
Advancements in rationally designed PGM-free fuel cell catalysts derived from metal–organic frameworks
Heather M.
Barkholtz
ab and
Di-Jia
Liu
 *a
*a
aArgonne National Laboratory, Chemical Sciences and Engineering, 9700 Cass Ave., Lemont, IL 60439, USA. E-mail: djliu@anl.gov
bSandia National Laboratory, Energy Storage Technology and Systems Group, PO box 5800, Albuquerque, NM 87158, USA
First published on 14th November 2016
Abstract
Over the past several years, metal–organic framework (MOF)-derived platinum group metal free (PGM-free) electrocatalysts have gained considerable attention due to their high efficiency and low cost as potential replacement for platinum in catalyzing oxygen reduction reaction (ORR). In this review, we summarize the recent advancements in design, synthesis and characterization of MOF-derived ORR catalysts and their performances in acidic and alkaline media. We also discuss the key challenges such as durability and activity enhancement critical in moving forward this emerging electrocatalyst science.
 Heather M. Barkholtz | Dr Heather Barkholtz is currently a postdoctoral appointee at Sandia National Laboratories. She received her PhD in Chemistry from Northern Illinois University in 2016. During her graduate study, she worked on the synthesis and characterization of nanoalloys and non-precious metal catalysts for energy and environmental applications, including MOF-derived PGM-free catalyst for PEM fuel cell. Her work was recognized by the first place poster presentation award at The Electrochemical Society annual meeting in 2015. Her current research interests lie in the safety and reliability of lithium-ion batteries for grid-level energy storage systems. |
 Di-Jia Liu | Dr Di-Jia Liu is a Senior Chemist at the Chemical Sciences and Engineering Division of Argonne National Laboratory. He received his BSc degree in Chemistry from Peking University (China) in 1982, PhD degree in Physical Chemistry from The University of Chicago in 1987 and postdoctoral training at The University of California at Berkeley from 1988 to 1990. His current research interests include rationally designed materials for the applications in fuel cell, battery, hydrogen storage, catalysis, and their structural investigation using synchrotron X-ray characterization techniques. He has over 100 publications and 21 granted US patents. |
Introduction
Driven by increasing demand for clean energy and concerns over global climate change, researchers worldwide have been actively exploring new sources of sustainable energy and efficient energy conversion technologies.1 Fuel cells, which electrochemically convert energy to electricity, are considered highly desirable technology solutions as they are environmentally benign, afford high energy and power density, and have greater fuel utilization efficiencies for both renewable and fossil fuels than traditional combustion engines.2Proton exchange membrane fuel cells (PEMFCs) directly convert the chemical energy in hydrogen and oxygen into electrical energy through the hydrogen oxidation reaction (HOR) at the anode and the oxygen reduction reaction (ORR) at the cathode. To date, platinum group metals (PGMs) are the materials of choice for making catalysts for HOR and ORR in PEMFCs. The high cost and limited reserve of PGMs, however, significantly hinder the broad-scale commercialization of PEMFCs. Finding low-cost, earthly abundant materials as replacements for PGMs in fuel cell catalysts has been a perpetual goal in the electrocatalyst material community. Among many different approaches, transition metals (TM, typically Fe or Co) and nitrogen doped materials have gained a lot of traction in recent years. These materials are generally derived from the high temperature treatment of mixtures of transition metal sources and nitrogen precursors. Since carbon is a high surface area support with excellent conductivity and can be fabricated in different engineered forms.3 The TM and N precursors are often applied over carbon supports to form TM/N/C composites after heat treatment. Many review articles have been devoted to the topic of Fe/N/C and Co/N/C ORR electrocatalysts.4–12 Furthermore, heteroatom-doped carbons without TM doping were also found active toward the ORR and have been extensively reviewed.13–18
Since the initial discovery of ORR activity on cobalt phthalocyanine by Jasinski over fifty years ago,19 macromolecules containing Co–N4 and Fe–N4 ligation with square-planar structures, such as phthalocyanine and porphyrin, were often used as the precursors for preparing PGM-free ORR catalysts based on perceived similarity in the active site structure to that in biological systems.6,20–23 These macromolecules, however, offer little surface area to self-expose themselves; therefore needing to be dispersed over high surface area conductive supports such as amorphous carbon. It was further discovered that the ORR activity of transition metal–carbon composites could be improved via pyrolysis in inert atmosphere24 prompting many reports of ORR activity from TM/N/C materials derived from organometallic compounds on a carbon support.24–41 These carbon supported TM/N/C materials were still plagued by limited ORR activity due to the relatively low turn-over frequency (TOF) compared to PGM catalysts.42,43 The carbon support, by itself, is catalytically inert, effectively diluting the TM–Nx active site density. In a fuel cell application, an electrode containing the catalyst of lower active site density cannot be compensated for by using more catalyst since it will lead to a thicker electrode layer with poor mass and charge transports.44 It is therefore desirable to prepare a TM/N/C material with a high density of evenly distributed active sites throughout the entire catalyst without any inert support. One solution is to use a self-support precursor which already contains transition metal, nitrogen, and carbon sources. Since there is no a priori requirement that the TM–N ligation in the precursor has to be coordinated in the planar structure before heat-treatment, there is a wide variety of N-ligated complexes to be explored.
Metal–organic frameworks (MOFs) are a relatively new class of porous crystalline materials containing repeating units of metal centers coordinated to electron donating organic ligands.45–49 A wide range of MOFs can be synthesized through judiciously selected transition metals and organic ligands, rendering them amenable to rational design at a molecular level.50,51 Additionally, MOFs can be designed with high surface area and well-defined channel/pore structures. All of these characteristics make them exciting systems for preparing TM/N/C catalysts for ORR. Pyrolysis of rationally designed MOF precursors can yield TM/N/C catalysts in which the transition metal–nitrogen coordination active sites are uniformly distributed throughout porous structures, increasing active site density and subsequently ORR activity. In addition, the surface area and pore structure of MOF precursors can propagate to the catalysts in various degrees through controlled pyrolysis and activation steps; offering tailored morphology for catalysing ORR. The MOF-derived TM/N/C offers a new paradigm in PGM-free catalyst design. Since no heterogeneous support is introduced, the catalyst is homogenous without distinction between the support and the catalyst. Maximizing surface area and active site utilizations directly translates to maximizing the catalyst performance.
In this review, we aim at offering a brief introduction on the preparations and performance of ORR catalysts derived from monometallic, bimetallic, and low-level metal-doped MOF precursors for fuel cell application. Some recent highlights from various laboratories worldwide in this rapidly developing field will be described. We will also discuss our perspectives in further improvements in design and synthesis, as well as better structure–function understanding needed to drive these catalyst performances closer to that of platinum at the fuel cell level.
PGM-free electrocatalysts derived from various metal–organic frameworks precursors
Catalysts derived from monometallic MOF precursors
The very first MOF-based ORR catalysts was derived from cobalt zeolitic imidazolate framework, Co-ZIF or Co(Im)2, at Argonne National Laboratory.52–54 In Co(Im)2 each cobalt atom is coordinated with four nitrogen atoms from adjacent 3,5-imidazolate ligands in tetrahedral direction (Fig. 1a) and each 3,5-imidazolate ligand connects with two cobalt atoms to form a three-dimensional porous structure with Co–Co distance of ∼8.1 Å along the [100] direction (Fig. 1b). The CoN4 moieties are regularly distributed within the porous frameworks and the number of CoN4 moieties in the single crystal of Co(Im)2 can reach as high as 3.6 × 1021 cm−3. Co(Im)2 can be synthesized using solvothermal reaction between the 3,5-imidazolate and Co(NO3)2·6H2O in N,N′-dimethylacetamide (DMA) solution. Resulting Co-ZIF powders were thermally activated at different temperatures (500–900 °C) and activity towards ORR was demonstrated using rotating ring-disk electrode (RRDE) at 1600 rpm in 0.1 M HClO4 electrolyte. Pyrolysis at 750 °C afforded the most promising ORR activity with an onset potential of 0.83 V and a half-wave potential of 0.77 V vs. RHE. Chronoamperometric measurements were also performed by alternating between 0.35 V and 0.72 V and the current density was measured as a function of time. No appreciable catalyst deactivation was observed during the measurement. The new catalyst structure was characterized using various analytic tools to address the structure–function relationship. For example, X-ray adsorption spectroscopy (XAS) revealed that during the pyrolysis at temperatures above 500 °C the Co2+ ions starts to transfer into finely dispersed metallic Co and carbide/nitride particles, whose diameters grow with increasing pyrolysis temperature. After applying an acid wash to remove the majority of metallic cobalt, XAS further revealed the presence of Co–Nx complexes which had been previously overshadowed by Co–Co scattering. Complementary structural information on the electronic states of C and N was also gathered using X-ray photoelectron spectroscopy (XPS). XPS showed that carbon atoms were converted into carbonaceous structures above 500 °C and then to graphitic forms at 900 °C during thermal activation. Additionally, organic nitrogen was converted to a mixture dominated by pyridinic-form with minor pyrrolic moieties at 750 °C. Further increasing the activation temperature produced an overall reduction in nitrogen content. Combined XAS and XPS findings suggested that a fraction of Co2+ ions were stabilized by coordinating with pyrrolic or pyridinic nitrogens in carbon to form Co–Nx during thermal activation. Further increasing pyrolysis temperatures to 900 °C led to the formation of lower surface area graphitic carbon and the loss of nitrogen, which correlated to decreased ORR performance. This study demonstrated for the first time of preparing PGM-free ORR catalysts with promising activity in the acidic medium, yielding a new direction of design and synthesis of new catalyst via rationally designed MOF.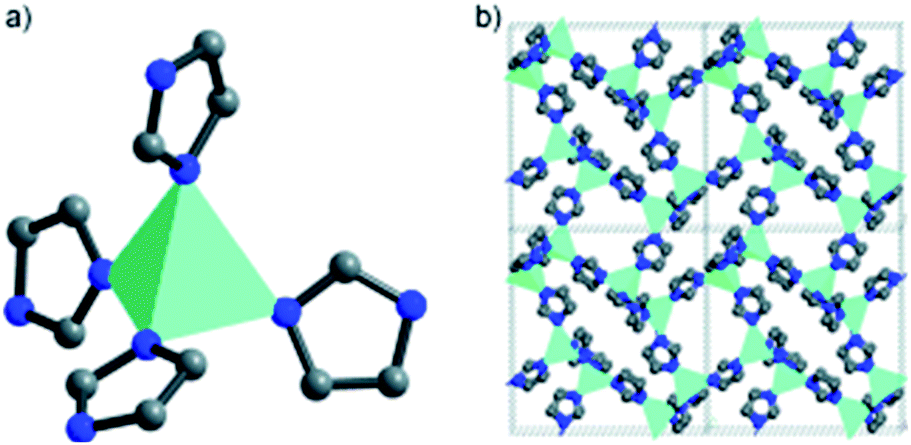 | ||
| Fig. 1 Co-ZIF schematic representation demonstrating (a) Co–N4 coordination and (b) structure packing along the [100] direction showing inherent porosity and long-range order. Reproduced with permission from ref. 54, © 2011, Wiley-VCH. | ||
A natural extension of monometallic Co(Im)2 system would be Co(mIm)2 (also known as ZIF-67) where the imidazole is replaced by 2-methyl-imidazole and a higher specific surface area than Co(Im)2.55 Furthermore, a substitution of alkyl group to the five member ring of imidazole presumably could facilitate the conversion to the six member ring of a carbonaceous moiety upon thermal activation without fragmenting the imidazole ring. Multiple groups including ours investigated this system.52,56 Particularly, Xia et al. investigated the effect of ZIF-67 crystal size on resulting ORR activity using RDE method.57 ZIF-67 crystals were synthesized with diameters ranging from 300 nm to several micrometers. Thermal activation of ZIF-67 crystals occurred at 750 °C for two hours and the MOF-derived catalyst retained the cubic morphology similar to that of the pristine ZIF-67. A direct correlation between ZIF-67 crystallite size and BET surface area after pyrolysis was established with smaller particles yielding higher surface area, as expected. When tested for ORR activity using RDE in O2 saturated 0.1 M HClO4, the catalyst derived from the smallest ZIF-67 precursor particle diameters yielded the best onset and half-wave potentials as well as the electron transfer numbers. Durability of the catalyst was also tested using the chronoamperometry method at a constant voltage of 0.65 V and it was found to be better than a commercial Pt/C standard.
