
Metal–organic frameworks: a novel host platform for enzymatic catalysis and detection
Effrosyni
Gkaniatsou
a,
Clémence
Sicard
*a,
Rémy
Ricoux
b,
Jean-Pierre
Mahy
b,
Nathalie
Steunou
 *a and
Christian
Serre
ac
*a and
Christian
Serre
ac
aInstitut Lavoisier de Versailles, UMR CNRS 8180, Université de Versailles St Quentin en Yvelines, Université Paris Saclay, 45 avenue des Etats-Unis, 78035 Versailles Cedex, France. E-mail: clemence.sicard@uvsq.fr; nathalie.steunou@uvsq.fr; Fax: +33-1-39-25-44-52; Tel: +33-1-39-25-43-71 Tel: +33-1-39-25-43-73
bLaboratoire de Chimie Bioorganique et Bioinorganique, Institut de Chimie Moléculaire et des Matériaux d’Orsay, UMR 8182, Université Paris Sud, Université Paris Saclay, rue du doyen Georges Poitou, 91405 Orsay, France
cInstitut des Matériaux Poreux de Paris, FRE CNRS 2000, Ecole Normale Supérieure, Ecole Supérieure de Physique et de Chimie Industrielles de Paris, Paris Research University, Paris, France
First published on 22nd November 2016
Abstract
The use of metal–organic frameworks (MOFs) as immobilization matrices for enzymes as a platform for emerging applications is reported. In addition to an overview of strategies developed to prepare enzyme–MOF biocomposites, the features that render MOFs interesting matrices for bio-immobilization are highlighted along with their potential benefits beyond a solid-state support in the design of innovative biocomposites.
1. Introduction
Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are an attractive class of porous crystalline hybrid solids, built from almost endless combinations of inorganic nodes and organic linkers with complexing groups (carboxylates, azolates, phosphonates…). Chemical and structural features are easily tunable, resulting in a vast array of micro- or meso-porous solids displaying regular porosity (typically SBET within the 100–6000 m2 g−1 range). These tailored porous materials show great promise for numerous applications such as gas storage, separation, catalysis, sensing, biomedicine, among others.1–3A new range of potential applications has recently emerged using MOFs as immobilization matrices for biomolecules, most notably enzymes. Enzymes are well known as being efficient and selective catalysts, ubiquitous in a wide range of areas from chemical synthesis to drug delivery. However, their use often requires immobilization on a solid support for ease of separation and recovery (improving cost-effectiveness), and may enhance enzyme activity.4,5 The immobilization method must retain maximal enzyme activity while not hindering the diffusion of analytes to and from the enzyme active site. To date, no method has been developed that entirely fulfills these requirements, therefore tailored solutions are needed for each unique enzyme–support-application.6 The development of novel immobilization matrices that can stabilize enzymes under a wide range of operating conditions remains of key interest for enzymatic applications.
As a result of the fascinating physico-chemical properties of MOFs (e.g. large and tuneable pore size, pore shape, pore volume, and polar/apolar balance), they are perfectly suited to create a stabilizing microenvironment for enzymes through specific host–guest interactions and/or confinement effect, as already observed with other organic molecules.7 Since the first report in 2006,8 the number of studies dealing with enzyme–MOF composites per year has increased exponentially and various combination approaches have now been developed.
In this review, we describe the current methods for enzyme immobilization in MOF materials and provide examples of enzyme–MOF bioreactors and biosensors. The main objective of this review is to outline key features of MOFs that improve the overall performance of biocomposites, as well as highlight the numerous remaining challenges. An extensive list of enzyme–MOF biocomposites reported to date is provided in Table 1, and more detailed descriptions of these materials can be found in recently published reviews.9–12
| Method | Enzyme | MOF | Application/remarks | Ref. |
|---|---|---|---|---|
| a Cross-linking corresponds to the reticulation of the protein, whereas covalent binding corresponds to a chemical bonding between the enzyme and the support material. | ||||
| Adsorption | Lipase | UiO-66(Zr), carbonized MIL-53(Al) | Bioreactor (warfarin synthesis) | 52 |
| Adsorption | Lipase | HKUST-1 | Bioreactor (esterification) | 53 |
| Adsorption | Laccase | MMU (Zr-MOF) | Bioreactor (proof of concept) | 54 |
| Adsorption | Trypsin | HKUST-1 | Bioreactor (magnetic separation) | 40 |
| Adsorption | Glucose dehydrogenase | ZIF-70, ZIF-7, ZIF-8, ZIF-67, ZIF-68 | Electrochemical biosensing of glucose | 20 |
