component I shares two His ligands with each of the three possible Cu(I) sites while component II contains only one His ligand; (ii) the main Cu(II) ligands for component I are flexible amino acid sidechains while the dominant Cu(II) ligands for component II are derived from the peptide backbone with rigid fused chelate rings; (iii) a Cu–N− bond arising from coordination to a negatively charged amide N− is highly covalent and any reactions involving such a chemical bond are usually slow.6,21,33,43 Consequently, component I is more reactive than component II, not only in redox catalysis but also in ligand substitution.5,6 These conclusions are consolidated by the observed reactivity orders for related peptides in ligand substitution6,21,33 and redox turnover (Table 6, eqn (10)).5 In particular, substitution of Asp1 with His1 not only locks the Cu(II) binding mode to the IA component within the pH range 6–9 (Fig. 4a(i) and 6b), but introduces a new Cu(I) ligand in the N-terminus to be shared with the Cu(II) binding site. This leads to a considerable increase in the redox reactivity of the Cu centre. Interestingly, removal of the extra non-N-terminal ligand His14 from Aβ16-D1H leads to a further increase in redox activity (Table 6), presumably attributable to the increased similarity between the binding sites for both Cu(I) and Cu(II) in the Aβ16-D1H/H14A peptide.
Summary and concluding remarks
Monomeric Aβ peptides are flexible and unstructured. Both metal binding ligands and binding modes change dynamically with the Cu oxidation state and with reaction conditions. A range of chemical and spectroscopic studies were carried out on copper complexes of several Aβ16 peptides in which the two key residues for Cu(II) binding in the N-terminal region were varied. Key conclusions are as follows:(i) At lower pH, the Aβ16 peptide binds Cu(II) predominantly as component IA in equilibrium with less stable component IB; at higher pH, these are converted to component II with transition pKa ∼ 7.8 (Fig. 1b and 4b);
(ii) Component I sites involve flexible amino acid sidechains common to the Cu(I) binding ligands, but the metal sites for component II are dictated by a deprotonated backbone amide nitrogen that cannot act as a Cu(I) ligand and is slow to dissociate.5,6,43 Consequently, component I is more reactive than component II in both metal substitution and redox catalysis.
(iii) Substitution of Asp1 with His provides an extra imidazole ligand for Cu(I) and also allows the N-terminal His1 to be part of a stable six-membered chelate ring for Cu(II) coordination. Consequently, the Cu(II) centre in Aβ16-D1H is locked into component IA with a dramatic increase in its redox activity.
(iv) Replacement of Ala2 with Pro suppresses component II relative to component I and increases the reactivity of the bound copper.
(v) Substitution of Asp1 with Ala restricts component I to the less stable IB form that is readily converted to component II at physiological pH with decreased catalytic reactivity.
This study provides insights into determinants that regulate the reactivity of copper toward Aβ peptides and may guide development towards a better understanding of the factors contributing to oxidative stress present within Alzheimer's disease.
Abbreviations
| Asc | Ascorbate |
| Bca | Bicinchoninic anion |
| Bcs | Bathocupröine disulfonate |
| Ches | N-Cyclohexyl-2-aminoethanesulfonic acid |
| DP2 | Dansyl peptide 2 (DAE(KDNS)RHDH) |
| DP3 | Dansyl peptide 3 (HP(KDNS)DHDH) |
| DP4 | Dansyl peptide 4 (H(KDNS)HH) |
| eqn | Equation |
| equiv. | Equivalent |
| ESI-MS | Electrospray ionization mass spectrometry |
| Fs | Ferene S |
| Fz | Ferrozine |
| Hepes | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| His | Histidine |
| Mes | 2-(N-Morpholino)ethanesulfonic acid |
| Mops | 3-(N-Morpholino)propanesulfonic acid |
Acknowledgements
This work was supported by funding from the Australian Research Council Grant DP130100728. We thank Prof. Ashley Bush for comments and discussions and Mrs Sioe See Volaric for providing technical support for recording EPR spectra.References
- A. I. Bush, The metallobiology of Alzheimer's disease, Trends Neurosci., 2003, 26, 207–214 CrossRef CAS PubMed.
