
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Age modulates the injury-induced metallomic profile in the brain†
Stuart D.
Portbury
a,
Dominic J.
Hare
 ab,
Charlotte J.
Sgambelloni
a,
David P.
Bishop
b,
David I.
Finkelstein
a,
Philip A.
Doble
b and
Paul A.
Adlard
*a
ab,
Charlotte J.
Sgambelloni
a,
David P.
Bishop
b,
David I.
Finkelstein
a,
Philip A.
Doble
b and
Paul A.
Adlard
*a
aThe Florey Institute of Neuroscience and Mental Health, Kenneth Myer Building, The University of Melbourne, 30 Royal Parade, Parkville, Victoria 3052, Australia. E-mail: paul.adlard@florey.edu.au; Tel: +61 3 9035 6775
bElemental Bio-imaging Facility, University of Technology Sydney, Thomas Street, Broadway, New South Wales 2007, Australia
First published on 1st February 2017
Abstract
The biological transition metals iron (Fe), copper (Cu) and zinc (Zn) are thought to contribute to the neuronal pathologies that occur following traumatic brain injury (TBI), and indeed our previously published work in young (3 month-old) mice clearly demonstrates a significant spatiotemporal modulation of metals following TBI. Of note, however, is the literature observation that there is both an apparent detrimental effect of aging on TBI outcomes and an alteration in metals and their various transporters with normal advancing age. Therefore, to determine whether there was an interaction between aging, metals and TBI, we have utilised laser ablation-inductively coupled plasma-mass spectrometry to examine the spatial and temporal distribution of Fe, Zn and Cu following an acute controlled cortical impact brain injury in aged (24 months) rodents. The relative abundance of metals in corresponding regions within the ipsilateral and contralateral hemispheres as well as the hippocampus was assessed. Substantial region and time point specific alterations in Fe, Zn and Cu were identified immediately and up to 28 days post-TBI. The data from this follow-up study has also been compared to our previous data from young animals, and aged mice exhibit an appreciably enhanced and persistent elevation of all metals in every region surveyed, with individual metal disparities at various time points observed post-injury. This may potentially contribute to the acceleration in the onset of cognitive decline and neurological disease that has been observed in the aged population following head trauma.
Significance to metallomicsMetals play critical roles in brain health. The spatial changes we have observed in the levels of iron, zinc and copper in the aged brain, both in naïve and brain injured animals, highlight the potential interaction of these metals in both age- and injury-related functional deficits. This suggests the use of metal-targeted therapeutics could be used to improve outcomes post-injury. |
Introduction
Traumatic brain injury (TBI) is a rapidly growing international health concern that is complicated by the diversity in severity, injury type and epidemiological patterns of TBI incidence.1,2 Along with suicide, it is a major cause of death and disability in young people,1,3 with injury modalities including falls, assaults, motor vehicle accidents and blunt force trauma.4 In its ‘World report on traffic injury prevention’, the World Health Organization (WHO) estimates that by 2020, road traffic accidents would be within the top three leading causes of global burden of disease, surpassing even HIV and tuberculosis.5 Although the form of TBI varies on a near case-by-case basis, there is a shared stereotypical response to the injury that includes, amongst other features, inflammation,6 release of serum and cerebrospinal fluid-based markers,7 and axonal injury.8 The age at which injury is sustained is an additional factor that indirectly contributes to morbidity and mortality and is now recognised as a risk factor for dementia.9 A recent comprehensive study by Gardner et al. assessed 164![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 611 patients with trauma, aged ≥55 years at the time of injury, and clearly demonstrated the influence of age on dementia risk following TBI.10 Moreover, even a single mild TBI conferred an elevated dementia risk in patients aged ≥65 at the time of injury. This study is supportive of earlier reports indicating a significant association between age and TBI leading to increased mortality, as well as unfavourable clinical outcomes in elderly patients with either mild11 or severe TBI;12,13 including the potential for TBI to proceed or initiate the onset of debilitating and fatal disorders such as Alzheimer's disease (AD),14 amyotrophic lateral sclerosis (ALS),15 and Parkinson's disease (PD).16,17 Consistent with this, widespread tau and amyloid-beta pathology, the characteristic hallmarks of AD, have been shown to persist for many years after a single TBI18 and chronic inflammation and ongoing white matter degeneration have also been observed as an enduring legacy of a single TBI many years after the initial insult.19 Similarly, in TBI experiments with mice, aged animals have a demonstrated inferior performance in behavioural tests, exhibit exacerbated chronic inflammation with prolonged activation of both astrocytes and microglia, and display pathological iron deposits co-localised with T2 hypointensities via magnetic resonance imaging when compared to younger adult mice.20–22 The latter relates to a common biochemical feature of TBI and age associated dementias such as AD, where there is a disordered regulation of the biologically critical transition metals iron (Fe), copper (Cu) and zinc (Zn).23 These observations have led to the metal dyshomeostasis theory, which implicates metals in the cellular cascades that contribute to AD, PD, ALS and other conditions.26,27 Whether metals are critical in the onset and/or progression of disease, or even normal aging, is yet to be definitively established, although it is likely that an age-related modulation of metal ions and their transporters are a contributing factor.
611 patients with trauma, aged ≥55 years at the time of injury, and clearly demonstrated the influence of age on dementia risk following TBI.10 Moreover, even a single mild TBI conferred an elevated dementia risk in patients aged ≥65 at the time of injury. This study is supportive of earlier reports indicating a significant association between age and TBI leading to increased mortality, as well as unfavourable clinical outcomes in elderly patients with either mild11 or severe TBI;12,13 including the potential for TBI to proceed or initiate the onset of debilitating and fatal disorders such as Alzheimer's disease (AD),14 amyotrophic lateral sclerosis (ALS),15 and Parkinson's disease (PD).16,17 Consistent with this, widespread tau and amyloid-beta pathology, the characteristic hallmarks of AD, have been shown to persist for many years after a single TBI18 and chronic inflammation and ongoing white matter degeneration have also been observed as an enduring legacy of a single TBI many years after the initial insult.19 Similarly, in TBI experiments with mice, aged animals have a demonstrated inferior performance in behavioural tests, exhibit exacerbated chronic inflammation with prolonged activation of both astrocytes and microglia, and display pathological iron deposits co-localised with T2 hypointensities via magnetic resonance imaging when compared to younger adult mice.20–22 The latter relates to a common biochemical feature of TBI and age associated dementias such as AD, where there is a disordered regulation of the biologically critical transition metals iron (Fe), copper (Cu) and zinc (Zn).23 These observations have led to the metal dyshomeostasis theory, which implicates metals in the cellular cascades that contribute to AD, PD, ALS and other conditions.26,27 Whether metals are critical in the onset and/or progression of disease, or even normal aging, is yet to be definitively established, although it is likely that an age-related modulation of metal ions and their transporters are a contributing factor.
For instance, Fe storage increases with age in the human brain in the cortex, hippocampus and basal ganglia, and the main cell types shown to accumulate Fe are microglia and astrocytes.28 The labile iron pool can increase to potentially harmful levels whereby elevated and unregulated reactive oxygen species (ROS) can lead to oxidative damage and cell death. Accumulation of Fe in specific brain regions occurs in many neurodegenerative disorders of ageing and is usually associated with oxidative stress and subsequent cellular injury and death.
