
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionothermal template transformations for preparation of tubular porous nitrogen doped carbons†
J.
Pampel
 *a,
A.
Mehmood
ab,
M.
Antonietti
a and
T.-P.
Fellinger
*ac
*a,
A.
Mehmood
ab,
M.
Antonietti
a and
T.-P.
Fellinger
*ac
aDepartment of Colloid Chemistry, Max-Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: jonas.pampel@mpikg.mpg.de; tim.fellinger@mpikg.mpg.de
bDepartment of Energy and Materials Engineering, Dongguk University-Seoul, 04620 Seoul, Republic of Korea
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Kemigården 4, 41296 Gothenburg, Sweden
First published on 5th April 2017
Abstract
A facile approach for the Zn-free ionothermal synthesis of highly porous nitrogen doped carbons possessing tubular transport pores is demonstrated employing adenine as biomass derived precursor and surfactant together with calcium or magnesium chloride hydrates as combined solvent-porogens. The overall process can be regarded as a combination of liquid templating by means of sol–gel synthesis with hard templating via in situ transformation of the melt into solid fibrous salt crystals. The employment of MgCl2·6H2O results in tubular nitrogen doped carbons showing anisotropic porosity and very high specific surface areas up to 2780 m2 g−1 and total pore volumes up to 3.86 cm3 g−1. The formation of the tubular porosity can be connected to a cooperative effect between in situ formed, solid hydrate phases and their modulation with adenine and its polycondensation products. The combination of high SSA with the channel-like porosity generates a highly accessible structure making those carbon materials appealing for applications that demand good mass transport. The obtained materials were exemplarily employed as supercapacitor electrodes resulting in high specific capacitances up to 238 F g−1 at a low scan rate of 2 mV s−1 and up to 144 F g−1 at a high scan rate of 200 mV s−1.
Conceptual insightsThe introduction of well-developed transport pores in highly porous carbon materials is a crucial research field to maximize the performance in the final application. In the present work, it has been demonstrated that the concerted process of directed crystallization of an inorganic salt phase and condensation/carbonization of an organic phase can lead to carbon materials with anisotropic porosity in one step. Previously, such geometries could be obtained by soft templating with micelles. Because pore collapse is a serious issue here, hard templating strategies are often employed, coming with the downsides of multiple and often harsh steps. The presented process combines soft templating and hard templating with similarities to the so-called evaporation-induced self-assembly (EISA) soft templating, however leaving a hard salt template phase behind. The solid template is herein easily removed, which results in a facile and green one-pot approach. The research may pave the way to salt nanostructures becoming the hard templates of future porous carbon preparation. |
1 Introduction
Facile and inexpensive preparation of highly porous carbon materials is increasingly relevant due to their widespread applicability especially in emerging energy related fields.1–3 Among other interesting uses, porous carbons are very appealing electrode materials and catalyst supports, where surface roughness and mass-transport contribute crucially to performance.4–9 The introduction of heteroatoms, such as nitrogen, into the carbon framework can further improve their properties leading to higher performances and allowing for their employment as electrocatalytically active material itself.10–13 There are multiple approaches to synthesize porous carbon materials. Classically utilized activation methods are rather inexpensive and easy to employ. However, those methods mainly lead to microporous materials coupled with bottle-neck or erratically branched pore systems, restricting the mass transport through the materials.14,15 Furthermore, pores are isotropically distributed and do not enforce directional mass flow. Hard and soft templating methods allow the introduction of hierarchical porosity, i.e., additional meso- and/or macroporosity, coupled with a high control over the size and shape as well as orientation of the present pores.16,17 Therein the utilization of ordered hard templates, e.g., KIT-6 or SBA-15, delivers the possibility to obtain ordered mesoporous carbons possessing well defined porosity and additional features such as anisotropic pore channels.18,19 Nonetheless, those methods typically include multiple steps (including the preparation of templates) as well as the employment of harsh chemicals leading to high costs, hampering commercialization and conflicting with the aims of a sustainable synthesis.The recently introduced ionothermal approach offers the possibility to synthesize micro- and mesoporous carbon materials in a rather inexpensive and experimentally simple one-step fashion.20 In the ionothermal synthesis a molten salt phase simultaneously serves as solvent and porogen during the carbonization of a precursor at high temperatures under inert gas atmosphere. After the synthesis, the porogen can be easily removed by aqueous washing to yield the pores. Moreover, the unique solvent properties allow for a sustainable conversion of biomass derived precursors or even directly biomass such as cellulose or lignin.21–29 The salt phase classically consists largely of ZnCl2, similar to the so-called ZnCl2 activation.30,31 Mostly however, mixtures of ZnCl2 and other alkali chlorides are used to match the solvent properties with the selected precursors. Generally, the aim is to effectively swell, disperse or ideally even dissolve the precursor molecules and/or the respective derivatives in the molten salt to obtain highly dispersed carbons after carbonization. Obviously, this can be promoted by the employment of more soluble low molecular weight precursors. At very high salt to precursor ratios the process can be described as a sol–gel carbonization, leading to aerogel like carbons, although also morphologies originating from a dispersed salt phase were observed.22,23,32 The general composition of the salt mixture influences the phase separation behavior from the carbonizing precursor and thereby the obtained porosity.20 Salt mixtures far away from the eutectic composition (e.g. with very high alkali chloride amounts) also caused additional macrotemplating due to remaining solid, dispersed salt particles in the carbonizing reaction mixture.23,33 This recent finding inspired us to investigate the utilization of intermediately crystallizing salt phases as structured templates. In situ formation of templates is well-known from soft templating, e.g. the evaporation-induced self-assembly (EISA) of block co-polymers, and a similar one-pot process would extend ionothermal carbonization by another structuration mechanism operative at the nano-lengthscale, perspectively allowing for ordered nanoporous ionothermal carbon materials.34,35
Herein, instead of ZnCl2 based salt mixtures the use of hydrated CaCl2 and MgCl2 as porogens for the synthesis of hierarchical porous nitrogen doped carbons (NDCs) is examined. Adenine is chosen as small molecular precursor due to its high thermal stability, high initial nitrogen content, and pre-formed aromatic structure ideally supporting the formation of nitrogen-rich graphitic networks. Intriguingly, the employment of CaCl2·2H2O results in very interesting anisotropic morphology with tubular pores. The resulting channel-like carbon morphology is even more pronounced when MgCl2·6H2O is used as salt phase, leading to highly porous NDCs with extended oriented porosity with channels of the aspect ratio of up to ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000. The products show very high specific surface areas (SSAs) and total pore volumes (TPVs) and are obtained in a one-step procedure. The channel-like porosity generates an easily accessible structure making the carbon materials appealing for applications that are demanding good mass transport. The synthesized materials were exemplarily employed as supercapacitor electrodes showing high specific capacitance.