In a separate study, Wang et al. reported the investigation of different monometallic MOFs for ORR catalyst conversion.58 They included ZIF-67, ZIF-8 or Zn(mIm)2, and Co2(bdc)2(dabco) where (bdc) = 1,4-benzenedicarboxylate, (dabco) = 1,4-diazabicyclo[2,2,2]-octane and Co2+ is coordinated by four oxygens from bdc and one nitrogen from dabco. These MOFs were thermally activated at 900 °C. Physical and electrochemical characterizations of the pyrolyzed MOFs showed a positive correlation between N/C ratios and ORR activity. The activation steps of ZIF-67 is schematically shown in Fig. 2 which also represents the general processes used by others. Excellent electrocatalytic activities of pyrolyzed ZIF-67 were found by RDE in O2 saturated alkaline and acidic media. The ZIF-8 based catalysts also showed ORR activity but at lower performance, indicating the importance of a transition metal such as cobalt. Co2(bdc)2(dabco) based materials retained minimal nitrogen content post thermal activation and displayed negligible ORR activity, reiterating the effectiveness of aromatic nitrogen instead of aliphatic nitrogen in the MOF precursor to the formation of the catalytic active center.
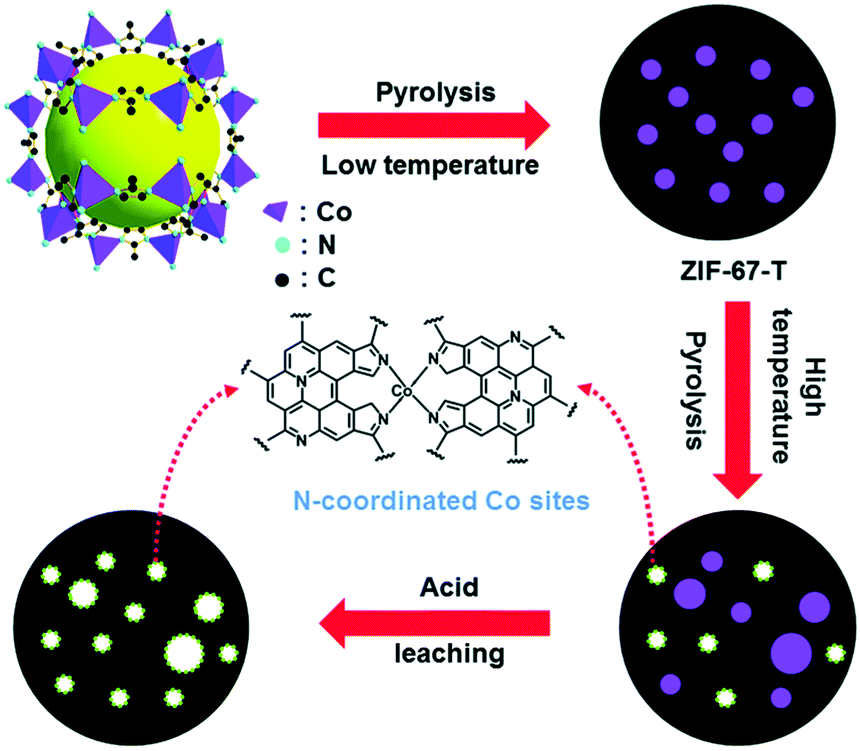 | ||
| Fig. 2 Schematic representation of synthesis and activation of ZIF-67-900 catalyst. Reproduced with permission from ref. 58, © 2014, The Royal Society of Chemistry. | ||
Zhao et al. reported an ORR catalyst prepared using an iron-based monometallic MOF, Fe3O(H2N-BDC)3 (H2N-BDC = 2-aminoterephtalic acid) as a template.59 Fe3O(H2N-BDC)3 has a uniform spindle-like shape with diameters ca. 50 nm and lengths ca. 140 nm. Much of the size and shape characteristics of the Fe-MOF were retained after thermal activation at 900 °C. Electrocatalytic activity investigated via RDE demonstrated onset and half-wave potentials of 1.03 V and 0.92 V in 0.1 M KOH electrolyte, respectively. The catalyst durability was investigated using chronoamperometry at 0.84 V for 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 seconds which retained 79% of the initial current density. The catalyst was also tested in an alkaline membrane direct methanol fuel cell at 60 °C. The cell reached an open circuit potential of 0.81 V, a peak power density of 22.7 mW cm−2, and a peak current density of 93.9 mA cm−2, surpassing the performance of a commercial Pt/C cathode.
000 seconds which retained 79% of the initial current density. The catalyst was also tested in an alkaline membrane direct methanol fuel cell at 60 °C. The cell reached an open circuit potential of 0.81 V, a peak power density of 22.7 mW cm−2, and a peak current density of 93.9 mA cm−2, surpassing the performance of a commercial Pt/C cathode.
In addition to TM and nitrogen, MOFs have been used to incorporate TM and sulfide, specifically cobalt sulfide, as ORR catalysts.60–64 Chen et al. first reported the use of ZIF-67 to prepare cobalt sulfide/N,S co-doped porous carbon core–shell composites which demonstrated good ORR activity when tested in alkaline RDE conditions.65 These catalysts were prepared through pyrolysis of ZIF-67 crystals at elevated temperature under Ar flow before H2S gas was introduced. After pyrolysis, the material displayed distorted rhombic dodecahedron morphology with cobalt sulfide particles surrounded by a porous carbon shell about 5 nm thick. Nitrogen and sulfur dopants were confirmed within the carbonaceous matrix via XPS; however, it was difficult to separate their respective contribution to the overall ORR activity. Almost concurrently, Hu et al. reported a cobalt nanoparticle-carbon@Co9S8 in double-shelled nanocage (Co-C@Co9S8 DSNC) by first depositing CoS shells on the ZIF-67 crystals followed by pyrolysis in N2 atmosphere.66 Resulting materials had an inner Co-C hollow shell about 30–60 nm thick with an outer 10–20 nm thick Co9S8 shell. The catalytic activity of Co-C@Co9S8 was tested in alkaline medium and it was found comparable to that of a commercial Pt/C. In addition to ZIF-67, ZIF-9 in which Co2+ is coordinated with two neighbouring benzimidazoles, was also explored for preparing the catalyst. Bai et al. reported synthesis of CoS1.097 embedded in a porous carbon matrix by simply combining ZIF-9 with solid sulfur followed by pyrolysis at elevated temperature.67 The resulting CoS1.097 nanocomposites were found ORR active when tested in O2 saturated 0.1 M KOH. Due to its high porosity, ZIF-67 was also used as microporous host of additive to prepare the composite catalysts. For example, Zhang et al. added thioacetamide into ZIF-67 as added nitrogen and sulfur sources before pyrolysis.68 They found thus obtained catalyst had excellent ORR activity in both acidic and alkaline conditions. Sulfur doping increased the nitrogen concentration and the percentage of pyridinic-N, which is believed to be responsible for ORR. Doping also influenced the material conductivity. A balance must be struck between adding enough dopant to create a high density of defect sites and maintaining low impedance at the same time.69
Catalysts with new carbon morphologies and ORR activity can also be produced through high temperature pyrolysis of MOFs. For example, under the high temperature, carbon nanotubes (CNT) can be formed from MOFs due to the co-existence of key ingredients, TM (namely Co or Fe) as catalyst and organic ligands as hydrocarbon source. Xia et al. reported an interesting N-doped carbon nanotube frameworks (NCNTFs) in which the pyrolyzed ZIF-67 crystallite maintained its cubic morphology entangled with nanotubes (Fig. 3).70 Some Co nanoparticles are found to be encapsulated by the graphitic shells, predominantly at the tip of carbon nanotubes of 5 nm in diameter. Such structure is consistent with a typical TM-catalyzed CNT growth mechanism. ORR activity was tested by RDE in O2 saturated 0.1 M KOH and 0.5 M H2SO4 electrolytes. Good activities, particularly in alkaline medium, were found. Similarly, good catalyst durability was also found in both media under multiple potential cycling. Contrary to the thin nanotubes, MOF was also employed to synthesize large graphene tube with ORR activity. Li et al. demonstrated the preparation of graphene tubes with diameter as high as 500 nm using MOF of cobalt coordinated by 4′-(4-pyridyl)-4,2′,6′,4′-terpyridine.71 Through Fe doping, the graphene tubes delivered good ORR activity in oxygen saturated KOH and H2SO4 electrolytes.
 | ||
| Fig. 3 FESEM image of ZIF-67 pyrolyzed at 900 °C, the scale bar is 500 nm. Reproduced with permission from ref. 70, © 2016, Nature Publishing Group. | ||
Monometallic MOFs have also been incorporated with different engineered carbons in efforts to further enhance the catalytic activity. Hou et al.72 reported a hybrid nitrogen-doped graphene/cobalt-embedded porous carbon electrocatalyst (N/Co-doped PCP//NRGO) derived from graphene oxide (GO) and ZIF-67. Preparation involved dispersing ZIF-67 in the GO solution followed by thermal activation at 900 °C under inert atmosphere. The resulting catalyst contained a mesoporous amorphous carbon network with embedded Co nanoparticles surrounded by a carbon shell. ORR activity was evaluated in oxygen saturated 0.1 M KOH solution and excellent onset and half-wave potentials were found. In a separate study, Xia et al. demonstrated a Co@Co3O4@C core@bishell nanoparticles (NPs) as ORR catalyst through in situ encapsulation of a cobalt–benzimidazolate MOF into a highly ordered porous carbon matrix (CM),69 followed by pyrolysis in Ar and oxidation in air (Fig. 4). The catalyst showed very good ORR activity in oxygen saturated alkaline solution in a RDE measurement. In addition to carbon, inorganic material has been explored as the templates to prepare MOF based catalysts. Li et al. grew ZIF-67 crystals on both sides of a layered double hydroxide (LDH) nanoplatelet, which served as a 2D inorganic matrix, followed by high temperature pyrolysis to achieve a highly porous honeycomb like structure.73 When tested for ORR activity in O2 saturated 0.1 M KOH electrolyte using RRDE, the catalyst produced an onset potential of 0.94 V and the half-wave potential of 0.83 V. Recently, Lu et al.74 used Co-MOFs to prepare Co nanoparticles embedded in an N-doped porous carbon matrix, which displayed bifunctional ORR/OER activity and stability in acid electrolyte.
 | ||
| Fig. 4 Schematic representation of the synthesis of Co@Co3O4@C-CM. Reproduced with permission from ref. 69, © 2015, The Royal Society of Chemistry. | ||
A summary of the ORR catalytic performances of representative catalysts discussed in this review are listed in Table 1.