| Adsorption (chitosan film) | Tyrosinase | Cu-MOF Cu(bdc)(ted)0.5 | Electrochemical biosensing of bisphenol | 41 |
| Adsorption (chitosan film) | Tyrosinase | Cu-MOF Cu(bdc)(ted)0.5 | Electrochemical biosensing of bisphenol | 55 |
| Indirect adsorption | Trypsin–NDC | UiO-66 (Zr), CYCU-4(Al) | Bioreactor (proteolysis) | 21 |
| Indirect adsorption | Trypsin–FTIC | CYCU-4(Al), MIL-101(Cr) | Bioreactor (proteolysis) | 56 |
| Glutaraldehyde cross-linkinga | Beta-glucosidase | MIL-53(Al)-NH3 | Prove interest of method | 57 |
| Glutaraldehyde cross-linkinga | Glucose oxidase | MIL-100(Fe), MIL-100(Cr, Al), MIL-127(Fe) | Electrochemical biosensing of glucose | 45 |
| Glutaraldehyde cross-linkinga | Laccase | MIL-100(Fe) | Oxygen reduction reaction | 47 |
| Glutaraldehyde cross-linkinga | Hydrolase | UiO-66-NH2 | Bioreactor (enantiopure octanediol) | 58 |
| Covalent binding (EDC/NHS) | Hemoglobin | MIL-125(Ti)-NH2 | Oxygen binding (proof of concept) | 48 |
| Covalent binding (EDC/NHS) | Trypsin | ZIF-8 monolith | Bioreactor (proteolysis) | 59 |
| Covalent binding (DCC) | Trypsin | MIL-88B(Cr)-NH2 MIL-88B(Cr), MIL-101(Cr) | Bioreactor (proteolysis) | 22 |
| Covalent binding (DCC/EDC) | EGFP, Lipase | IRMOF-3 | Bioreactor (transesterification) | 60 |
| Cage inclusion | Vitamin B12, GFP, myoglobin | IRMOF-74-Y | Proof of concept | 28 |
| Cage inclusion | Microperoxidase 11 | Tb-TATB | Bioreactor (proof of concept) | 26 |
| Cage inclusion | HRP, Cyt c, microperoxidase 11 | PCN-333(Al) | Bioreactor (proof of concept) | 29 |
| Cage inclusion | Cytochrome c | Tb-TATB | Mechanism comprehension | 27 |
| Cage inclusion | Microperoxidase 11 | Tb-TATB | Mechanism comprehension | 38 |
| Cage inclusion | Myoglobin | Tb-TATB | Bioreactor (size selectivity) | 42 |
| Cage inclusion | Vitamin B12, Cyt c, myoglobin, horseradish peroxidase | POST-66(Y) | Bioreactor (proof of concept) | 31 |
| In situ synthesis (with polyvinylpyrrolidone) | Cytochrome c horseradish peroxidase, lipase | ZIF-8, ZIF-10 | Biosensor for organic peroxides | 32 |
| In situ synthesis | Bovine serum albumin, OVA, lipase, hemoglobin, lysozyme, insulin, HRP, trypsin, urease… | ZIF-8, HKUST-1, Eu/Tb-BDC, MIL-88A | Bioreactor (proof of concept), controlled release | 33 |
| In situ synthesis | Catalase | ZIF-90 | Bioreactor (size sheltering) | 61 |
| In situ synthesis (microcapsule) | Enhanced green fluoresecent protein, lipase, beta-galactosidase | ZIF-8 | Bioreactor (size selectivity, magnetic separation) | 37 |
| In situ synthesis (micelles) | Horseradish peroxidase | ZIF-8 | Bioreactor (proof of concept) | 62 |
| In situ synthesis (capsules) | Glucose oxidase | ZIF-8 | Electrochemical biosensing of glucose | 63 |
| In situ synthesis | Bovine serum albumin | ZIF-8 | Patterning, fingerprint detection | 64 |
| In situ synthesis (PVP) | Glucose oxidase | ZIF-8 | Colorimetric biosensing of glucose | 65 |
| In situ synthesis | Glucose oxidase | ZIF-8 (+ polydopamine) | Bioreactor (proof of concept) | 66 |
| In situ synthesis | Glucose oxidase + HRP | ZIF-8 | Multi-enzyme systems | 34 |
| In situ synthesis | Dehydrogenase, HRP, AchE | MIL-88A(Fe) hollow sphere | Bioreactor (proof of concept) | 36 |
| In situ synthesis | Urease | ZIF-8 | Comparison of 2 strategies | 35 |
2. Strategies for enzyme immobilization with MOFs
Enzyme–MOF biocomposites are designed to retain active enzymes with maximal loading and minimal leaching, and are typically produced either by surface adsorption, covalent binding, cage inclusion or in situ synthesis (Fig. 1). While the first three methods involve a similar immobilization process to that reported for other porous or dense solid-state support material (e.g. biopolymers, layered double hydroxides, oxides, mesoporous oxides),4,5,13,14in situ synthesis is specific to MOF materials due to the possible use of gentle synthesis conditions that prevent enzyme degradation.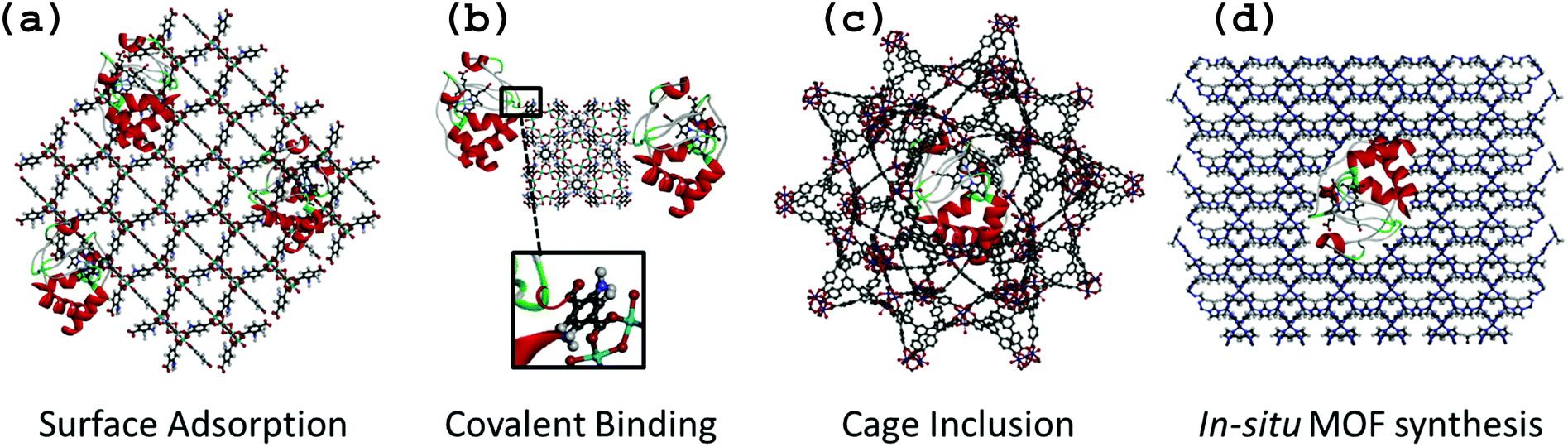 | ||
| Fig. 1 Schematic representation of the different immobilization methods for cytochrome c (water molecules were omitted for clarity): (a) surface adsorption on MIL-125-NH2 (view along 010 direction); (b) covalent binding on MIL-125-NH2 (view along 001 direction); (c) cage inclusion in a cage of MIL-100-Fe-BTB (BTB = 1,3,5-Tris(4-carboxyphenyl)benzene) (view on the 5 member ring window); (d) in situ MOF synthesis with ZIF-8 (view along 110 direction). | ||
It is noteworthy to mention the remarkable approach proposed by Sontz et al.,15 in which a modified protein (Ferritin) was used as the MOF metallic building block with preserved bioactivity. However, this work will not be discussed further since the methodology is relatively complex and beyond the scope of the present review.