:1000. The products show very high specific surface areas (SSAs) and total pore volumes (TPVs) and are obtained in a one-step procedure. The channel-like porosity generates an easily accessible structure making the carbon materials appealing for applications that are demanding good mass transport. The synthesized materials were exemplarily employed as supercapacitor electrodes showing high specific capacitance.
2 Results and discussion
In fact, a number of inorganic salts show melting points only above the onset of pyrolysis/carbonization of organic species. Such salts were utilized as reaction medium with precursor injection but not for standard ionothermal salt melt carbonization.36 However, a number of such salts have hydrated forms that melt at lower temperatures and can act as ionothermal liquid reaction media as long as the presence of water is not harmful. Throughout the thermal treatment, the dehydration of the molten salt hydrate, e.g., by means of evaporation, can lead to intermediate crystallization followed by another melting event. In particular, calcium and magnesium chloride show melting points of 772 °C and 708 °C, respectively.37 CaCl2·2H2O is melting at 175 °C and by dehydration can also form the monohydrate that has a melting point at 260 °C.38 Thus, the liquid molten salt hydrates may be employed as solvents for homogeneous polymerization/carbonization of organic compounds. It has to be considered that melting and the elimination of crystal water of salt hydrates may be superimposed leading to a complex mechanistic pathway.39 This, and the additional interactions with the carbonizing phase can lead to the dynamic formation/depletion of solid salt throughout the carbonization process. A similar situation is expected for magnesium chloride. The hexahydrate MgCl2·6H2O is a promising precursor due to the low melting point (Tm = 117 °C) and its ability to melt/dehydrate in multiple steps.40 Sugimoto et al. studied the thermal decomposition of MgCl2·6H2O in detail using in situ synchrotron powder pattern data recorded in a dry helium gas stream.41 They thereby obtained different decomposition reactions of MgCl2·6H2O dehydration steps, but also the formation of Mg(OH)Cl and MgO at higher temperatures. However, the actual phase transitions/reactions will strongly depend on the chosen reaction conditions, e.g., heating rate and applied gas stream. If water cannot immediately evaporate throughout the dehydration, a hydrate melt will form instead of the occurrence of a solid phase. Important in this context is that also crystallization of lower hydrates from liquid phase can be expected and can strongly modify the ionothermal carbonization mechanism by introducing a secondary templating effect that is the in situ formation of very small crystals as hard templates.There are already few reports in literature introducing the use of alkaline earth metal chlorides such as CaCl2 and MgCl2 for preparation of biomass derived activated carbons, mainly for cost reasons.42–47 The use of in situ generated CaCl2 particles with diameters of ∼5 nm as mesopore template was shown by Huang et al. in 2011.48 They obtained a micro- and mesoporous nitrogen doped carbon with a SSA of ∼1300 m2 g−1. The possibility to obtain hierarchical NDCs with CaCl2 from biomass was shown by Liu et al.49 Although MgCl2 is discussed in context with metal chloride “activation” agents, there is a lack of detailed investigations of the obtained porosity/morphology and the porogenic mechanism.47,50,51 Valix et al. obtained a porous carbon material pyrolyzing bagasse at 500 °C which was wet-impregnated with MgCl2.47 The SSA however stayed below 200 m2 g−1. Mise et al. obtained higher SSAs up to 620 m2 at a pyrolysis temperature of 850 °C using fruit shells as precursors.51 Currently, the role assigned to CaCl2 and MgCl2 is to act as dehydrating agent enhancing the crosslinking reaction of the biomass resulting in higher yields. The mechanism of the porogenesis, however, is hardly discussed, while the commonly used term “activating agent” anticipates a destructive leaching of the carbon matrix. However, carbon etching by carbothermal reduction of possibly formed metal oxides thermodynamically requires temperatures higher than 1500 °C, clearly exceeding the applied reaction temperatures.52 On the other hand, the melting points of the single salts are also too high to act as molten salt porogens as in the case of ZnCl2-based salt mixtures. Impregnation in form of aqueous salt solutions and the thermal dehydration of biomass however give rise to quantities of water that allow formation of the respective metal hydrate melts. Hence, the molten hydrate state can explain the successful formation of internal surface area as well as porosity and is thereby interesting to examine.
2.1 Salt hydrates as ionothermal reaction mediums
Adenine is herein utilized as precursor as it showed promising features in the conventional ionothermal synthesis using ZnCl2 containing salt mixtures.23 The morphology and porosity in dependence of the used salt phase and synthesis temperature are analyzed by N2-physisorption, scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Those results in combination with the salt phase(s) present after the synthesis, as analyzed by PXRD, give further insights on the origin of the secondary templating effect. If not otherwise mentioned, the adenine to salt wt-ratio was adjusted to 1:10 and the final synthesis temperature to 900 °C. The obtained samples are specified as Ade-S-X-Y where S represents the salt phase, X represents the adenine to salt wt.-ratio (1:X) and Y the final pyrolysis temperature. Pure adenine starts to sublime at T ∼ 320 °C according to the thermogravimetric analysis (TGA) measurements (Fig. S1, ESI†). In the presence of tenfold excess of pristine CaCl2 a highly porous NDCs with high SSA of 1530 m2 g−1 and large TPV of 2.24 cm3 g−1 is obtained (Fig. S2 and Table S1, ESI†). Apparently, volatile species can be trapped by the presence of salt and undergo crosslinking, and complete evaporation of adenine, as observed in the TGA measurements, is prevented already using solid CaCl2. Like previously reported for the carbonization of glucose in the presence of LiCl/KCl mixtures, a high salt to precursor ratio ensures a small size of the crosslinked intermediates which finally leads to the formation of graphene like nitrogen doped carbon (Fig. S3a and b, ESI†).53 A clear advantage of the molten hydrated state of CaCl2 on the carbonization of adenine is however found in the carbon yields and the obtained product morphologies. Employment of CaCl2·2H2O results in a 65% increased carbon yield as compared to the use of anhydrous CaCl2 (Table S1, ESI†). The NDC synthesized in the presence of CaCl2·2H2O has a high SSA of 1700 m2 g−1 and TPV of 1.60 cm3 g−1. SEM images of the CaCl2·2H2O derived product show the presence of tubular carbon domains indicating the discussed secondary templating effect (Fig. 1a). The liquid (intermediate) state of CaCl2·2H2O supports homogeneous reaction conditions at the onset of carbonization. However, the continuous dehydration of the salt phase causes its resolidification with the carbonization reaction being pushed to the surface around the crystallizing salt. The shape and size of the in situ forming templates as well as their resulting carbon replicates is rather non-uniform, but extended hollow tube-like carbon morphologies are characteristic when comparing Ade-CaCl2·2H2O-10-900 to Ade-CaCl2-10-900 (Fig. S3c and d, ESI†).
