DOI:
10.1039/C7MD90012K
(Editorial)
Med. Chem. Commun., 2017,
8, 820-822
The structure-guided discovery of osimertinib: the first U.S. FDA approved mutant selective inhibitor of EGFR T790M†
Received
21st March 2017
, Accepted 21st March 2017
Small molecule EGFR inhibitors such as gefitinib and erlotinib have demonstrated clinical efficacy in the treatment of EGFR mutation positive non-small cell lung cancer (NSCLC) after disappointing earlier results with these agents in unselected patient populations. These mutations of EGFR are commonly referred to as sensitising or activating mutations. However, resistance to these drugs is commonly observed through an acquired EGFR T790M secondary mutation, known as the gatekeeper mutation. Osimertinib is the first inhibitor approved by the U.S. FDA to selectively target the T790M resistant mutant whilst sparing the wild-type form of the enzyme. Essential to the discovery of this drug was the focused structure-driven computational and medicinal chemistry design strategy and synthetic chemistry innovation. The focused design work and clear clinical hypothesis resulted in rapid progress of the project with identification of the clinical candidate less than three years from project start, and U.S. FDA approval in a record 2 years and 9 months after the first dose in man.
The initiation of the project that led to the discovery of osimertinib at AstraZeneca was triggered in May 2009 at the Alderley Park research site, after forty compounds were tested in biochemical assays against the EGFR T790M mutant and wild-type forms of EGFR. The rationale for this evaluation was the expectation that interaction with the more hydrophobic methionine gatekeeper residue could selectively be targeted over the more hydrophilic threonine of wild-type EGFR. Encouragingly, compounds were identified that were indeed more active against the T790M resistant mutant; in contrast to compounds such as gefitinib which were more potent against wild-type.1 These compounds were selected for testing using the experience of in-house kinase projects utilising alternative chemical scaffolds which were known to favorably interact with methionine gatekeeper residues. Although these data indicated a ‘T790M mutant selective’ profile was feasible, the compounds showed poor activity in cellular EGFR phosphorylation assays due to the high ATP affinity of the T790M mutant, which is a primary mechanism of resistance to ATP competitive EGFR inhibitors such as gefitinib. In an attempt to overcome this challenge, computational modeling identified an appropriate vector to position an acrylamide ‘warhead’ on the identified molecule to covalently target Cys-797. If successful, this would result in an irreversible, non ATP competitive inhibitor, which might overcome the impact of the T790M resistant mutation. Compounds such as these were the first to show the targeted T790M mutant selective profile in our cellular EGFR assays, and of particular note was the observation that they also maintained activity against the EGFR sensitizing mutants targeted by gefitinib and erlotinib. Although these compounds were a significant step forward, they required further evolution to improve both potency and physicochemical properties to be considered as clinical candidates. Ultimately, these improvements were made using binding mode knowledge to guide variations of the fused ‘head-group’ at C-4 of the pyrimidine and the addition of a basic substituent into the ‘solvent channel’ aniline2 (Fig. 1). Another essential facet to the early development of the series was the understanding of the inherent reactivity of such compounds using an assay measuring the half-life of reaction of compounds with glutathione (GSH).