- K. P. Kepp, Bioinorganic Chemistry of Alzheimer's Disease, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- C. Hureau, Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 1: an overview, Coord. Chem. Rev., 2012, 256, 2164–2174 CrossRef CAS.
- C. Hureau and P. Dorlet, Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 2: dependence of Cu(II) binding sites with Aβ sequences, Coord. Chem. Rev., 2012, 256, 2175–2187 CrossRef CAS.
- L. G. Trujano-Ortiz, F. J. González and L. Quintanar, Redox Cycling of Copper–Amyloid β 1–16 Peptide Complexes Is Highly Dependent on the Coordination Mode, Inorg. Chem., 2015, 54, 4–6 CrossRef CAS PubMed.
- T. Branch, P. Girvan, M. Barahona and L. Ying, Introduction of a Fluorescent Probe to Amyloid-β to Reveal Kinetic Insights into Its Interactions with Copper(II), Angew. Chem., Int. Ed., 2015, 54, 1227–1230 CrossRef CAS PubMed.
- C. Hureau, V. Balland, Y. Coppel, P. L. Solari, E. Fonda and P. Faller, Importance of dynamical processes in the coordination chemistry and redox conversion of copper amyloid-beta complexes, J. Biol. Inorg. Chem., 2009, 14, 995–1000 CrossRef CAS PubMed.
- T. R. Young, A. Kirchner, A. G. Wedd and Z. Xiao, An Integrated Study of the Affinities of the Aβ16 Peptide for Cu(I) and Cu(II): Implications for the Catalytic Production of Reactive Oxygen Species, Metallomics, 2014, 6, 505–517 RSC.
- C. Hureau, Y. Coppel, P. Dorlet, P. L. Solari, S. Sayen, E. Guillon, L. Sabater and P. Faller, Deprotonation of the Asp1-Ala2 peptide bond induces modification of the dynamic copper(II) environment in the amyloid-beta peptide near physiological pH, Angew. Chem., Int. Ed., 2009, 48, 9522–9525 CrossRef CAS PubMed.
- S. C. Drew, C. J. Noble, C. L. Masters, G. R. Hanson and K. J. Barnham, Pleomorphic copper coordination by Alzheimer's disease amyloid-beta peptide, J. Am. Chem. Soc., 2009, 131, 1195–1207 CrossRef CAS PubMed.
- C. A. Damante, K. Osz, Z. Nagy, G. Pappalardo, G. Grasso, G. Impellizzeri, E. Rizzarelli and I. Sovago, The metal loading ability of beta-amyloid N-terminus: a combined potentiometric and spectroscopic study of copper(II) complexes with beta-amyloid(1-16), its short or mutated peptide fragments, and its polyethylene glycol (PEG)-ylated analogue, Inorg. Chem., 2008, 47, 9669–9683 CrossRef CAS PubMed.
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Pulse EPR Spectroscopy Reveals the Coordination Sphere of Copper(II) Ions in the 1–16 Amyloid-β Peptide: A Key Role of the First Two N-Terminus Residues, Angew. Chem., Int. Ed., 2009, 48, 9273–9276 CrossRef CAS PubMed.
- S. C. Drew, C. L. Masters and K. J. Barnham, Alanine-2 carbonyl is an oxygen ligand in Cu2+ coordination of Alzheimer's disease amyloid-beta peptide – relevance to N-terminally truncated forms, J. Am. Chem. Soc., 2009, 131, 8760–8761 CrossRef CAS PubMed.
- C. L. Masters, G. Multhaup, G. Simms, J. Pottgiesser, R. N. Martins and K. Beyreuther, Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels, EMBO J., 1985, 4, 2757–2763 CAS.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Amyloid plaque core protein in Alzheimer disease and Down syndrome, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245–4249 CrossRef CAS.