| Sample ID | Ref. | Electrolytea | Onset potentialb (V vs. RHE) | Half-wave potentialb (V vs. RHE) | Durability (type, % loss, time) | Current density at 0.9 ViR-free | Current density at 0.8 ViR-free | Power density |
|---|---|---|---|---|---|---|---|---|
| a Electrolyte was purged with O2 prior to measurement and linear sweep voltammetry data was recorded at room temperature and 1600 rpm, unless otherwise noted. b Potential is reported vs. RHE unless otherwise noted. c Alkaline direct methanol fuel cell (ADMFC) cathode testing; see reference or bulk text for ADMFC testing conditions. | ||||||||
| Monometallic metal–organic framework precursors | ||||||||
| 1 | 54 | 0.1 M HClO4 | 0.83 | 0.77 | Chronoamperometry, 0%, 10800 seconds |
|||
| MDC | 57 | 0.1 M HClO4 | 0.86 | 0.71 | Chronoamperometry, 58%, 15000 seconds |
|||
| ZIF-67-900 | 58 | 0.1 M KOH | 0.91 | 0.85 | Chronoamperometry, 21%, 40000 seconds |
|||
| ZIF-67-900 | 58 | 0.5 M H2SO4 | 0.85 | 0.71 | ||||
| CNP | 59 | 0.1 M KOH | 1.03 (900 rpm) | 0.92 (900 rpm) | Chronoamperometry, 21%, 40000 seconds |
22.7c mW cm−2 @ Pmax | ||
| N/Co-doped PCP//NRGO | 72 | 0.1 M KOH | 0.97 | 0.86 | Chronoamperometry, 14.4%, 20000 seconds |
|||
| CoxSy@C-1000 | 65 | 0.1 M KOH | 0.92 | Chronoamperometry, 8%, 18000 seconds |
||||
| Co-C@Co9S8 DSNC | 66 | 0.1 M KOH | 0.96 | Chronoamperometry, 4%, 18000 seconds |
||||
| PC-Co1.097 NC | 67 | 0.1 M KOH | Chronoamperometry, 12%, 10000 seconds |
|||||
| NCNTF | 70 | 0.1 M M KOH | 0.97 | 0.87 | Chronoamperometry, 4%, 100000 seconds |
|||
| Potential cycling, 7 mV E1/2, 5000 cycles |
||||||||
| LDH@ZIF-67-800 | 73 | 0.1 M KOH | 0.94 | 0.83 | Chronoamperometry, 1%, 20000 seconds |
|||
| ZIF-TAA-p | 68 | 0.1 M HClO4 | 0.78 | Chronoamperometry, 65%, 10800 seconds |
||||
| 0.1 M KOH | 0.88 | Chronoamperometry, 5%, 86400 seconds |
||||||
| Potential cycling, 0 mV E1,2, 5000 cycles |
||||||||
| Co-MOFs-1000 | 74 | 0.1 M KOH | 0.88 | |||||
| Bimetallic metal–organic framework precursors | ||||||||
| FeIM/ZIF-8 | 75 | 0.1 M HClO4 | 0.915 | PEMFC voltage hold, <50%, 100 hours | 12 A cm−3 | 0.287 W cm−2 @ Pmax | ||
| NCNT-20 | 174 | 0.1 M KOH | −0.183 V vs. SCE | |||||
| (Fe,Co)@NGC NCs | 76 | 0.1 M KOH | 0.91 | 0.85 | Chronoamperometry, 9%, 20000 seconds |
|||
| P-CNCo-20 | 77 | 0.1 M KOH | −0.04 V vs. Ag/AgCl | −0.12 V vs. Ag/AgCl | Potential cycling, 5 mV E1/2, 10000 cycles |
|||
| Co–N–C | 78 | 0.1 M KOH | 0.982 | 0.871 | Potential cycling, 5 mV E1/2, 5000 cycles |
|||
| 0.1 M HClO4 | 0.761 | Potential cycling 4 mV E1/2, 5000 cycles |
||||||
| CPCs | 80 | 0.1 M KOH | 0.93 | 0.78 | Chronoamperometry, 5%, 40000 seconds |
|||
| 1% Fe/IRMOF-3-900 | 81 | 0.1 M NaOH | 1.02 | 0.88 | Chronoamperometry, 8.5%, 10000 seconds |
|||
| Potential cycling, 7 mV E1,2, 5000 cycles |
||||||||
| Co/Zn(mIm)2-P | 56 | 0.1 M HClO4 | 0.93 | 0.76 | PEMFC voltage hold, 45%, 100 hours | 28 mA cm−2 | 0.25 W cm−2 @ Pmax | |
| Transition metal-doped metal–organic framework precursors | ||||||||
| 1/20/80-Z8-1050 °C – 15 min | 22 | PEMFC voltage hold, >50%, 100 hours | 60 A cm−3 | 0.9 W cm−2 @ Pmax | ||||
| 230 A cm−3 extrapolated | 0.75 W cm−2 @ 0.6 V | |||||||
| FeNC-70 | 82 | 0.5 M HClO4 | 0.80 V vs. NHE | 0.58 V vs. NHE | Potential cycling, 20 mV E1/2, 1000 | |||
| Fe/TPTZ/ZIF8 | 84 | 3.5 mA cm−2 | 190 mA cm−2 @ 0.6 V | |||||
| NC-Ar(F0) | 175 | PEMFC chronoamperometry, varies, 100 hours | 0.8 mA mg−1 | 98.3 mA mg−1 @ 0.6 V | ||||
| NC-Ar(F90) | PEMFC chronoamperometry, varies, 100 hours | 0.7 mA mg−1 | 98.7 mA mg−1 @ 0.6 V | |||||
| N-Fe-MOF | 71 | 0.1 M KOH | 0.88 | |||||
| 0.5 M H2SO4 | 0.79 | |||||||
| Zn(Im)2TPIP | 83 | 0.1 M HClO4 | 0.881 | 0.73 | Potential cycling, varies | 5.9 A cm−3 | 0.080 W cm−2 @ Pmax | |
| Zn(mIm)2TPIP | 0.1 M HClO4 | 0.902 | 0.76 | Potential cycling, varies | 67.0 A cm−3 | 0.620 W cm−2 @ Pmax | ||
| Zn(eIm)2TPIP | 0.1 M HClO4 | 0.914 | 0.78 | Potential cycling, varies | 88.1 A cm−3 | 0.500 W cm−2 @ Pmax | ||
| 146.1 A cm−3 extrapolated | ||||||||
| Zn(4abIm)2TPIP | 0.1 M HClO4 | 0.904 | 0.76 | Potential cycling, varies | 39.4 A cm−3 | 0.460 W cm−2 @ Pmax | ||
| NCPor_0.8-1050Ar + NH3 | 85 | PEMFC chronoamperometry, >50%, 50 hours | 19.5 mA cm−2 | 664 mA cm−2 @ 0.6 V | 0.6 W cm−2 @ Pmax | |||
| NCPor_0.8-1150Ar + NH3 | PEMFC chronoamperometry, 27.5%, 90 hours | 0.51 W cm−2 @ Pmax | ||||||
| FePhen@MOF-ArNH3 | 125 | 0.1 M KOH | 1.03 | 0.86 | ||||
| 0.1 M HClO4 | 0.93 | 0.77 | PEMFC cycling, 100 mA cm−2 at 0.6 V, 10000 cycles |
50 mA cm−2 – air > 100 mA cm−2 – O2 | 0.38 W cm−2 @ Pmax – air | |||
| Zn(mIm)2Fe(Ac)2 | 87 | 64.7 mA cm−2 | 0.2415 W cm−2 @ Pmax | |||||
| Zn(mIm)2Fe(Ac)2(Phen)6 | 136.9 mA cm−2 | 0.4119 W cm−2 @ Pmax | ||||||
| Zn(mIm)2TPI | 221.9 mA cm−2 | 0.6033 W cm−2 @ Pmax | ||||||
| Z8-100-100 rpm-R | 88 | 5.3 mA cm−2 | 910 mA cm−2 @ 0.5 V | 0.500 W cm−2 @ Pmax | ||||
| Fe/N/CF | 89 | 0.5 M H2SO4 | 0.93 | 0.80 | Chronoamperometry, 7.9 mV E1/2, 35000 cycles |
250 mA cm−2 | 0.900 W cm−2 @ Pmax | |
| PEMFC chronoamperometry, varies, 100 hours | 60 A cm−3 | |||||||
| PEMFC cycling, >50%, 15000 cycles |
450 A cm−3 extrapolated | |||||||
| Co,N-CNF | 90 | 0.1 M KOH | −0.082 V vs. Ag/AgCl | −0.155 V vs. Ag/AgCl | ||||
| 0.5 M H2SO4 | 0.045 V vs. Ag/AgCl | Potential cycling, 10 mV E1/2, 5000 cycles | ||||||
| Zn(mIm)2TPIP | 92 | 0.1 M HClO4 | 0.91 | 0.778 | 240.7 mA cm−2 | 0.5179 W cm−2 @ Pmax | ||
| Zn(Im)2TPIP | 0.1 M HClO4 | 0.88 | 0.726 | 15.6 mA cm−2 | 0.0570 W cm−2 @ Pmax | |||
| 0.85:1 |
176 | 147.6 mA cm−2 – Air | 0.3257 W cm−2 @ 0.6 V | |||||
| C-Fe-Z8_Ar | 93 | 0.1 M HClO4 | 0.95 | 0.82 | Potential cycling, 40 mV, 10000 cycles |
|||
| ”Transition metal-free” metal–organic framework precursors | ||||||||
| Carbon-L | 98 | 0.1 M KOH | 0.8610 | 0.6972 | Chronoamperometry, 25%, 25000 seconds |
|||
| Carbon-S | 0.1 M KOH | 0.8441 | 0.6782 | |||||
| NS(3:1)-C-MOF-5 |
105 | 0.1 M KOH | 0.192 V vs. NHE | Chronoamperometry, 20%, 20000 seconds |
||||
| MOFCN900 | 106 | 0.1 M KOH | 0.035 V vs. Hg/HgO | Potential cycling, 30 mV, 1000 cycles | ||||
| NGPC-1000-10 | 99 | 0.1 M KOH | −0.017 V vs. Ag/AgCl | −0.198 V vs. Ag/AgCl | Potential cycling, 10 mV E1/2, 1000 cycles | |||
| NC900 | 100 | 0.1 M KOH | 0.83 (2500 rpm) | |||||
| NPS-C-MOF-5 | 107 | 0.1 M KOH | −0.006 V vs. Ag/AgCl | Chronoamperometry, 20%, 20000 seconds |
||||
| GNPC-800 | 101 | 0.1 M KOH | 0.957 | Chronoamperometry, 6%, 28000 seconds |
33.8c mW cm−2 @ Pmax | |||
| C-CZ-4-1000 | 104 | 0.1 M KOH | 1.03 | 0.887 | Potential cycling, minimal, 5000 cycles | |||
| CIRMOF-3-950 | 109 | 0.1 M KOH | −0.08 V vs. Ag/AgCl | Chronoamperometry, 21.5%, 20000 seconds |
||||
| NGPC/NCNT-900 | 108 | 0.1 M KOH | −0.051 V vs. Ag/AgCl | −0.171 V vs. Ag/AgCl | Chronoamperometry, 14%, 43200 seconds |
|||
| P–N–carbon-950 | 110 | 0.1 M KOH | 0.80 V vs. Ag/AgCl | Chronoamperometry, 0%, 20000 seconds |
||||
| Potential cycling, 20 mV E1/2, 5000 cycles | ||||||||
Catalysts derived from bimetallic MOF precursors
Although monometallic MOFs were the first ones applied as a viable precursor to prepare ORR catalysts,54 it carried some significant limitations. The major deficiency is that the TM content is simply too high. For example, the cobalt or iron content in the zeolitic imidazolate frameworks is typically around 30 wt% which can increase to >60% after pyrolysis. This value is simply too high compared to the optimal transition metal loading in the PGM-free catalysts (<∼5 wt%). To overcome such deficiency, a bimetallic MOF concept was introduced.75 Combining multiple metals in a MOF or multiple MOF systems allow one to fine tune the desirable catalyst characteristics through rational design of MOF template.We first reported the preparation of ORR catalyst using a mixture of Fe zeolitic imidazolate framework (FeIM) and Zn zeolitic methyl-imidazolate framework (Zn(mIm)2 or ZIF-8) as the precursor.4 FeIM is comprised of Fe2+ coordinated both tetrahedrally and octahedrally to imidazolate ligands. The MOF by itself exhibits reasonably good ORR activity after thermal activation.75 By varying the ratios of FeIM/ZIF-8, significantly higher activity than that of FeIM alone was observed by RDE measurement. In such case, the zinc in the mixture was nearly completely vaporized under elevated temperature, leaving 5.29 wt% of Fe in the optimized FeIM/ZIF-8 catalyst after heat activation. The MOF mixture also retained 4.5 wt% nitrogen, which were predominantly in the pyridinic form, promoting the perceived Fe/N/C active site formation. The catalyst was fabricated into the cathode and tested in a single PEM fuel cell in fully humidified H2/O2. A maximum power density of 0.287 W cm−2 and a volumetric current density of 12 A cm−3 measured at 0.8 ViR-free were achieved, making FeIM/ZIF-8 templated electrocatalysts among the well-performed PGM-free materials at that time.