Similarly, a disruption in Zn levels can cause profound neuronal damage and disruption of synaptic activity. Zn is readily dissociated from its binding proteins in cells undergoing changes in redox state due to unregulated production of reactive oxygen species,29 and the liberated Zn ions have been established as a potent inducer of neuronal injury.30 The increased and unregulated production of ROS is a major contributor to the pathological changes associated with normal ageing and particularly age-related disorders of the brain. Conversely, however, adequate concentrations of Zn are required for normal neurotransmission, and synaptic vesicular Zn is required for normal memory function,31 and the Zn transporter responsible for loading presynaptic vesicles with Zn (ZnT3) has been shown to decrease with age in both rodents and humans.31
Finally, Cu is an essential element in the central nervous system (CNS). However, its propensity to form ROS renders it toxic in situations where an increase in labile species is observed.32–35 Increased Cu levels have been observed in rodent models of human aging.34,36 However, diminished Cu levels are reported in the frontal, occipital and parietal lobes37 amygdala and hippocampus in AD.38 Thus, the age and disease-dependent modulation of metals provides a complex landscape on which TBI can occur, and which TBI can influence.
We recently reported a time-course analysis of changes in cerebral metals post-TBI in young adult mice utilising laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS). Our results demonstrated a potentially injurious sustained elevation of non-heme associated Fe in the ipsilateral cortex.25 In this study we sought to provide clarity around the age-related changes in metal levels that occurs in the CNS, under both baseline conditions and following a TBI. These studies may provide insight into mechanisms underlying the inter-relationships between TBI, ageing, neurodegeneration and functional outcomes.
Methods and materials
Ethics
All procedures were carried out in accordance with protocols approved by the Howard Florey Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes as described by the National Health and Medical Research Council of Australia.Controlled cortical impact model
Controlled cortical impact (CCI) was carried out using a Hatteras PCI 3000 precision cortical impactor (Hatteras Instruments, Cary, NC). 24 month-old male C57Bl6 mice were anesthetised via intraperitoneal injection of 100 mg kg−1 ketamine and 10 mg kg−1 xylazine. Anesthesia was monitored via respiration and pedal retraction reflexes. Upon adequate anesthesia the surgical site was clipped and shaved and cleansed with 70% ethanol. A 10 mm mid-line incision was made over the skull, and the skin and fascia were reflected to make a 4 mm craniotomy on the central aspect of the right parietal bone using a motorised drill (AP-2.25 mm, lateral 2.5). Excised bone was placed in sterile PBS until resituating post-injury. The animal was then positioned in a stereotaxic frame. The 3 mm impact tip of the CCI was lowered to the surface of the exposed dura, signified by an audible contact alarm, and the injury subsequently delivered (3 m s−1 velocity with a 1.5 mm penetration depth). The excised bone was then replaced and glued in position with super glue, the