- J. W. Karr, H. Akintoye, L. J. Kaupp and V. A. Szalai, N-Terminal deletions modify the Cu2+ binding site in amyloid-beta, Biochemistry, 2005, 44, 5478–5487 CrossRef CAS PubMed.
- J. W. Karr and V. A. Szalai, Role of aspartate-1 in Cu(II) binding to the amyloid-beta peptide of Alzheimer's disease, J. Am. Chem. Soc., 2007, 129, 3796–3797 CrossRef CAS PubMed.
- C. Ghosh and S. G. Dey, Ligand-field and ligand-binding analysis of the active site of copper-bound Abeta associated with Alzheimer's disease, Inorg. Chem., 2013, 52, 1318–1327 CrossRef CAS PubMed.
- B. Alies, H. Eury, C. Bijani, L. Rechignat, P. Faller and C. Hureau, pH-Dependent Cu(II) coordination to amyloid-beta peptide: impact of sequence alterations, including the H6R and D7N familial mutations, Inorg. Chem., 2011, 50, 11192–11201 CrossRef CAS PubMed.
- C. Harford and B. Sarkar, Amino Terminal Cu(II)- and Ni(II)-Binding Motif of Proteins and Peptides: Metal Binding, DNA Cleavage, and Other Properties, Acc. Chem. Res., 1997, 30, 123–130 CrossRef CAS.
- T. R. Young, C. J. K. Wijekoon, B. Spyrou, P. S. Donnelly, A. G. Wedd and Z. Xiao, A Set of Robust Fluorescent Peptide Probes for Quantification of Cu(II) Binding Affinities in the Micromolar to Femtomolar Range, Metallomics, 2015, 7, 567–578 RSC.
- M. Mital, N. E. Wezynfeld, T. Frączyk, M. Z. Wiloch, U. E. Wawrzyniak, A. Bonna, C. Tumpach, K. J. Barnham, C. L. Haigh, W. Bal and S. C. Drew, A Functional Role for Aβ in Metal Homeostasis? N-Truncation and High-Affinity Copper Binding, Angew. Chem., Int. Ed., 2015, 54, 10460–10464 CrossRef CAS PubMed.
- N. E. Wezynfeld, E. Stefaniak, K. Stachucy, A. Drozd, D. Płonka, S. C. Drew, A. Krężel and W. Bal, Resistance of Cu(Aβ4–16) to Copper Capture by Metallothionein-3 Supports a Function for the Aβ4–42 Peptide as a Synaptic CuII Scavenger, Angew. Chem., Int. Ed., 2016, 55, 8235–8238 CrossRef CAS PubMed.
- K. Y. Djoko, Z. Xiao, D. L. Huffman and A. G. Wedd, Conserved Mechanism of Copper Binding and Transfer. A Comparison of the Copper-Resistance Proteins PcoC from Escherichia coli and CopC from Pseudomonas syringae, Inorg. Chem., 2007, 46, 4560–4568 CrossRef CAS PubMed.
- W. M. Kok, J. M. Cottam, G. D. Ciccotosto, L. A. Miles, J. A. Karas, D. B. Scanlon, B. R. Roberts, M. W. Parker, R. Cappai, K. J. Barnham and C. A. Hutton, Synthetic dityrosine-linked [small beta]-amyloid dimers form stable, soluble, neurotoxic oligomers, Chem. Sci., 2013, 4, 4449–4454 RSC.
- Z. Xiao, L. Gottschlich, R. van der Meulen, S. R. Udagedara and A. G. Wedd, Evaluation of quantitative probes for weaker Cu(I) binding sites completes a set of four capable of detecting Cu(I) affinities from nanomolar to attomolar, Metallomics, 2013, 5, 501–513 RSC.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1 and related proteins: detection probes and affinity standards, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef CAS PubMed.
- M. I. Karayannis, D. N. Samios and C. H. P. Gousetis, A study of the molar absorptivity of ascorbic acid at different wavelengths and pH values, Anal. Chim. Acta, 1977, 93, 275–279 CrossRef CAS PubMed.