Most bimetallic MOF based precursors developed later on, however, were synthesized through co-crystallization or co-precipitation. Xi et al. combined Fe and Co within a Prussian blue analogue MOF of the formula MII3[MIII(CN)6]2 where MII = Fe and MIII = Co.76 The Fe3[Co(CN)6]2 were further coated by a layer of polydopamine to serve as a nitrogen doped carbon source. After pyrolysis, iron and cobalt nanoparticles surrounded by graphitic carbon shells, (Fe,Co)@NGC NCs, were formed. A good ORR activity and durability were found when tested in O2 saturated 0.1 M KOH solution.
The early successes of ZIF-8 and ZIF-67 derived catalysts also led the investigations of their bimetallic MOF mixture. The approach is based on the assumption that adding second metal with identical charge and stereo chemistry will lead to uniform distribution of both metals within the MOF lattice. This is often, however, difficult to confirm; particularly when one metal content is significantly lower than the other. Since Zn2+ and Co2+ have the same oxidation state and coordination chemistry with imidazoles, one would expect that they can interexchange with each other in the zeolitic imidazolate frameworks. Chen et al. combined Co and Zn in various ratios to synthesize bimetallic ZIFs as high surface area precursors to prepare Co/N/C based ORR electrocatalysts.77 Furthermore, these authors found that the addition of a heteroatom dopant, phosphorus, further increased the ORR activity compared to straight bimetallic MOF-derived catalyst measured by improvement in the onset and half-wave potentials. Additionally, TEM imaging did not show Co0 aggregation into nanoparticles, indicating that the Co species are atomically distributed through the catalyst material as possibly Co–Nx active sites. You et al. also studied the bimetallic Zn/Co zeolitic methylimidazolate system.78 By altering the ratio of the two metals therefore the spatial isolation of Co2+ by Zn2+ in MOF, they were able to control the Co nanoparticle size as well as the overall surface area after pyrolysis. Similar to others, they found the new catalyst with excellent ORR activity and durability in oxygen saturated alkaline, acidic, as well as neutral electrolytes measured by RDE method. We at Argonne also studied Zn/Co ZIF mixtures as a natural extension from our earlier study on monometallic Co-ZIF54 and bimetallic FeIM/ZIF-8.75 Our interest lies mainly in the catalyst performance in acidic environment, particularly, under PEM fuel cell operating condition. An advantage of Co over Fe based PGM-free catalyst is the lower peroxide production through Fenton reaction,79 a process detrimental to lifespan of the fuel cell's membrane. We prepared Zn/Co zoelitic methylimidazolate framework precursor using an ion substitution synthesis with approximately 5% Co doping in ZIF-8 structure.56 After thermal activation, acid leaching and high temperature activation in NH3, a very active catalyst with BET surface area of 1563 m2 g−1 was achieved. The membrane electrode assembly (MEA) containing this catalyst demonstrated an areal current density of 28 mA cm−2 at 0.8 ViR-free and a peak power density of 374 mW cm−2 in a fuel cell test under one-bar oxygen (Fig. 5).
 | ||
| Fig. 5 Single fuel cell ORR activity of Co,Zn(mIm)2-P derived from Co doped ZIF-8 structures. Reproduced with permission from ref. 56, © 2016, John Wiley and Sons. | ||
Very limited MOFs beyond zoelitic methylimidazolate frameworks were investigated for ORR application. Wu et al. chose an isoreticular MOF prepared from Zn2+ coordinated to 2-aminoterephthalic acid (IRMOF-3) into which Fe3+ was selectively doped through ion-exchange with Zn2+ to yield a bimetallic MOF.80 After pyrolysis in Ar at 800 °C, the catalyst thus formed maintained the cube-like structure in which the cubes were composed of porous iron core graphitic carbon shell nanoparticles. A good ORR activity was demonstrated using RDE in O2 saturated 0.1 M KOH electrolyte. Similarly, Sun et al. prepared the same bimetallic IRMOF-3 at much lower Fe loading of ca. 1%.81 Such reduction of ion usage led to improved ORR catalytic performance with an onset potential of 1.02 V and a half-wave potential of 0.88 V when tested in O2 saturated 0.1 M KOH electrolyte using RDE, comparable to that of a commercial Pt/C benchmark.
Catalysts derived from transition metal-doped MOF precursors
Perhaps the most significant enhancement in MOF-based ORR catalytic activity comes from the method of controlled doping transition metals (Fe,Co) in the form of ligated metal complexes. The first representative work of this approach was introduced by Proietti et al.22 who reported unprecedented ORR activity and PEMFC performance of a PGM-free catalyst derived from an iron-doped ZIF-8. Iron(II) acetate was mixed with 1,10-phenanthroline and ZIF-8 in a 1:20:80 mass ratio before being pyrolyzed in inert atmosphere to form Fe/N/C composite. The resulting catalyst has a high BET surface area of 964 m2 g−1, which is very important in MOF-based PGM-free catalysts since the active site is generally uniformly distributed inside of such catalyst and there is no distinction between the “catalyst” portions from that of “support”. The higher specific surface area generally leads to higher overall catalytic activity. The catalyst was tested as the cathode in a single cell PEMFC under fully humidified H2/O2 at absolute pressure of 1.5 bar and 80 °C and reached a peak power density of 0.91 W cm−2 and a power density of 0.75 W cm−2 at 0.6 V. In addition, a measured specific volumetric activity of 60 A cm−3 at 0.8 ViR-free was achieved, see Fig. 6. This study set up a benchmark for PGM-free ORR catalyst and triggered several follow-up investigations by various groups since.
 | ||
| Fig. 6 Single fuel cell data comparing the ZIF-8 derived Fe–N–C material (empty blue stars) and this group's previously most active carbon supported iron salt Fe–N–C material (empty red circles). An impressive volumetric current density was found to be 230 A cm−3 at 0.8 ViR-free using extrapolation. The U.S. DOE target of 130 A cm−3 (grey circle) for 2010 was surpassed and the U.S. DOE target of 300 A cm−3 for 2015 was nearly achieved. Figure reproduced with permission from ref. 22, © 2011, Nature Publishing Group. | ||
Palaniselvam et al.82 studied the effect of tuneable porosity of MOF templates by doping three different MOF materials, ZIF-68, ZIF-69, and ZIF-70 with iron–phenanthroline complexes in their cavities. The Zn2+ ion is coordinated to 2-nitroimidazole and benzimidazole (pore diameter of 7.5 Å) in ZIF-68; to 2-nitroimidazole and 5-chlorobenzimidazole (pore diameter of 4.4 Å) in ZIF-69; and to imidazole and 2-nitroimidazole (pore diameter of 13.1 Å) in ZIF-70, respectively. All three materials were subjected to thermal activation at 900 °C in inert atmosphere and tested via RDE in O2 saturated 0.5 M HClO4. The researchers discovered that, among three resulting catalysts, the one derived from ZIF-70 gave the highest BET surface area, nitrogen content as well as the best ORR activity; perhaps due to a better incorporation of iron–phenanthroline complexes inside the ZIF's pores, whereas the pores in ZIF-68 and ZIF-69 are not large enough to accommodate these iron complexes. Along the same line of thinking, Li et al. developed large 3D polyhedral MOF cages from a divergent tridentate ligand, 2,4,6-tris(4-pyridyl)-1,3,5-triazine ligand coordinated to Co metal centers.71 These MOF cages served as high surface area porous hosts for dicyandiamide and iron(II) acetate which was subsequently pyrolyzed to yield graphene and graphene nanotube rich N- and Fe-doped carbon material. The high concentration of N-doped graphene nanotubes is believed to contribute to the good ORR activity in both acidic and alkaline electrolytes.
MOFs have been perceived as expensive precursors for PGM-free catalyst preparation based on their off-the-shelf prices from chemical vendors. Under most circumstances, however, the costs are associated with the synthesis processes instead of raw materials. For example, the 2-methylimidazole and zinc salt used for ZIF-8 synthesis are all very low-cost commodity chemicals. If one could eliminate the use of solvent and related separation process from the conventional solvothermal reaction, one could significantly simplify synthesis therefore lowering the associated cost. To this end, Zhao et al.83 demonstrated an all solid-state, “one pot” method of synthesizing ZIF-based catalyst templates. It combines ZnO, imidazole ligand and iron additive in a solid mixture and converts them to iron doped ZIF through a one-step reaction at mild temperature (<200 °C). Four MOFs were prepared using imidazole (Im), 2-methylimidazole (mIm), 2-ethylimidazole (eIm), and 4-azabenzimidazole (4abIm) ligands to form Zn(Im)2, Zn(mIm)2, Zn(eIm)2, and Zn(4abIm)2, respectively. Additionally, 1,10-phenanthroline iron(II) perchlorate (TPI) complex was added to the one pot mixture as the iron dopant. Resulting MOFs were converted to catalysts through pyrolysis and all four samples showed high BET surface areas. The catalytic activities were evaluated by rotating ring-disk electrode (RRDE) methods in O2 saturated 0.1 M HClO4 electrolyte and in single cell PEMFC at 80 °C under fully humidified H2/O2. Fuel cell and RRDE tests demonstrated the identical trend in activity with Zn(eIm)2TPI > Zn(mIm)2TPI > Zn(4abIm)2TPI ≫ Zn(Im)2TPI. Zn(eIm)2TPI yielded an impressive volumetric current density of 88.1 A cm−3 at 0.8 ViR-free. These catalysts were further tested in the oxygen saturated KOH solution. Excellent activities surpassing that of a Pt/C benchmark catalyst were achieved.
Tian et al.84 explored the effect of different Fe2+–ligand coordination in ZIF-8 to the resulting ORR activity. 2,4,6-Tris(2-pyridyl)-s-triazine (TPTZ) and 1,10-phenanthroline (Phen) were applied to ligate with Fe2+ of iron(II) acetate inside ZIF-8 to form templates Fe/TPTZ/ZIF-8 and Fe/Phen/ZIF-8 for preparing Fe/N/C electrocatalysts through pyrolysis. For comparison, an iron(II) acetate and ZIF-8 template, Fe/ZIF-8, in the absence of ligand was also prepared. They found that the Fe2+ from Fe(TPTZ)2 had displaced some Zn2+ ions from ZIF-8 framework while a Zn(TPTZ)2 layer was formed surrounding the Fe-doped ZIF-8 particles. Such phenomenon was not observed when the ligand was changed from TPTZ to Phen, suggesting a higher binding strength between Fe2+ and Phen precluded the exchange of Fe2+ with Zn2+ in ZIF-8. Activity testing in single PEMFC found that Fe/Phen/ZIF-8 catalyst had the best ORR performance among the three, while Fe/ZIF-8 and Fe/TPTZ/ZIF-8 samples had nearly identical activity, suggesting the ion-exchange between Fe dopant and Zn in ZIF framework is unfavourable and should be avoided.
Similarly, Yang et al. replaced the iron(II) acetate with iron porphyrin decorated on a ZIF-8 microporous host inspired from their early study of the catalyst derived from pyrolyzed vitamin B12 on carbon black.85,86 Since vitamin B12 is simply a cobalt porphyrin derivative, replacing it with iron containing porphyrin represented a reasonable extension. They optimized the activation conditions and tested the catalyst in the PEMFC. A very good fuel cell performance of areal current density of 19.5 mA cm−2 at 0.9 V, 664 mA cm−2 at 0.6 V, and a maximum power density of 0.60 W cm−2 at 0.32 V was achieved. They also investigated the catalytic durability and found that it correlated to the level of graphitization. Unfortunately, graphitization also led to the reduction in pyridinic N content hence the lower initial activity.
Barkholtz et al.87 also investigated the use of different iron precursors in a one-pot solid state synthesis method.83 1,10-Phenanthroline iron(II) perchlorate (TPI), iron(II) acetate (Fe(Ac)2), and a combination of iron(II) acetate and 1,10-pehanthroline (Phen) in a 1:6 molar ratio were used in the synthesis of Fe-doped ZIF-8, followed by the thermal activation and post-treatment before tested in fuel cell. The best catalyst was derived from ZIF-8 and TPI mixture with an areal specific current density of 221.9 mA cm−2 at 0.8 ViR-free and a peak power density of 603.3 mW cm−2 when tested under fully humidified one-bar O2. Other catalysts also performed well although at somewhat lower activities. The study suggested that controlling the initial coordination chemistry around ferrous ion impacts the catalytic property after pyrolysis.