skin similarly glued together and the animal then placed in a 37 °C heated cage until completely ambulatory. Animals did not receive any analgesia post-surgery.Animals were euthanised at 24 h (n = 4), 72 h (n = 3), 7 day (n = 3), 14 day (n = 4) and 28 day-post surgeries (n = 4), and animals without injury (n = 3) were euthanised on day 28 only. For the 3 month-old ESI,† animals were euthanised at 24 h (n = 5), 72 h (n = 4), 7 day (n = 4), 14 day (n = 5) and 28 days’ post surgeries (n = 4). Animals were anesthetised with Nembutal, before a 50 mL pericardial perfusion with ice-cold 4% paraformaldehyde in PBS. Brains were rapidly dissected and further incubated in PFA (4% in PBS at 4 °C), followed by immersion in a cryoprotective solution for 7 days (30% sucrose in PBS, 4 °C). Sections were subsequently cryostat sectioned at 30 μm intervals at −20 °C and mounted on standard microscope slides. Sections were stored at −80 °C prior to analysis, when they were air-dried at room temperature.
LA-ICP-MS imaging
Analysis was performed using a New Wave Research UP213 laser ablation system with a two-volume large format cell (ablation area 15 × 15 cm). This system was hyphenated to an Agilent Technologies 7500ce ICP-MS fitted with ‘cs’ lenses for enhanced sensitivity. Quantitative data was obtained by representative ablation of matrix-matched tissue standards produced according to the protocol previously reported.39Hydrogen was used as a reaction gas at a flow rate of 3 mL min−1 to remove potential interferences arising from trace oxygen impurities in the argon gas used, particularly 40Ar16O+ on 56Fe+.40 The specificity of hydrogen for reducing interference on Fe was the main determinant of its selection; non-specific collision gases such as helium reduce signal to noise ratios and do not attenuate interferences effectively in LA-ICP-MS.41 We have recently shown that matrix-based interference formation at biologically-relevant concentrations are negligible using these experimental conditions.42 Measured masses included m/z = 56 for Fe, 62 for Cu and 66 for Zn with an integration time of 0.08 seconds per mass. Limits of detection (LOD) and quantification (LOQ) were 0.55 μg g−1 and 1.58 μg g−1 for Fe; 1.20 μg g−1 and 1.89 μg g−1 for Cu; and 0.39 μg g−1 and 1.08 μg g−1 for Zn.
Image construction and region of interest selection
Horizontal lines of ablation were drawn across both ipsilateral and contralateral upper quadrants of the selected section. Lines of ablation were spaced apart the same distance as the laser beam diameter. A beam diameter of 30 μm was used, traversing the section at a speed of 120 μm s−1, laser fluence of 0.3 J cm−2 and repetition rate of 20 Hz. The ICP-MS was configured to collect 4 data points per second40 thus the resultant images had pixel dimensions was equivalent to 30 × 30 μm2 (total pixel area = 900 μm2). Images were produced by reducing multiple ablation lines into ASCII data files via a Python script for importing into ENVI 6.0 (Exelis Visual Information Solutions, Boulder, CO, USA), from which regions of interest (ROIs) were extracted and statistically analysed. Each ROI represents a rectangular quadrant that radiates outwards from the impact site towards the centre of the brain, or the equivalent region on the contralateral side of the brain (Fig. 1).25 One