- G. Milazzo and S. Caroli, Tables of Standard Electrode Potentials, Wiley, New York, 1978 Search PubMed.
- C. Reinhard, S. S. Hebert and B. De Strooper, The amyloid-beta precursor protein: integrating structure with biological function, EMBO J., 2005, 24, 3996–4006 CrossRef CAS PubMed.
- A. I. Bush, C. L. Masters and R. E. Tanzi, Copper, beta-amyloid, and Alzheimer's disease: tapping a sensitive connection, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11193–11194 CrossRef CAS PubMed.
- M. W. Beck, S. B. Oh, R. A. Kerr, H. J. Lee, S. H. Kim, S. Kim, M. Jang, B. T. Ruotolo, J.-Y. Lee and M. H. Lim, A rationally designed small molecule for identifying an in vivo link between metal–amyloid-β complexes and the pathogenesis of Alzheimer's disease, Chem. Sci., 2015, 6, 1879–1886 RSC.
- C. J. K. Wijekoon, T. R. Young, A. G. Wedd and Z. Xiao, The CopC Protein from Pseudomonas Fluorescens SBW25 Features A Conserved Novel High Affinity Cu(II) Binding Site, Inorg. Chem., 2015, 54, 2950–2959 CrossRef CAS PubMed.
- L. Zhang, M. Koay, M. J. Maher, Z. Xiao and A. G. Wedd, Intermolecular transfer of copper ions from the CopC protein of Pseudomonas syringae. Crystal structures of fully loaded Cu(I)Cu(II) forms, J. Am. Chem. Soc., 2006, 128, 5834–5850 CrossRef CAS PubMed.
- T. J. Lawton, G. E. Kenney, J. D. Hurley and A. C. Rosenzweig, The CopC Family: Structural and Bioinformatic Insights into a Diverse Group of Periplasmic Copper Binding Proteins, Biochemistry, 2016, 55, 2278–2290 CrossRef CAS PubMed.
- C. Kállay, K. Várnagy, G. Micera, D. Sanna and I. Sóvágó, Copper(II) complexes of oligopeptides containing aspartyl and glutamyl residues. Potentiometric and spectroscopic studies, J. Inorg. Biochem., 2005, 99, 1514–1525 CrossRef PubMed.
- I. Sovago and K. Osz, Metal ion selectivity of oligopeptides, Dalton Trans., 2006, 3841–3854, 10.1039/B607515K.
- X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. A. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, Cu(II) potentiation of Alzheimer Aβ neurotoxicity: correlation with cell-free hydrogen peroxide production and metal reduction, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS PubMed.
- R. A. Himes, G. Y. Park, G. S. Siluvai, N. J. Blackburn and K. D. Karlin, Structural Studies of Copper(I) Complexes of Amyloid-β Peptide Fragments: Formation of Two-Coordinate Bis(histidine) Complexes, Angew. Chem., Int. Ed., 2008, 47, 9084–9087 CrossRef CAS PubMed.
- J. Shearer and V. A. Szalai, The Amyloid-β Peptide of Alzheimer's Disease Binds CuI in a Linear Bis-His Coordination Environment: Insight into a Possible Neuroprotective Mechanism for the Amyloid-β Peptide, J. Am. Chem. Soc., 2008, 130, 17826–17835 CrossRef CAS PubMed.
- S. Furlan, C. Hureau, P. Faller and G. La Penna, Modeling the Cu+ binding in the 1-16 region of the amyloid-beta peptide involved in Alzheimer's disease, J. Phys. Chem. B, 2010, 114, 15119–15133 CrossRef CAS PubMed.
- L. Guilloreau, S. Combalbert, A. Sournia-Saquet, H. Mazarguil and P. Faller, Redox chemistry of copper-amyloid-beta: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state, ChemBioChem, 2007, 8, 1317–1325 CrossRef CAS PubMed.