To understand the influence of MOF particle size to catalytic activity and mass transport, Armel et al. investigated ZIF-8 derived Fe–N–C catalysts by altering ZIF-8 crystals with diameters ranging from 1800 nm to 80 nm using an excess ligand method to control the particle size.88 Using previously reported activation conditions including ball milling at 400 rpm, they found that larger ZIF-8 crystals yielded smaller Fe–N–C particles than smaller ZIF-8 precursors. They postulated that the high rpm ball milling caused aggregation of finer particles, leading to overall larger diameter crystals. Furthermore, they found that smaller diameter ZIF-8 precursors yielded materials with higher ORR activity at lower potentials due to increased surface area. Specifically, the best catalyst was derived from ZIF-8 crystals of diameter 100–280 nm.
A common thread among MOF-derived TM–N–C materials is the importance of micropores, which dominate the overall catalyst surface area and house the majority of TM–Nx active sites. Given relatively low TOF per PGM-free catalytic site, it is crucial that these TM–Nx sites are as accessible to reactants (O2), electrons and protons as possible so that the overall electrode performance can be compensated. The mass-transport in the conventional amorphous carbon based electrode catalyst is accomplished through tiered cavities from macro → meso → micro-pores, each with added impedance. The charge-transfer in such carbon is percolated through the contact between these particles which can be interrupted when the carbon is electrochemically oxidized. Therefore, a new catalyst morphology tailored for cathodic electrocatalytic reaction needs to be developed to overcome such shortcomings. To this end, Shui et al. developed a nano-network catalyst in which the pyrolyzed TPI/ZIF-8 are embedded inside of interconnected porous carbon nanofibers.89 The nanofibrous catalyst was prepared by the electrospinning method. The micropores are uniformly distributed in the “strings” and “knots” of the nanofibers whereas the macroporous voids between the fibres permit rapid mass transport to the active sites in the micropores. The interconnected nanofibers allow efficient electron mobility and low electrode impedance and reduce the conductivity loss by the oxidative corrosion at only fibre surface. See Fig. 7. As such, a high volumetric ORR activity of 60 A cm−3 measured at 0.8 ViR-free was achieved in a fuel cell test. Accelerated stress tests were performed by cycling voltage from 0.6 to 1.0 V followed by measuring fuel cell current density at 0.5 V. The relative ORR activity lost after 15000 cycles was smaller than that of a commercial Pt/C benchmark.
 | ||
| Fig. 7 Schematic representation of the electrospun Fe–N–C material highlighting the microporous nature of the nanofiber as well as the macroporous voids allowing for efficient mass transport within the electrode. Below is the resulting single fuel cell activity test demonstrating unprecedented ORR activity surpassing the DOE 0.8 ViR-free target for 2017 when extrapolation was used. Reproduced with permission from ref. 89, © 2015, The National Academy of Sciences of the United States of America. | ||
Although MOFs have been used as templates to prepare desired morphology for ORR catalysts, effort has been made to incorporate another templating precursor such as a silica coating to prevent aggregation. Shang et al. prepared cobalt and nitrogen co-doped carbon nanoframe (Co,N-CNF) by coating Zn/Co ZIFs with a mesoporous SiO2 shell followed by thermolysis and HF etching.90 The resulting Co,N-CNF material has a hierarchical pore structure and a BET surface a–ea of 1170 m2 g−1. Authors attributed such high surface to be a result of preservation of ZIF morphology by the SiO2 protective layer during pyrolysis. Compared to Pt/C, the catalyst demonstrated higher activity in O2 saturated alkaline solution and promising activity in acidic medium.
In addition to MOF template and additive, post-synthesis and pyrolysis treatment could make significant difference to the final catalytic activity. For example, through thermolysis optimization, Liu and Barkholtz recently reported an unprecedented performance over iron-doped ZIF-8 system. A specific areal activity of 29.5 mA cm−2 at 0.9 ViR-free and a specific volumetric activity of 108 A cm−3 at 0.8 ViR-free in fuel cell tests under one bar fully humidified oxygen/hydrogen were achieved.91,92 An ORR catalyst should be ultimately evaluated under the fuel cell operating condition since such test exposes the effectiveness of the catalyst properties including surface, porosity, conductivity, etc. that are not fully revealed in RDE test. Fuel cell test also adds another layer of complexity which requires the optimization of MEA fabrication to maximize catalyst–ionomer–reactant triple-phase boundary (TPB). By design, MOF derived PGM-free ORR catalysts have catalytic sites evenly distributed throughout the specific surface, and the catalyst surface and the support are indistinguishable. Therefore, the coverage of ionomer has to be as high as possible without hindering the access of gas reactant. A practical fuel cell vehicle runs on air as the oxidant instead of pure O2, which often requires minor variation in MEA preparation. We recently conducted a systematic study on the Nafion® ionomer to catalyst weight ratio (I/C) in preparing MEA containing TPI/ZIF-8 derived cathode catalyst. In this work, I/Cs were changed and air fed fuel cell performance monitored. We found that the cathode with I/C = 0.85:1 delivered the best fuel cell performance under fully humidified one-bar air, achieving specific current density of 147.6 mA cm−2 at 0.8 ViR-free and power density of 325.7 mW cm−2 at 0.6 V, respectively. In a similar fashion, Wang et al.93 found that the pyrolysis environment was crucial for preparing a highly active ORR catalyst from Fe-doped ZIF-8. RRDE testing of these materials showed good ORR activity.
At present, TM-doped MOF precursors may be the most promising systems in delivering high performing ORR catalyst. Their ORR activities have been vetted not only by RDE methods but also, more importantly, at the fuel cell level. Majority of highly active catalysts so far are derived from ferrous ion doped ZIF-8. Investigating new MOFs with alternative ligands and framework structures,50,80,82,83,92 could potentially lead to further advancement of the ORR activity comparable to that of PGM catalysts.
Metal-free catalysts derived from MOF precursors
Although metal-free catalysts derived from MOF appears to be a misnomer, the PGM-free electrocatalysts discussed in this section refer to those containing no transition metals such as iron, cobalt, etc. which are believed to be part of ORR active site centers. All the MOFs discussed in this section use moieties of transition metals, particularly of zinc, as the secondary building unit (SBU) which are removed during the catalyst processing. Introducing nitrogen and other heteroatoms into carbonaceous materials can produce desirable electronic modifications, resulting in enhanced catalytic activity based on previous report.94 Considerable effort has been dedicated to the search for metal-free electrocatalysts for the ORR reaction.60,95–97 Metal-free electrocatalysts have displayed relatively good stability in fuel cell conditions in addition to reasonable ORR activity.95 MOFs have been used as porous templates to host metal-free, heteroatom dopants before being converted to ORR catalysts through pyrolysis and post-treatment.Similar to the TM-dope PGM-free catalyst approach, zeolitic imidazolate frameworks (ZIFs) are also widely used in preparing metal-free ORR catalyst due to their existing nitrogen functional groups in the organic ligands. P. Zhang et al.98 prepared a metal-free porous ORR catalyst by impregnating ZIF-7 (Zn2+ coordinated to benzimidazolate ligands) with glucose, followed by a programmed high temperature pyrolysis. The nitrogen in the final catalyst was originated from ZIF-7. The catalyst thus obtained was evaluated in oxygen saturated KOH solution and reasonably good activity and durability were achieved when compared to Pt/C benchmark material. L. J. Zhang et al.99 applied ZIF-8 as a precursor to prepare nitrogen-doped porous carbon electrocatalysts by first preparing nanocrystalline ZIF-8 followed by pyrolysis at 1000 °C under N2. The catalyst retained much of the ZIF-8 morphology and demonstrated reasonably good ORR activity and durability when tested in alkaline medium. Inspired by the strategy of incorporating more N and higher surface area for better ORR activity, Aijaz et al.100 infiltrated ZIF-8 cavities with furfuryl alcohol (FA) and NH4OH and activated the precursor in a two-step process before the carbonization at 900 °C. The nitrogen doped carbonaceous electrocatalyst thus obtained yielded a remarkable BET surface area of 2747 m2 g−1. The catalytic activity towards ORR was evaluated via RDE using O2 saturated 0.1 M KOH and achieved a very good onset potential compared to a commercial Pt/C catalyst.
The loss of Zn under high temperature treatment increases the porosity of pyrolyzed catalyst, which often leads to lower electro-conductivity. To increase the conductivity for better charge transfer, Zhong et al. applied graphene oxide (GO) as a support onto which ZIF-8 was grown.101 They first enriched the GO with poly(vinyl pyrrolidone) and speculated that its amide carbonyl functional groups may coordinate with Zn2+ ions to facilitate nucleation of ZIF-8 crystals. After pyrolysis and acid leaching, the ZIF-8 precursor became graphene-based nitrogen-doped porous carbon. XPS study identified the composite with high level of nitrogen doping, dominated by pyridinic and graphitic N-moieties, the alleged source of ORR activity in metal-free catalysts.102,103 Very good ORR activity was found by RDE test in O2 saturated 0.1 M KOH. More importantly, the catalyst was evaluated in an alkaline direct methanol fuel cell where an onset potential of 0.71 V and a peak power density of 33.8 mW cm−2 were observed, outperforming a commercial Pt/C cathode. Authors argued that the synergistic effect between the GO and nitrogen-doped carbon were responsible for high conductivity and good ORR performance. In a similar study, Ge et al. prepared a 3D structured N-doped carbon/carbon nanotube composite derived from pyrolyzed ZIF-8 on a CNT backbone104 in an attempt to generate better structural stability and electronic conductivity. The CNTs were modified with carboxyl functional groups which were presumably able to coordinate to Zn2+ as nucleation sites for ZIF-8 crystals growth. After pyrolysis, TEM study revealed that the ZIF-8 decomposed into nanoporous graphene sheets evenly decorated on the surface of the CNTs. The catalyst showed ORR onset and half-wave potentials approaching to that of Pt/C when screened by RDE in O2 saturated 0.1 M KOH.
Other than ZIFs, MOFs that do not contain nitrogen in the organic ligands, such as MOF-5, are also investigated for synthesis of PGM-free ORR catalysts. Li et al.,105 infiltrated MOF-5 (Zn4O(BDC)3, BDC = 1,4-benzenedicarboxylate) with urea and DMSO as N- and S-sources, respectively. After pyrolysis and acid wash, the electrocatalysts prepared with a 3:1 molar ratio of N to S resulted in the best ORR activity in O2 saturated 0.1 M KOH and a BET surface area of 1091 m2 g−1. Pandiaraj et al.106 also investigated MOF-5 as a template to host melamine as a nitrogen reservoir through in situ polymerization and they produced a graphitic C3N4 (g-C3N4). During pyrolysis, g-C3N4 decomposed to release nitrogen into the surrounding carbon structure of the pyrolyzed MOF-5. Both mechanical mixing and wet chemistry were used and the resulting catalysts were tested by RDE in O2 saturated 0.1 M KOH electrolyte through voltage cycling. A reasonably good activity and excellent durability were observed. Li et al.107 reported for the first time a ternary-doped porous carbon derived from MOF-5 for ORR. Ternary-doped carbonaceous catalyst was prepared by impregnating MOF-5 with dicyandiamide (DCDA), triarylphosphine (TPP), and dimethyl sulfoxide (DMSO) as N, P, and S sources, respectively, followed by pyrolysis in N2 at 900 °C. The resulting powder was investigated with XPS and N, P, and S were successfully identified in the carbonaceous matrix. The ORR activity of ternary-doped electrocatalyst was found to approach that of a commercial Pt/C when tested via RDE in O2 saturated 0.1 M KOH. The authors suggested that the embedded N, P, and S with different electro-negativities may break the electro-neutrality of the carbon framework and change the asymmetric spin density, leading to enhanced O2 adsorption and weakened O–O bonding, therefore better ORR activity. Similar to ZIF-8/nano-carbon composites,101,104 Zhang et al. prepared nitrogen doped graphitic porous carbon and carbon nanotube hybrids (NGPC/NCNTs) derived from MOF-5 using a multiple-step process involving heat-treatment, acid leaching, nickel addition and urea treatment, see Fig. 8.108 TEM revealed a very complex morphology consisting of graphitic carbon particles connected by bamboo-like CNT structures. Excellent ORR catalytic activities approaching to that of Pt/C were found when the catalyst was evaluated in both O2 saturated KOH and H2SO4 solutions.
 | ||
| Fig. 8 Schematic representation of the synthesis and activation of NGPC/NCNT catalysts. Reproduced with permission from ref. 108, © 2015, The American Chemical Society. | ||
Besides of ZIFs and MOF-5, a limited number of other MOFs were also investigated. S. Fu et al. prepared nitrogen-doped carbon materials by pyrolyzing IRMOF-3 at 950 °C.109 The carbonized IRMOF-3 (CIRMOF-3-950) was found to have a respectable surface are of 553 m2 g−1 and reasonable ORR activity when tested in O2 saturated 0.1 M KOH by RDE. Y. Fu et al. employed UiO-66 in which Zr instead of Zn ligates with N-containing 2-aminoterephthalic acid. A phosphorus compound was added to serve as a precursor for preparing N–P-doped carbon.110 It is predicted that the presence of both N and P dopants will introduce defects within the carbon matrix creating a highly localized state close to the Fermi level of the ORR active catalysts.111 The authors attempt to optimize the P-content and uniformity via post-synthesis reaction of amino group in 2-aminoterephthalic acid with glyphosine. The pyrolyzed MOF was washed in HF acid to remove Zr. The catalyst achieved reasonably good ORR activity and durability when tested by RRDE in 0.1 M KOH solution.