brain slice per animal underwent ablation and subsequent analysis. Additionally, the hippocampus was manually extracted as an individual ROI using the Zn map43 for analysis without adjacent tissue of any ROI region (Fig. 1). ROIs had average areas of: ROI1 (cortex) 835.1 ± 326 mm2; ROI2 (cortex, hippocampus and white matter) = 3129 ± 486 mm2 (minus the area of ROI1); ROI3 (thalamus and cortex) = 6116 ± 885 mm2 (minus ROI 1 and 2). The mean area of the entire dorsal hemisphere was 10.08 ± 1.46 cm2. Total areas of extracted ROIs were equivalent between ipsilateral and contralateral sides.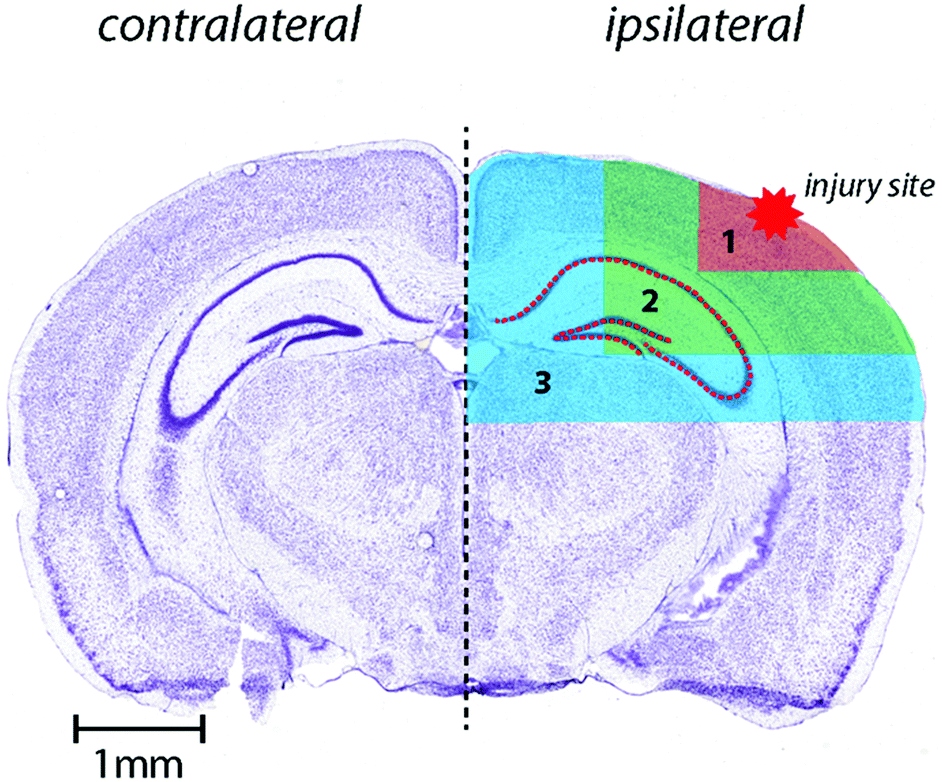 | ||
| Fig. 1 Schematic demonstrating regions of interest selection for assessment of changes in metal levels radial to the site of impact including hippocampus as outlined. Selected ROI's were applied to each measured metal and extracted for statistical comparisons. Whole-hemisphere metal concentration and equivalent ROI's on the contralateral side were also extracted. Nissl stained reference section (bregma-2.255 mm) used from Allen Brain atlas24 and modified from our previous publication.25 | ||
Statistical analysis
Statistical analysis was carried out in Prism 6.0 h (Graph-Pad, La Jolla, CA, USA). Analysis was carried out using either a two-tailed t test or a two-way ANOVA as appropriate, with significance recognised as p < 0.05.Results
Representative examples of time course LA-ICPMS images of the left and right hemispheres are shown in Fig. 2.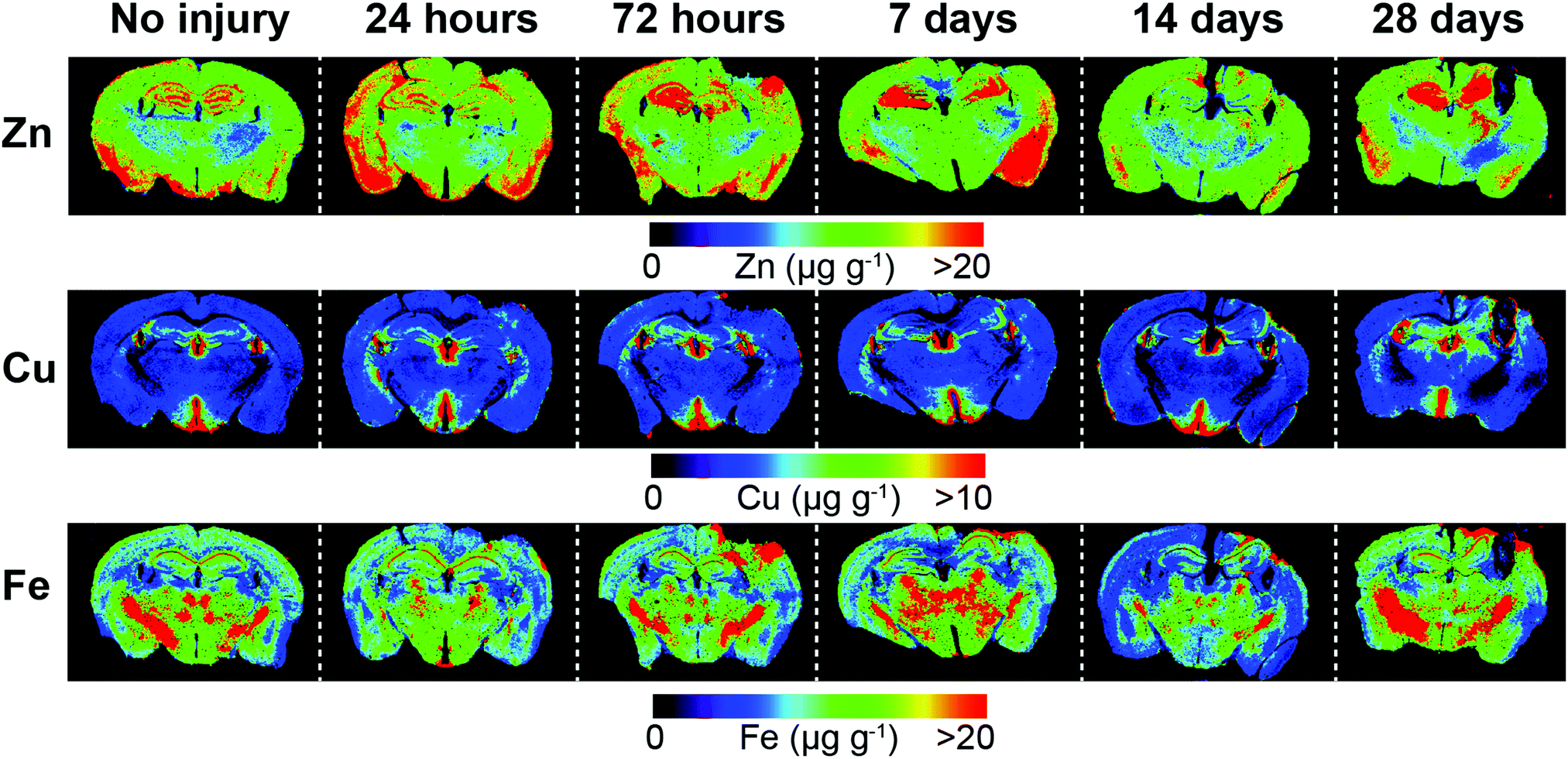 | ||
| Fig. 2 Representative LA-ICP-MS images of both hemispheres including uninjured and time course examples from 24 hours to 28 day post-lesion. Images were scanned at a spatial resolution of 30 μm. | ||
Iron levels are significantly altered after brain injury
Analysis of spatial distribution and concentration of Fe across the time course following TBI revealed a sustained and continually increasing elevation in each region examined (Fig. 3). A comparison of ipsilateral to contralateral hemispheres revealed that, after TBI, the Fe levels were significantly increased within the immediate vicinity of the impact site (ROI1 p < 0.0001; Fig. 3a) and in the two additional ROIs extending radially from the lesion site (ROI2 p < 0.0001; ROI3 p < 0.01; Fig. 3b and c) leading to a significant overall increase in iron over the entire ipsilateral hemisphere (p < 0.001; Fig. 3d). Intra-day comparisons were also made between ipsilateral and contralateral sides, revealing a significant increase in ROI 1 at 28 days (Fig. 3a). There was also a significant increase in ipsilateral with respect to the contralateral sides in ROI2 at days 14 and 28 day (Fig. 3b). The entire hemisphere revealed a significant intra-day increase between ipsilateral and contralateral sides at day 28 (Fig. 3d). However, no significant difference in Fe concentration was observed in the hippocampus (Fig. 3e).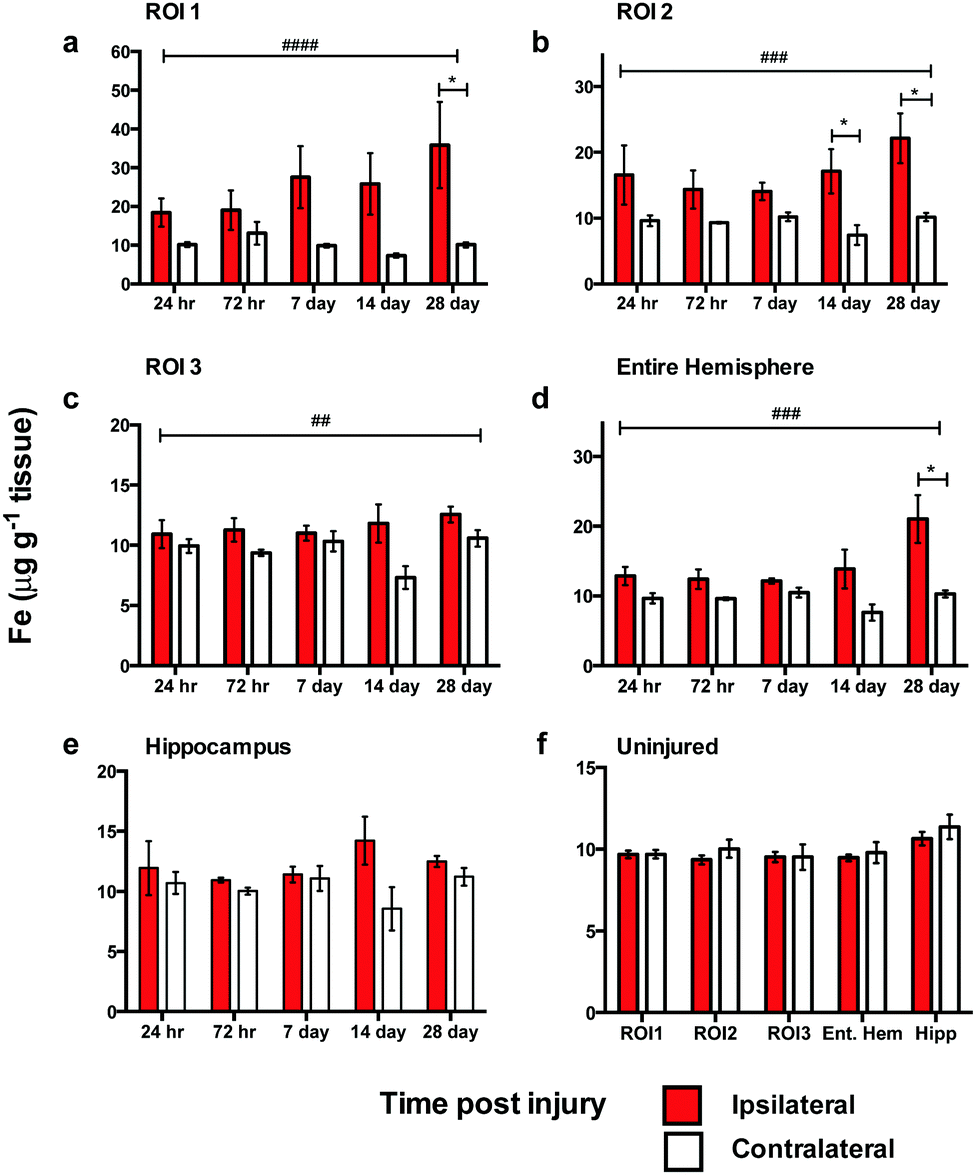 | ||
