DOI:
10.1039/C6MT00220J
(Paper)
Metallomics, 2017,
9, 391-401
Detection of Zn2+ release in nitric oxide treated cells and proteome: dependence on fluorescent sensor and proteomic sulfhydryl groups
Received
30th September 2016
, Accepted 29th November 2016
First published on 29th November 2016
Abstract
Nitric oxide (NO) is both an important regulatory molecule in biological systems and a toxic xenobiotic. Its oxidation products react with sulfhydryl groups and either nitrosylate or oxidize them. The aerobic reaction of NO supplied by diethylamine NONOate (DEA-NO) with pig kidney LLC-PK1 cells and Zn-proteins within the isolated proteome was examined with three fluorescent zinc sensors, zinquin (ZQ), TSQ, and FluoZin-3 (FZ-3). Observations of Zn2+ labilization from Zn-proteins depended on the specific sensor used. Upon cellular exposure to DEA-NO, ZQ sequestered about 13% of the proteomic Zn2+ as Zn(ZQ)2 and additional Zn2+ as proteome·Zn–ZQ ternary complexes. TSQ, a sensor structurally related to ZQ with lower affinity for Zn2+, did not form Zn(TSQ)2. Instead, Zn2+ mobilized by DEA-NO was exclusively bound as proteome·Zn–TSQ adducts. Analogous reactions of proteome with ZQ or TSQ in vitro displayed qualitatively similar products. Titration of native proteome with Zn2+ in the presence of ZQ resulted in the sole formation of proteome·Zn–ZQ species. This result suggested that sulfhydryl groups are involved in non-specific proteomic binding of mobile Zn2+ and that the appearance of Zn(ZQ)2 after exposure of cells and proteome to DEA-NO resulted from a reduction in proteomic sulfhydryl ligands, favoring the formation of Zn(ZQ)2 instead of proteome·Zn–ZQ. With the third sensor, FluoZin-3, neither Zn–FZ-3 nor proteome·Zn–FZ-3 was detected during the reaction of proteome with DEA-NO. Instead, it reacted independently with DEA-NO with a modest enhancement of fluorescence.
Significance to metallomics
A significant fraction of Zn2+ bound within the proteome other than zinc-metallothionein becomes reactive with zinc sensors upon exposure to NO, presumably as a result of sulfhydryl ligand modification. The native cell proteome also contains large numbers of adventitious binding sites for Zn2+ that hypothetically involve thiol ligands. In the presence of NO, the affinity of these sites is also reduced. As a result, the reactivity of various sensors with proteomic Zn2+ labilized by NO depends on their metal binding affinity in relation to the reduced affinity of the proteome, both native and adventitious binding sites, for Zn2+.
|
Introduction
Numerous zinc fluorescent sensors have been designed to provide researchers with tools to observe pools of intracellular Zn2+ that hypothetically participate in transient cellular events induced by normal or pathological conditions.1–3 In one of the well described reactions, elevated Zn2+ activates the MTF-1 transcription factor, leading to the induction of mRNA and the subsequent synthesis of metallothionein (MT) that sequesters excess intracellular Zn2+ and ZnT1 that transports Zn2+ from the cytosol into the extracellular medium.4–6 In addition, it has been proposed that Cd2+ stimulates MTF-1 binding to its cognate DNA by displacing Zn2+ from basal MT, upregulating MTF-1, and eventually leading to the sequestration of Cd2+ by elevated concentrations of MT.6
Nitric oxide (NO) serves both as a regulatory and toxic biomolecule, depending on the situation.7–10 Because NO and its oxidation products such as NO2 and N2O3 display strong preference for reacting with sulfhydryl groups, past studies have investigated the reactivity of Zn–MT with these compounds, recognizing that MT contains 20 cysteinyl thiolate groups and uses them to bind up to 7 Zn2+ ions.11–13 For example, Pitt and coworkers hypothesized that NO exposure of lung endothelial cells releases Zn2+ from MT that subsequently causes vasoconstriction.14 Experiments supporting mobilization of Zn2+ were based on enhancement of fluorescence of the Zn2+ sensors zinquin and FluoZin-3 (FZ-3) in the presence of elevated NO production (Fig. 1).14–16
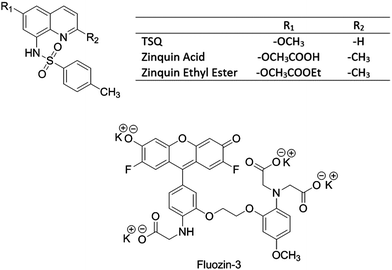 |
| | Fig. 1 Structures of Zn2+ sensors. | |
Many fluorescent probes for Zn2+ have been synthesized during the past two decades.1–3 Commonly, the design, synthesis, and photophysics of new sensors are reported together with a generic test of their ability to detect labile cellular Zn2+. For example, some cells have been exposed to NO in order to render Zn2+, possibly from the MT pool, available to determine whether the sensor is an effective sensor for Zn2+.14–16 We began to study the reactions of NO with cells because of an interest in the chemical reactivity of Zn–MT with NO and its oxidation products.17 When it became evident that Zn–MT is not particularly reactive with NO in comparison with sulfhydryl groups throughout the proteome, other questions arose: what is the source of Zn2+ that Zn2+ sensors are imaging and what is the underlying bioinorganic chemistry that is responsible for the fluorescence increase?
Initial studies showed that ZQ and its close relative, TSQ, image different sets of Zn-proteins within the proteome in the form of ternary adducts between the probe and Zn-proteins:18–21
| | | ZQ/TSQ + Zn-proteins ⇌ ZQ/TSQ–Zn-proteins | (1) |
This finding immediately expanded the possibilities for reaction of sensors beyond simply binding Zn
2+:
| | | Zn2+ + 2ZQ/TSQ ⇌ Zn(ZQ/TSQ)2 | (2) |
For example, studies have shown that Zn
2+ associates with the general proteomic pool,
| | | Zn2+ + proteome ⇌ proteome·Zn | (3) |
leading to the possibility that ZQ and TSQ and perhaps other sensors react with proteome·Zn to generate fluorescent ternary species:
19| | | ZQ/TSQ + proteome·Zn ⇌ proteome·Zn(ZQ/TSQ) | (4) |
In light of the above results, this study was undertaken to assess how ZQ, TSQ, and FZ-3 interact with the cell's complement of Zn-proteins and proteome after mobilization of Zn
2+ from native proteomic binding sites by NO. ZQ and TSQ were compared as closely related sensors that differ primarily at the 2 position of the isoquinoline ring with ZQ bearing a methyl group instead of a hydrogen that leads to its larger affinity for Zn
2+ (
Fig. 1). FZ-3 was chosen as a distinctly different molecular structure that is commonly used as a probe for cellular Zn
2+ including in studies of the interaction of NO with cells.
14,22
Materials and methods
Chemicals and reagents
TSQ was purchased from AnaSpec, Inc., zinquin from Enzo Life Sciences, FluoZin-3 from Molecular Probes, and DEA-NONOate from Cayman Chemical. All sensors were dissolved in DMSO and stored in the dark at −20 °C. DEA-NONOate was dissolved in 0.01 M NaOH and stored in aliquots at −80 °C until use. All the other chemicals and reagents were purchased either from Fisher Scientific or Sigma-Aldrich at the highest grade available.
Cell culture
The LLC-PK1 (pig kidney cells) cell line was purchased from the American Tissue Culture Company. Cells were grown in Medium 199 with HEPES modification (Sigma) supplemented with 2.2 g L−1 NaHCO3, 50 mg L−1 penicillin G, 50 mg L−1 streptomycin and 4% fetal calf serum (FCS). The cells were incubated at 37 °C in the presence of 5% CO2. Media was changed every 2–3 days until confluence. Cells were subdivided by trypsin treatment to free cells from plates.
Fluorescence spectroscopy of LLC-PK1 cells
Cultured cells were grown in 100 mm culture plates until confluence was obtained. Culture media was discarded and the plates were washed three times with cold cholate buffer, prepared by dissolving 2.47 g Na2HPO4, 0.53 g NaH2PO4, 17.0 g NaCl and 13.33 g choline chloride in 2 L MilliQ water. Cells were then gently removed from the plates using a rubber cell scraper and pooled in Dulbecco's phosphate buffered saline (DPBS) (0.1 g L−1 MgCl2·6H2O, 0.2 g L−1 KCl, 0.2 g L−1 KH2PO4, 8.0 g L−1 NaCl, 1.15 g L−1 anhydrous Na2HPO4). Cells were collected by centrifugation at 680 g for 5 min and resuspended in DPBS. An aliquot of 1 mL cell suspension (ca. 107 cells) was placed in a cuvette and treated with 20 μM zinquin ethyl ester or TSQ and incubated for 30 min at 25 °C followed by 500 μM DEA-NO for another 25 min. Using an excitation wavelength of 370 nm, emission spectra were recorded between 400 nm and 600 nm with a Hitachi F-4500 fluorescence spectrophotometer. Afterwards, 10 μM N,N,N′,N′-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN) was added and the fluorescence spectrum monitored for another 10 min.
Separation of cell lysate following the reaction of LLC-PK1 cells and DEA-NO in the presence of zinquin or TSQ
Cells were grown and harvested as above and resuspended in 10 mL DPBS. The cell suspension (ca. 108 cells) was incubated with 20 μM zinquin ethyl ester or TSQ for 30–45 min followed by 500 μM DEA-NO for another 40 min in the dark. In control experiments, cells were reacted with 20 μM sensor for 70–85 min. Subsequently, cell suspensions were taken through the cycle of centrifugation and resuspension in fresh DPBS three times to remove extracellular sensor. The last cell pellet was resuspended in 1 mL of cold MilliQ water. Cells were then lysed by sonication and centrifuged at 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 20 min at 4 °C to collect the cell lysate or supernatant.
000 g for 20 min at 4 °C to collect the cell lysate or supernatant.