Surface adsorption
Surface adsorption is commonly used for the preparation of bioelectrodes relying on the physical adsorption of enzymes onto the material via van der Waals forces and hydrophobic and electrostatic interactions (Fig. 1a). Due to possible leaching of proteins, this method can be coupled to a reticulation step of the protein by using cross-linking agents such as glutaraldehyde.4,16,17 As previously reported for other supports, surface adsorption offers the advantage of entrapping a large and precisely known amount of enzyme at the surface of a material, and therefore is particularly well suited for classical electrodes (Pt, glassy carbon, ITO…) as well as for interdigitated microelectrodes.18,19 By mixing a solution of pre-formed MOF with enzyme, Ma et al. studied the surface adsorption of glucose dehydrogenase (GDH) and methylene green (MG) on a series of ZIFs (Zeolitic Imidazole Frameworks) with various surface areas and functional groups.20 They evaluated their GDH adsorption capacities and showed that ZIF-68 [Zn(benzimidazole)(nitroimidazole)] and ZIF-70 [Zn(imidazole)1.13(nitroimidazole)0.87] led to the highest adsorbed amount of GDH. This was attributed to their large external surface area as well as to their strong hydrophobic character. By FT-IR spectroscopy, the presence of donor–acceptor and hydrogen bonding interactions between GDH and ZIF-70 was observed, showing that GDH adsorption on ZIF-70 was not only due to physical interactions, but also to chemical interactions. This example is a good illustration of the importance of the hybrid nature and tunability of the functional groups of MOFs when used as immobilization matrices.An alternative strategy exploits the porosity of MOFs using an enzyme covalently attached to a small molecule. The size of the small molecule allows diffusion through the pores of the MOF, while the much larger enzyme remains adsorbed on the outer surface. Liu and co-workers used this technique to immobilize Trypsin (Try), an enzyme commonly used for protein digestion, through covalent coupling with the dye 4-chloro-7-nitrobenzofurazan (NBD).21 Enzyme loading and distribution could be monitored from the fluorescence properties of the dye, and multipoint anchoring could be achieved by attaching two or more dye molecules per enzyme. The resulting trypsin-based bioreactor demonstrated high efficiency for protein digestion. In another study, the topical UiO-66(Zr) (Zr6O4(OH)4[benzenedicarboxylate]6) was used to produce a NBD-Try-UiO-66 material exhibiting high proteolytic activity that is stable over several cycles. The authors compared the performance of NBD-Try-UiO-66(Zr) to either a purely surface adsorbed sample (Try-UiO-66) or a composite prepared with a larger dye, fluorescein isothiocyanate (FITC-Try-UiO-66) for which significant enzyme leaching was observed. Thus, if the dye is carefully chosen to match the window size, this method is a relatively easy way to achieve increased stability.
Covalent binding
The chemical bonding of biomolecules to solid matrices has been extensively studied,5 so it is not surprising that this method is also used to immobilize enzymes on MOF surfaces. Furthermore, MOF surfaces present a wide array of functional groups (e.g. free carboxyl, amino, hydroxyl groups etc.) that can be activated for coupling with reactive groups on the surface of the enzyme (Fig. 1b).A typical example of covalent immobilization is the work of Shih et al., who used the highly stable mesoporous MIL-101(Cr) (Cr3O(OH)(H2O)2[benzenedicarboxylate]3) and microporous MIL-88B(Cr) (polymorph of MIL-101(Cr)) and its amino-functionalized analogue MIL-88B-NH2(Cr) (Cr3O(OH)(H2O)2[benzenedicarboxylate-NH2]3) for the grafting of Try.22 The goal was to determine how the functionalization and structural properties of MOFs may impact the stabilization of enzyme activity. Due to the harmful effect of Cr on the environment, such materials may not however be considered for any biotechnological application. The MOFs were first activated using dicyclohexyl carbodiimide (DCC), which allowed them to react with the amino groups of Try to form the MOF–Try conjugate. The various Try–MOFs were used for the digestion of Bovine Serum Albumine (BSA), and it was found that Try-MIL-88B-NH2(Cr) showed an enzymatic activity similar to that of the free Try, whereas activity of Try-MIL-88B(Cr) and Try-MIL-101(Cr) was reduced by almost half. The authors argued that the amino functionalization increased the surface hydrophilicity, which helped the stabilization of the enzyme (or BSA molecules) through the formation of additional hydrogen bonds. This highlights how varying the functionalization of MOFs can be an important tool for improving biocomposite performance. The authors also performed simple physical adsorption of Try onto MIL-88B-NH2, which resulted in minimal activity, demonstrating that despite a rather complicated approach with multiple steps for activation of functional groups (during which the enzyme can be deactivated), covalently bonding enzyme molecules to MOFs can be helpful to obtain robust biocomposites with minimal leaching.
Cage inclusion