| Fig. 3 Iron concentrations across the time course following controlled cortical impact TBI. In ROI1 there was a significant difference in Fe levels between the ipsilateral and contralateral sides across the time course (####, ANOVA, p = 0.0001) (a). ROI1 intra-day comparisons also revealed a significant difference between ipsilateral and contralateral sides at 28 days (*p < 0.05) (a). There was also a significant increase of Fe in ROI2 (###, ANOVA, p = 0.0001) (b), ROI3 (##, ANOVA, p = 0.0032) (c), and total hemisphere (###, ANOVA p = 0.0002) (d). ROI2 intra-day analysis revealed a significant difference between ipsilateral and contralateral sides at days 14 (*p = 0.0380) and 28 (*p = 0.0203) (b). The entire hemisphere revealed a significant intra-day difference between ipsilateral and contralateral sides at day 28 (*p = 0.0212) (d). Hippocampus revealed no significant overall or intra-day differences (e). Uninjured 28 day regional analysis showed no significant changes and reflected a stable concentration in all regions examined (f). | ||
Zinc remains unchanged after brain injury
In contrast to the iron results, there were no significant differences in Zn levels between the ipsilateral and contralateral sides in ROI1, ROI2, ROI3, or the entire hemisphere (Fig. 4a–c). Intra-day analysis in each region also revealed no significant time point concentration differences. In the hippocampus, there was no significant difference between the ipsilateral and contralateral sides, however, a whole brain decrease in Zn concentration was observed between days 7 to 14 in all ROIs (Fig. 4a–d) and hippocampus (Fig. 4e).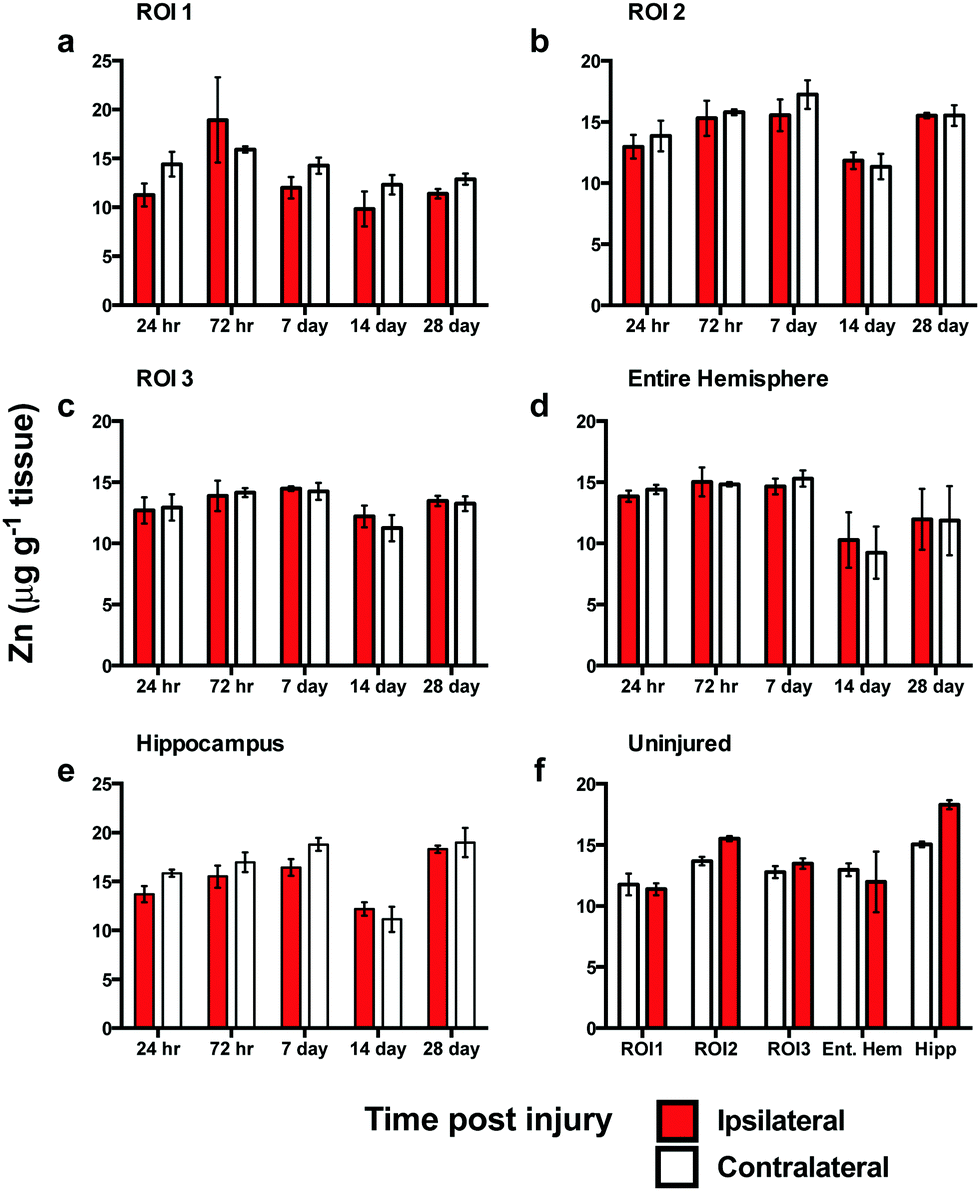 | ||
| Fig. 4 Zinc concentrations across the time course following controlled cortical impact TBI. There was no significant difference in Zn levels between the ipsilateral and contralateral sides or intra-day in ROI1 (a), ROI2 (b), ROI3 (c), total hemisphere (d), or hippocampus (e). Analysis of uninjured tissue revealed no regional changes in Zn concentrations at 28 days (f). | ||
Copper levels are altered after brain injury
In ROI1 there was a significant increase in Cu levels between the ipsilateral and contralateral sides across the time course (p < 0.01) (Fig. 5a), however, this was not observed in ROI2, ROI3, or the entire hemisphere. ROI1 intra-day comparisons also revealed a significant increase between ipsilateral and contralateral sides at day 14 and day 28, while ROI2 intra-day comparison revealed a significant intra-day increase between the ipsilateral and contralateral sides on day 14 (Fig. 5b). No significant difference was observed in the hippocampus between the ipsilateral and contralateral sides (Fig. 5e).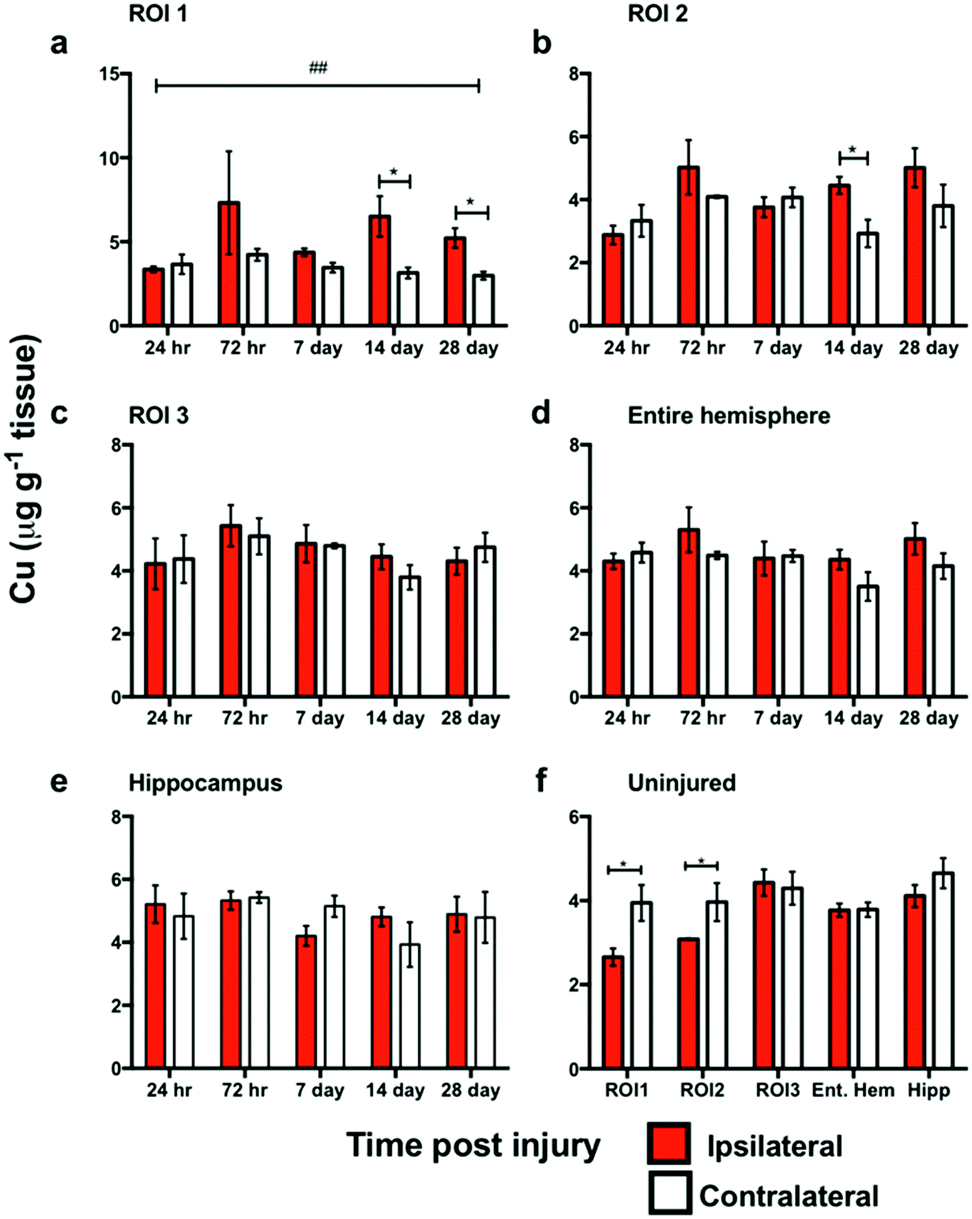 | ||
| Fig. 5 Copper concentrations across the time course following controlled cortical impact TBI. In ROI1 there was a significant difference in Cu levels between the ipsilateral and contralateral sides across the time course (##, ANOVA, p = 0.0053), and intra-day comparisons also revealed a significant difference between ipsilateral and contralateral sides at day 14 (*p < 0.0354) and day 28 (*p = 0.0118) (a). ROI2 intra-day comparison revealed a significant intra-day difference between the ipsilateral and contralateral sides on day 14 (*p = 0.0255) (b). ROI3 (c) entire hemisphere (d), and hippocampus (e) revealed no significant overall or intra-day differences. Analysis of uninjured tissue revealed a significant regional copper concentration changes at 28 days in ROI1 (*p = 0.015) & ROI2 (*p = 0.046) that was not seen in ROI3, the entire hemisphere or the hippocampus (f). | ||
Comparison of metal changes in 3 month and 24 month-old mice.