In the short time since metal-free ORR catalysts from MOF templates were first introduced, significant improvements in ORR activities have been achieved. Nearly all reports used a Zn2+ ion based MOF material as the precursor as Zn can be completely removed during pyrolysis above 900 °C. Furthermore, most reports relied on ZIF-8 with100,101,104 or without additives99 due to the high surface area and favourable pore structure. Although these catalysts seem quite promising, they demonstrated ORR activity mostly in alkaline media. Their activities toward ORR are also generally lower than those prepared using transitional metal as additives. Future work should emphasize MOF-derived metal-free catalysts for acidic media and tested in real fuel cell to better understand their potential as fuel cell catalysts. To truly demonstrate the benefit of TM-free precursors, one has to be extremely careful in controlling the MOF synthesis and post-treatment since it is known that ORR activity can be altered even with a trace amount of Fe or Co.
Understanding the active site of MOF derived PGM-free catalysts
Elucidation of the active site is critical to the rational design of PGM-free TM/N/C ORR catalysts. Although much work has been devoted to the subject in recent years,41,68,112–141 a molecular-level understanding of the active site remains elusive. A survey of the literature reveals two predominant hypotheses on the active site structures: nitrogen defects within the carbonaceous support95,142–144 and transition metal–nitrogen coordination on/in the carbonaceous material.41,112,127,143,145 Inspired by its counterpart in the biological system, most of these studies suggested the active site structures in which transition metal ions are ligated by the surrounding Nx (x = 2, 3, 4). For example, Lefèvre et al. demonstrated experimentally the presence of Fe/Nx/C and Co/Nx/C in the ORR catalysts using a time-of-flight secondary ion mass spectrometry method.21,146 Fe/N4/C with iron ligated by four nitrogens from a pair of phenanthroline-liked moieties in a micropore was proposed as the catalytic site in a highly active, iron-doped nitrogen–carbon composite catalyst.41 The Fe/N4/C site, of which the Fe was ligated by four Ns embedded on a curved graphitic plane, was also identified in an iron and nitrogen decorated CNT ORR catalyst by X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopic method.92,140,147In situ EXAFS investigations also suggested that ORR catalytic mechanism involves a sequential electron transfer from the carbon matrix to oxygen chemisorbed on Fe/N4 site.141,147 On the other hand, studies on TM-free catalysts suggested that nitrogen embedded in carbon surface plays a critical role in ORR catalysis. For example, Gao et al. proposed the active site in N-doped carbon materials are carbon atoms with Lewis basicity next to pyridinic N demonstrated by highly oriented pyrolitic graphite model catalysts with well-defined π-conjugation and well-controlled doping of N species.148 Similar to other PGM-free catalysts, the MOF-derived catalysts are generally subject to high temperature conversion which often erases the memory of the precursor's coordination structure completely. The resulting catalytic sites could be chaotic without long-range or uniform short-range structures to be investigated by the traditional characterization techniques. However, since TMs inside of a MOF usually have the identical coordination structure which is uniformly distributed throughout the framework, it is possible to produce one (or a very limited number of) active site(s) under a well-controlled thermolysis condition. In this regard, MOF is similar to the precursors of organometallic macromolecule but different from those of simple ligand–TM mixtures in which the initial TM coordination are often not clearly defined. For MOFs with Zn2+ as SBU, zinc is vaporized during thermolysis which could interfere the fusion of ligands during organics to carbon conversion, leading to porous but less graphitized carbon of lower conductivity. Such process could result in morphologic differences between the catalysts derived from MOFs with those converted from the carbon supported precursors. 57Fe Mössbauer spectroscopy, which is highly sensitive the coordination structure surrounding iron, has emerged as a powerful tool in studying Fe–Nx centers, particularly when used in combination with XPS.127,128,130,149–153 The investigations have led to a direct correlation between the existence of Fe–Nx centers and kinetically controlled ORR activity. For example, Kramm et al. studied correlations between mass activity and physicochemical properties of Fe/N/C catalysts derived from Fe/Phen in ZIF-8 precursor using 57Fe Mössbauer spectroscopy and other techniques. They found that the catalyst contained the same FeN4-like species existed in the materials prepared by impregnating iron acetate on carbon black followed by high temperature heat-treatment in NH3.129 Among all FeN4-like species, only D1 sites assigned to FeN4/C and D3 sites assigned to N-FeN2+2/C were correlated to the ORR catalysis. The difference from MOF-derived catalyst with that prepared by Fe–ammonia treated carbon catalyst was simply that the former contained many more D1 and D3 sites than the latter. More recently, Zitolo et al. combined 57Fe Mössbauer spectroscopy with XANES and EXAFS methods and investigated Fe/Phen doped ZIF-8 under both ammonia and argon high temperature treatment.123 Since XANES and EXAFS are bulk spectroscopic techniques, it is critically important that the PGM-free catalyst prepared has identical active sites throughout the entire phase. They controlled the treatment conditions and confirmed that both catalysts indeed contained nearly the same active site structure uniformly distributed through the bulk phase, established by an exclusive D1 plus D2 band feature associated with Fe–N4 species in Mössbauer spectroscopy. Through X-ray absorption spectral analysis and DFT simulation, they obtained two most likely structures in which iron(II) was ligated by four equilateral Ns on the same plane whereas two out-of-plane oxygens were bonded to Fe either on the same or the opposite sides. See Fig. 9. Interestingly, this 4+2 EXAFS feature was also observed in Fe/N/C composited prepared by doping low concentration iron in high surface area carbon under the ammonia flow during high temperature pyrolysis.154 Similar to the catalysts derived from Fe-containing MOF, the studies on the active site in the Co-containing catalysts derived from MOFs also exist such as the X-ray absorption investigation for catalyst prepared from Co(Im)2.55 However, the investigation on Co-based catalysts lacks behind that of Fe-containing materials primarily due to (a) the Co catalysts are generally less active and (b) powerful tool such as Mössbauer spectroscopy does not apply to cobalt. | ||
| Fig. 9 K-edge XANES spectrum of Fe0.5950 (black dashed line) and the theoretical spectrum of the depicted structures (red line). Reproduced with permission from ref. 123, © 2015, Nature Publishing Group. | ||
Obviously, the investigation of active site structure is far from complete for the MOF-derived catalysts. New physical characterizations beyond the currently available tools are needed to address the active site at the ultra-small and ultra-fast scales. We anticipate also that rational design of various MOF precursors with known initial metal–ligand coordination will also facilitate the understanding of active site conversion under high temperature treatment.
Conclusions and perspectives
The search for low-cost, highly active and durable ORR electrocatalysts is of great importance for fuel cell energy technology. TM/N/C materials derived from MOFs break away from an over fifty-year tradition of inert carbon-supported PGM-free catalyst and offer a promising solution in preparing high active site density and high surface area catalysts made of earthly-abundant elements. The well-defined pore structure, repeating TM–ligand coordination and wide selection of TM–ligand combinations render MOFs as unique precursors in rational design of the ORR catalysts with uniform distributed catalytic sites throughout the entire bulk phase. Furthermore, the correlation between pre-defined coordination structure and the final catalyst morphology and activity sheds the light in the conversion chemistry between the precursor and the final product. Since the initial reports on MOF-derived PGM-free catalysts,22,52–54 there has been an explosive growth of the research in this area. As we discussed in this review, many MOF-derived catalysts with controllable size, morphology, pore structure, surface area, and active site components were reported within the last few years. We have highlighted their electrocatalytic activity towards ORR as well as any reported single fuel cell testing and/or durability studies. This review is intended to provide a survey of the field to aid researchers in choosing future directions.Several general observations based on the current literature can be summarized as the following: among all MOF derived PGM-free catalysts, bimetallic or TM-doped MOFs with controllable, low level Fe or Co produced the best ORR catalysts, instating the importance of TM doping learned from early PGM-free catalyst development. When iron or cobalt incorporated into the same MOF, higher ORR catalytic activity is often achieved with Fe-doping. For the catalysts derived from the same MOF, TM-doped catalyst generally outperforms that of “TM-free” system in both acidic and alkaline media. In comparison, the ORR catalytic activity for the same PGM-free catalyst usually performs better against Pt/C in the alkaline medium than the acidic medium. It is not unusual for PGM-free catalyst having onset or half-wave potential higher than that of Pt/C in KOH electrolyte in RDE measurement. That being said, one should be mindful when comparing with Pt/C ORR potential which is sensitive to the platinum loading. For example, a correct half-wave potential for Pt/C should be 0.88 V (RHE). Although RDE and RRDE have been the popular methods in measuring ORR catalytic activity, they should really be considered only as the first level of screening. Ultimately, the usefulness of the catalyst can only be defined by its performance under fuel cell operating condition. Importance of the physical properties pertaining to the catalyst performance such as surface area, porosity, active site accessibility, etc. will become more apparent after the catalyst is integrated into the MEA and evaluated in fuel cell.
Although significant advancements have been made in a short period of time since the first MOF-derived ORR catalyst was discovered, the field is still at its infancy from the perspective of practical application, especially in the area of activity and durability measured at the fuel cell level. Although TM and, to a lesser degree, heteroatom doping demonstrated improved electronic and catalytic properties,155–163 the electrocatalytic activities of these new catalysts are still inferior to PGM-based materials.43,164,165 Although MOF offer a new venue in preparing “support-free” catalyst with the high active site density, this alone could not guarantee a practical catalyst since the overall activity is the product of site density and TOF. To enhance TOF through rational design of MOF precursor is a challenging proposition at the moment as the nature of the ORR active sites in these PGM-free catalysts is still hotly contested.123,132,147,166–170 In TM/N/C composite, TM–Nx motifs are most likely responsible for ORR activity through adsorption of intermediates (O2, HOO, H2O2, O, OH, etc.)171 although their exact structure, location, and interaction with intermediates are currently difficult to characterize. Coherent experimental and computational studies are needed to better understand the ORR site and mechanism in PGM-free catalyst and the result could serve as the foundation for the design of new MOF-derived catalysts. Durability, particularly under the acidic operating environment, represents a bigger challenge and the next frontier in PGM-free catalyst research. Many high performance catalysts included in this review article showed very limited stability when tested under the PEMFC operating condition. Similar to the activity improvement, the prerequisite for improving durability resides on the basic understanding of the active site structure and its deactivation mechanism. The information is very limited on both at present. All these represent ample amount of opportunities for the research and breakthrough in MOF-derived fuel cell catalysts.