Isolation of proteome using Sephadex G-75 chromatography
Cell supernatant prepared as described above in the absence of DEA-NO was loaded onto an 80 cm × 0.75 cm gel filtration column of Sephadex G-75 (GE Healthcare) equilibrated with 20 mM Tris buffer (pH 7.4) at room temperature to separate the high and low molecular weight fractions. During gel-filtration chromatography, the column was eluted with degassed 20 mM Tris-Cl (pH 7.4) and fifty fractions were collected. Fluorescence emission spectra of all fractions were obtained as described above. The zinc content in each fraction was also measured by flame atomic absorption spectroscopy (AAS). The high molecular weight fractions were combined and termed the ‘proteome’. The fraction of the proteome that binds Zn2+ was termed ‘Zn-proteins’. It is noted that metallothionein, migrating as a 10 kDa band of Zn2+ was not detected in these cells nor was any putatively oxidized and polymerized metallothionein protein observed when cell supernatant was preincubated with 1 mM 2-mercaptoethanol to reduce disulfide bonds in polymeric protein and 50 μM Zn2+ was added to reconstitute any reduced protein as Zn–MT.
Reaction of proteome with DEA-NO in the presence of sensor
Proteome in 20 mM degassed Tris buffer (pH 7.4) was incubated with 20 μM of the sensor, namely, zinquin acid (45 min), TSQ (40 min) or FluoZin-3 tetrapotassium salt (20 min). Time dependent fluorescence spectra were recorded using an excitation wavelength of 370 nm and emission wavelength scan ranging from 400 nm to 600 nm. Subsequently, the reaction mixture was treated with 500 μM DEA-NO for an hour. The final reaction mixture was loaded onto a Sephadex G-75 column, and the fractions were eluted with 20 mM degassed Tris buffer (pH 7.4). All the collected fractions were analyzed for both fluorescence and zinc content.
Quantification of Zn2+ by atomic absorption spectrophotometry
The concentration of Zn2+ in solutions was determined by flame atomic absorption spectrophotometry. A GBC model 904 instrument employed an acetylene torch to atomize samples using an 80:20 mixture of compressed air and acetylene. Zinc measurements were made with deuterium background correction. The instrument was calibrated before each run with standard Zn2+ solutions.
Quantification of sulfhydryl groups
The concentration of sulfhydryl groups was determined with Ellman's reagent.23 Specifically, 60 μL of proteome sample was diluted with 540 μL 20 mM Tris-Cl, pH 7.4 and then incubated with 60 μL of 10 mM 5,5-dithiobis-2-nitrobenzoic acid (DTNB) for 30–60 min before the absorbance at 412 nm was obtained. An extinction coefficient of 13600 cm−1 M−1 was used to determine the concentration of reactive thiol groups in the samples.
Results
Reactions of LLC-PK1 cells with DEA-NO in the absence or presence of zinquin
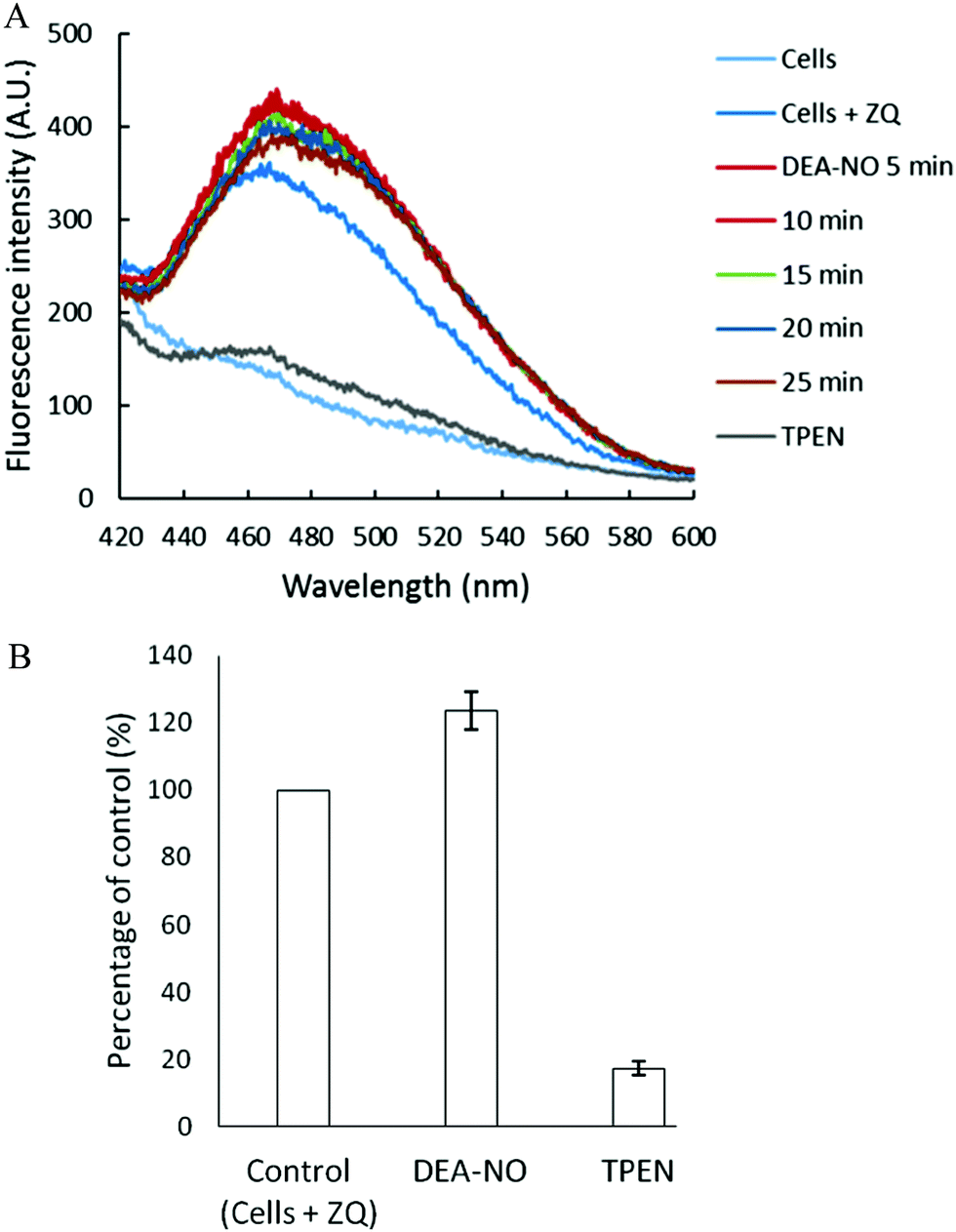
Pig kidney LLC-PK1 cells were used to investigate the impact of nitric oxide on the availability of intracellular Zn2+ to react with fluorescent zinc sensors. DEA-NO, which releases NO with a half time of about 20 min at room temperature, served as the source of nitric oxide.24–26 Initially, 1 × 107 LLC-PK1 cells were incubated with 20 μM zinquin ethyl ester (ZQee) for 30 min. During this time the charge neutral probe diffused into the cells and began to undergo ester hydrolysis. A fluorescence emission spectrum centered at 470 nm was observed, indicative of the formation of ternary adduct species, ZQ–Zn-proteins (Fig. 2A and reaction (1)).19,21 Based on previous estimates, the amount of protein-bound zinc participating in adduct formation represents about 10–15% of the total.18,20,21 The introduction of 500 μM DEA-NO to the cells for another 25 min caused a gradual increase of fluorescence at 470 nm, along with a noticeable shoulder at 490 nm (Fig. 2A). Compared with the reaction of cells with ZQee alone, a 25% increment in fluorescence intensity was observed (Fig. 2B). Subsequently, 10 μM TPEN, a cell permeable, strong zinc chelator, quenched the fluorescence to 17% of the intensity observed with control cells treated with 20 μM ZQee (Fig. 2B), confirming that enhancement of fluorescence following incubation with ZQee and DEA-NO involved the participation of Zn2+. Moreover, the reaction of cells with DEA-NO reduced its total sulfhydryl concentration from 780 nmol/106 cells to 440 nmol/106 cells as measured after isolation of supernatant from sonicated cells.
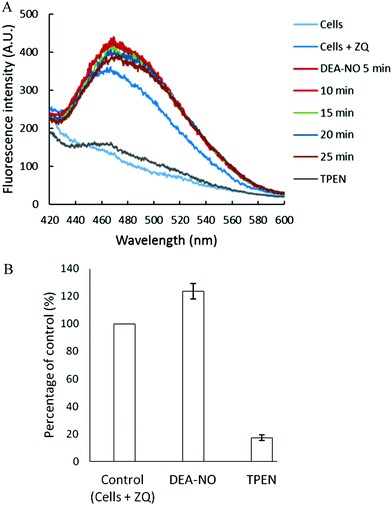 |
| | Fig. 2 Reaction of LLC-PK1 cells with DEA-NO in the presence of zinquin ethyl ester (ZQee). (A) Fluorescence spectra of the reaction between 107 LLC-PK1 cells and 20 μM ZQee for 30 min followed by the treatment with 500 μM DEA-NO for another 25 min. (B) Percent enhancement of fluorescence upon addition of 500 μM DEA-NO and its reversal by 10 μM TPEN. Error bars represent standard errors for at least three measurements and A.U. indicates arbitrary units. | |
The appearance of the shoulder at 490 nm suggested the mobilization of Zn2+ from the proteome (Zn-proteins) by NO or one of its oxidation products and, in turn, the production of the Zn(ZQ)2 complex (reactions (5)–(8)), which fluoresces with an emission maximum of 490 nm.19,21 It was hypothesized that oxidation products of NO modified Zn2+-bound thiol ligands, thus releasing Zn2+.27
| | | 2˙NO + ½O2 + Zn–S-proteins ⇌ Zn2+ + NO–S-proteins + NO2− + H+ | (5) |
| | | ˙NO + ½O2 + Zn–S-proteins → Zn2+ + ˙S-proteins + NO2− + H+ | (6) |
| | | 2˙S-proteins → proteins-S–S-proteins | (7) |
Another contributor to the fluorescence increase might be Zn
2+ mobilized from Zn-proteins that non-specifically rearranges to adventitious binding sites within the proteome (proteome·Zn) (
reaction (3)). In turn, proteome·Zn may react with ZQ to generate proteome·Zn–ZQ that fluoresces at 470 nm (
reaction (4)).