We compared our previously published data in three month-old mice to our current data in 24 month-old animals as presented in the current manuscript. A direct comparison between both age groups revealed that Fe, Zn and Cu all increase in both the contralateral and ipsilateral hemispheres with ageing (Fig. S1a and b, ESI†). A comparison of ROI's in the ipsilateral and contralateral hemispheres between 3 and 24 month-old mice post-TBI revealed that Fe is elevated in the uninjured contralateral hemisphere in 24 month-old mice in each ROI, and that each ROI shows a proportionally larger ipsilateral increase in the concentration of Fe for the 24 month-old mice when compared to the 3 month old mice from day 7 to day 28 (Fig. S2, ESI†). Ipsilateral and contralateral regions of each ROI also show a proportionally larger increase in concentration of Zn for the 24 month-old mice when compared to the 3 month old mice. In 24 month-old mice ROI1, ROI2, ROI3 and total hemisphere shows a reduction in Zn in both ipsilateral and contralateral hemispheres from day 7 onwards, representing a whole-brain zinc reduction from 7–28 days’ post TBI that is not evident in the three month-old mice (Fig. S3, ESI†). Ipsilateral and contralateral regions of each ROI show a proportionally larger increase in concentration of Cu for the 24 month-old mice when compared to the 3 month old mice. In 24 month-old mice ROI1 reveals a sustained increase in Cu concentration from 72 hours’ post TBI. ROI2, and total hemisphere of the 24 month-old mice reveals a proportionally larger increase in Cu in the ipsilateral hemisphere when compared to contralateral hemisphere from day 7 onwards that is not evident in the three month-old mice (Fig. S4, ESI†).Discussion
The risk of neurodegeneration after TBI is influenced not just by the severity of the initial insult, but also by the patient's age at the time of injury.44 Multiple studies of TBI confirm a progressive rise in mortality and a decline in favourable outcomes including dementia with each decade over 50 years of age.11–13,16,45 As previously outlined, metals have long been implicated in the neuropathology of aging and neurodegenerative diseases. We therefore assayed Fe, Zn and Cu in the brains of aged mice after a controlled cortical impact TBI to assess the changes in the metallomic profile that may contribute to or influence age-related TBI outcomes.The regions of interest presented herein were selected to allow the differentiation of discrete functional brain areas surrounding the injury site that may be differentially affected by the TBI, and to describe the extent of the metal response in relation to the site of impact, as many studies have indicated metal dyshomeostasis not just immediately located at the site of injury but also in deeper tissue.20,25,46–48
We have previously shown that there is a sustained elevation in Fe in the brains of young adult mice after TBI25 independent of haem associated Fe caused by blood infiltration at the injury site verified by Perls Prussian blue staining for non-haem Fe. Moreover, it has been demonstrated with ICP-MS analysis that cortical Fe 24 hours’ post injury is the same between ipsilateral and contralateral regions, after an observed elevation at six hours in the ipsilateral region, suggesting a mechanism may exist to handle heme-associated Fe at the acute stage of injury.49 the data presented herein shows a similar phenomenon in aged mice, however the magnitude of increase is much greater in the aged mice than that exhibited in 3 month old mice after TBI.25 Brain Fe elevation has been demonstrated in TBI50 in conjunction with a simultaneous increase in the Fe handling protein ferritin.51 Ferritin is a ubiquitous intracellular protein that safely stores Fe and releases it in a controlled fashion acting as a buffer against Fe deficiency and overload. However, excess Fe unbound to ferritin or haem associated proteins can initiate neurotoxicity through the formation of reactive oxygen species (ROS) potentiating oxidative stress and associated cell death. In AD patients Fe accumulation occurs without an age associated increase in ferritin, increasing the risk of oxidative stress.52 Significantly, both TBI20,25,46 and mild TBI47,48,50 are characterised by non-haem Fe associated intracellular accumulation. Moreover, Fe deposition has been shown to be positively related to cognitive impairment in patients with chronic mild TBI.53 Given these observations, it is plausible that excess Fe because of TBI or mild TBI could contribute to the onset or acceleration of neurodegenerative conditions. Notably, excess Fe also associates with aggregation prone proteins that initiate cell death pathways in several neurodegenerative diseases of aging such as AD, PD and ALS.54 Additionally, intracellular Fe accumulation has also recently been associated with a specific cell death pathway termed ferroptosis.55 Notably, the ferroptosis pathway is independent from its capacity to initiate ROS production, and hence presents another autonomous mechanism by which elevated Fe can contribute to neurodegeneration. Our results indicate that Fe accumulates in elevated concentrations after TBI in aged animals, and as such may present as an influential age associated accelerant to the now recognised increased risk of the elderly developing dementia after TBI.
Zinc is an essential element required for efficient brain function,56 however under injurious states Zn has been proposed to be an influential factor in TBI pathogenesis with the literature denoting both neuroprotective and neurotoxic roles.57–61 Surprisingly then, our data revealed no change in Zn concentration when comparing ipsilateral to contralateral hemispheres. While this result is consistent with our previous study utilising young mice,25 it should be noted that the overall level of Zn in the brain was elevated in the aged mice, which may impact a variety of cellular processes relevant to ageing. Zinc chelation after TBI has been demonstrated to be protective in 6 month-old rats62 whereby the Zn chelation induced the expression of several neuroprotective genes in conjunction with decreased cell death after 7 days. However, Zn chelation has also been shown to be detrimental to outcomes post-TBI in mice (age not designated), evidenced by a significant increase in necrotic and apoptotic cells within 24 hours.63 Two further studies suggest that oral zinc intake before TBI in 10 week-old rats64 and oral Zn intake post-TBI in young adult male rats65 ameliorates the cognitive decline observed post-TBI. These and other studies not cited herein are confounding due to age differences, strain of animals and data collection time points. While no difference in Zn concentration was observed between specific ROIs in the ipsilateral and contralateral hemispheres, it is possible that there is cellular translocation of Zn distribution, as observed by several other studies, that leads to either neurotoxicity66 or neuroprotection.67 However, to reveal such cellular translocation would require a more refined delineation of analysed regions, at the sub-μm level via synchrotron XFM analysis or micro-particle induced X-ray emission (μ PIXE) analysis. Previous studies have shown that spatial Zn concentrations in the mouse brain are highly compartmentalised and heterogeneous.43,68 High levels of Zn in the hippocampus are a significant source of biological variation,69 and this may account for the variance we observed in our measurements. In this experiment the scanning parameters used were optimal for a large sample size whilst still obtaining adequate spatial resolution to delineate brain regions at the mesoscale.70 In future experiments, we will undertake higher resolution mapping by LA-ICPMS using a 5 × 5 μm pixel size to examine changes within the injury site.
Of potential interest, however, is the observation of the whole brain reduction of Zn concentration in the aged animals from day 7 onwards post-TBI. Clinical studies and experimental work with animal models have both revealed a correlation between Zn status and neuropsychological disorders whereby Zn deficiency has been shown to induce depression-like and anxiety-like behaviours.71 As depression and mood disorders are a common yet non-immediate outcome after TBI and mild TBI, especially in the elderly,72 it would be of interest to determine whether Zn is decreased beyond the 28 day assessment period observed herein. Our results may reflect an altered capacity of the aged brain to effectively modulate Zn under injurious circumstances, as this effect was not observed in our previous study utilising young mice.25
Cu in the brain is involved in mitochondrial respiration, neurotransmitter biosynthesis, and as a cofactor for antioxidant enzymes.73 It is a critical and essential element that also functions in the regulation of synaptic transmission and ion channel function. However, Cu is a transition metal that is a remarkably potent pro-oxidant, having the ability to form hydrogen peroxide and subsequent hydroxyl radicals at an excessive pace.74,75 Our results revealed an overall elevation of brain Cu associated with aging and an increased Cu concentration from day 7 to day 28 around the impact site that was not observed in our previous study utilising younger mice.25 An elevation of Cu could potentiate Cu toxicity in the form of oxidative stress leading to neuronal impairment. Additionally, the collective resulting metal profile generated by our data may be of broader relevance to the observed heightened onset of PD after TBI45 in that elevation of Fe and Cu in the presence of Zn depletion, has been shown in aged human brain, with a consequence of compromised DNA integrity altering gene expression.76 It is noteworthy that our results reveals a similar profile of Fe, Cu and Zn, and that this profile leads to DNA fragmentations that have been clearly demonstrated as a component of the age related neurodegenerative disorders PD and AD.77–80
Conclusion
This study demonstrates that a controlled cortical impact injury results in an altered brain metal profile in aged mice. Several of our observations are consistent with and supported by both animal studies, and human clinical data in the literature. However, we further reveal that there is both an age-dependent alteration in baseline metal levels in the brain, and more importantly, there is an exaggerated alteration in metal levels seemingly reserved for the aging brain following injury. Given that the elderly are more likely to experience negative outcomes following a TBI, our data may provide insight around our understanding of the mechanisms involved in cognitive/functional decline in elderly TBI victims.Acknowledgements
PAA is supported by Australian Research Council (ARC) Future Fellowship. DJH wishes to acknowledge support from the Ramaciotti Foundation. DJH and PAD were also supported by an ARC Linkage Project with Agilent Technologies and ESI Ltd. In addition, the Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.Notes and references
- A. I. Maas, N. Stocchetti and R. Bullock, Lancet Neurol., 2008, 7, 728–741 CrossRef PubMed.
- B. Roozenbeek, A. I. Maas and D. K. Menon, Nat. Rev. Neurol., 2013, 9, 231–236 CrossRef PubMed.
- J. Ghajar, Lancet, 2000, 356, 923–929 CrossRef CAS.