 | ||
| Fig. 1 SEM images of (a) Ade-CaCl2·2H2O-10-900, (b) Ade-Mg-5-900, and (c) Ade-Mg-10-900 depicting the secondary templating effect. | ||
The reaction medium was then changed to MgCl2·6H2O (shortened to Mg in the sample name), and two different adenine-to-salt ratios were studied (1:5, and 1:10). A moderate salt-to-precursor ratio of 5:1 (Ade-Mg-5-900) again reveals the presence of channel like linear pores (Fig. 1b). The porogenesis is likely to be linked to the aimed secondary templating effect as previously observed for CaCl2·2H2O. The material shows a very high SSA of 2780 m2 g−1, a N-content of 2.3 wt% and a large TPV of 2.83 cm3 g−1 (Fig. S4 and Table S2, ESI†). Adenine and its oligomeric decomposition products can mix well with the initially liquid MgCl2·6H2O salt phase, but apparently also act as a high temperature “surfactant” which prevents the formation of large salt domains leading to a high interface between the carbonizing precursor and the salt. The obtained carbon yields of 25.7 wt% for Ade-CaCl2·2H2O-10-900 and 25.6% for Ade-Mg-5-900 are low compared to other ionothermal pathways (Table S2, ESI†), but can be related to the high nitrogen content on the monomer and the coupled mass loss throughout denitrification. Interestingly, reduction of the relative salt amount (Ade-Mg-1-900) results in an even lower carbon yield (17.0%) implying that the almost threefold increased SSA of Ade-Mg-5-900 is indeed caused by a templating effect and not by a promoted carbon or nitrogen leaching (activation) mechanism (Fig. S4, ESI†). The statement is confirmed by the very similar carbon yield (26.4%) of the sample obtained with highest investigated salt to precursor ratio of 10:1. The large excess of salt in the case of Ade-Mg-10-900 causes enhanced separation of the carbon domains resulting in a very large TPV of 3.60 cm3 g−1 and an only slightly decreased, still very high SSA of 2480 m2 g−1 (Fig. S4, ESI†). These findings are in line with the typically trends observed for salt templating in ZnCl2-containing salt melts.32 It is to mention that the TPV of Ade-Mg-10-900 exceeds the one of Ade-CaCl2·2H2O-10-900 and commercial activated carbons by a factor of 2 and 3, respectively. Intriguingly, the formation of anisotropic pores by in situ template transformation is even more pronounced for Ade-Mg-10-900, resulting in the very interesting morphological features depicted in Fig. 1c. The majority of the NDC sample is made up of parallel aligned tubular carbon domains. Moreover, the tubular domains already exist in the unwashed material (Fig. S5, ESI†). The tubular channels of the washed material show a width of ∼50 nm and lengths of up to ∼4 μm leading to an aspect ratio of ∼1:80 (Fig. 2c and d). Hence, the method allows for the creation of 1-dimensional transport tubes with diameters in the range of large mesopores and/or small macropores. This structure is supported by the high N2-uptake at high pressures (Fig. S4a, ESI†). Furthermore, the corresponding pore size distributions (PSD) of Ade-Mg-10-900 confirms the existence of pores in the range of 50 nm (Fig. S6, ESI†). However, no clear maximum of the PSD is visible at 50 nm indicating that larger transport pores also exist. It has to be mentioned that the tubular structure is not always perfectly aligned, as depicted in Fig. S7 (ESI†). Nevertheless, this self-organized templating scheme is interesting, and many parameters such as the size and shape uniformity of the precursor salt particles are left to be optimized. The very high SSA points to an additional high internal microporosity of the materials. The material prepared with the highest salt content combines very high SSA, high TPV with an anisotropic open pore structure, which should allow a high accessibility of the microporous surface sites.
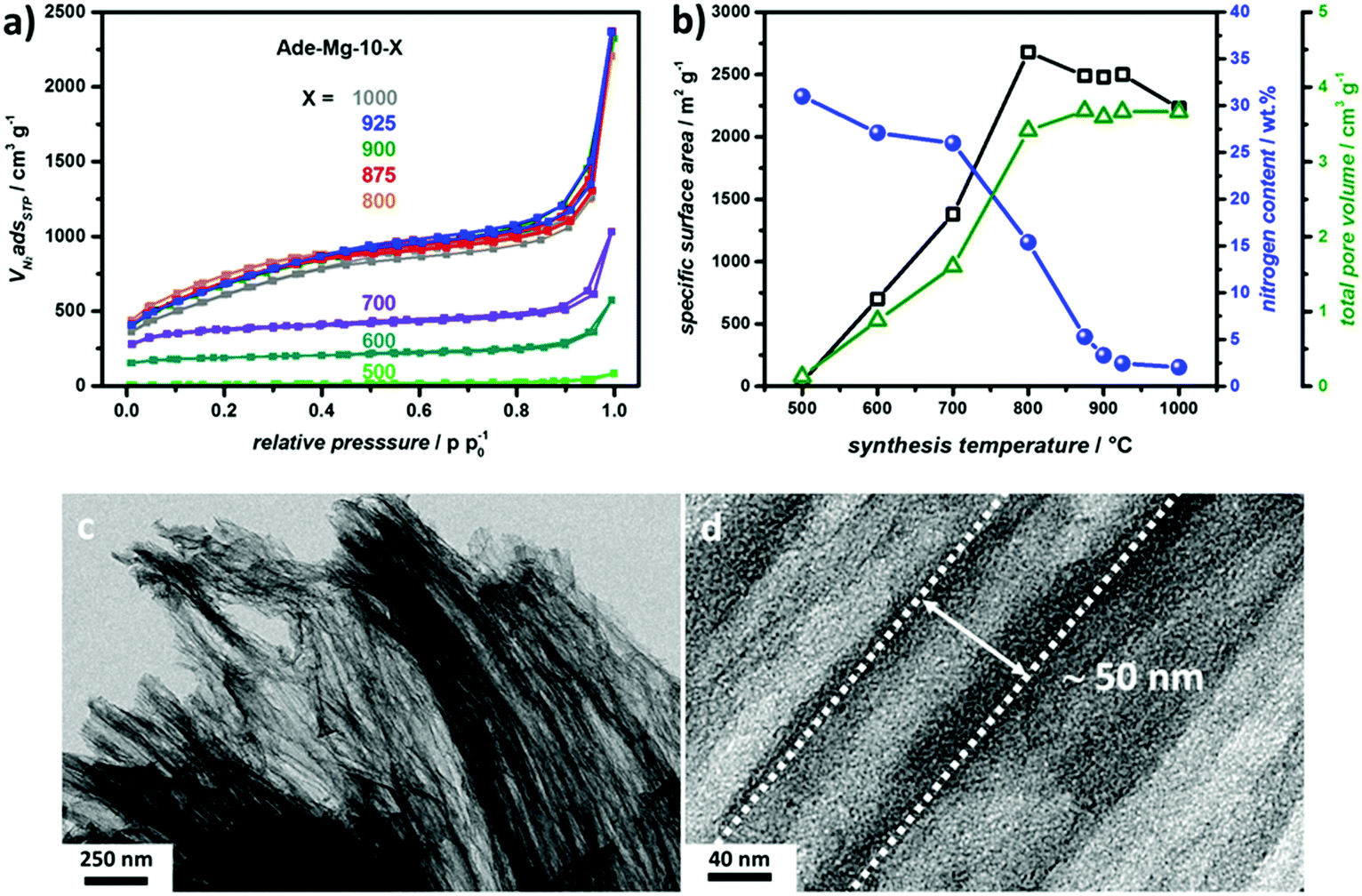 | ||
| Fig. 2 (a) Nitrogen physisorption isotherms of the Ade-Mg-10 samples prepared at different synthesis temperatures. (b) SSA, N-content, and TPV of the different Ade-Mg-10-X samples with respect to the synthesis temperature applied. (c and d) Representative TEM images of Ade-Mg-10-900 depicting its aligned tubular pore structure. | ||
2.2 Investigation of the porogenesis
To investigate the formation of the product morphology, different Ade-Mg-10 samples were synthesized at temperatures between 500 and 1000 °C. Ade-Mg-10-500 shows only negligible SSA. Increasing synthesis temperatures lead to an almost linear increase of the SSA which reaches a maximum value of 2680 m2 g−1 at 800 °C. Between temperatures of 800 °C and 1000 °C only a small decrease in SSA is observed and the N2-physisorption isotherms are very similar (Fig. 2a and Table S3, ESI†). Consequently, those samples can be considered very alike in porosity as also seen in the corresponding PSDs (Fig. S8, ESI†). Moreover, the tubular pore domains are present in all samples (Fig. S9, ESI†). The TPV shows a very similar trend to the SSA pointing to an associated formation mechanism. The TPV is increasing from 