Finally, we should point out that the MOF is not the only new generation template for PGM-free catalyst design. Other high surface area, metallated organic materials could also serve as precursors for ORR catalyst preparation. In particular, TM metallated porous organic polymer (POP) represents another emerging class of precursors that can be used for PGM-free ORR catalyst synthesis.172,173 POPs are typically synthesized through a crosslinking reaction between monomers containing N-ligated TM with or without a contorted core. Similar to MOF, TM–N site in POP is well-defined and uniformly distributed throughout high area micropores. High surface area can also be maintained after the thermal activation. Differentiating from MOF, POP does not contain transition metal based SBU such as Zn therefore can be activated at significantly lower temperature with higher yield and possibly better preservation of TM–N active site. Similar to MOF, we expect that POP could also serve as an important venue for rationally designed PGM-free catalyst and model system in understanding the ORR active site formation.
Acknowledgements
We wish to acknowledge the financial support by the U.S. Department of Energy's Office of Science and the Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office.References
- P. Trogadas, V. Ramani, P. Strasser, T. F. Fuller and M. O. Coppens, Angew. Chem., Int. Ed., 2016, 55, 122–148 CrossRef CAS PubMed.
- S. Guo and E. Wang, Acc. Chem. Res., 2011, 44, 491–500 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- C. Johnston, P. Piela and P. Zelenay, Handb. Fuel Cells, 2009, 48–70 CAS.
- F. Jaouen, J. Herranz, M. Lefevre, J.-P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka and A. Garsuch, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CAS.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- D. S. Su and G. Sun, Angew. Chem., Int. Ed., 2011, 50, 11570–11572 CrossRef CAS PubMed.
- U. I. Kramm, Encyclopedia of Applied Electrochemistry, Springer, 2014, pp. 909–918 Search PubMed.
- D. C. Higgins and Z. Chen, Can. J. Chem. Eng., 2013, 91, 1881–1895 CrossRef CAS.
- K. N. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212–1249 CAS.
- F. Jaouen, Non-Noble Met. Fuel Cell Catal., 2014, 29–118 CAS.
- G. Liu, X. Li, J.-W. Lee and B. N. Popov, Catal. Sci. Technol., 2011, 1, 207–217 CAS.
- J.-P. Dodelet, Electrocatalysis in Fuel cells, Springer, 2013, pp. 271–338 Search PubMed.
- D. Singh, J. King and U. S. Ozkan, Non-Noble Met. Fuel Cell Catal., 2013, 119–156 CAS.
- N. Daems, X. Sheng, I. F. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 CAS.
- K. Mamtani and U. S. Ozkan, Catal. Lett., 2015, 145, 436–450 CrossRef CAS.
- S. Zhang, K. Gong and L. Dai, Electrocatalysis in Fuel Cells, Springer, 2013, pp. 375–389 Search PubMed.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- F. Jaouen, M. Lefèvre, J.-P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed.
- M. Lefèvre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2000, 104, 11238–11247 CrossRef.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416–425 CrossRef PubMed.
- M. Lefèvre, J. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705–8713 CrossRef.
- G. Wu, Z. Chen, K. Artyushkova, F. H. Garzon and P. Zelenay, ECS Trans., 2008, 16, 159–170 CAS.
- T. S. Olson, K. Chapman and P. Atanassov, J. Power Sources, 2008, 183, 557–563 CrossRef CAS.
- H. T. Chung, C. M. Johnston, F. H. Garzon and P. Zelenay, ECS Trans., 2008, 16, 385–391 CAS.
- U. Koslowski, I. Herrmann, P. Bogdanoff, C. Barkschat, S. Fiechter, N. Iwata, H. Takahashi and H. Nishikori, ECS Trans., 2008, 13, 125–141 CAS.
- S. Pylypenko, S. Mukherjee, T. S. Olson and P. Atanassov, Electrochim. Acta, 2008, 53, 7875–7883 CrossRef CAS.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, J. Zhang, Z. Shi, A. L. Marques, E. P. Marques, S. Wu and J. Zhang, Electrochim. Acta, 2008, 53, 7703–7710 CrossRef CAS.
- H.-J. Zhang, X. Yuan, W. Wen, D.-Y. Zhang, L. Sun, Q.-Z. Jiang and Z.-F. Ma, Electrochem. Commun., 2009, 11, 206–208 CrossRef CAS.
- H. T. Chung, C. M. Johnston and P. Zelenay, ECS Trans., 2009, 25, 485–492 CAS.
- H.-J. Zhang, X. Yuan, L. Sun, X. Zeng, Q.-Z. Jiang, Z. Shao and Z.-F. Ma, Int. J. Hydrogen Energy, 2010, 35, 2900–2903 CrossRef CAS.
- H. T. Chung, C. M. Johnston, K. Artyushkova, M. Ferrandon, D. J. Myers and P. Zelenay, Electrochem. Commun., 2010, 12, 1792–1795 CrossRef CAS.
- Y. Ma, H. Zhang, H. Zhong, T. Xu, H. Jin, Y. Tang and Z. Xu, Electrochim. Acta, 2010, 55, 7945–7950 CrossRef CAS.
- H.-J. Zhang, Q.-Z. Jiang, L. Sun, X. Yuan, Z. Shao and Z.-F. Ma, Int. J. Hydrogen Energy, 2010, 35, 8295–8302 CrossRef CAS.
- A. Velázquez-Palenzuela, L. Zhang, L. Wang, P. L. Cabot, E. Brillas, K. Tsay and J. Zhang, Electrochim. Acta, 2011, 56, 4744–4752 CrossRef.
- T. Onodera, S. Suzuki, T. Mizukami and H. Kanzaki, J. Power Sources, 2011, 196, 7994–7999 CrossRef CAS.
- A. Y. Tsivadze, M. Tarasevich, A. Kuzov, L. Kuznetsova, O. Lozovaya and E. Davydova, Dokl. Phys. Chem., 2012, 442, 45–48 CrossRef CAS.
- H.-J. Zhang, X. Yuan, L. Sun, J. Yang, Z.-F. Ma and Z. Shao, Electrochim. Acta, 2012, 77, 324–329 CrossRef CAS.
- S.-T. Chang, H.-C. Hsu, H.-C. Huang, C.-H. Wang, H.-Y. Du, L.-C. Chen, J.-F. Lee and K.-H. Chen, Int. J. Hydrogen Energy, 2012, 37, 13755–13762 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- F. Jaouen and J.-P. Dodelet, Electrochim. Acta, 2007, 52, 5975–5984 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- S. Komini Babu, H. T. Chung, G. Wu, P. Zelenay and S. Litster, ECS Trans., 2014, 64, 281–292 CrossRef CAS.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- M. O'Keeffe, Chem. Soc. Rev., 2009, 38, 1215–1217 RSC.
- G. Goenaga, S. Q. Ma, S. W. Yuan and D. J. Liu, ECS Trans., 2010, 33, 579–586 CAS.
- D. J. Liu, G. Goenaga, S. Q. Ma, S. W. Yuan and J. L. Shui, ECS Trans., 2011, 30, 97–104 Search PubMed.
- S. Q. Ma, G. A. Goenaga, A. V. Call and D. J. Liu, Chem. – Eur. J., 2011, 17, 2063–2067 CrossRef CAS PubMed.
- A. F. Gross, E. Sherman and J. J. Vajo, Dalton Trans., 2012, 41, 5458–5460 RSC.
- L. Chong, G. A. Goenaga, K. Williams, H. M. Barkholtz, L. R. Grabstanowicz, J. A. Brooksbank, A. B. Papandrew, R. Elzein, R. Schlaf, T. A. Zawodzinski, J. Zou, S. Ma and D.-J. Liu, ChemElectroChem, 2016, 3, 1541–1545 CrossRef CAS.
- W. Xia, J. Zhu, W. Guo, L. An, D. Xia and R. Zou, J. Mater. Chem. A, 2014, 2, 11606–11613 CAS.
- X. J. Wang, J. W. Zhou, H. Fu, W. Li, X. X. Fan, G. B. Xin, J. Zheng and X. G. Li, J. Mater. Chem. A, 2014, 2, 14064–14070 CAS.
- S. L. Zhao, H. J. Yin, L. Du, L. C. He, K. Zhao, L. Chang, G. P. Yin, H. J. Zhao, S. Q. Liu and Z. Y. Tang, ACS Nano, 2014, 8, 12660–12668 CrossRef CAS PubMed.
- S. B. Yang, X. L. Feng, X. C. Wang and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed.
- Y.-X. Zhou, H.-B. Yao, Y. Wang, H.-L. Liu, M.-R. Gao, P.-K. Shen and S.-H. Yu, Chem. – Eur. J., 2010, 16, 12000–12007 CrossRef CAS PubMed.
- P. Ganesan, M. Prabu, J. Sanetuntikul and S. Shanmugam, ACS Catal., 2015, 5, 3625–3637 CrossRef CAS.
- N. Mahmood, C. Zhang, J. Jiang, F. Liu and Y. Hou, Chem. – Eur. J., 2013, 19, 5183–5190 CrossRef CAS PubMed.
- Q. Liu and J. Zhang, CrystEngComm, 2013, 15, 5087–5092 RSC.
- B. Chen, R. Li, G. Ma, X. Gou, Y. Zhu and Y. Xia, Nanoscale, 2015, 7, 20674–20684 RSC.
- H. Hu, L. Han, M. Yu, Z. Wang and X. W. Lou, Energy Environ. Sci., 2016, 9, 107–111 CAS.
- F. Bai, H. Huang, C. Hou and P. Zhang, New J. Chem., 2016, 40, 1679–1684 RSC.
- Y. Zhu, B. Zhang, Z. Feng and D. S. Su, Catal. Today, 2016, 260, 112–118 CrossRef CAS.
- W. Xia, R. Zou, L. An, D. Xia and S. Guo, Energy Environ. Sci., 2015, 8, 568–576 CAS.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006–15014 CrossRef.
- Q. Li, P. Xu, W. Gao, S. Ma, G. Zhang, R. Cao, J. Cho, H.-L. Wang and G. Wu, Adv. Mater., 2014, 26, 1378–1386 CrossRef CAS PubMed.
- Y. Hou, Z. H. Wen, S. M. Cui, S. Q. Ci, S. Mao and J. H. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- Z. Li, M. Shao, L. Zhou, R. Zhang, C. Zhang, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2016, 28, 2337–2344 CrossRef CAS PubMed.
- H.-S. Lu, H. Zhang, R. Liu, X. Zhang, H. Zhao and G. Wang, Appl. Surf. Sci., 2017, 392, 402–409 CrossRef CAS.
- D. Zhao, J. L. Shui, C. Chen, X. Q. Chen, B. M. Reprogle, D. P. Wang and D. J. Liu, Chem. Sci., 2012, 3, 3200–3205 RSC.
- J. Xi, Y. Xia, Y. Xu, J. Xiao and S. Wang, Chem. Commun., 2015, 51, 10479–10482 RSC.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- I. C. Halalay, B. Merzougui and A. Mance, ECS Trans., 2008, 16, 969–981 CAS.
- Y. Wu, S. Zhao, K. Zhao, T. Tu, J. Zheng, J. Chen, H. Zhou, D. Chen and S. Li, J. Power Sources, 2016, 311, 137–143 CrossRef CAS.
- H. Sun, H. Su, X. Ma, P. Zhang, X. Zhang, X. Dai, J. Gao, C. Chen and S.-G. Sun, Electrochim. Acta, 2016, 205, 53–61 CrossRef CAS.
- T. Palaniselvam, B. P. Biswal, R. Banerjee and S. Kurungot, Chem. – Eur. J., 2013, 19, 9335–9342 CrossRef CAS PubMed.
- D. Zhao, J. L. Shui, L. R. Grabstanowicz, C. Chen, S. M. Commet, T. Xu, J. Lu and D. J. Liu, Adv. Mater., 2014, 26, 1093–1097 CrossRef CAS PubMed.
- J. Tian, A. Morozan, M. T. Sougrati, M. Lefevre, R. Chenitz, J. P. Dodelet, D. Jones and F. Jaouen, Angew. Chem., Int. Ed., 2013, 52, 6867–6870 CrossRef CAS PubMed.