- M. Faul, L. Xu, M. M. Wald, V. Coronado and A. M. Dellinger, Inj. Prev., 2010, 16, A268–A268 CrossRef.
- World report on traffic injury prevention, World Health Organization, Geneva, 2004.
- K. N. Corps, T. L. Roth and D. B. McGavern, JAMA Neurol., 2015, 72, 355–362 CrossRef PubMed.
- J. R. De Kruijk, P. Leffers, P. P. Menheere, S. Meerhoff, J. Rutten and A. Twijnstra, J. Neurol., Neurosurg. Psychiatry, 2002, 73, 727–732 CrossRef CAS.
- J. M. Meythaler, J. D. Peduzzi, E. Eleftheriou and T. A. Novack, Arch. Phys. Med. Rehabil., 2001, 82, 1461–1471 CrossRef CAS PubMed.
- V. E. Johnson and W. Stewart, Nat. Rev. Neurol., 2015, 11, 128–130 CrossRef PubMed.
- R. C. Gardner, J. F. Burke, J. Nettiksimmons, A. Kaup, D. E. Barnes and K. Yaffe, JAMA Neurol., 2014, 71, 1490–1497 CrossRef PubMed.
- M. Susman, S. M. DiRusso, T. Sullivan, D. Risucci, P. Nealon, S. Cuff, A. Haider and D. Benzil, J. Trauma, 2002, 53, 219–223 CrossRef PubMed.
- C. W. Hukkelhoven, E. W. Steyerberg, A. J. Rampen, E. Farace, J. D. Habbema, L. F. Marshall, G. D. Murray and A. I. Maas, J. Neurosurg., 2003, 99, 666–673 CrossRef PubMed.
- A. C. Mosenthal, R. F. Lavery, M. Addis, S. Kaul, S. Ross, R. Marburger, E. A. Deitch and D. H. Livingston, J. Trauma, 2002, 52, 907–911 Search PubMed.
- C. Van Den Heuvel, E. Thornton and R. Vink, Prog. Brain Res., 2007, 161, 303–316 CAS.
- H. Chen, M. Richard, D. P. Sandler, D. M. Umbach and F. Kamel, Am. J. Epidemiol., 2007, 166, 810–816 CrossRef PubMed.
- P. K. Crane, L. E. Gibbons, K. Dams-O'Connor, E. Trittschuh, J. B. Leverenz, C. D. Keene, J. Sonnen, T. J. Montine, D. A. Bennett, S. Leurgans, J. A. Schneider and E. B. Larson, JAMA Neurol., 2016, 73, 1062–1069 CrossRef PubMed.
- S. Jafari, M. Etminan, F. Aminzadeh and A. Samii, Mov. Disord., 2013, 28, 1222–1229 CrossRef PubMed.
- V. E. Johnson, W. Stewart and D. H. Smith, Brain Pathol., 2012, 22, 142–149 CrossRef CAS PubMed.
- V. E. Johnson, J. E. Stewart, F. D. Begbie, J. Q. Trojanowski, D. H. Smith and W. Stewart, Brain, 2013, 136, 28–42 CrossRef PubMed.
- G. Onyszchuk, Y. Y. He, N. E. Berman and W. M. Brooks, J. Neurotrauma, 2008, 25, 153–171 CrossRef PubMed.
- G. Onyszchuk, S. M. LeVine, W. M. Brooks and N. E. Berman, Neurosci. Lett., 2009, 452, 204–208 CrossRef CAS PubMed.
- R. Sandhir and N. E. Berman, Neurochem. Int., 2010, 56, 188–193 CrossRef CAS PubMed.
- S. Bolognin, L. Messori and P. Zatta, NeuroMol. Med., 2009, 11, 223–238 CrossRef CAS PubMed.
- E. S. Lein, M. J. Hawrylycz, N. Ao, M. Ayres, A. Bensinger, A. Bernard, A. F. Boe, M. S. Boguski, K. S. Brockway, E. J. Byrnes, L. Chen, L. Chen, T. M. Chen, M. C. Chin, J. Chong, B. E. Crook, A. Czaplinska, C. N. Dang, S. Datta, N. R. Dee, A. L. Desaki, T. Desta, E. Diep, T. A. Dolbeare, M. J. Donelan, H. W. Dong, J. G. Dougherty, B. J. Duncan, A. J. Ebbert, G. Eichele, L. K. Estin, C. Faber, B. A. Facer, R. Fields, S. R. Fischer, T. P. Fliss, C. Frensley, S. N. Gates, K. J. Glattfelder, K. R. Halverson, M. R. Hart, J. G. Hohmann, M. P. Howell, D. P. Jeung, R. A. Johnson, P. T. Karr, R. Kawal, J. M. Kidney, R. H. Knapik, C. L. Kuan, J. H. Lake, A. R. Laramee, K. D. Larsen, C. Lau, T. A. Lemon, A. J. Liang, Y. Liu, L. T. Luong, J. Michaels, J. J. Morgan, R. J. Morgan, M. T. Mortrud, N. F. Mosqueda, L. L. Ng, R. Ng, G. J. Orta, C. C. Overly, T. H. Pak, S. E. Parry, S. D. Pathak, O. C. Pearson, R. B. Puchalski, Z. L. Riley, H. R. Rockett, S. A. Rowland, J. J. Royall, M. J. Ruiz, N. R. Sarno, K. Schaffnit, N. V. Shapovalova, T. Sivisay, C. R. Slaughterbeck, S. C. Smith, K. A. Smith, B. I. Smith, A. J. Sodt, N. N. Stewart, K. R. Stumpf, S. M. Sunkin, M. Sutram, A. Tam, C. D. Teemer, C. Thaller, C. L. Thompson, L. R. Varnam, A. Visel, R. M. Whitlock, P. E. Wohnoutka, C. K. Wolkey, V. Y. Wong, M. Wood, M. B. Yaylaoglu, R. C. Young, B. L. Youngstrom, X. F. Yuan, B. Zhang, T. A. Zwingman and A. R. Jones, Nature, 2007, 445, 168–176 CrossRef CAS PubMed.
- S. D. Portbury, D. J. Hare, C. Sgambelloni, D. I. Finkelstein and P. A. Adlard, Metallomics, 2015, 8, 193–200 RSC.
- P. A. Adlard and A. I. Bush, J. Alzheimer's Dis., 2006, 10, 145–163 Search PubMed.
- K. J. Barnham and A. I. Bush, Chem. Soc. Rev., 2014, 43, 6727–6749 RSC.
- L. Zecca, M. Gallorini, V. Schunemann, A. X. Trautwein, M. Gerlach, P. Riederer, P. Vezzoni and D. Tampellini, J. Neurochem., 2001, 76, 1766–1773 CrossRef CAS PubMed.
- W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3478–3482 CrossRef CAS.
- E. Aizenman, A. K. Stout, K. A. Hartnett, K. E. Dineley, B. McLaughlin and I. J. Reynolds, J. Neurochem., 2000, 75, 1878–1888 CrossRef CAS PubMed.
- P. A. Adlard, J. M. Parncutt, D. I. Finkelstein and A. I. Bush, J. Neurosci., 2010, 30, 1631–1636 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed.
- H. R. Massie, V. R. Aiello and A. A. Iodice, Mech. Ageing Dev., 1979, 10, 93–99 CrossRef CAS PubMed.
- C. J. Maynard, R. Cappai, I. Volitakis, R. A. Cherny, A. R. White, K. Beyreuther, C. L. Masters, A. I. Bush and Q. X. Li, J. Biol. Chem., 2002, 277, 44670–44676 CrossRef CAS PubMed.
- S. Takahashi, I. Takahashi, H. Sato, Y. Kubota, S. Yoshida and Y. Muramatsu, Biol. Trace Elem. Res., 2001, 80, 145–158 CrossRef CAS PubMed.