The cage inclusion approach involves the encapsulation of small enzymes inside the cages of mesoporous MOFs by diffusion (Fig. 1c). This method has been extensively used to encapsulate active ingredients, inorganic nanoparticles or molecular complexes within MOF pores.7,23 In contrast to adsorption, the encapsulation of enzymes may significantly enhance their stabilization, even under harsh conditions or in unnatural environments. The isolation of protein molecules not only prevents their aggregation and thereby deactivation, but also provides a protective environment that limits the influence of denaturation factors (e.g. toxic solvents, strongly acidic or basic media, temperature). Moreover, the confinement of enzymes in 3D microenvironments is known to increase stability and minimize protein unfolding.24 This entrapment of enzymes also reduces leaching of enzymes from the support, which can be regarded as one of the main shortcomings of numerous other immobilization methods.25 On the other hand, a dense host matrix may hinder the accessibility to the entrapped biomolecules and thus the mass-transfer efficiency of analytes may be strongly limited.25 Due to their high porosity, MOFs should be very interesting host matrices although encapsulation is not feasible for large enzymes due to size limitations.The entrapment approach is simple and rapid, and only requires mixing pre-formed MOF with an enzyme in aqueous solution and stirring at low temperatures (RT-37 °C). Given the typical dimensions of enzymes – even microenzymes – one shall consider here only MOFs with large pores (cages < 4 nm) or ultra-large pores (cages > 4 nm) which are quite rare, compared to the vast number of common microporous MOFs reported in the literature. One of the earliest reports was by Lykourinou et al., based on the mesoporous Tb-TATB MOF (TATB = triazine-1,3,5-tribenzoate), with cage diameters of 3.9 and 4.7 nm, accessible by windows of 1.3 and 1.7 nm diameters, respectively, to immobilize the Microperoxidase-11 (MP-11, 3.3 × 1.7 × 1.1 nm).26 The resulting composite showed better peroxidase activity and reusability than free MP-11, which tends to aggregate in aqueous solutions thus losing its activity. It is important to note that enzyme encapsulation within MOF cages requires enzyme diffusion through windows that are smaller in size than the cavity itself. The same group demonstrated the possibility of encapsulating cytochrome c, a protein that is significantly larger (2.6 × 3.2 × 3.3 nm) than the Tb-MOF windows (1.3 and 1.7 nm).27 A mechanism similar to translocation process was proposed, where the enzyme undergoes conformational changes while migrating into the MOF cavities. The enzyme conformation within the cages is neither its native, nor its denatured form.27
The use of extended ligand is the easiest way to obtain expanded isoreticular structures of already known MOFs as shown in Fig. 2. Deng et al. reported the expansion of MOF-74 (M2(2,5-dioxidoterephthalate, M = Zn2+, Mg2+)) by increasing the number of phenyl rings present on the organic spacer. Vitamin B12 (2.7 nm), myoglobin (Mb, 2.1 × 3.5 × 4.4 nm) and Green Fluorescent Protein (GFP, 3.4 × 4.5 nm) were encapsulated in various extended IRMOF-74-Y (Y = number of phenyl rings on the ligand).28 Feng et al.29 developed a series of MOFs that can be described as extended isoreticular forms of the MIL-100 topology, (MIL-100: M3O(OH)(H2O)2[benzenetricarboxylate]2, with M = Cr3+, Al3+, Fe3+…) as previously reported by using the BTB ligand30 (BTB = 1,3,5-tris(4-carboxylphenyl)benzene). PCN-333(Al) (Al3O(OH)(H2O)2[TATB]2, TATB = triazine-1,3,5-tribenzoate) exhibits two types of mesoporous cages – smaller cages of 4.2 nm, accessible through pentagonal windows of 2.6 nm, and larger cages of 5.5 nm with two types of windows (i.e. pentagonal windows of 2.6 nm and hexagonal windows of 3 nm). It was chosen as a host matrix for horseradish peroxidase (HRP, 4.0 × 4.4 × 6.8 nm), cytochrome c (Cyt c, 2.6 × 3.2 × 3.3 nm) and MP-11. Immobilized HRP and Cyt c showed better catalytic activity in water media compared to that of the free enzymes. In contrast, MP-11 showed decreased activity after its immobilization, likely because of the presence of several enzyme molecules within a cage that may lead to aggregation. The composites exhibited experimental loadings in agreement with the maximum theoretical capacities and maintained their activity after several catalytic cycles, showing almost no leaching.29
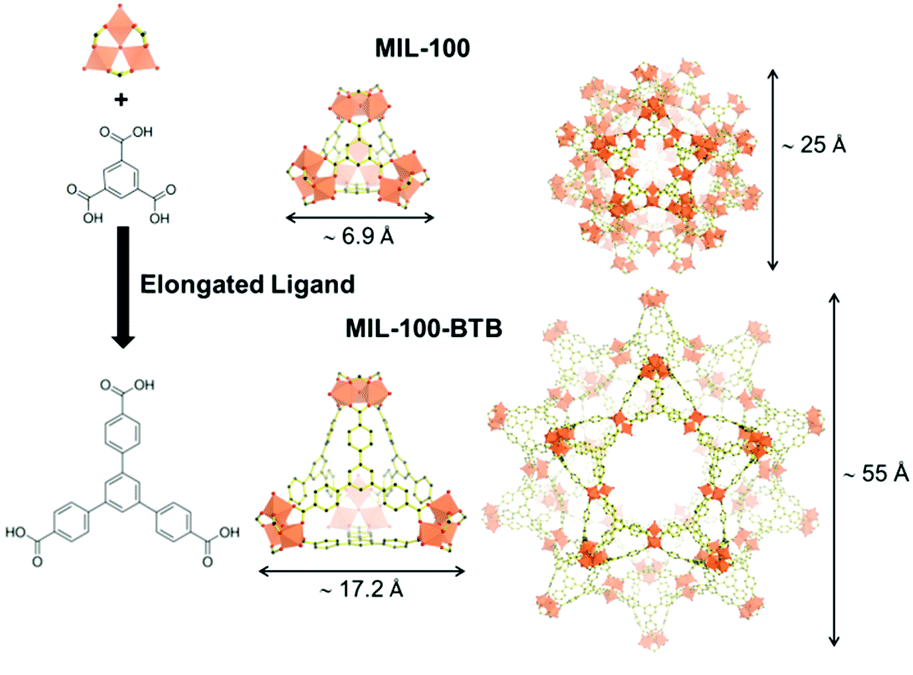 | ||
| Fig. 2 Schematic representation of the elongated ligand approach to increase the pores size of MIL-100 (top) into MIL-100-BTB (bottom). The assembly of the organic ligand (right) with oxo-centered metal trimers (top left hand corner) is forming the supertetrahedron sub-unit (ST, middle). Small mesoporous cages (left) result from the combination of 20 ST. Dimensions of the ST and cages are included. Iron polyhedra, carbon and oxygen are represented in orange, black and red, respectively (hydrogen atoms are omitted for clarity). Adapted from Horcajada et al.30 | ||
Despite recent promising results obtained with mesoporous MOFs based composites, the vast majority of these ultra-large mesoporous MOFs, with the exception of MIL-100/101 series, remain relatively unstable under various conditions (solvents, pH, air etc.) for a prolonged period of time. This drastically hampers their practical use as enzyme immobilization matrices under real conditions. Therefore, improvement in the stability of ultra-large mesoporous MOF is needed to fully exploit the potential of this immobilization strategy and produce industrially viable bio-composite materials.
An alternative was recently proposed by Kim et al., who synthesized a microporous MOF, POST-66(Y) ({Y4(H2O)}3[methyl substituted truxene tricarboxylate]8(OH)6(NO3)6), which was then transformed into a micro/mesoporous structure via a post-synthetic procedure involving the partial hydrolysis and degradation of the MOF in water.31 Mesopores ranging from 3 to 20 nm are then formed and coexist with the micropores of the initial MOF structure. In contrast to untreated POST-66(Y), several guest molecules (vitamin B12, Cyt c, Mb and HRP) were successfully encapsulated in the post-treated MOF. The catalytic activity of entraped HRP was maintained and the composite showed better stability in organic solvents (e.g. DMSO) than free HRP and good recyclability as well. This strategy is a valuable approach for enzyme entrapment in a range of microporous MOFs, whereas the cage inclusion method is restricted to only ultra-large mesoporous MOFs. However, there are several key limitations such as long-term stability of the water unstable MOF, minimal control over the transformation of the framework, and the lack of reproducibility of mesopore distribution.