- J. Alí-Torres, A. Mirats, J.-D. Maréchal, L. Rodríguez-Santiago and M. Sodupe, 3D Structures and Redox Potentials of Cu2+–Aβ(1–16) Complexes at Different pH: A Computational Study, J. Phys. Chem. B, 2014, 118, 4840–4850 CrossRef PubMed.
- V. Balland, C. Hureau and J.-M. Savéant, Electrochemical and homogeneous electron transfers to the Alzheimer amyloid-β copper complex follow a preorganization mechanism, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113–17118 CrossRef CAS PubMed.
- D. Jiang, L. Men, J. Wang, Y. Zhang, S. Chickenyen, Y. Wang and F. Zhou, Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathalogical relevance, Biochemistry, 2007, 46, 9270–9282 CrossRef CAS PubMed.
- X. Zhu, B. Su, X. Wang, M. A. Smith and G. Perry, Causes of oxidative stress in Alzheimer disease, Cell. Mol. Life Sci., 2007, 64, 2202–2210 CrossRef CAS PubMed.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS PubMed.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS PubMed.
- C. Cheignon, P. Faller, D. Testemale, C. Hureau and F. Collin, Metal-catalyzed oxidation of A[small beta] and the resulting reorganization of Cu binding sites promote ROS production, Metallomics, 2016, 8, 1081–1089 RSC.
- C. S. Atwood, G. Perry, H. Zeng, Y. Kato, W. D. Jones, K.-Q. Ling, X. Huang, R. D. Moir, D. Wang, L. M. Sayre, M. A. Smith, S. G. Chen and A. I. Bush, Copper Mediates Dityrosine Cross-Linking of Alzheimer's Amyloid-β, Biochemistry, 2004, 43, 560–568 CrossRef CAS PubMed.
- R. A. Grünewald, Ascorbic acid in the brain, Brain Res. Rev., 1993, 18, 123–133 CrossRef.
- M. E. Rice, Ascorbate regulation and its neuroprotective role in the brain, Trends Neurosci., 2000, 23, 209–216 CrossRef CAS PubMed.
- G. Eskici and P. H. Axelsen, Copper and Oxidative Stress in the Pathogenesis of Alzheimer's Disease, Biochemistry, 2012, 51, 6289–6311 CrossRef CAS PubMed.
- K. Reybier, S. Ayala, B. Alies, J. V. Rodrigues, S. Bustos Rodriguez, G. La Penna, F. Collin, C. M. Gomes, C. Hureau and P. Faller, Free Superoxide is an Intermediate in the Production of H2O2 by Copper(I)-Aβ Peptide and O2, Angew. Chem., Int. Ed., 2016, 55, 1085–1089 CrossRef CAS PubMed.
- B. Alies, I. Sasaki, O. Proux, S. Sayen, E. Guillon, P. Faller and C. Hureau, Zn impacts Cu coordination to amyloid-[small beta], the Alzheimer's peptide, but not the ROS production and the associated cell toxicity, Chem. Commun., 2013, 49, 1214–1216 RSC.
- B. E. Conway, Electrochemical Data, Greenwood Press, New York, 1969 Search PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, New York, 2004 Search PubMed.
- R. J. P. Williams, Metallo-enzyme catalysis: the entatic state, J. Mol. Catal., 1985, 30, 1–26 CrossRef CAS.
- S. L. Leong, T. R. Young, K. J. Barnham, A. G. Wedd, M. G. Hinds, Z. Xiao and R. Cappai, Quantification of Copper Binding to Amyloid Precursor Protein Domain 2 and its Caenorhabditis Elegans Ortholog. Implications for Biological Function, Metallomics, 2014, 6, 105–116 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Conditional Cu(II) dissociation constants (Table S1), pH-dependent EPR spectra (Fig. S1–S7). See DOI: 10.1039/c6mt00299d |
| ‡ Present address: The Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Vic 3010, Australia. |
| This journal is © The Royal Society of Chemistry 2017 |