- T. Tarohda, M. Yamamoto and R. Amamo, Anal. Bioanal. Chem., 2004, 380, 240–246 CrossRef CAS PubMed.
- L. O. Plantin, U. Lying-Tunell and K. Kristensson, Biol. Trace Elem. Res., 1987, 13, 69–75 CrossRef CAS PubMed.
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS PubMed.
- D. J. Hare, J. Lear, D. Bishop, A. Beavis and P. A. Doble, Anal. Methods, 2013, 5, 1915–1921 RSC.
- J. Lear, D. J. Hare, F. Fryer, P. A. Adlard, D. I. Finkelstein and P. A. Doble, Anal. Chem., 2012, 84, 6707–6714 CrossRef CAS PubMed.
- P. R. D. Mason and W. J. Kraan, J. Anal. At. Spectrom., 2002, 17, 858–867 RSC.
- D. J. Hare, F. Fryer, B. Paul, D. P. Bishop and P. A. Doble, Anal. Methods, 2016, 8, 7552–7556 RSC.
- D. J. Hare, J. K. Lee, A. D. Beavis, A. van Gramberg, J. George, P. A. Adlard, D. I. Finkelstein and P. A. Doble, Anal. Chem., 2012, 84, 3990–3997 CrossRef CAS PubMed.
- D. H. Smith, V. E. Johnson and W. Stewart, Nat. Rev. Neurol., 2013, 9, 211–221 CrossRef CAS PubMed.
- R. C. Gardner, J. F. Burke, J. Nettiksimmons, S. Goldman, C. M. Tanner and K. Yaffe, Ann. Neurol., 2015, 77, 987–995 CrossRef PubMed.
- H. Bayir, P. M. Kochanek and V. E. Kagan, Dev. Neurosci., 2006, 28, 420–431 CrossRef CAS PubMed.
- C. Bouras, P. Giannakopoulos, P. F. Good, A. Hsu, P. R. Hof and D. P. Perl, Eur. Neurol., 1997, 38, 53–58 CrossRef CAS PubMed.
- Q. G. Zhang, M. D. Laird, D. Han, K. Nguyen, E. Scott, Y. Dong, K. M. Dhandapani and D. W. Brann, PLoS One, 2012, 7, e34504 CAS.
- S. Ayton, M. Zhang, B. R. Roberts, L. Q. Lam, M. Lind, C. McLean, A. I. Bush, T. Frugier, P. J. Crack and J. A. Duce, Free Radical Biol. Med., 2014, 69, 331–337 CrossRef CAS PubMed.
- E. Raz, J. H. Jensen, Y. Ge, J. S. Babb, L. Miles, J. Reaume, R. I. Grossman and M. Inglese, AJNR Am. J. Neuroradiol., 2011, 32, 1851–1856 CrossRef CAS PubMed.
- H. D. Liu, W. Li, Z. R. Chen, M. L. Zhou, Z. Zhuang, D. D. Zhang, L. Zhu and C. H. Hang, Neurol. Sci., 2013, 34, 1173–1180 CrossRef PubMed.
- J. R. Connor, B. S. Snyder, J. L. Beard, R. E. Fine and E. J. Mufson, J. Neurosci. Res., 1992, 31, 327–335 CrossRef CAS PubMed.
- L. Lu, H. Cao, X. Wei, Y. Li and W. Li, BioMed Res. Int., 2015, 2015, 470676 Search PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison, 3rd and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- B. Szewczyk, Front. Aging Neurosci., 2013, 5, 33 Search PubMed.
- D. R. Morris and C. W. Levenson, Curr. Opin. Clin. Nutr. Metab. Care, 2013, 16, 708–711 CrossRef CAS PubMed.
- M. Capasso, J. M. Jeng, M. Malavolta, E. Mocchegiani and S. L. Sensi, J. Alzheimer's Dis., 2005, 8, 93–108 CAS.
- E. C. Cope, D. R. Morris and C. W. Levenson, Nutr. Rev., 2012, 70, 410–413 CrossRef PubMed.
- C. J. Frederickson, W. Maret and M. P. Cuajungco, Neuroscientist, 2004, 10, 18–25 CrossRef CAS PubMed.
- S. D. Portbury and P. A. Adlard, J. Alzheimer's Dis., 2015, 46, 297–311 CrossRef CAS PubMed.
- H. L. Hellmich, C. J. Frederickson, D. S. DeWitt, R. Saban, M. O. Parsley, R. Stephenson, M. Velasco, T. Uchida, M. Shimamura and D. S. Prough, Neurosci. Lett., 2004, 355, 221–225 CrossRef CAS PubMed.
- P. Doering, M. Stoltenberg, M. Penkowa, J. Rungby, A. Larsen and G. Danscher, PLoS One, 2010, 5, e10131 Search PubMed.
- E. C. Cope, D. R. Morris, A. G. Scrimgeour, J. W. VanLandingham and C. W. Levenson, Physiol. Behav., 2011, 104, 942–947 CrossRef CAS PubMed.
- E. C. Cope, D. R. Morris, A. G. Scrimgeour and C. W. Levenson, Neurorehabil. Neural Repair, 2012, 26, 907–913 CrossRef PubMed.
- S. W. Suh, J. W. Chen, M. Motamedi, B. Bell, K. Listiak, N. F. Pons, G. Danscher and C. J. Frederickson, Brain Res., 2000, 852, 268–273 CrossRef CAS PubMed.
- P. Doering, G. Danscher, A. Larsen, M. Bruhn, C. Sondergaard and M. Stoltenberg, Neuroscience, 2007, 150, 93–103 CrossRef CAS PubMed.
- D. J. Hare, J. L. George, R. Grimm, S. Wilkins, P. A. Adlard, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. Doble, Metallomics, 2010, 2, 745–753 RSC.
- A. Matusch, C. Depboylu, C. Palm, B. Wu, G. U. Höglinger, M. K.-H. Schäfer and J. S. Becker, J. Am. Soc. Mass Spectrom., 2010, 21, 161–171 CrossRef CAS PubMed.
- J. Lear, D. J. Hare, P. Adlard, D. Finkelstein and P. Doble, J. Anal. At. Spectrom., 2012, 27, 159 RSC.
- E. C. Cope and C. W. Levenson, Curr. Opin. Clin. Nutr. Metab. Care, 2010, 13, 685–689 CrossRef CAS PubMed.
- J. C. Menzel, Brain Inj., 2008, 22, 375–380 CrossRef PubMed.
- M. M. Pena, J. Lee and D. J. Thiele, J. Nutr., 1999, 129, 1251–1260 CAS.
- X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS PubMed.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa and A. I. Bush, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS PubMed.
- P. Vasudevaraju, Bharathi, T. Jyothsna, N. M. Shamasundar, S. K. Rao, B. M. Balaraj, K. Rao and T. S. Sathyanarayana Rao, Indian J. Psychiatry, 2010, 52, 140–144 CrossRef CAS PubMed.
- Z. I. Alam, A. Jenner, S. E. Daniel, A. J. Lees, N. Cairns, C. D. Marsden, P. Jenner and B. Halliwell, J. Neurochem., 1997, 69, 1196–1203 CrossRef CAS PubMed.
- M. L. Hegde, V. B. Gupta, M. Anitha, T. Harikrishna, S. K. Shankar, U. Muthane, K. Subba Rao and K. S. Jagannatha Rao, Arch. Biochem. Biophys., 2006, 449, 143–156 CrossRef CAS PubMed.
- A. Suram, K. S. Rao, K. S. Latha and M. A. Viswamitra, NeuroMol. Med., 2002, 2, 289–297 CrossRef CAS.
- W. G. Tatton and C. W. Olanow, Biochim. Biophys. Acta, 1999, 1410, 195–213 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mt00260a |
| This journal is © The Royal Society of Chemistry 2017 |