- L. Yang, N. Larouche, R. Chenitz, G. Zhang, M. Lefèvre and J.-P. Dodelet, Electrochim. Acta, 2015, 159, 184–197 CrossRef CAS.
- S.-T. Chang, C.-H. Wang, H.-Y. Du, H.-C. Hsu, C.-M. Kang, C.-C. Chen, J. C. S. Wu, S.-C. Yen, W.-F. Huang, L.-C. Chen, M. C. Lin and K.-H. Chen, Energy Environ. Sci., 2012, 5, 5305–5314 CAS.
- H. Barkholtz, L. Chong, Z. Kaiser, T. Xu and D.-J. Liu, Catalysts, 2015, 5, 955–965 CrossRef CAS.
- V. Armel, J. Hannauer and F. Jaouen, Catalysts, 2015, 5, 1333–1351 CrossRef CAS.
- J. Shui, C. Chen, L. Grabstanowicz, D. Zhao and D.-J. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10629–10634 CrossRef CAS PubMed.
- L. Shang, H. Yu, X. Huang, T. Bian, R. Shi, Y. Zhao, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 1668–1674 CrossRef CAS PubMed.
- D. J. Liu, http://https://www.hydrogen.energy.gov/pdfs/progress15/v_f_12_liu_2015.pdf, 2015.
- H. Barkholtz, L. Chong, Z. B. Kaiser and D.-J. Liu, Fuel Cells, 2016, 16, 428–433 CrossRef CAS.
- X. Wang, H. Zhang, H. Lin, S. Gupta, C. Wang, Z. Tao, H. Fu, T. Wang, J. Zheng, G. Wu and X. Li, Nano Energy, 2016, 25, 110–119 CrossRef.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- W. Wei, H. W. Liang, K. Parvez, X. D. Zhuang, X. L. Feng and K. Mullen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- P. Zhang, F. Sun, Z. H. Xiang, Z. G. Shen, J. Yun and D. P. Cao, Energy Environ. Sci., 2014, 7, 442–450 CAS.
- L. J. Zhang, Z. X. Su, F. L. Jiang, L. L. Yang, J. J. Qian, Y. F. Zhou, W. M. Li and M. C. Hong, Nanoscale, 2014, 6, 6590–6602 RSC.
- A. Aijaz, N. Fujiwara and Q. Xu, J. Am. Chem. Soc., 2014, 136, 6790–6793 CrossRef CAS PubMed.
- H.-x. Zhong, J. Wang, Y.-w. Zhang, W.-l. Xu, W. Xing, D. Xu, Y.-f. Zhang and X.-b. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- L. Ge, Y. Yang, L. Wang, W. Zhou, R. De Marco, Z. Chen, J. Zou and Z. Zhu, Carbon, 2015, 82, 417–424 CrossRef CAS.
- J. S. Li, Y. Y. Chen, Y. J. Tang, S. L. Li, H. Q. Dong, K. Li, M. Han, Y. Q. Lan, J. C. Bao and Z. H. Dai, J. Mater. Chem. A, 2014, 2, 6316–6319 CAS.
- S. Pandiaraj, H. B. Aiyappa, R. Banerjee and S. Kurungot, Chem. Commun., 2014, 50, 3363–3366 RSC.
- J. S. Li, S. L. Li, Y. J. Tang, K. Li, L. Zhou, N. Kong, Y. Q. Lan, J. C. Bao and Z. H. Dai, Sci. Rep., 2014, 4, 5130–5138 CAS.
- L. Zhang, X. Wang, R. Wang and M. Hong, Chem. Mater., 2015, 27, 7610–7618 CrossRef CAS.
- S. Fu, C. Zhu, Y. Zhou, G. Yang, J.-W. Jeon, J. Lemmon, D. Du, S. K. Nune and Y. Lin, Electrochim. Acta, 2015, 178, 287–293 CrossRef CAS.
- Y. a. Fu, Y. Huang, Z. Xiang, G. Liu and D. Cao, Eur. J. Inorg. Chem., 2015, 2100–2105 Search PubMed.
- E. Cruz-Silva, D. A. Cullen, L. Gu, J. M. Romo-Herrera, E. Muñoz-Sandoval, F. López-Urías, B. G. Sumpter, V. Meunier, J.-C. Charlier, D. J. Smith, H. Terrones and M. Terrones, ACS Nano, 2008, 2, 441–448 CrossRef CAS PubMed.
- F. Charreteur, F. Jaouen, S. Ruggeri and J.-P. Dodelet, Electrochim. Acta, 2008, 53, 2925–2938 CrossRef CAS.
- K. Wiesener, Electrochim. Acta, 1986, 31, 1073–1078 CrossRef CAS.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2004, 108, 11375–11383 CrossRef CAS.
- P. H. Matter, E. Wang, J.-M. M. Millet and U. S. Ozkan, J. Phys. Chem. C, 2007, 111, 1444–1450 CAS.
- N. P. Subramanian, X. Li, V. Nallathambi, S. P. Kumaraguru, H. Colon-Mercado, G. Wu, J.-W. Lee and B. N. Popov, J. Power Sources, 2009, 188, 38–44 CrossRef CAS.
- C. E. Szakacs, M. Lefèvre, U. I. Kramm, J.-P. Dodelet and F. Vidal, Phys. Chem. Chem. Phys., 2014, 16, 13654–13661 RSC.
- E. F. Holby, G. Wu, P. Zelenay and C. D. Taylor, J. Phys. Chem. C, 2014, 118, 14388–14393 CAS.
- E. Holby and C. Taylor, Appl. Phys. Lett., 2012, 101, 064102 CrossRef.
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- H. Tributsch, U. I. Koslowski and I. Dorbandt, Electrochim. Acta, 2008, 53, 2198–2209 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- E. F. Holby and P. Zelenay, Nano Energy, 2016 DOI:10.1016/j.nanoen.2016.05.025.
- K. Strickland, E. Miner, Q. Jia, U. Tylus, N. Ramaswamy, W. Liang, M.-T. Sougrati, F. Jaouen and S. Mukerjee, Nat. Commun., 2015, 6, 7343–7348 CrossRef CAS PubMed.
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2015, 138, 635–640 CrossRef PubMed.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CAS.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach and S. Mukerjee, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
- U. I. Kramm, M. Lefèvre, N. Larouche, D. Schmeisser and J.-P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978–985 CrossRef CAS PubMed.
- G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More and D. J. Myers, J. Mater. Chem., 2011, 21, 11392–11405 RSC.
- M. Hamzehie, L. Samiee, M. Fattahi, A. Seifkordi, F. Shoghi and A. Maghsodi, Renewable Energy, 2015, 77, 558–570 CrossRef CAS.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J.-P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618–8619 CrossRef CAS PubMed.
- K. Artyushkova, A. Serov, S. Rojas-Carbonell and P. Atanassov, J. Phys. Chem. C, 2015, 119, 25917–25928 CAS.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby and P. Zelenay, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- J. Herranz, F. Jaouen, M. Lefevre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach and P. Bertrand, J. Phys. Chem. C, 2011, 115, 16087–16097 CAS.
- A. Muthukrishnan, Y. Nabae, T. Okajima and T. Ohsaka, ACS Catal., 2015, 5, 5194–5202 CrossRef CAS.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- D.-S. Yang, M. Y. Song, K. P. Singh and J.-S. Yu, Chem. Commun., 2015, 51, 2450–2453 RSC.
- J. Yang, D.-J. Liu, N. N. Kariuki and L. X. Chen, Chem. Commun., 2008, 329–331 RSC.
- D.-J. Liu, J. Yang and D. J. Gosztola, ECS Trans., 2007, 5, 147–154 CAS.
- E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. C, 2010, 114, 15306–15314 CAS.
- R. Franke, D. Ohms and K. Wiesener, J. Electroanal. Chem. Interfacial Electrochem., 1989, 260, 63–73 CrossRef CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- J. Van Veen, H. Colijn and J. Van Baar, Electrochim. Acta, 1988, 33, 801–804 CrossRef CAS.
- M. Lefèvre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2005, 109, 16718–16724 CrossRef PubMed.
- U. Tylus, Q. Jia, K. Strickland, N. Ramaswamy, A. Serov, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2014, 118, 8999–9008 CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- N. Cheng, C. Kemna, S. Goubert-Renaudin and A. Wieckowski, Electrocatalysis, 2012, 3, 238–251 CrossRef CAS.
- K. Artyushkova, S. Levendosky, P. Atanassov and J. Fulghum, Top. Catal., 2007, 46, 263–275 CrossRef CAS.
- K. Artyushkova, S. Pylypenko, T. S. Olson, J. E. Fulghum and P. Atanassov, Langmuir, 2008, 24, 9082–9088 CrossRef CAS PubMed.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- U. Kramm, I. Abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 158, B69–B78 CrossRef CAS.
- J.-L. Shui, N. K. Karan, M. Balasubramanian, S.-Y. Li and D.-J. Liu, J. Am. Chem. Soc., 2012, 134, 16654–16661 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang and D. S. Su, ChemSusChem, 2014, 7, 1240–1250 CrossRef CAS PubMed.
- L. Zhang, X. Chen, J. Guan, Y. Jiang, T. Hou and X. Mu, Mater. Res. Bull., 2013, 48, 3485–3491 CrossRef CAS.
- Y. Marco, L. Roldán, S. Armenise and E. García-Bordejé, ChemCatChem, 2013, 5, 3829–3834 CrossRef CAS.
- S. Dey, P. Chithaiah, S. Belawadi, K. Biswas and C. Rao, J. Mater. Res., 2014, 29, 383–391 CrossRef CAS.
- Z. Xiang, D. Cao, L. Huang, J. Shui, M. Wang and L. Dai, Adv. Mater., 2014, 26, 3315–3320 CrossRef CAS PubMed.
- Y. Lin, Y. Zhu, B. Zhang, Y. A. Kim, M. Endo and D. S. Su, J. Mater. Chem. A, 2015, 3, 21805–21814 CAS.
- S. Azevedo, C. Chesman and J. Kaschny, Eur. Phys. J. B, 2010, 74, 123–128 CrossRef CAS.
- S. R. Stoyanov, A. V. Titov and P. Král, Coord. Chem. Rev., 2009, 253, 2852–2871 CrossRef CAS.
- M. G. Mashapa and S. S. Ray, J. Nanosci. Nanotechnol., 2010, 10, 8180–8184 CrossRef CAS PubMed.
- Z. Liu, L. Ma, J. Zhang, K. Hongsirikarn and J. G. Goodwin Jr, Catal. Rev., 2013, 55, 255–288 CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- W. Vielstich, A. Lamm and H. A. Gasteiger, Handbook of fuel cells: fundamentals, technology, and applications, John Wiley & Sons, 2009 Search PubMed.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS.
- V. Nallathambi, J.-W. Lee, S. P. Kumaraguru, G. Wu and B. N. Popov, J. Power Sources, 2008, 183, 34–42 CrossRef CAS.
- R. Zhou, M. Jaroniec and S. Z. Qiao, ChemCatChem, 2015, 7, 3808–3817 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. Yuan, J.-L. Shui, L. Grabstanowicz, C. Chen, S. Commet, B. Reprogle, T. Xu, L. Yu and D.-J. Liu, Angew. Chem., Int. Ed., 2013, 52, 8349–8353 CrossRef CAS PubMed.
- Z.-S. Wu, L. Chen, J. Liu, K. Parvez, H. Liang, J. Shu, H. Sachdev, R. Graf, X. Feng and K. Müllen, Adv. Mater., 2014, 26, 1450–1455 CrossRef CAS PubMed.
- P. P. Su, H. Xiao, J. Zhao, Y. Yao, Z. G. Shao, C. Li and Q. H. Yang, Chem. Sci., 2013, 4, 2941–2946 RSC.
- N. Larouche, R. Chenitz, M. Lefevre, E. Proietti and J. P. Dodelet, Electrochim. Acta, 2014, 115, 170–182 CrossRef CAS.
- H. Barkholtz, L. Chong, Z. B. Kaiser, T. Xu and D.-J. Liu, Int. J. Hydrogen Energy, 2016 DOI:10.1016/j,ijhydene.2016.08.193.
| This journal is © The Royal Society of Chemistry 2017 |