0.13 to 1.60 cm3 g−1 between 500 and 700 °C. At 800 °C there is a step increase to more than double the TPV resulting in a high value of 3.42 cm3 g−1 before it almost stagnates at ∼3.60 cm3 g−1 in between 800 and 1000 °C. Representative TEM images of Ade-Mg-10-900 are depicted in Fig. 2c and d. The carbon yield stays almost constant during the whole set of experiments indicating that the high porosity is also not related to carbon etching (Table S3, ESI†). Instead, the gain in SSA and TPV above 700 °C is accompanied by a large loss in the nitrogen content. The very high N-content of 26.0% at 700 °C means nitrogen provides two out of seven atoms of the carbonizing organic phase. At lower synthesis temperatures, most N-sites are present as edge sites (e.g. pyrrolic or pyridinic N) that are terminating the carbon framework, as revealed by X-ray photon spectroscopy (XPS) measurements (Fig. S10 and S11, ESI†). Hence the chemical composition of Ade-Mg-10-700 restricts the crosslinking, just like high heteroatom contents restrict graphitizability.54 At 800 °C, only every sixth atom is a nitrogen atom, including graphitic N-sites, which allows for extended carbon structures. Apparently, the strongly decreasing N-content between 700 and 800 °C, is connected to a largely enhanced crosslinking of the precursor phase, which is preventing pore collapse throughout the aqueous work-up. This is further reflected by the higher N2-uptake at low pressures as well as an increased uptake in the high pressure region (0.85 < p/p0−1 < 1.00) indicating increased microporosity and larger amount of large tubular pores, respectively. A moderate decrease in SSA occurs between 800 and 900 °C which is accompanied by a further distinct loss in nitrogen content. We ascribe this to the common reorganizational dynamics at these temperatures that lead to closure of micropores.55The identification of the secondary templating phase is very difficult, because the underlying decomposition of MgCl2·6H2O is a multistep process leading to a complex self-organization effect. We carried out PXRD measurements on unwashed samples (indicated by the suffix “as-prepared”) prepared at temperatures down to 300 °C to relate salt phase transformation to the morphology generation (Fig. S12b, ESI†). Our observations do not fully reflect the previous results by Sugimoto et al. pointing to a large influence of the reaction conditions on the resulting salt transformations.41 In absence of any organic phase, we detect a mixture of Mg(OH)Cl and different MgCl2 hydrates up to 400 °C, while at higher temperatures MgCl2 and increasingly MgO are the dominant phases. In presence of the organic phase the first interesting observation is the occurrence of the ammonium salt MgCl2NH4Cl·6H2O at 300 °C that indicates reaction of adenine with the magnesium salt. Moreover, the evolution of NH3 at relatively low temperatures points to a catalytic effect of the salt on the polymerization of the adenine molecules. Else, we also have a mixture of Mg(OH)Cl and MgCl2 hydrates, while the overall water content is higher as compared to the bare inorganic samples. In contrast to the purely inorganic samples, the hydrated salts also remain at temperatures above 400 °C and co-exist with the upcoming MgCl2 and MgO phases. Although the organic phase is apparently stabilizing the hydrates, the water content is decreasing with increasing temperatures. In contrast to the pure salts the dehydration is less connected to the formation of MgO, so that MgCl2 is becoming the dominant phase. For Ade-Mg-10-700_as-prepared no hydrates are remaining and water-free MgCl2 is the main phase over MgO (Fig. S13, ESI†).
Jung et al. explained porosity formation in their carbon fibers by thermal transformation of MgCl2 into MgO, which was acting as the hard template.56 Huang et al. obtained a fiber-like Mg(OH)Cl structure by the decomposition of MgCl2HH.57 The characteristic fibrous morphology for the Ade-Mg-10-X samples is observed for all samples above 500 °C, where both MgCl2 hydrates and MgO are detected (Fig. S12a, ESI†). However, the fibrous structure is not observed in any of the purely inorganic samples. It can be concluded that the interaction of the organic phase is crucial to obtain the fibrous structure. Intriguingly, it was shown in literature that adenine can form a crystalline phase with MgCl2·6H2O.58 In fact, first fibrous structures are already observed by SEM for Ade-Mg-10-400, where MgO is not yet formed (Fig. S12a, ESI†). Energy dispersive X-ray (EDX) mapping on Ade-Mg-10-700_as-prepared reveals that the tubular structure indeed contains magnesium, oxygen and chlorine (Fig. S14, ESI†). Besides the tubular structures a spherical morphology was observed that was only composed of MgO. Generally, the N-signal is homogeneously distributed throughout the material, indicating that both MgCl2 and MgO may act as templates; however, the tubular porosity is arising from the interaction of the organic phase with intermediately transforming MgCl2 hydrates.