19
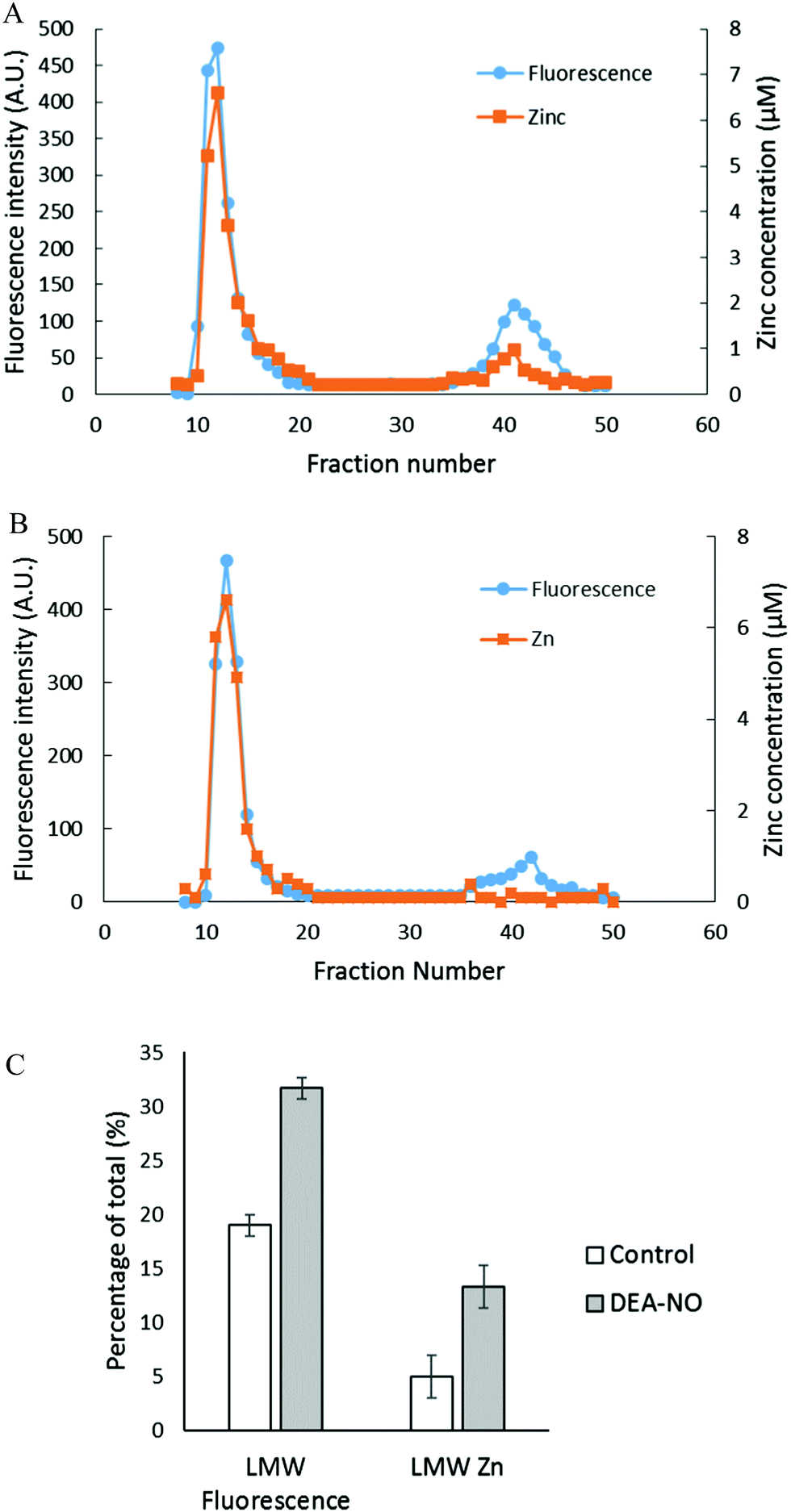
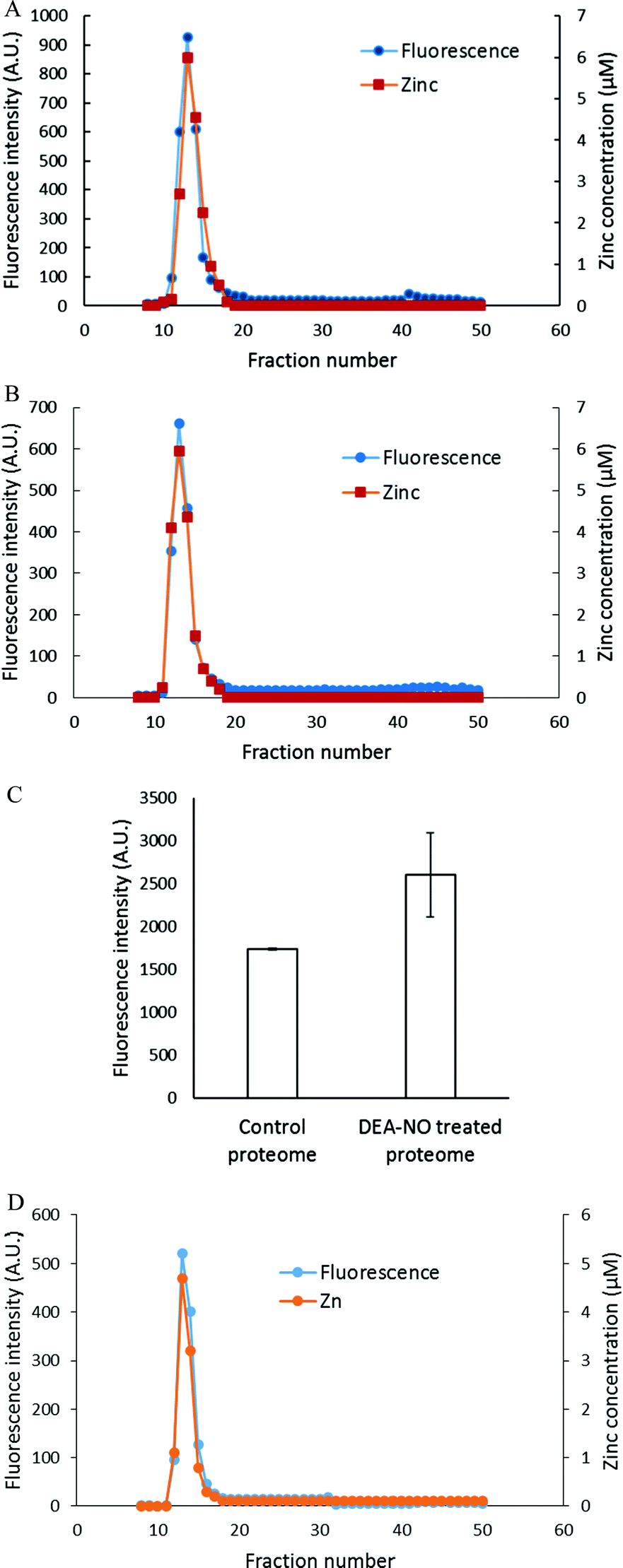
To investigate further the basis for the above observations, 1.25 × 108 LLC-PK1 cells in 10 mL were reacted with 20 μM ZQee for 30 min at room temperature before incubation with 500 μM DEA-NO for another 40 min. The cells were then lysed, centrifuged, and the resultant supernatant separated by Sephadex G-75 gel filtration. When the fractions were analyzed for fluorescence and zinc content, proteomic and low molecular weight (LMW) bands of both fluorescence and zinc were found for both control (cells plus ZQee) and DEA-NO exposed (cells and ZQee plus DEA-NO) cells (Fig. 3A and B). Proteome-associated fluorescence that centered at 470 nm increased by 16% in DEA-NO treated cells compared to control, unexposed cells. Evidently, DEA-NO induced the formation of more proteome·Zn–ZQ adducts. Plausibly, the modification of SH groups by DEA-NO either made available altered native Zn-proteomic sites (Zn-*proteins) for reaction with ZQ or Zn2+ at such sites moved to non-specific proteomic sites where ternary adducts were generated (reactions (9)–(11)).