In situ MOF synthesis
For the “in situ MOF synthesis” approach, nucleation and growth of MOF formation and enzyme immobilization take place simultaneously in a single step. The enzyme and MOF precursors (ligands and metal ions, and eventually additives) are mixed together in solution, resulting in the crystallization of MOF particles with embedded enzymes, either at the particle surface or as a core–shell material (Fig. 1d). Similar to the cage inclusion protocol, this method provides a 3D microenvironment for the enzyme with minimal leaching, but a major advantage of “in situ MOF synthesis” is that a greater number of enzymes can be considered since immobilization is not restrained by the size of the pores of the MOF. The major limitation of the “in situ MOF synthesis” is that it is restricted to MOFs capable of forming under mild conditions (preferably aqueous solutions and low temperature) to prevent enzyme denaturation. This is why Zeolitic Imidazolate Frameworks (ZIFs), which are known in most cases to form easily under mild biocompatible conditions, have been used. The first example with the “in situ MOF synthesis” approach was reported by Lyu et al., who successfully immobilized Cyt c in ZIF-8 (Zn[imidazole]2).32 It is important to note that Cyt c was mixed with the polymer polyvinylpyrrolidone (PVP) before being mixed with the ZIF's components in order to protect the enzyme from denaturation and aggregation, and enabling the use of methanol as a solvent. The peroxidase activity of Cyt c–ZIF-8 showed a 10-fold increase as compared to the free enzyme.Liang et al., reported an interesting alternative method for the preparation of bovine serum albumine–ZIF-8 composites using water as the sole solvent and thus removing the need for protective PVP.33 It is suggested that crystallization of MOF particles is induced by the affinity between MOF molecular precursors and functional groups on the biomolecules via intermolecular hydrogen bonding, electrostatic and hydrophobic interactions, leading to local accumulation and thus nucleation and growth of the particles. The authors demonstrated the versatility of this approach through the use of a wide range of biomolecules (ovalbumin, ribonuclease A, human serum albumin, pyrroloquinoline quinone-dependent glucose dehydrogenase, lipase, hemoglobin, lysozyme, insulin, HRP, trypsin, urease and oligonucleotide) and also numerous MOFs, including the metal polycarboxylates HKUST (Cu3[(H2O)3benzenetricarboxylate]2), MIL-88A(Fe) (Fe3O(OH)(H2O)2[fumarate]3) and Eu/Tb2[1,4-(benzenedicarboxylate)]3(H2O)4. The nature of the biomolecule strongly impacted the morphogenesis of the MOF particles, leading to a series of ZIF-8 crystals with varying morphologies. Such observation is consistent with a “biomineralization” or biocrystallization process for the nucleation and growth of MOFs. Enzyme leaching was negligible and the composites were shown to maintain enzymatic activity, even under harsh conditions.
The work of Wu et al. was also based on this approach and described a multi-enzymatic system with ZIF-8 particles.34 Glucose oxidase (GOx) and HRP were mixed with the ZIF-8 precursors at room temperature, to yield the final product GOx&HRP@ZIF-8. The unique aspect of this approach is the possibility to perform enzymatic cascade reactions within the particle. GOx converts glucose into D-glucono-1,5-lactone that further hydrolyses into gluconic acid, generating H2O2, which is then used by HRP to oxidise the ABTS substrate. Catalytic tests showed that GOx&HRP@ZIF-8 had higher activity than GOx@ZIF-8 or HRP@ZIF-8, which was attributed to the immediate use by HRP of the H2O2 produced by the GOx reaction, confirming thus the importance of confining bi-enzymatic systems in close proximity.
Although these preliminary studies appear promising, they rely on the use of MOFs that can be prepared under simple and biocompatible synthetic conditions. Therefore, unless drastic improvements to synthesis conditions of MOFs are reached in a near future, this immobilization method will be limited to only a few MOF candidates. In addition, many aspects need further investigation and optimization, such as the enzyme spatial localization and dispersion within the MOF particles and the control of MOF coating thickness, which are critical parameters for biocatalytic applications. Enzyme spatial localization within the MOFs has recently been investigated by electron microscopy studies.31,35 The effect of PVP on the location of enzymes was clearly evidenced, showing the presence of enzymes at the outer surface of MOFs in the presence of PVP or the homogeneous distribution of enzymes in the MOF crystal in the absence of PVP.
An alternative “indirect” in situ MOF synthesis approach was proposed by Jeong et al. through the synthesis of MIL-88A(Fe) hollow spheres via interfacial reaction in-droplet microfluidics.36 These spheres, with controlled size ranging from 35 to 2000 μm, were prepared by an in situ approach in which an aqueous phase containing the metal precursor and poly(vinyl alcohol) (to stabilize droplets) in water, and an organic phase containing the organic linker (fumaric acid) and tributylamine (to deprotonate the ligands) in octanol, were separately introduced into a microfluidic channel. This resulted in the formation of iron chloride microdroplets within a stream of organic solvent containing the fumaric acid that finally yielded the hollow spheres of MIL-88A. The spheres were used as encapsulation matrices for three different enzymes (glycerol dehydroxygenase, HRP and acetylcholinesterase) and results showed the entrapped enzymes maintained activity while having better recyclability than free ones. This method offers the opportunity of solubilizing the ligand in a non-aqueous solvent without enzyme deterioration, which allows a wider range of MOF materials to be synthesized.
In another “indirect” in situ MOF synthesis approach reported by Huo et al., MOF particles were also formed in situ but around a pre-formed microcapsule.37 A magnetic MOF bioreactor was prepared, containing Candida antarctica lipase B (CALB), using inversed phase Pickering emulsions. First, the emulsion containing CALB (1.5% agarose hydrogel droplets in a continuous paraffin oil phase) was stabilized with UiO-66 nanoparticles, acting also as a surface for further MOF nucleation and growth. Afterwards, a ZIF-8 shell was synthesized in situ around the capsule's core in order to stabilize it and to provide size selectivity toward various substrates. It is worth noting that Fe3O4 nanoparticles were also introduced in the initial emulsion to induce magnetic separation of the bioreactor. This method is not as straightforward as the direct in situ MOF growth, since several steps are needed, but the pre-stabilization of the enzyme within microdroplets enables the use of synthetic conditions that are not biocompatible (e.g. butanol) and the incorporation of additional nanoparticles (Fe3O4). It therefore opens up the possibility of designing more complex enzyme–MOF composites.
These indirect in situ MOF synthesis methods overcome some of the limitations of direct in situ synthesis approaches (higher number of MOF–enzyme couples possible, control of enzymes loading and localization), but these methods are undeniably more complicated to implement experimentally.