Based on the results of the present work and the detailed investigation of Sugimoto et al. concerning the thermal decomposition of pristine MgCl2·6H2O, we come up with a more handy, however still speculative, model for the tubular porogenesis via the described secondary templating effect in Fig. 3.41 The interaction of adenine with MgCl2 hydrates can be explained as a surface coordination via hydrogen bonding that leads to the stabilization of certain crystal facets and therefore enforcing unidirectional crystal growth. With increasing temperature the organic phase is solidifying by means of carbonization whereas delayed dehydration/decomposition of the salt is occurring. A sufficiently crosslinked organic/carbonizing phase, enclosing the salt fiber during MgCl2·XH2O decomposition, preserves the one-dimensional shape of the intermediate salt phase, finally leading to highly porous NDC materials possessing tubular aligned pores. The overall process is comparable to the directed growth of inorganic crystals based on face selective adsorption of hydrophilic block copolymers.59 Moreover, the final higher arrangement of the precursor and salt phase may be similar to the cooperative formation of the tubular morphology of the well-known SBA-15.60
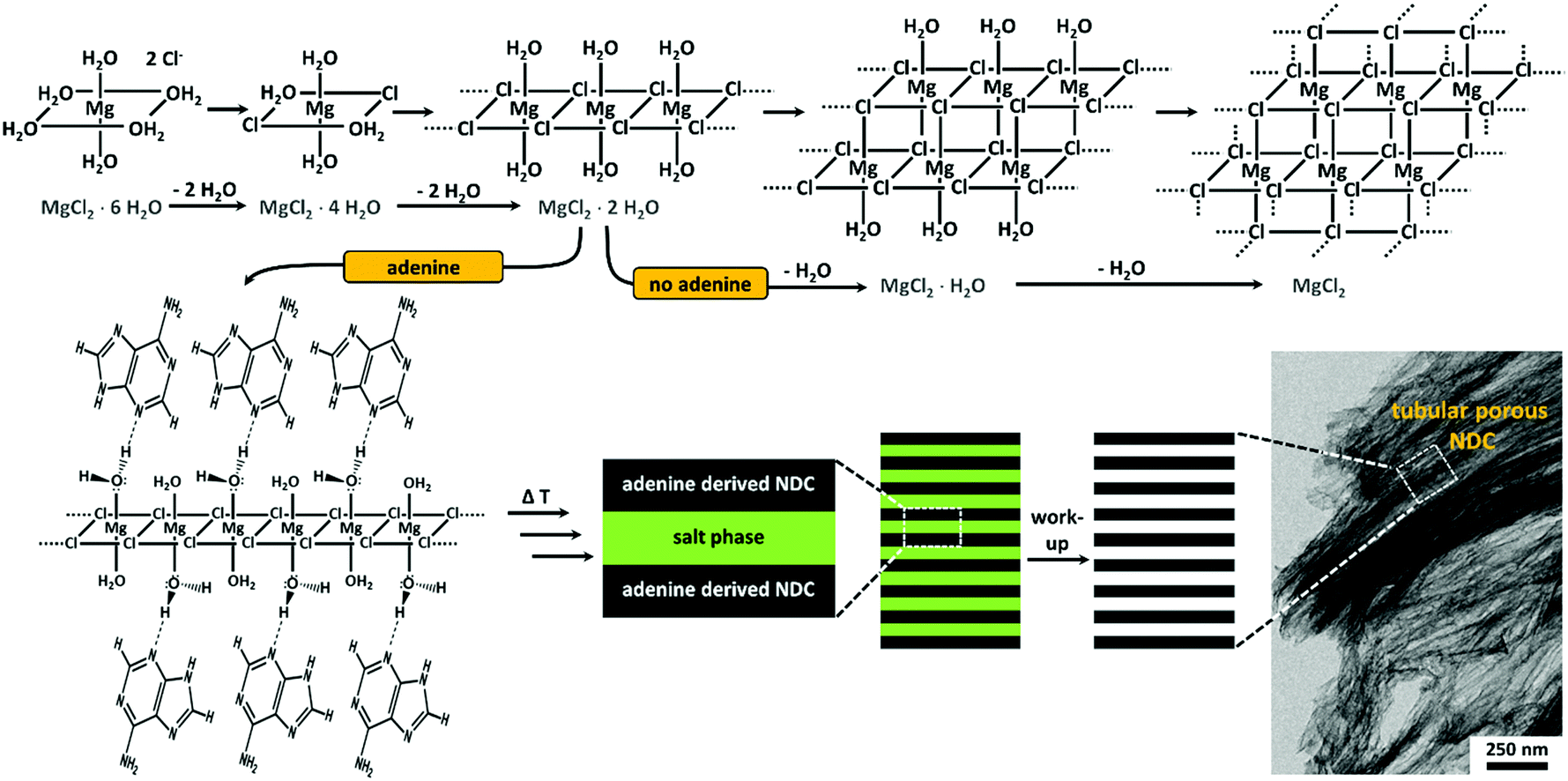 | ||
| Fig. 3 Schematic illustration of the speculative model for the cooperative secondary templating during the synthesis of adenine derived NDCs obtained with MgCl2·6H2O as reaction medium. The reactions occurring in the absence of adenine are adapted from ref. 41. | ||
As mentioned before, it should be possible to improve the degree of “structuring” (i.e., appearance and length of the parallel aligned tubular pores) by further adjustment of the synthesis conditions. This hypothesis was supported by the synthesis of an additional Ade-Mg-10-900 sample obtained with a slightly modified procedure. On the base of the described kinetic formation model, an additional dwelling step at 400 °C for 10 h was introduced. The resulting material possessed NDC-domains (particles) with aligned tubular pores, here in the range of up to 60 μm, corresponding to a width-length aspect ratio larger than 1:1000 (Fig. S15, ESI†).
2.3 Supercapacitor performance of the tubular porous NDCs
All the tubular porous NDCs synthesized in a 1:10 precursor to salt ratio at temperatures higher than 800 °C combine a highly accessible structure with large SSA, making those materials highly appealing for the employment as supercapacitor (SC) electrodes. The favorable transport pores should especially support the performance at fast charge and discharge rates, while the presence of nitrogen in the carbon framework enhances the performance due to pseudocapacitive effects.12 The SC performance of Ade-Mg-10-800, Ade-Mg-10-875, Ade-Mg-10-900 and Ade-Mg-10-1000 was analyzed by means of cyclic voltammetry (CV) measurements in a symmetrical two electrode set-up employing 1 M sulfuric acid and PVDF as electrolyte and as binder, respectively.