| | | Zn-*proteins + ZQ ⇌ ZQ–Zn-*proteins | (9) |
| | | Zn-*proteins + proteome ⇌ *proteins + proteome·Zn | (10) |
| | | Proteome·Zn + ZQ ⇌ proteome·Zn–ZQ | (11) |
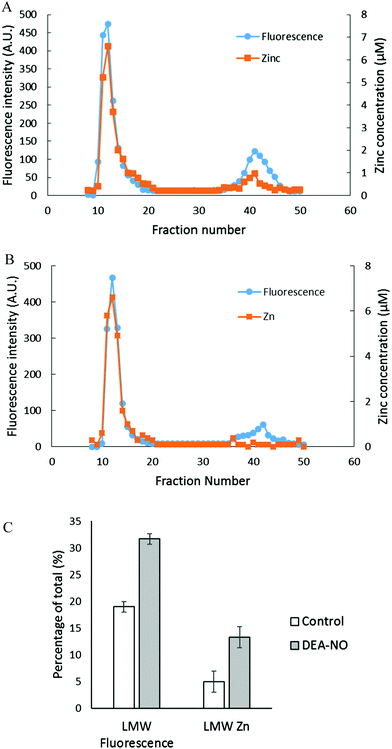 |
| | Fig. 3 Sephadex G-75 gel filtration of LLC-PK1 cell supernatant incubated with 20 μM ZQee followed by 500 μM DEA-NO. (A) 108 LLC-PK1 cells were reacted with 500 μM DEA-NO for 40 min following 20 μM ZQee for 30 min. The cells were lysed, centrifuged, and the supernatant separated using Sephadex G-75 column chromatography. The fractions were analyzed for both fluorescence and zinc. (B) Control: a parallel reaction was run under identical conditions without DEA-NO. (C) Comparison of the low molecular weight (LMW) fluorescence and zinc content between control and DEA-NO exposed cells. Error bars represent standard errors for at least three measurements. | |
The low molecular weight fluorescence pool of control reaction constituted 19% of the total fluorescence (high molecular weight and low molecular weight), whereas that of DEA-NO treated reaction represented 32% (Fig. 3C). The spectra of low molecular weight fractions centered near 490 nm, the wavelength maximum of Zn(ZQ)2. Consistent with the fluorescence data, after DEA-NO treatment the low molecular weight zinc content was calculated to be 13% of the total zinc, whereas that of control reaction was only 5% (Fig. 3C). The significant increase of both the low molecular weight zinc and its accompanying fluorescence emission centered at 490 nm indicated the formation of Zn(ZQ)2 during the reaction of NO with cells. Possibly, ZQ extracted Zn2+ from native Zn2+ binding sites that had been modified and weakened (Zn-*proteins) by nitric oxide to generate Zn(ZQ)2 (reaction (12)). Alternatively, Zn2+ labilized at such sites may have shifted to non-specific sites of binding within the proteome (reaction (10)) and then reacted with ZQ to generate Zn(ZQ)2 (reactions (11) and (13)):
| | | Zn-*proteins + 2ZQ ⇌ *proteins + Zn(ZQ)2 | (12) |
| | | Proteome·Zn–ZQ + ZQ ⇌ proteome + Zn(ZQ)2 | (13) |
Reaction of isolated proteome with DEA-NO in the presence of zinquin acid (ZQacid)
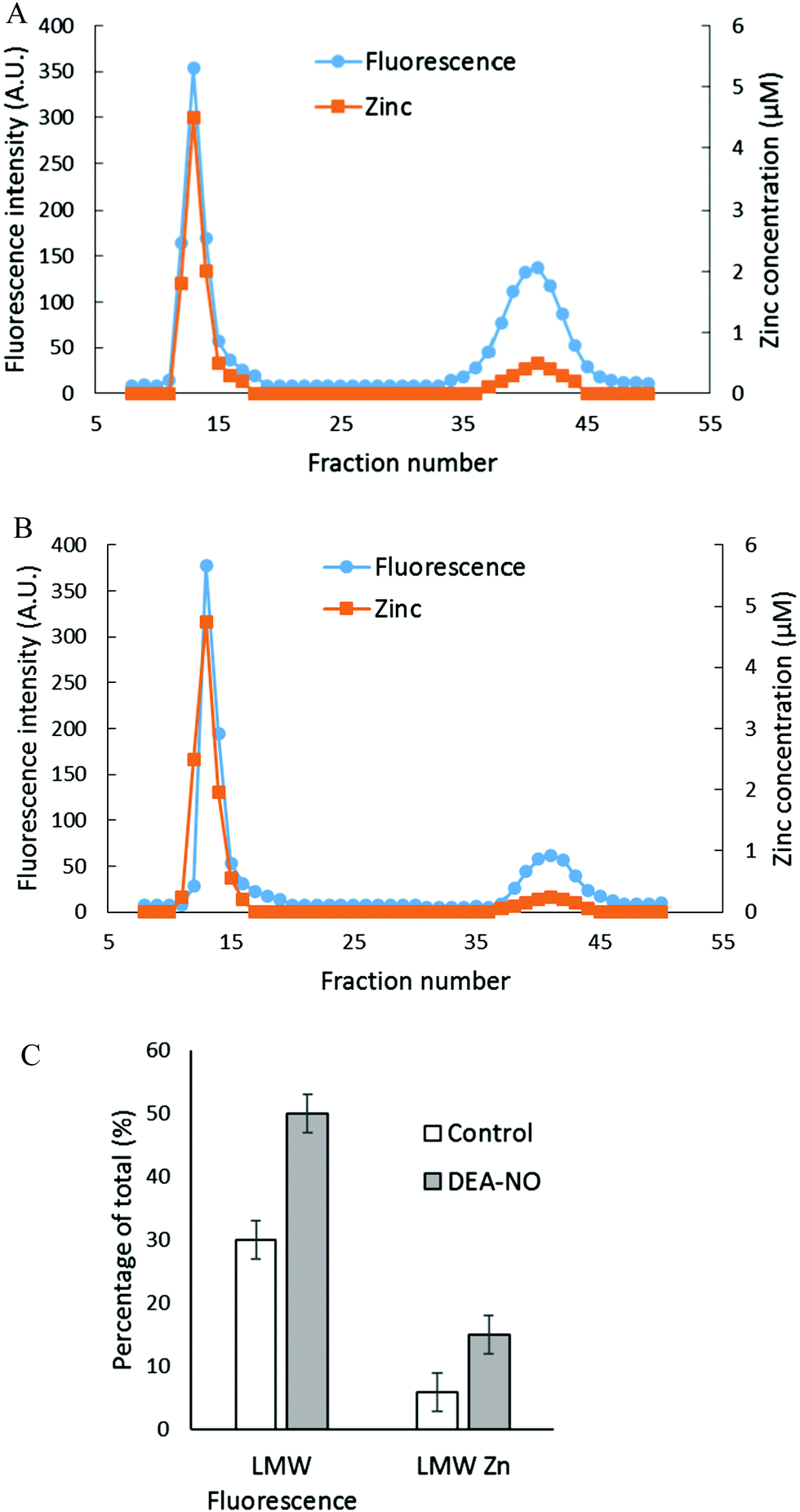
The reactions of NO and ZQ with LLC-PK1 cells were investigated in a simpler system that contained isolated proteome and was not complicated by membrane barriers and plentiful glutathione that are present in cells. Proteome containing 10 μM native protein-bound Zn2+ was first reacted with 20 μM ZQacid for 45 min at room temperature. As in whole cells, a gradual increase of fluorescence with the emission maximum of 470 nm was observed, indicative of the formation of ZQ–Zn-protein ternary adducts.21 Subsequently, 500 μM DEA-NO was added for an hour. When the final reaction mixture was separated by Sephadex G-75 chromatography, a noticeable low molecular weight pool of fluorescence and zinc, larger than that of the control sample, was detected (Fig. 4A and B). The LMW fluorescence pool (emission maximum of 490 nm) was measured to be 50% of total fluorescence, which is 1.7 times that of the control (30% of total fluorescence) (Fig. 4C) and displayed an emission maximum at 490 nm. In addition, the LMW zinc pool was determined to be three times larger than that of control proteome (15% of total zinc content vs. 5%). The reduction of sulfhydryl content by 41% (from 212 μM to 126 μM) following the treatment with DEA-NO demonstrated the reaction between proteomic sulfhydryl groups and nitric oxide. As in whole cells, the low molecular weight fluorescence and zinc pool in the reaction of isolated proteome indicated the production of Zn(ZQ)2. Moreover, proteomic fluorescence intensity increased by 19% in comparison with the control, suggesting the generation of new ternary adduct sites that resulted from the labilization of Zn2+. Overall, the in vitro results agreed qualitatively with those derived from the exposure of cells to DEA-NO.