3. Discussion
MOFs have recently emerged as an important family of porous hybrid materials owing to their unique structural and functional properties. Among their most attractive features are crystalline structure, high pore volume and highly modular nature, related to a wide choice of possible organic linkers and inorganic building units. This allows modification of the physical environment of the pores, thereby tuning the interactions with guest species. Functionalization of the framework may also strongly affect the chemical stability of the MOF structure under different conditions. As highlighted by the many examples discussed in this review,20,22 the hybrid nature of MOFs plays a key role in enzyme stabilization. The ability to easily tune the hydrophilic–hydrophobic balance of a MOF using ligands with specific functional groups can improve compatibility of a MOF with a given enzyme. Furthermore, the orientation of the enzyme active site can be modulated through specific interactions between both components, thus increasing catalytic activity of the composite.Overcoming enzyme leaching and thus improving the performance, stability and reusability of a biocomposite is a key point that appears to be successfully addressed by MOF-based matrices following however model conditions over a few cycles only. Functional groups on organic linkers in the framework can be selected to provide specific interaction sites for the enzyme and thus minimize its leaching and improve its stability in the matrix. This hypothesis was confirmed in the work of Chen et al., where they studied the interactions between MP-11 molecules and Tb-mesoMOF by Raman spectroscopy.38 The study revealed that MP-11 molecules interact with the framework of Tb-mesoMOF, through π–π interactions between the heme of MP-11 and the conjugated triazine and benzene rings of the linkers. These interactions resulted in the retention of MP-11 molecules within the pores. In contrast, for a mesoporous silica matrix such as MCM-41, a severe leaching of the enzyme was observed as a result of the lack of specific interactions between the biomacromolecules and the silica phase. No comparison to organo-functionalized silica was reported in this work.
The reusability and recyclability of MOF–enzyme based catalysts can be further improved by the introduction of magnetic moieties to allow magnetic separation from reaction mixtures, as previously reported for other multi-components materials.39 Zhao et al. used Fe3O4 nanoparticles, treated with polydopamine (PDA), as a magnetic core onto which a MOF shell (HKUST-1: Cu3[benzenetricarboxylate]2) was grown.40 The magnetic nanoMOFs were then used for immobilization of Trypsin and exhibited high magnetic responsiveness, allowing easy removal from the reaction mixture. Fe3O4 magnetic particles were also used by Huo et al., which resulted in similar recyclability.37
Beyond the solid-support matrices
In most previous studies of MOF-based enzymatic immobilization, MOFs have been considered mostly as improved solid matrices without fully considering their intrinsic properties (e.g. separation) from a view of potential applications.Following this concept, Wang et al. constructed an electrochemical tyrosinase-based sensor for the detection of bisphenol A (BPA).41 The presence of CuMOF ([Cu(benzenedicarboxylic)(triethyelediamine)0.5]·2DMF·0.2H2O) on the bioelectrodes led to improved sensitivity for the detection of BPA, attributed to the high surface area of the MOF that not only helped enzyme immobilization but also BPA adsorption. The authors proposed that the preconcentration of BPA at the electrode surface may be due to π–π stacking interactions between the aromatic ring of BPA and the MOF constitutive organic ligand.
The porous character of MOF-based matrices has also been exploited to gain size selectivity. For example, the peroxidase activity of myoglobin (Mb) encapsulated in Tb-mesoMOF was evaluated by using two substrates, 2,2′-azinobis(3-ethyl-benzthiazoline)-6-sulfonate (ABTS) with molecular dimensions: 1.01 × 1.73 nm and 1,2,3-trihydroxybenzene (THB) with molecular dimensions of 0.57 × 0.58 nm.42 This resulted in size-selective catalytic activity, as the larger substrate (ABTS) could not pass through the remaining pores of the Tb-mesoMOF (0.8 nm) showing almost no conversion. In contrast, for the smaller substrate, THB, the composite demonstrated enhanced catalytic activity compared to the free enzyme. Similar results were reported by Huo et al., using ZIF-8 shell microcapsules for the encapsulation of Candida antarctica lipase B (CALB).37 The biocatalytic activity was evaluated through transesterification reactions using small and large substrates. When small substrates (1-butanol + vinyl acetate) were used, activity was higher than that for the free enzyme, although 100% conversion was reached more slowly (12 h and 48 h for free and encapsulated CALB, respectively) suggesting diffusion limitations of reactants and products through the narrow micropores of the ZIF-8 capsule's shell. When larger substrates were used (3-(4-hydroxyphenyl) propan-1-ol + vinyl laurate), an extremely slow conversion rate was observed reflecting the physical barrier imposed by the microporous MOF.
Expanding this concept further, even more careful combination of properties from both enzyme and MOF should lead to improved biocomposites. For example, the catalytic properties of a wide range of MOFs have been extensively reported over the last years based on the presence of a large number of spatially controlled acid/base sites (Lewis, Brønsted), redox, and/or functionalized organic linkers within their pores.43,44 One can thus envision a system for MOF-based immobilization, in which the MOF contributes to the overall catalytic activity of the composite by way of cascade reactions or synergistic effects.
This idea is nicely illustrated by the recent work of Patra et al. which exploits the redox nature of MIL-100(Fe) for building enzymatic bioelectrodes that can be used either for glucose sensing or for oxygen reduction reactions (ORR).45 First, several MIL-100 solids based on various metal(III) cations (Fe, Cr, Al), in addition to the microporous iron(III) tetracarboxylate MIL-127(Fe) (Fe3O(OH)0.66Cl0.33(C16N2O8H6)1.5(H2O)3·nH2O), were tested as matrices for the immobilization of glucose oxidase (GOx). The resulting biocomposites based on MIL-100(Fe) exhibited much higher catalytic performance than Cr or Al analogues or the MIL-127(Fe) solid. This was attributed to the strong synergism between GOx and the catalytic/redox properties of Fe3+ centers of MIL-100(Fe) that exhibit intrinsic peroxidase-like activity.46 Recently, MIL-100(Fe) was first exploited as a novel and efficient matrix for the co-immobilization of laccase and ABTS.47 This particular bioelectrode presents a high electrocatalytic current density of ORR and a reproducible electrochemical response characterized by a high stability over a long period of time (3 weeks). These results represent a significant leap forward in the field of laccase-based bioelectrocatalysts for the design of biofuel cells. Wang et al. also proposed a GOx/ZIF-8 system for the electrochemical detection of glucose that incorporates graphene nano-sheets to increase the electron transfer rate at the electrode.48 Similarly, Hou et al. designed a GOx/ZIF-8 system for colorimetric detection of glucose by taking advantage of the peroxidase activity of ZIF-8 to enhance the detection performance of the biosensor.65
4. Conclusion and perspectives
MOFs are an emerging class of viable host matrices for enzymes. Robust methods such as surface adsorption, covalent binding, cage inclusion, and new in situ synthesis processes have been developed, all of which have shown promising results to immobilize enzymes in a manner that retains biological function with minimal leaching. The examples covered within this review emphasize how the multimodal porosity and hybrid nature of MOFs are certainly key advantages for bio-immobilization and biocatalysis applications. The structural and chemical diversity and functionality of MOFs, combined with their high specific surface area, can produce a wide range of biocatalysts with suitable properties in terms of catalytic activity, stability or recyclability. Recently, MOF–enzyme based bioelectrodes have shown great promise for glucose sensing and ORR due to their high sensitivity, catalytic efficiency, and long-term stability. Biocatalysts reported in the literature clearly reveal that MOFs are appealing host matrices to create stabilizing microenvironments for enzymes through specific host–guest interactions. Immobilization can be achieved through different approaches depending on the specific MOF–enzyme couple and the targeted catalytic process. While surface adsorption, covalent binding, and cage inclusion were classically reported for other enzyme host matrices, the “in situ MOF synthesis” methods are more specific to MOF chemistry. Cage inclusion and “in situ MOF synthesis” enable 3D encapsulation of enzymes in a microenvironment created either by the porosity of MOFs or by the aggregation of MOFs particles. These methods are particularly attractive for enzymatic devices since they improve long-term stability by preventing leaching and denaturation of entrapped enzymes. However, cage inclusion is restricted to a few enzyme/MOFs couples with appropriate size matching (i.e. microenzymes and mesoporous MOFs) while the “in situ MOF synthesis” approach is limited to MOFs that have biocompatible synthesis conditions (room temperature, non toxic reactants or solvents).Independent of the immobilization strategy used, the synthesis and processing of enzyme–MOFs composites suffer from the same issues as those of pure MOFs. Although a huge family of MOFs has already been described in the literature, practical application of these matrices is often hampered by inadequate chemical stability (especially in water), and difficulty of controlling the diameter, morphology, structure and defects of particles. Overcoming these drawbacks is imperative to implementing MOF-based devices in different fields and is also crucial for the scale-up of laboratory syntheses into reliable manufacturing procedures. Moreover, it is worth noting that only a limited number of studies has investigated the long term stability of MOFs based composites under enzymatic operating conditions while a few MOFs in aqueous or buffer solutions (PBS) have demonstrated limited stability.