At the lowest scan rate investigated (2 mV s−1), the sample synthesized at 800 °C delivers the highest gravimetric capacitance of 238 F g−1 (Fig. 4a). Regarding the same scan rate, higher synthesis temperatures lead to a drop in capacitance which can be connected to the decreasing nitrogen content resulting in smaller pseudocapacitive effects. Next to the absolute nitrogen content, the different type of N-sites lead to varied intensities of the pseudocapacitive effect. Lower synthesis temperatures mainly lead to pyridinic and pyrrolic nitrogen which can be protonated and deprotonated increasing the capacitance.61–64 Contrarily, quaternary nitrogen, representing the most temperature-stable site, should have a minor influence on the pseudocapacitance but enhances the electron transport through the carbon material supporting improved capacitance retention at faster charge/discharge rates.62,64 Indeed, XPS measurements reveal a very high relative amount of pyridinic (42%) and pyrrolic (38%) N-sites for Ade-Mg-10-800 whereas at synthesis temperature higher than 900 °C graphitic N is the dominant species (Fig. S10 and S11, ESI†). Correspondingly, the performance of Ade-Mg-10-800 quickly drops with increased scan rates delivering a relative low capacitance of 115 F g−1 at 200 mV s−1. All other samples generally show only slight capacitance fading for scan rates higher than 50 mV s−1 and reveal overall similar drops in capacitance. At the highest scan rate, the largest capacitance is obtained for the sample synthesized at 875 °C delivering a value of 144 F g−1. The influence of an improved conductivity is further clarified by the CVs depicted in Fig. 4b. At a medium scan rate of 50 mV s−1, both materials, Ade-Mg-10-800 and Ade-Mg-10-875, show a rectangular-like shape. The overall area of the sample synthesized at 800 °C is slightly larger due to pseudocapacitance related to a larger nitrogen content. Ade-Mg-10-800 clearly loses its rectangular shape when the rate is increased to 200 mV s−1 indicating a lower electric conductivity of the material, while the shape of the CV stays almost unchanged for Ade-Mg-10-875 during the same increase in scan rate. This indeed is typical for the combination of a high electronic conductivity with a high accessibility of the surface.
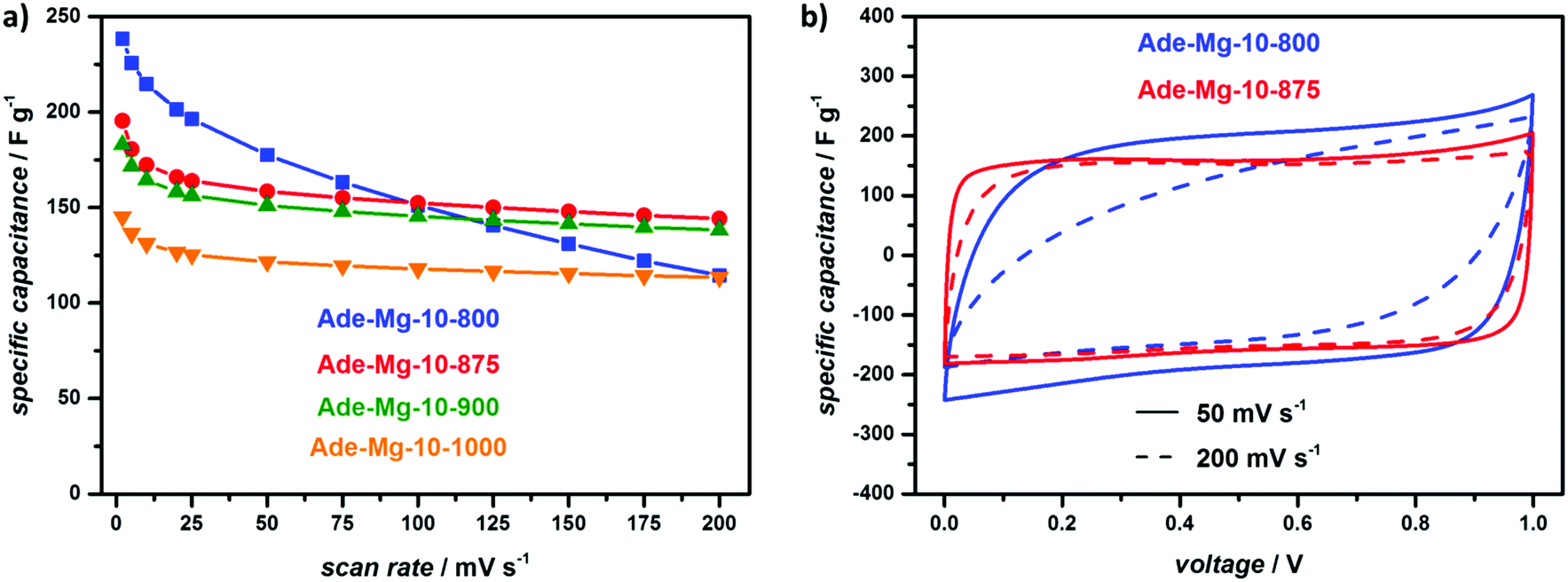 | ||
| Fig. 4 (a) Specific capacitance of the adenine derived NDCs synthesized with MgCl2·6H2O at different temperatures (all samples were synthesized in a precursor to salt wt.-ratio of 1:10) obtained by CV measurements in a two electrode set-up with varied scan rate in 1 M sulfuric acid; (b) corresponding CVs of Ade-Mg-10-800 and Ade-Mg-10-875 obtained with a scan rate of 50 mV s−1 and 200 mV s−1. | ||
In order to study the cycling stability Ade-Mg-10-900 was exemplarily investigated revealing a very high cycle life (Fig. S16, ESI†). After an initial loss of 5.2% over the first 100 cycles, the capacitance was very stable further decreasing only by 0.7% over the following 900 cycles, which leads to a final capacitance retention of 94.1% after 1000 cycles.
In future studies, the synthesis temperature (between 800 and 900 °C) should be optimized in more detail to determine the optimal nitrogen content for the developed material, which can be understood as a trade-off between pseudocapacitance leading to high energy densities and electronic conductivity allowing high power applications.