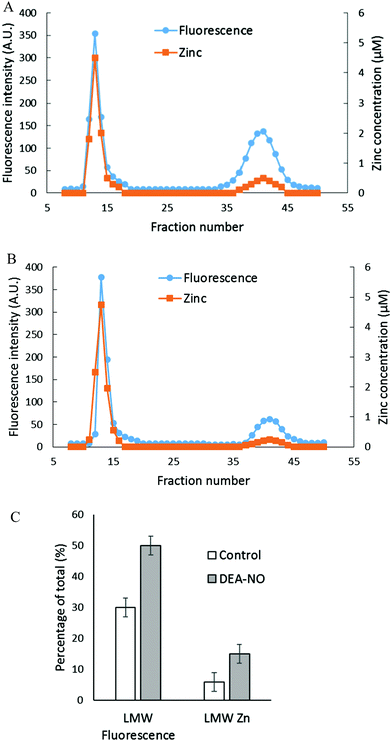 |
| | Fig. 4 Sephadex G-75 gel filtration of isolated proteome incubated with 20 μM zinquin acid (ZQacid) followed by 500 μM DEA-NO. (A) Isolated proteome (10 μM Zn2+) was reacted with 20 μM ZQacid for 45 min followed by 500 μM DEA-NO for another 1 hour. The final reaction mixture was separated using Sephadex G-75 column chromatography, and the fractions were analyzed for both fluorescence and zinc content. (B) Control: a parallel reaction was run under identical conditions in the absence of DEA-NO. (C) Comparison of the low molecular weight fluorescence and zinc content. Error bars represent standard errors for at least three measurements. | |
Reaction of LLC-PK1 cells with DEA-NO in the presence of TSQ
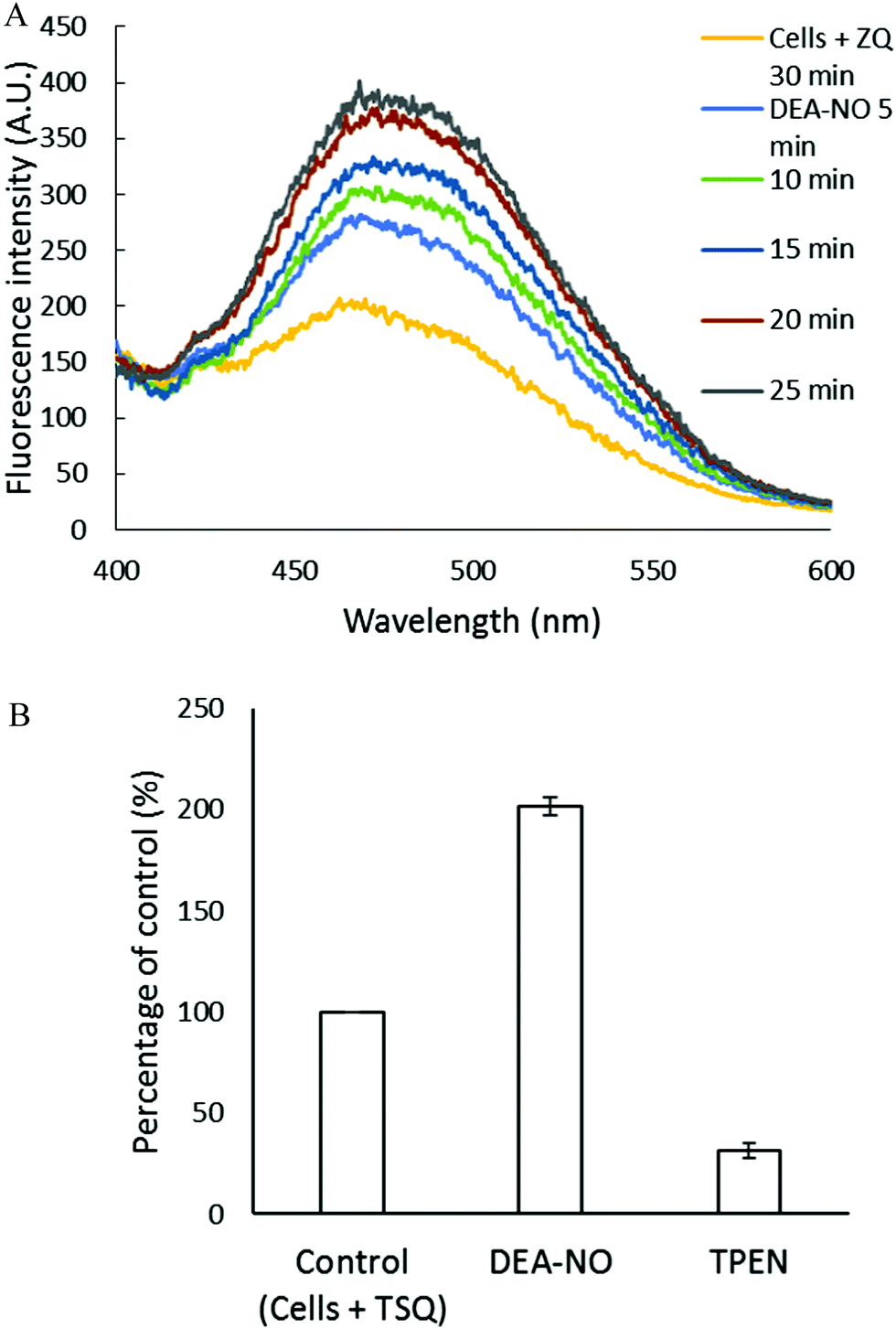
The experiments involving ZQ were repeated substituting TSQ, its closely related analog (Fig. 1). First, TSQ was reacted with LLC-PK1 cells to form TSQ–Zn-protein ternary adducts (reaction (1)) as was evident from the fluorescence spectrum centered at around 470 nm (Fig. 5A).18 In this bound form, TSQ like ZQacid is firmly fixed within the cells even though it is a neutral molecule. Subsequent incubation with 500 μM DEA-NO for 40 min caused a two-fold increase of fluorescence, substantially more than obtained with zinquin, and a shift of the emission maximum to 480 nm, which suggested that some Zn(TSQ)2 had formed. As with ZQ, 10 μM TPEN was able to reduce the fluorescence to 32% of the control value, signaling the participation of Zn2+ in the enhancement of fluorescence in DEA-NO treated as well as control cells (Fig. 5B). Consistent with the fluorescence enhancement, the sulfhydryl concentration declined from 740 nmol/106 cells to 450 nmol/106 cells as measured after isolation of lysate from sonicated cells.
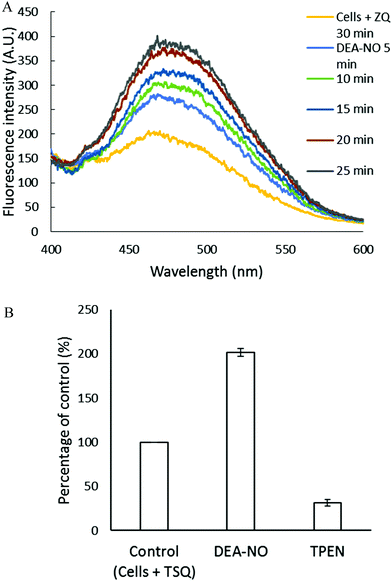 |
| | Fig. 5 Reaction of LLC-PK1 cells with DEA-NO in the presence of TSQ. (A) Fluorescence spectra of the reaction between 107 LLC-PK1 cells and 20 μM TSQ for 30 min followed by the treatment with 500 μM DEA-NO for another 25 min. (B) Percent enhancement of fluorescence upon addition of 500 μM DEA-NO and its reversal by 10 μM TPEN. Error bars represent standard errors for at least three measurements. | |
To characterize the fluorescent species, LLC-PK1 cells were incubated with 20 μM TSQ for 40 min followed by 500 μM DEA-NO for another 45 min. Cells were then washed, lysed and centrifuged, and the resultant supernatant fractionated using Sephadex G-75 column chromatography. Compared with the results with zinquin, no detectable pool of low molecular weight fluorescence and Zn2+ as Zn(TSQ)2 was found in either DEA-NO treated or control cells (Fig. 6A and B). Almost all the fluorescence and zinc were detected in the high molecular weight proteome fractions. The fluorescence intensity of the high molecular weight fractions was 50% greater than that of control (Fig. 6C) and was aligned with the Zn2+ concentrations in the fractions. Moreover, the emission maxima of the proteomic fractions were located at about 470 nm, consistent with the presence of ternary adduct species. In contrast to the results in Fig. 5A, these findings indicate that Zn2+ mobilized by incubation with DEA-NO remained bound completely by the proteome in the presence of TSQ, forming ternary adducts instead of Zn(TSQ)2. If some Zn(TSQ)2 was formed in vivo as suggested by Fig. 5A, perhaps proteome·Zn–TSQ species became favored during chromatography as proteome and TSQ were separated and the reaction equilibrium, as in reaction (13), shifted to the left. That is, possibly because of its lower affinity for Zn2+ relative to ZQ, TSQ cannot compete with the adduct species to retain Zn(TSQ)2 during gel filtration.20 However, a rapid one step separation of proteome from low molecular weight molecules using centrifugal filtration (3 kDa cut off Centricon filter) also did not demonstrate any Zn(TSQ)2. Thus, it was concluded that mobilized Zn2+ was present only as proteome·Zn–TSQ.
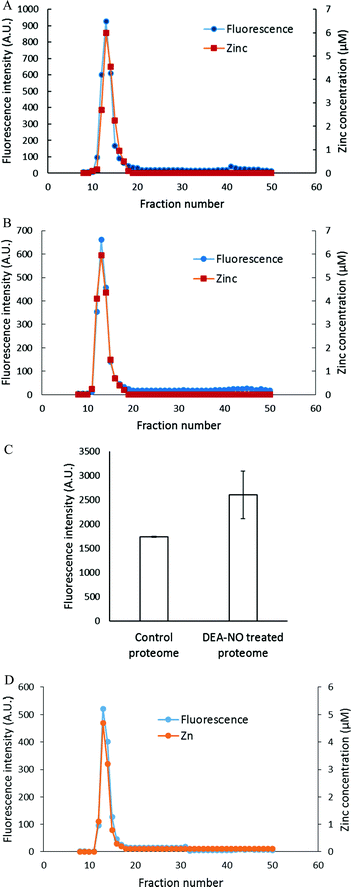 |
| | Fig. 6 Sephadex G-75 chromatography of LLC-PK1 cell supernatant or isolated proteome incubated with TSQ followed by 500 μM DEA-NO. (A) 108 LLC-PK1 cells were reacted with 500 μM DEA-NO for 45 min following 20 μM TSQ for 40 min. The cells were lysed, centrifuged, and the supernatant was separated using a Sephadex G-75 column. The fractions were analyzed for both fluorescence and zinc. (B) Control: a parallel reaction was run at identical condition with no DEA-NO added. (C) Comparison of the integrated fluorescence intensity of proteome. (D) Isolated proteome (10 μM Zn2+) was reacted with 20 μM TSQ for 40 min followed by 500 μM DEA-NO for another 1 hour. The final reaction mixture was separated using a Sephadex G-75 column, and the fractions were analyzed for both fluorescence and zinc content. Error bars represent standard errors for at least three measurements. | |
Reaction of isolated proteome with DEA-NO in the presence of TSQ
The reaction of DEA-NO and TSQ with proteome was also examined (Fig. 6D). Isolated proteome containing 10 μM Zn2+ was reacted with 20 μM TSQ for 40 min before 500 μM DEA-NO was added for one hour. The reduction of sulfhydryl concentration by about 41% (from 340 μM to 200 μM) indicated a significant reaction between nitric oxide or its oxidation products and proteomic sulfhydryl groups. When the reaction mixture was subjected to Sephadex G-75 gel filtration, all of the fluorescence and Zn2+ resided in proteomic fractions; Zn(TSQ)2 was not formed in either DEA-NO treated and control reactions.