The design of MOF–enzyme biocomposites is still in its infancy and MOFs have not yet reached the maturity of other immobilization matrices such as oxides or clays. However, research into novel MOFs–enzyme composites is rapidly growing, exploring new compositions and materials processing. Recent progress in the synthesis of multi-component materials by combining MOFs, magnetic particles and/or polymers was reported illustrating the potential for this class of materials in the development of sophisticated and multi-functional devices.
One remaining critical issue for this family of biohybrid materials concerns the chemical and thermodynamic compatibility between enzyme and MOF particle that governs the loading, dispersion and orientation of enzymes in MOF matrices. The performance of biocatalytic and biosensing devices might be driven by the functionality and defects of the framework, and the surface chemistry of MOF particles. Therefore, the characterization and/or modeling of the microstructural properties of MOFs49–51 is a necessary step towards enhancing the interfacial properties of MOF–enzyme composites and could finally provide a useful guide for the design of optimized bionanocomposites.
Acknowledgements
The authors acknowledge the help of Dr Farid Nouar for the graphical representations and Dr Meghan J. McFadden for correcting the English of the manuscript. The authors also thank the ANR-11-LABEX-0039 for funding.References
- S. Kaskel, The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications, Wiley, Weinheim, 2016 Search PubMed.
- Themed issue, Chem. Soc. Rev., 2014, 43, 5415–6176.
- Themed issue, Chem. Rev., 2012, 2, 673–1268.
- Themed issue, Chem. Soc. Rev., 2013, 42, 6211–6568.
- L. Cao, Carrier-bound Immobilized Enzymes: Principles, Application and Design, Wiley, Weinheim, 2006 Search PubMed.
- E. Forsberg, C. Sicard and J. D. Brennan, Annu. Rev. Anal. Chem., 2014, 7, 337–359 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- T. J. Pisklak, M. Macías, D. H. Coutinho, R. S. Huang and K. J. Balkus, Top. Catal., 2006, 38, 269–278 CrossRef CAS.
- J. Mehta, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2016, 322, 30–40 CrossRef CAS.
- D. Raja, W.-L. Liu, H.-Y. Huang and C.-H. Lin, Comments Inorg. Chem., 2015, 35, 332–350 CrossRef CAS.
- X. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077–5085 CAS.
- Y. Chen and S. Ma, Dalton Trans., 2016, 45, 9744–9753 RSC.
- C. Mousty, Anal. Bioanal. Chem., 2010, 396, 315–325 CrossRef CAS PubMed.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- P. A. Sontz, J. B. Bailey, S. Ahn and F. A. Tezcan, J. Am. Chem. Soc., 2015, 137, 11598–11601 CrossRef CAS PubMed.
- N. Steunou, C. Mousty, O. Durupthy, C. Roux, G. Laurent, C. Simonnet-Jegat, J. Vigneron, A. Etcheberry, C. Bonhomme, J. Livage and T. Coradin, J. Mater. Chem., 2012, 22, 15291–15302 RSC.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, 3 Biotech., 2013, 3, 1–9 CrossRef.
- P. Torres-Salas, A. Monte-Martinez, B. Cutiño-Avila, B. Rodriguez-Colinas, M. Alcalde, A. O. Ballesteros and F. J. Plou, Adv. Mater., 2011, 23, 5275–5282 CrossRef CAS PubMed.
- D. Brady and J. Jordaan, Biotechnol. Lett., 2009, 31, 1639–1650 CrossRef CAS PubMed.
- W. Ma, Q. Jiang, P. Yu, L. Yang and L. Mao, Anal. Chem., 2013, 85, 7550–7557 CrossRef CAS PubMed.
- W.-L. Liu, C.-Y. Wu, C.-Y. Chen, B. Singco, C.-H. Lin and H.-Y. Huang, Chem. – Eur. J., 2014, 20, 8923–8928 CAS.
- Y.-H. Shih, S.-H. Lo, N.-S. Yang, B. Singco, Y.-J. Cheng, C.-Y. Wu, I.-H. Chang, H.-Y. Huang and C.-H. Lin, ChemPlusChem, 2012, 77, 982–986 CrossRef CAS.
- J. Juan-Alcañiz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC.
- (a) V. V. Vinogradov and D. Avnir, RSC Adv., 2015, 5, 10862–10868 RSC; (b) V. V. Vinogradov and D. Avnir, J. Mater. Chem. B, 2014, 2, 2868–2873 RSC; (c) D. T. Nguyen, M. Smit, B. Dunn and J. I. Zink, Chem. Mater., 2002, 14, 4300–4306 CrossRef CAS.
- (a) M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277–6289 RSC; (b) E. Magner, Chem. Soc. Rev., 2013, 42, 6211–6222 RSC.
- V. Lykourinou, Y. Chen, X.-S. Wang, L. Meng, T. Hoang, L.-J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed.