3 Conclusions
In conclusion, the present work introduced the in situ liquid-to-hard template transformation for the synthesis of tubular porous NDC-materials. CaCl2·2H2O and MgCl2·6H2O were shown to be useful salts for the ionothermal carbonization, in some regards generalizing the concept that was previously elaborated using ZnCl2 based salt mixtures. Herein the additional dehydration induced crystallization of CaCl2 and MgCl2 is a central curiosity that accounts for new perspectives of nanostructuration. The overall process be regarded as a combination of liquid templating by means of sol–gel synthesis with the in situ formation of solid, well-structured porogens. The employment of adenine as a biomass derived precursor and MgCl2·6H2O in a tenfold access results in a tubular NDC material showing very promising porosity characteristics with SSAs and TPVs up to 2780 m2 g−1 and 3.86 cm3 g−1, respectively. The investigation of the formation mechanism of the tubular pore structure points to a cooperative effect between in situ formed, solid hydrate phases and their modulation with adenine and its polycondensation products by means of hydrogen bonding. The high SSA, TPV, and tubular porosity of the adenine derived materials point to the material's suitability for electrochemical applications and catalysis or in topical applications such as lithium sulfur batteries and capacitive desalination. The synthesized materials were exemplarily employed as supercapacitor electrodes, nicely revealing the relation between nitrogen content and final performance. The material with high nitrogen content delivered a high energy density due to pseudocapacitive effects whereas a lower nitrogen content coupled with a higher synthesis temperature allows high power applications due to an improved electronic conductivity.4 Experimental part
4.1 Synthesis
In a typical experiment, 800 mg of adenine (≥99%, Sigma Aldrich) were thoroughly grinded (in a glove box) with the respective mass of CaCl2 (96%, Acros Organics), CaCl2·2H2O (99.5%, AppliChem) or MgCl2·6H2O (98+%, Acros Organics) to achieve the aimed precursor to salt wt.-ratio. The resulting mixture was transferred into a ceramic crucible (50 ml; VWR) and heat treated under nitrogen atmosphere. The sample was ramped with 2.5 K min−1 to the aimed synthesis temperature and the final dwelling was conducted for 1 h at the respective temperature. After cooling to RT, the mostly black monolithic structure was carefully grinded, washed twice with 1 L of deionized water and dried in vacuum at 60 °C overnight. The samples synthesized with MgCl2·6H2O were additionally washed in 1 L of 2 M HCl and neutralized again with deionized water before drying. The reference samples, just containing salt, were prepared in the same fashion, however, without any washing steps.4.2 Characterization
The samples were analyzed by scanning electron microscopy (SEM, Zeiss Gemini 1550) and transmission electron microscopy (TEM, Zeiss 912 Omega). Thermogravimetric analysis (TGA) of adenine was conducted with a Netzsch TG 209 F1 device under constant N2-flow applying a heating rate of 10 K min−1. N2-Physisorption measurements were performed at a Quantachrome Quadrasorb SI porosimeter. The SSAs were determined according to the multipoint Brunauer–Emmett–Teller (BET) model using the data points suggested by the micropore BET assistant of the software (however, not exceeding the range between 0.05 and 0.30 pp0−1). The TPV was calculated at pp0−1 = 0.99. Pore size distributions were calculated from the adsorption branch employing the quenched solid density functional theory model for slit, spherical and cylindrical pores provided by Quantachrome data reduction software QuadraWin Version 5.11. Elemental analysis was obtained by combustion analysis on a Vario Micro device. A Bruker D8 Advance device was used for powder X-ray diffraction (PXRD) measurements between 3.0° and 70.0° 2θ using a step of 0.05° and a measurement time of 4 s per step under Cu-Kα radiation (λ = 1.5418 Å). X-ray photon spectroscopy (XPS) was conducted with a PHI 5000 VersaProbe X-ray photoelectron spectrometer (Ulvac-PHI, Japan) using Al Kα X-ray source (1486.6 eV) and the carbon peak as internal reference. The N1 s spectra were used for the quantification of the different N sites.4.3 Electrochemical Analysis
The electrochemical characterization of the materials was carried out in a two electrode Swagelok-cell and recorded on a Gamry Ref3000 potentiostate. Two electrodes were prepared for one measurement in the same fabrication procedure. Accordingly, a freshly prepared and sonicated suspension of 90 wt% nitrogen doped carbon and 10 wt% polyvinylidenefluoride (PVDF) in 300 μL acetone were dropcasted on a platinum disc (1 cm diameter) to achieve a mass loading of approximately 2 mg cm−2 for one electrode. The electrode was first dried in air for 30 min and subsequently in an oven at 60 °C for 60 min. The supercapacitors were assembled with two electrodes of the same material as cathode and anode, respectively. A filter paper served as separator and 40 μL of 1 M sulfuric acid as electrolyte. The cyclovoltammetry tests were conducted with varied scan rates in a voltage window of 1 V. Stability measurements were performed with a constant current of 10 A g−1 employing the same potential range.The specific capacitances (Csp) of the investigated samples were calculated from the recorded cyclovoltammograms (CVs) according to eqn (1).
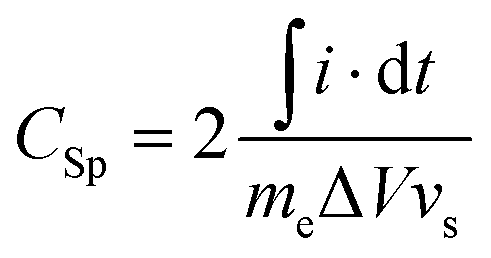 | (1) |
Acknowledgements
We thank the MPI technical staff for performing various service measurements and the Chez-Briel-Institution for the substantial support. JP especially acknowledges Mackenzie Field and the DAAD-RISE program for practical and financial support, respectively. Moreover, AM thanks the German Federal Ministry of Education and Research (BMBF) for financial support via the Green Talents Award. 7th framework project EUROLIS co-financed the research under grant agreement No. 314515. Open Access funding provided by the Max Planck Society.References
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- S. Subramoney, Adv. Mater., 1998, 10, 1157–1171 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Energy Mater., 2011, 1, 356–361 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- L. Dai, D. W. Chang, J.-B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- J. Deng , T. Xiong , H. Wang , A. Zheng and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3750–3756 Search PubMed.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- K. Y. Kang, B. I. Lee and J. S. Lee, Carbon, 2009, 47, 1171–1180 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., 2010, 122, 2619–2623 CrossRef.
- K. Kaneko, J. Membr. Sci., 1994, 96, 59–89 CrossRef CAS.
- T. Kyotani, Carbon, 2000, 38, 269–286 CrossRef CAS.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC.
- F. Schüth, Angew. Chem., Int. Ed., 2003, 42, 3604–3622 CrossRef PubMed.