Reaction of isolated proteome with added Zn2+ in the presence of ZQ or TSQ
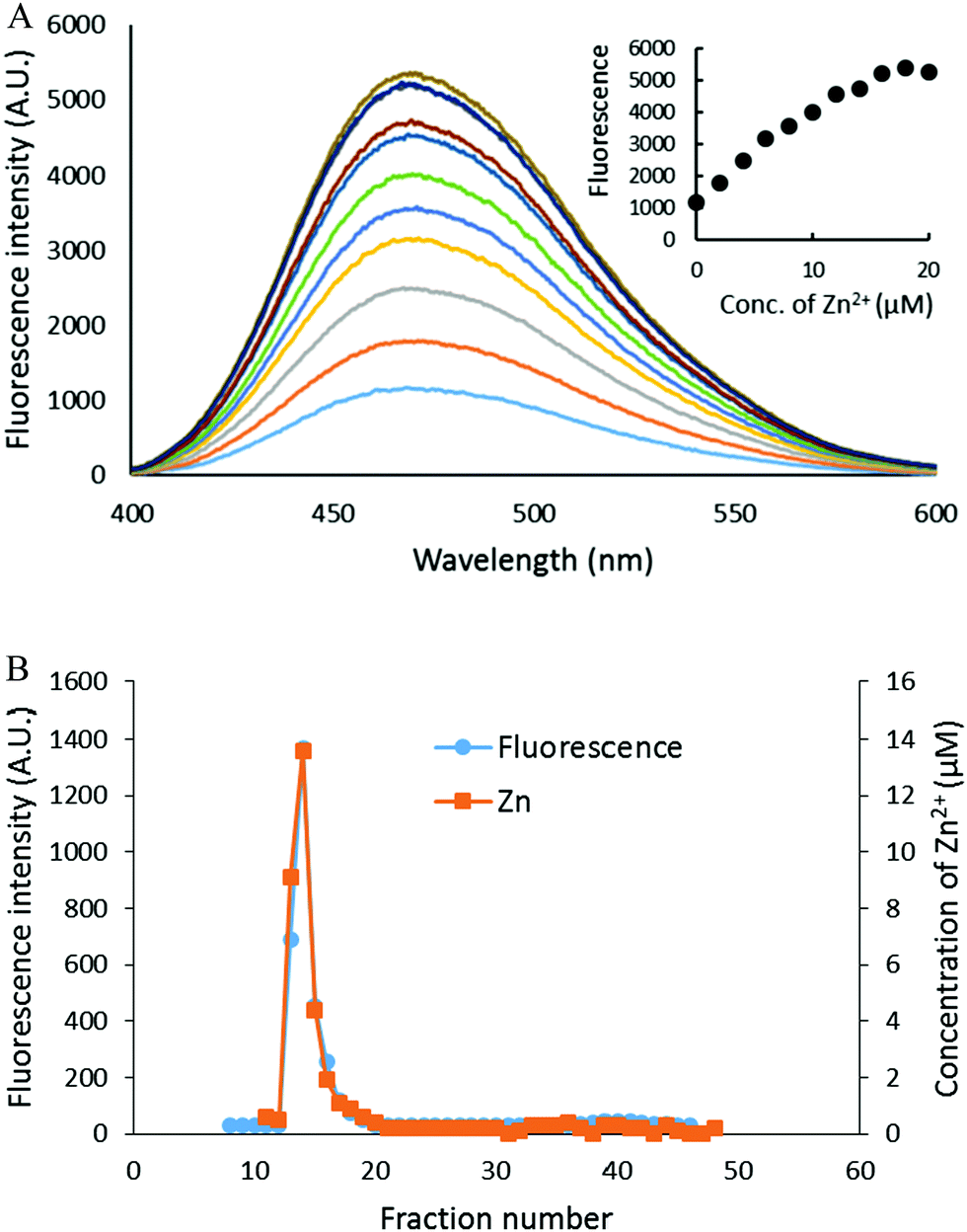
The capacity of the proteome to bind adventitious Zn2+ in the absence or presence of DEA-NO and ZQ or TSQ was also examined in order to probe the involvement of reactions (2)–(4). Control proteome (8 μM Zn2+) was titrated with Zn2+ in the presence of excess ZQ or TSQ. The fluorescence emission intensity centered at about 470 nm, due to the formation of proteome·Zn–ZQ/TSQ ternary adducts, increased in parallel with the concentration of added Zn2+ (Fig. 7A and 8A). In both cases, the emission maximum remained unchanged at 470 nm as Zn2+ was added to the proteome. Moreover, when the final reaction mixtures were fractionated by gel filtration with Sephadex G-75, no measurable low molecular weight fluorescence or Zn2+ was detected with either fluorophore (Fig. 7B and 8B). Thus, the proteome contained extensive capacity to bind Zn2+ and formed adducts with Zn2+ and either sensor (reactions (3) and (4)). These results differ from the findings about the reaction of cells or proteome with DEA-NO in the presence of zinquin. In those experiments, a red shifted emission maximum of the DEA-NO treated sample (Fig. 2) and a detectable low molecular weight fluorescence and zinc pool (Fig. 3 and 4) indicated that Zn(ZQ)2 was produced along with proteome·Zn–ZQ.
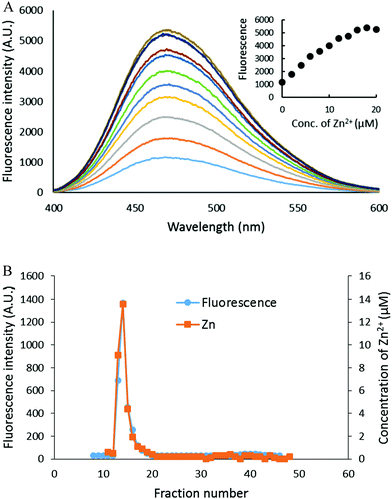 |
| | Fig. 7 Zn2+ titration of proteome pre-treated with ZQacid. (A) Isolated proteome (8 μM Zn2+) was reacted with 20 μM ZQacid for 30 min (the bottom most spectrum). Then, the reaction mixture was titrated with the increasing concentration of ZnCl2. (B) The final reaction mixture from (A) was fractionated using Sephadex G-75 gel filtration, and the eluted fractions were analyzed for both fluorescence and zinc content. | |
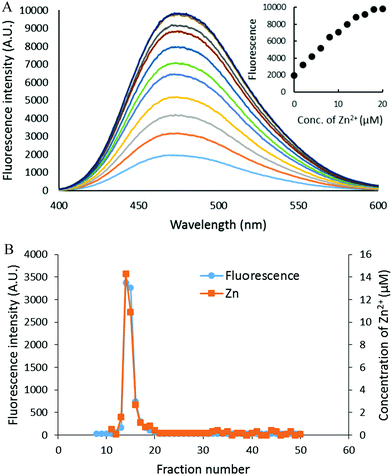 |
| | Fig. 8 Zn2+ titration of isolated proteome pre-treated with TSQ. (A) Isolated proteome (8 μM Zn2+) was reacted with 20 μM TSQ for 30 min (the bottom most spectrum). Following on, the reaction mixture was titrated with increasing concentrations of ZnCl2. The inset summarizes the fluorescence changes as a function of Zn2+. (B) The final reaction mixture from (A) was fractionated using Sephadex G-75 gel filtration, and the eluted fractions were analyzed for both fluorescence and zinc content. | |
The difference in outcome related to the absence or presence of DEA-NO. The results suggested the hypothesis that the behavior of ZQ depends on the existence and extent of modification of sulfhydryl groups that may participate in the adventitious proteomic binding of Zn2+ (reactions (14) and (15)).
| | | 2˙NO + ½O2 + proteome-S·Zn → Zn2+ + proteome-S–NO + NO2− + H+ | (14) |
| | | ˙NO + ½O2 + proteome-S·Zn → Zn2+ + ½proteome-S–S-proteome + NO2− + H+ | (15) |
As the number of adventitious binding sites for Zn
2+ declines through reaction with NO, the formation of a substantial concentration of Zn(ZQ)
2 becomes more favorable (
reaction (8)).