- Y. Chen, V. Lykourinou, C. Vetromile, T. Hoang, L.-J. Ming, R. W. Larsen and S. Ma, J. Am. Chem. Soc., 2012, 134, 13188–13191 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O’Keefe, O. Terasaki, J. F. Stoddart and O. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- D. Feng, T.-F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y.-P. Chen, X. Wang, K. Wang, X. Lian, Z.-Y. Gu, J. Park, X. Zou and H.-C. Zhou, Nat. Commun., 2015, 6, 5979 CrossRef PubMed.
- P. Horcajada, H. Chevreau, D. Heurtaux, F. Benyettou, F. Salles, T. Devic, A. Garcia-Marquez, C. Yu, H. Lavrard, C. L. Dutson, E. Magnier, G. Maurin, E. Elkaım and C. Serre, Chem. Commun., 2014, 50, 6872–6874 RSC.
- Y. Kim, T. Yang, G. Yun, M. B. Ghasemian, J. Koo, E. Lee, S. J. Cho and K. Kim, Angew. Chem., Int. Ed., 2015, 54, 13273–13278 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- K. Liang, C. J. Coghlan, S. G. Bell, C. Doonan and P. Falcaro, Chem. Commun., 2016, 52, 473–476 RSC.
- G.-Y. Jeong, R. Ricco, K. Liang, J. Ludwig, J.-O. Kim, P. Falcaro and D.-P. Kim, Chem. Mater., 2015, 27, 7903–7909 CrossRef CAS.
- J. Huo, J. Aguilera-Sigalat, S. El-Hankari and D. Bradshaw, Chem. Sci., 2015, 6, 1938–1943 RSC.
- Y. Chen, S. Han, X. Li, Z. Zhang and S. Ma, Inorg. Chem., 2014, 53, 10006–10008 CrossRef CAS PubMed.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- M. Zhao, X. Zhang and C. Deng, Chem. Commun., 2015, 51, 8116–8119 RSC.
- X. Wang, X. Lu, L. Wu and J. Chen, Biosens. Bioelectron., 2015, 65, 295–301 CrossRef CAS PubMed.
- Y. Chen, V. Lykourinou, T. Hoang, L.-J. Ming and S. Ma, Inorg. Chem., 2012, 51, 9156–9158 CrossRef CAS PubMed.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Zhang, Z.-Y. Gu, M. Bosch, Z. Perry and H.-C. Zhou, Coord. Chem. Rev., 2014, 293–294, 327–356 Search PubMed.
- S. Patra, T. H. Crespo, C. Serre, A. Permyakova, C. Sicard, A. Chaussé, N. Steunou and L. Legrand, J. Mater. Chem. B, 2015, 3, 8983–8992 RSC.
- J.-W. Zhang, H.-T. Zhang, Z.-Y. Du, X. Wang, S.-H. Yu and H.-L. Jiang, Chem. Commun., 2014, 50, 1092–1094 RSC.
- S. Patra, S. Sene, C. Mousty, C. Serre, A. Chaussé, L. Legrand and N. Steunou, ACS Appl. Mater. Interfaces, 2016, 8, 20012–20022 CAS.
- W. Wang, L. Wang, Y. Huang, Z. Xie and X. Jing, Chem. – Asian J., 2016, 11, 750–756 CrossRef CAS PubMed.
- H. Zhang, Y. Lv, T. Tan and D. Spoel, J. Phys. Chem. B, 2016, 120, 477–484 CrossRef CAS PubMed.
- R. Semino, N. A. Ramsahye, A. Ghoufi and G. Maurin, ACS Appl. Mater. Interfaces, 2016, 8, 809–819 CAS.
- M. Benzaqui, R. Semino, N. Menguy, F. Carn, T. Kundu, J.-M. Guigner, N. B. McKeown, K. J. Msayib, M. Carta, R. Malpass-Evans, C. Guillouzer, G. Clet, N. A. Ramsahye, C. Serre, G. Maurin and N. Steunou, ACS Appl. Mater. Interfaces, 2016, 8, 27311–27321 CAS.
- W.-L. Liu, N.-S. Yang, Y.-T. Chen, S. Lirio, C.-Y. Wu, C.-H. Lin and H.-Y. Huang, Chem. – Eur. J., 2015, 21, 115–119 CrossRef CAS PubMed.
- Y. Cao, Z. Wu, T. Wang, Y. Xiao, Q. Huoa and Y. Liu, Dalton Trans., 2016, 45, 6998–7003 RSC.
- S. Pang, Y. Wu, X. Zhang, B. Li, J. Ouyang and M. Ding, Process Biochem., 2016, 51, 229–239 CrossRef CAS.
- X. Lu, X. Wang, L. Wu, L. Wu, Dhanjai, L. Fu, Y. Gao and J. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16533–16539 CAS.
- W.-L. Liu, S.-H. Lo, B. Singco, C.-C. Yang, H.-Y. Huang and C.-H. Lin, J. Mater. Chem. B, 2013, 1, 928–932 RSC.
- C. M. Doherty, G. Grenci, R. Riccò, J. I. Mardel, J. Reboul, S. Furukawa, S. Kitagawa, A. J. Hill and P. Falcaro, Adv. Mater., 2013, 25, 4701–4705 CrossRef CAS PubMed.
- S.-L. Cao, D.-M. Yue, X.-H. Li, T. J. Smith, N. Li, M.-H. Zong, H. Wu, Y.-Z. Ma and W.-Y. Lou, ACS Sustainable Chem. Eng., 2016, 4, 3586–3595 CrossRef CAS.
- L. Wen, A. Gao, Y. Cao, F. Svec, T. Tan and Y. Lv, Macromol. Rapid Commun., 2016, 37, 551–557 CrossRef CAS PubMed.
- S. Jung, Y. Kim, S.-J. Kim, T.-H. Kwon, S. Huh and S. Park, Chem. Commun., 2011, 47, 2904–2906 RSC.
- F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu, K. C.-W. Wu and C.-K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed.
- P. Chulkaivalsucharit, X. Wu and J. Ge, RSC Adv., 2015, 5, 101293 RSC.
- Y. Wang, C. Hou, Y. Zhang, F. He, M. Liu and X. Li, J. Mater. Chem. B, 2016, 4, 3695–3702 RSC.
- K. Liang, C. Carbonell, M. J. Styles, R. Ricco, J. Cui, J. J. Richardson, D. Maspoch, F. Caruso and P. Falcaro, Adv. Mater., 2015, 27, 7293–7298 CrossRef CAS PubMed.
- C. Hou, Y. Wang, Q. Ding, L. Jiang, M. Li, W. Zhu, D. Pan, H. Zhu and M. Liu, Nanoscale, 2015, 7, 18770–18779 RSC.
- X. Wu, C. Yang, J. Ge and Z. Liu, Nanoscale, 2015, 7, 18883–18886 RSC.
| This journal is © The Royal Society of Chemistry 2017 |