- M. Oschatz, E. Kockrick, M. Rose, L. Borchardt, N. Klein, I. Senkovska, T. Freudenberg, Y. Korenblit, G. Yushin and S. Kaskel, Carbon, 2010, 48, 3987–3992 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- N. Fechler, T.-P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS PubMed.
- M. Graglia, J. Pampel, T. Hantke, T. P. Fellinger and D. Esposito, ACS Nano, 2016, 10, 4364–4371 CrossRef CAS PubMed.
- F. Schipper, A. Vizintin, J. Ren, R. Dominko and T. P. Fellinger, ChemSusChem, 2015, 8, 3077–3083 CrossRef CAS PubMed.
- J. Pampel and T.-P. Fellinger, Adv. Energy Mater., 2016, 6, 1502389 CrossRef.
- X. Deng, B. Zhao, L. Zhu and Z. Shao, Carbon, 2015, 93, 48–58 CrossRef CAS.
- G. Ma, Z. Zhang, K. Sun, H. Peng, Q. Yang, F. Ran and Z. Lei, RSC Adv., 2015, 5, 107707 RSC.
- Z. Ma, H. Zhang, Z. Yang, G. Ji, B. Yu, X. Liu and Z. Liu, Green Chem., 2016, 18, 1976–1982 RSC.
- Z. Ma, H. Zhang, Z. Yang, Y. Zhang, B. Yu and Z. Liu, J. Mater. Chem. A, 2014, 2, 19324–19329 CAS.
- L. Roldán, Y. Marco and E. García-Bordejé, Microporous Mesoporous Mater, 2016, 222, 55–62 CrossRef.
- H. Yuan, L. Deng, X. Cai, S. Zhou, Y. Chen and Y. Yuan, RSC Adv., 2015, 5, 56121–56129 RSC.
- F. Caturla, M. Molina-Sabio and F. Rodríguez-Reinoso, Carbon, 1991, 29, 999–1007 CrossRef CAS.
- A. Ahmadpour and D. D. Do, Carbon, 1996, 34, 471–479 CrossRef CAS.
- K. Elumeeva, N. Fechler, T. P. Fellinger and M. Antonietti, Mater. Horiz., 2014, 1, 588–594 RSC.
- J. Pampel, C. Denton and T.-P. Fellinger, Carbon, 2016, 107, 288–296 CrossRef CAS.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- Y. Chang, M. Antonietti and T. P. Fellinger, Angew. Chem., Int. Ed., 2015, 54, 5507–5512 CrossRef CAS PubMed.
- E. Riedel and C. Janiak, Anorganische Chemie, De Gruyter, 2011 Search PubMed.
- A. Lannung, Z. Anorg. Allg. Chem., 1936, 228, 1–18 CrossRef CAS.
- V. M. van Essen, J. Cot Gores, L. P. J. Bleijendaal, H. A. Zondag, R. Schuitema, M. Bakker and W. G. J. van Helden, ASME 2009 3rd International Conference on Energy Sustainability, 2009, 2, 825–830.
- E. Wiberg and N. Wiberg, Inorg. Chem. Commun., Academic Press, 2001 Search PubMed.
- K. Sugimoto, R. E. Dinnebier and J. C. Hanson, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 235–242 CAS.
- V. da Silva Lacerda, J. B. López-Sotelo, A. Correa-Guimarães, S. Hernández-Navarro, M. Sánchez-Báscones, L. M. Navas-Gracia, P. Martín-Ramos and J. Martín-Gil, J. Environ. Manage., 2015, 155, 67–76 CrossRef PubMed.
- J.-W. Kim, M.-H. Sohn, D.-S. Kim, S.-M. Sohn and Y.-S. Kwon, J. Hazard. Mater., 2001, 85, 301–315 CrossRef CAS PubMed.
- J. Sahira, A. Mandira, P. B. Prasad and P. R. Ram, Res. J. Chem. Sci., 2013, 3, 19–24 Search PubMed.
- S. Karthikeyan, P. Sivakumar and P. N. Palanisamy, E-J. Chem., 2008, 5, 409–426 CrossRef CAS.
- R. H. Gumus and I. Okpeku, Adv. Chem. Eng. Sci., 2015, 5, 51–61 CrossRef CAS.
- M. Valix, W. H. Cheung and G. McKay, Langmuir, 2006, 22, 4574–4582 CrossRef CAS PubMed.
- Y. a. Huang, F. Yang, Z. Xu and J. Shen, J. Colloid Interface Sci., 2011, 363, 193–198 CrossRef CAS PubMed.
- J. Liu, Y. Deng, X. Li and L. Wang, ACS Sustainable Chem. Eng., 2016, 4, 177–187 CrossRef CAS.
- R. C. Bansal, J. B. Donnet and F. Stoeckli, Active Carbon, Taylor & Francis, 1988 Search PubMed.
- S. R. Mise, J. IPHE, 2008, 4, 5 Search PubMed.
- H. J. T. Ellingham, J. Soc. Chem. Ind., 1944, 63, 125–160 CrossRef CAS.
- X. Liu, C. Giordano and M. Antonietti, Small, 2014, 10, 193–200 CrossRef CAS PubMed.
- A. Oberlin and M. Oberlin, J. Microsc., 1983, 132, 353–363 CrossRef CAS.
- E. R. Buiel, A. E. George and J. R. Dahn, Carbon, 1999, 37, 1399–1407 CrossRef CAS.
- M.-J. Jung, S. Im Ji, E. Jeong, H. Jin and Y.-S. Lee, Carbon Lett., 2009, 10, 217–220 CrossRef.
- Q. Huang, G. Lu, J. Wang and J. Yu, J. Anal. Appl. Pyrolysis, 2011, 91, 159–164 CrossRef CAS.
- T. F. Mastropietro, D. Armentano, N. Marino and G. De Munno, Polyhedron, 2007, 26, 4945–4954 CrossRef CAS.
- S.-H. Yu, H. Colfen, K. Tauer and M. Antonietti, Nat. Mater., 2005, 4, 51–55 CrossRef CAS PubMed.
- Q. Huo, D. I. Margolese, U. Ciesla, D. G. Demuth, P. Feng, T. E. Gier, P. Sieger, A. Firouzi and B. F. Chmelka, Chem. Mater., 1994, 6, 1176–1191 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- Y.-H. Lee, K.-H. Chang and C.-C. Hu, J. Power Sources, 2013, 227, 300–308 CrossRef CAS.
- X. Liu, Y. Zhou, W. Zhou, L. Li, S. Huang and S. Chen, Nanoscale, 2015, 7, 6136–6142 RSC.
- R. Arrigo, M. Havecker, R. Schlogl and D. S. Su, Chem. Commun., 2008, 4891–4893 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mh00592f |
| This journal is © The Royal Society of Chemistry 2017 |