Reaction of isolated proteome with DEA-NO in the presence of FluoZin-3
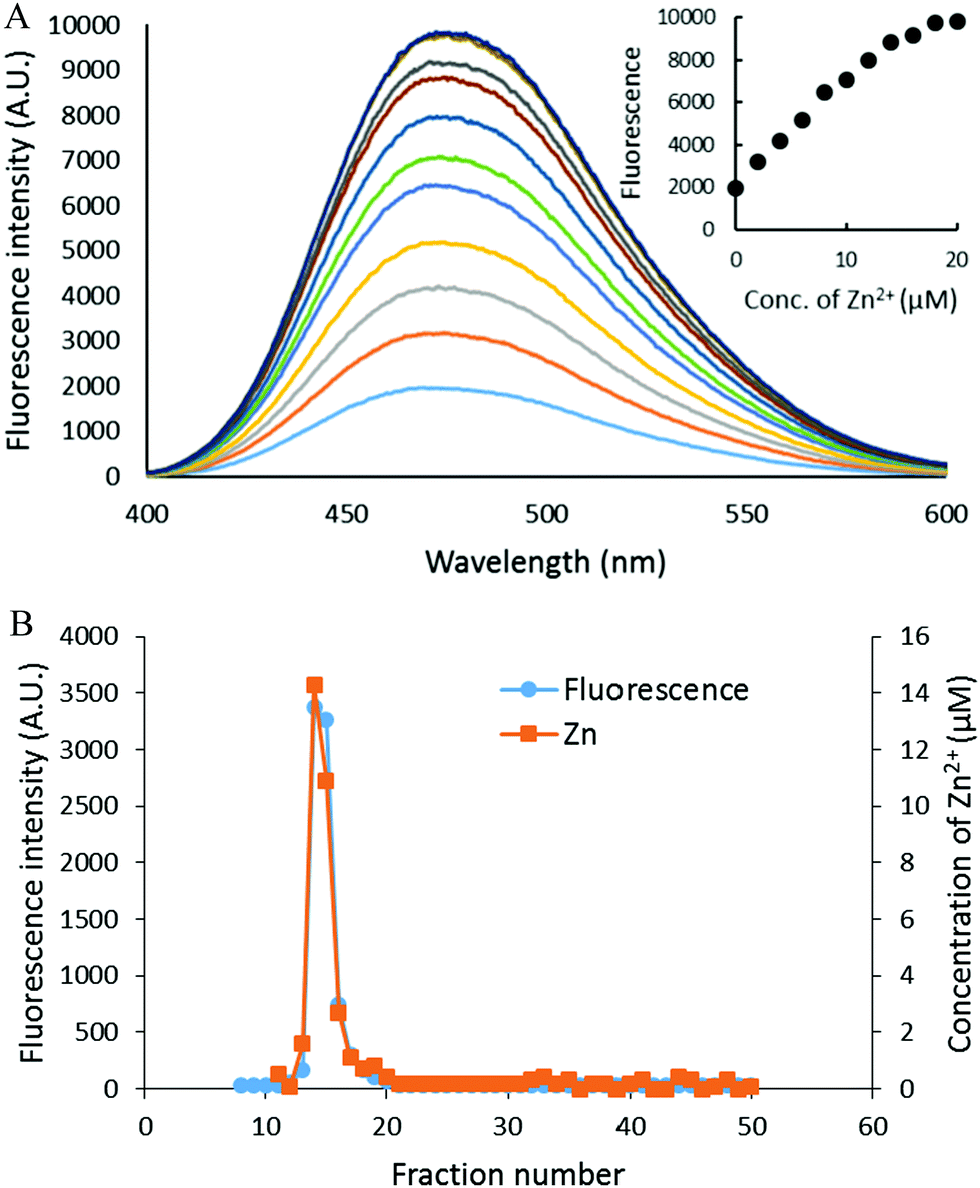
The reaction of isolated proteome (10 μM Zn2+) with 500 μM DEA-NO was also monitored using another Zn2+ sensor that does not form ternary complexes with Zn-proteins, FZ-3 (20 μM). The absence of any fluorescence signal during the reaction of proteome and FluoZin-3 for 20 min indicated that Zn-proteome did not react with FZ-3 to form ternary adducts. The introduction of DEA-NO to the reaction mixture of proteome and FZ-3 for 1 hour resulted in the reduction of proteomic sulfhydryl concentration by about 41% (from 342 μM to 202 μM). The presence of DEA-NO caused a gradual increase of fluorescence throughout this period (Fig. 9A and B). Interestingly, addition of 10 μM TPEN, a powerful Zn2+ chelator, at an intermediate time did not quench any of the enhanced fluorescence. Instead, the fluorescence signal continued increasing. This finding suggested that the increase of fluorescence was not caused by the formation of Zn–FluoZin-3 complex, because its fluorescence should have been quenched upon the addition of TPEN. In a separate experiment, upon fractionation of the final reaction mixture of proteome (10 μM Zn2+), 20 μM FZ-3 and 500 μM DEA-NO using a Sephadex G-75 column, no LMW Zn–FZ-3 complex was isolated (Fig. 9C) confirming that FluoZin-3 does not chelate proteomic Zn2+ after reaction of DEA-NO with the proteome. However, virtually all of the fluorescence was observed in the LMW region of the chromatogram, indicating that a FZ-3 related species was responsible for the fluorescence enhancement seen during the reaction.
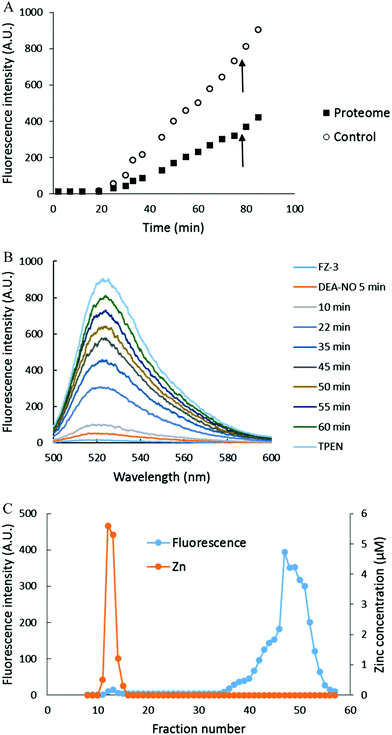 |
| | Fig. 9 Reaction of DEA-NO with isolated proteome preincubated with FluoZin-3. (A) Isolated proteome (10 μM Zn2+) was reacted with 20 μM FZ-3 for 20 min followed by 500 μM DEA-NO for another hour. Finally 10 μM TPEN was added for 10 min. In the control experiment, 20 μM FZ-3 in 20 mM Tris (pH 7.4) was reacted with 500 μM DEA-NO for an hour followed by 10 μM TPEN for another 10 min. The arrow indicates the time of TPEN addition. (B) Fluorescence spectra of the reaction between 20 μM FZ-3 and 500 μM DEA-NO. (C) Sephadex G-75 chromatographic fractionation of the reaction mixture of proteome (10 μM Zn2+) and 500 μM DEA-NO in the presence of 20 μM FZ-3 for an hour. | |
To investigate the source of this fluorescence enhancement, 20 μM FluoZin-3, itself, was reacted with 500 μM DEA-NO for 1 hour followed by 10 μM TPEN for another 10 min (Fig. 9A). A gradual increase of fluorescence larger than observed in the proteome experiment was recorded. Again, 10 μM TPEN did not reverse the intensification of the fluorescence. It was concluded that NO reacts with FZ-3 with an increase of fluorescence, independent of its reaction with Zn2+. FluoZin-3 was also reacted with S-nitrosyl-penicillamine under similar conditions except that the reactants were kept in the dark to prevent the photochemical cleavage of S–NO bond to release NO. No reaction with FluoZin-3 was observed, indicating that the reaction of NO donors with FZ-3 is specific for nitric oxide or its oxidation products.
Discussion
Fluorescent sensors of all kinds are in routine use to reveal structures, chemical changes, and ongoing processes in living cells.28–31 Major efforts have been made and are continuing to design probes that selectively detect essential and extraneous metal ions in cells and, thereby, to reveal their participation in normal and pathologic biochemistry.1,3,30,32,33 In the case of Zn2+, for example, the appearance of a fluorescence microscopic signal in the presence of a cell permeable sensor, followed by its diminution in the presence of the powerful membrane permeant chelator, TPEN, has been employed in many studies to conclude that labile Zn2+ is present in the cellular process under investigation. Similarly, the absence of such changes signals that mobile Zn2+ is not involved. In one early study, sheep pulmonary artery endothelial cells (SPAECs) exposed to the NO donor, S-nitroso-cysteine, and ZQ displayed a 25% increase in fluorescence compared with control cells.15 This result led to the conclusion that Zn2+ mobilized by NO plays a key role in NO-dependent vessel constriction.
The results of the present experiments involving cells and isolated proteome are comparable to those of the original study. Furthermore, though not discussed in the original paper, the high background fluorescence of control SPAEC cells incubated with ZQ is consistent with the initial formation of fluorescent ZQ–Zn-protein adducts.15 Originally, these results were explained by reaction (16):
| | | Zn–S-proteome + 2NO + ½O2 + 2ZQ ⇌ NO–S-proteome + Zn(ZQ)2 + NO2− + H+ | (16) |
Oxidation products of NO modify thiol groups at Zn
2+ binding sites, weakening the association of Zn
2+ with the remaining ligands. In this circumstance, ZQ captures Zn
2+ either by direct ligand substitution (
reaction (12)) or reacts with Zn
2+ that has dissociated from its original binding sites (
reaction (8)).
From a chemical point of view, recent studies and the present experiments reveal that the milieu in which ZQ undergoes reaction is more complex. In cells ZQ binds to a significant fraction of the unmodified Zn-proteome and forms ternary ZQ–Zn-protein adducts (reaction (1) and Fig. 2A, 3B).21 Depending on the cell type, it competes to sequester a small fraction of Zn2+ from Zn-proteins as Zn(ZQ)2 (Fig. 3B).21 The introduction of NO increases the fluorescence by ca. 20–25%, signaling the formation of additional adduct species at modified sites, ZQ–Zn-NO–S-proteins and/or adducts at adventitious Zn2+ binding sites within the proteome, proteome-S·Zn–ZQ. More Zn(ZQ)2 forms as well (Fig. 2B and 3C). When TSQ, a closely related Zn2+ probe, replaces ZQ, the results are different: Zn(TSQ)2 is not produced (Fig. 5 and 6). Turning to a third sensor, FluoZin-III, yet another response to NO was noted as fluorescence emission increased due to the modification of the fluorophore not because of the binding of Zn2+ (Fig. 9). No evidence was elicited demonstrating that NO-treated FluoZin-III served as an effective probe for Zn2+ associated with modified native sites or adventitious proteomic binding sites.
It has been proposed that aerobic NO releases Zn2+ preferentially from Zn–metallothionein (Zn–MT) because its plethora of sulfhydryl groups bind up to seven Zn2+ ions within the protein's two domain structure.11–13 A previous study on the reaction of DEA-NO with cells containing measurable Zn–MT demonstrated that the large pools of thiols in the proteomic and glutathione fractions readily react with oxidation products of NO and that Zn–MT is relatively unreactive with DEA-NO in the presence of other sources of thiol groups.17
The present study utilized a cell type that does not contain measurable Zn–MT, which would otherwise be observed midway between high and low molecular weight bands of Zn2+ in control Sephadex G-75 chromatograms (Fig. 3B). In this setting, it was clear that ZQ mobilized Zn2+ from Zn-proteins that had reacted with DEA-NO (Fig. 3 and 4). Thus, Zn-proteins are a major source of Zn2+ for reaction with fluorescent probes after Zn2+ was mobilized by the hypothetical reaction of DEA-NO with sulfhydryl, Zn-binding ligands.
The detailed understanding of the origin of the fluorescence enhancement observed after cells or proteome were incubated with ZQ and DEA-NO bears on the meaning of the results. Although ZQ and TSQ can form 2:1 complexes with Zn2+, the generation of ternary adduct species with control Zn-proteome is much more favorable (Fig. 3B and 4B). Upon exposure of isolated proteome to DEA-NO, this distinct preference remained for TSQ in the presence of the modified proteome (Fig. 6A and D). In contrast, the differential was lost in the cellular and proteomic reactions with ZQ as both the formation of ternary adducts and Zn(ZQ)2 increased in the presence of DEA-NO (Fig. 2A, 3 and 4).
A plausible explanation for the different behavior of ZQ and TSQ relies on the higher affinity of ZQ of Zn2+ due to the presence in its structure of an electron donating methyl group alpha to the isoquinoline's nitrogen ligand (unpublished information) (Fig. 1).19,20 The methyl group also sterically favors tetrahedral ligand coordination to coincide with a preferred Zn-ligand geometry. This difference apparently favors the formation of the 2 to 1 ligand to metal complex of ZQ relative to its ternary adducts. Zn2+ labilized by NO and detected as Zn(ZQ)2 might stem from metal ion still bound to sulfhydryl-modified native binding sites or from Zn2+ that has migrated to adventitious sites (reactions (12) and (13)). In either case it is not free or even loosely bound to cellular ligands within the cell. This conclusion is also consistent with the finding that another sensor with relatively low affinity for Zn2+, Newport Green, fails to sequester Zn2+ from Zn-proteins exposed to NO.34
These results qualitatively resemble the observed interactions of ZQ and TSQ with cells and proteome exposed to N-ethylmaleimide (NEM).20 Because NEM is a simple electrophile that is particularly reactive with thiol groups, the similarity of outcomes using DEA-NO and NEM supports the hypothesis that each targets sulfhydryl-containing proteins as well as glutathione. That these should include thiolate metal binding ligands of Zn-proteins as well as free sulfhydryl groups in cells treated with these reagents is intriguing because the thiolate nucleophile when bound to Zn2+ would seem to be a less reactive nucleophile than the free thiol at pH 7.
The capacity of proteome to bind Zn2+ in the presence of ZQ or TSQ was explored in titration experiments (Fig. 7 and 8). It sequesters at least 3–5 times more added Zn2+ than is present in the native Zn-proteome with sufficient stability that the formation of proteome·Zn-(ZQ/TSQ) is preferred exclusively to Zn(ZQ/TSQ)2. The result with ZQ differed from those observed with cells or proteome treated with ZQ and DEA-NO. Native proteome competed effectively with ZQ to form proteome·Zn–ZQ without the appearance of Zn(ZQ)2; NO-exposed proteome did not (Fig. 3 and 4). These divergent findings suggest that a substantial fraction of adventitious binding sites for Zn2+ involve sulfhydryl ligands. Once modified by DEA-NO, the proteome has less capability to associate with Zn2+ and ZQ (reactions (14) and (15)). Because TSQ binds Zn2+ with lower affinity than ZQ, the altered proteome still manages to outcompete TSQ for Zn2+ and no Zn(TSQ)2 forms (Fig. 6). Thus, depending on the fluorescent probe, the Zn2+ binding characteristics of the proteome under different cellular conditions play a significant role in how the sensor functions.
The functional implications of the apparent strong affinity of mobilizable Zn2+ for non-specific binding sites in the proteome have not been sorted out. For example, if the proteome provides a strong buffer for Zn2+, how does intracellular Zn2+ signaling take place? Signaling has been viewed as a process in which mobilized Zn2+ binds reversibly to specific sites that initiate signal cascades,
| | | Zn2+ + Ssignal ⇌ Zn–Ssignal (K, k1/k−1) | (17) |
such that the chemical basis of signaling are the variable concentration of Zn
2+, a modest stability constant (
K) for Zn–S
signaling, and, as a consequence, favorable rates for on and off steps (
k1/
k−1) of
reaction (17). This model will need to be rethought if the signaling reaction is better represented as
| | | Proteome·Zn + Ssignal ⇌ Zn–Ssignal + proteome (K, k1/k−1) | (18) |
in which the stability constant of Zn–S
signal is large so that S
signal can compete with proteome for bound Zn
2+.
4
Lastly, the reaction of FluoZin-3 with DEA-NO treated Zn-proteome was complicated by a side reaction of NO or its oxidation products with the probe that increased its fluorescence emission independent of the presence of Zn2+ (Fig. 9). A previous study of the properties of Newport Green as a sensor of proteomic Zn2+ released by DEA-NO also displayed a similar reaction with DEA-NO.34 Both sensors utilize a fluorescein-based fluorophore. In fact, NO sensors have been constructed based on the reaction of NO with this fluorophore.2,35,36 Not surprisingly, therefore, both Newport Green and FluoZin-3 act as weak NO sensors. Thus, when both Zn2+ and NO may be involved in cellular processes that are under study, particular care needs to be taken in analyzing results based on the use of these probes. Lastly, the results of Fig. 9 suggest that NO-modified FluoZin-3 has lost its Zn2+ sensing property. Experiments are ongoing to understand the impact of NO on the chemistry of this probe.
Acknowledgements
This research was supported by NIH grants ES-024509 and ES-04184 and by a Research Growth Initiative award from the University of Wisconsin–Milwaukee.
References
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef PubMed.
- M. D. Pluth, E. Tomat and S. J. Lippard, Annu. Rev. Biochem., 2011, 80, 333 CrossRef PubMed.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef PubMed.
-
A. Nowakowski, M. Karim and D. Petering, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2015, pp. 1–10 Search PubMed.
- D. H. Petering, Chemtracts: Inorg. Chem., 2004, 17, 569–580 Search PubMed.
- S. J. Langmade, R. Ravindra, P. J. Daniels and G. K. Andrews, J. Biol. Chem., 2000, 275, 34803–34809 CrossRef PubMed.
- J. S. Stamler, D. J. Singel and J. Loscalzo, Science, 1992, 258, 1898 Search PubMed.
- S. Habib and A. Ali, Indian J. Clin. Biochem., 2011, 26, 3–17 CrossRef PubMed.
- M. T. Gladwin, J. H. Crawford and R. P. Patel, Free Radical Biol. Med., 2004, 36, 707–717 CrossRef PubMed.
- D. A. Wink and J. B. Mitchell, Free Radical Biol. Med., 1998, 25, 434–456 CrossRef PubMed.
- K.-D. Kroncke, K. Fehsel, T. Schmidt, F. T. Zenke, I. Dasting, J. R. Wesener, H. Bettermann, K. D. Breunig and V. Kolbbachofen, Biochem. Biophys. Res. Commun., 1994, 200, 1105–1110 CrossRef PubMed.
- K. Katakai, J. Liu, K. Nakajima, L. K. Keefer and M. P. Waalkes, Toxicol. Lett., 2001, 119, 103–108 CrossRef PubMed.
- M. A. Schwarz, J. S. Lazo, J. C. Yalowich, W. P. Allen, M. Whitmore, H. A. Bergonia, E. Tzeng, T. R. Billiar, P. D. Robbins and J. R. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4452–4456 CrossRef.
- P. J. Bernal, K. Leelavanichkul, E. Bauer, R. Cao, A. Wilson, K. J. Wasserloos, S. C. Watkins, B. R. Pitt and C. M. S. Croix, Circ. Res., 2008, 102, 1575–1583 CrossRef PubMed.
- C. M. S. Croix, K. Wasserloos, K. Dineley, I. Reynolds, E. Levitan and B. Pitt, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2002, 282, L185–L192 CrossRef PubMed.
- A. Gow and H. Ischiropoulos, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2002, 282, L183–L184 CrossRef PubMed.
- J. Zhu, J. Meeusen, S. Krezoski and D. H. Petering, Chem. Res. Toxicol., 2010, 23, 422–431 CrossRef PubMed.
- J. W. Meeusen, H. Tomasiewicz, A. Nowakowski and D. H. Petering, Inorg. Chem., 2011, 50, 7563–7573 CrossRef PubMed.
- A. B. Nowakowski, J. W. Meeusen, H. Menden, H. Tomasiewicz and D. H. Petering, Inorg. Chem., 2015, 54, 11637–11647 CrossRef PubMed.
- A. Nowakowski and D. Petering, Metallomics, 2012, 4, 448–456 RSC.
- A. B. Nowakowski and D. H. Petering, Inorg. Chem., 2011, 50, 10124–10133 CrossRef PubMed.
- W. Lin, B. Mohandas, C. P. Fontaine and R. A. Colvin, BioMetals, 2007, 20, 891–901 CrossRef PubMed.
- G. Ellman and H. Lysko, Anal. Biochem., 1979, 93, 98–102 CrossRef CAS PubMed.
- W. Maret and Y. Li, Chem. Rev., 2009, 109, 4682–4707 CrossRef CAS PubMed.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, J. Org. Chem., 1993, 58, 1472–1476 CrossRef CAS.
- K. Schmidt, W. Desch, P. Klatt, W. R. Kukovetz and B. Mayer, Naunyn Schmiedeberg's Arch. Pharmacol., 1997, 355, 457–462 CrossRef CAS PubMed.
- D. Jourd'heuil, F. L. Jourd'heuil and M. Feelisch, J. Biol. Chem., 2003, 278, 15720–15726 CrossRef PubMed.
- X. Michalet, F. Pinaud, L. Bentolila, J. Tsay, S. Doose, J. Li, G. Sundaresan, A. Wu, S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- S. B. VanEngelenburg and A. E. Palmer, Curr. Opin. Chem. Biol., 2008, 12, 60–65 CrossRef CAS PubMed.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- I. Johnson, Histochem. J., 1998, 30, 123–140 CrossRef CAS PubMed.
- X. Peng, J. Du, J. Fan, J. Wang, Y. Wu, J. Zhao, S. Sun and T. Xu, J. Am. Chem. Soc., 2007, 129, 1500–1501 CrossRef CAS PubMed.
- X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef CAS PubMed.
- M. R. Karim and D. H. Petering, Metallomics, 2016, 8, 201–210 RSC.
- M. D. Pluth, M. R. Chan, L. E. McQuade and S. J. Lippard, Inorg. Chem., 2011, 50, 9385–9392 CrossRef CAS PubMed.
- M. H. Lim, Nat. Protoc., 2007, 2, 408–415 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a