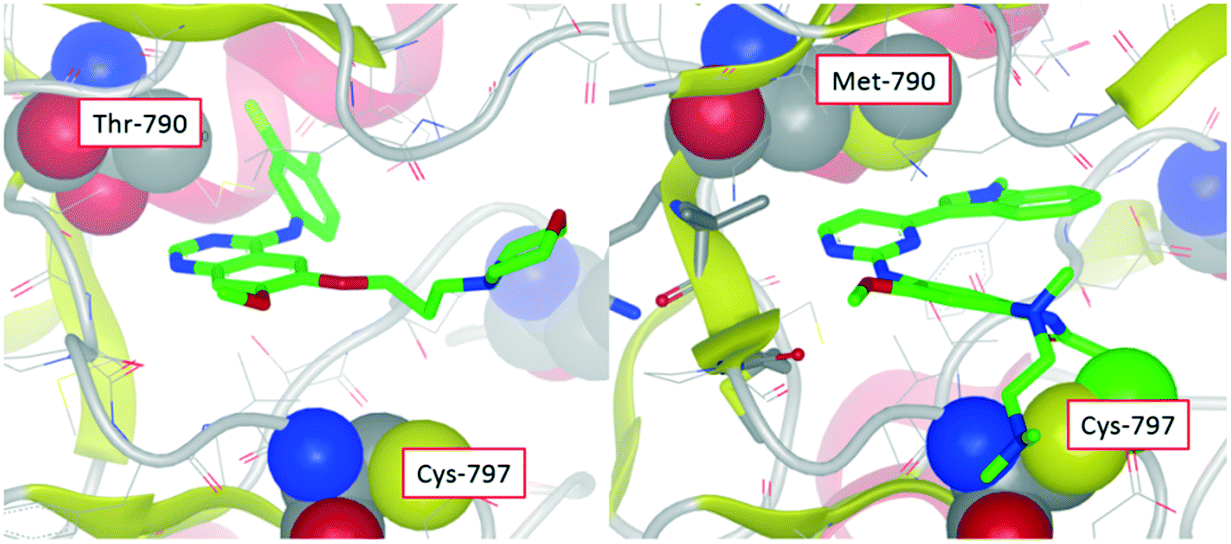
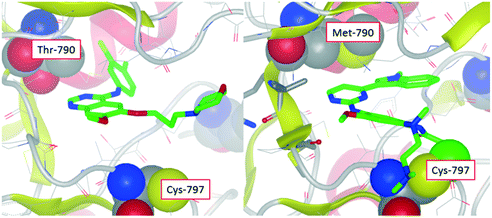 |
| | Fig. 1 X-ray crystal structure of gefitinib in wild-type EGFR showing close proximity of the compound with the threonine gatekeeper (left). Modelled structure of osimertinib in T790M mutant of EGFR showing close proximity with the methionine gatekeeper residue and also covalent bond to Cys-797 (right). | |
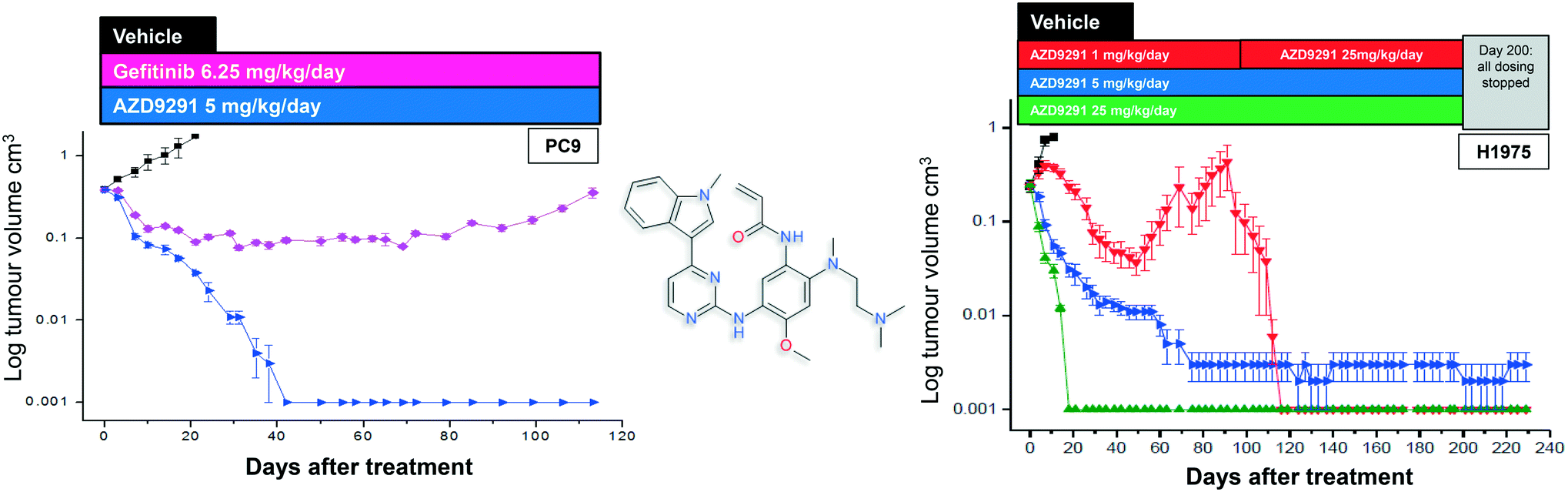
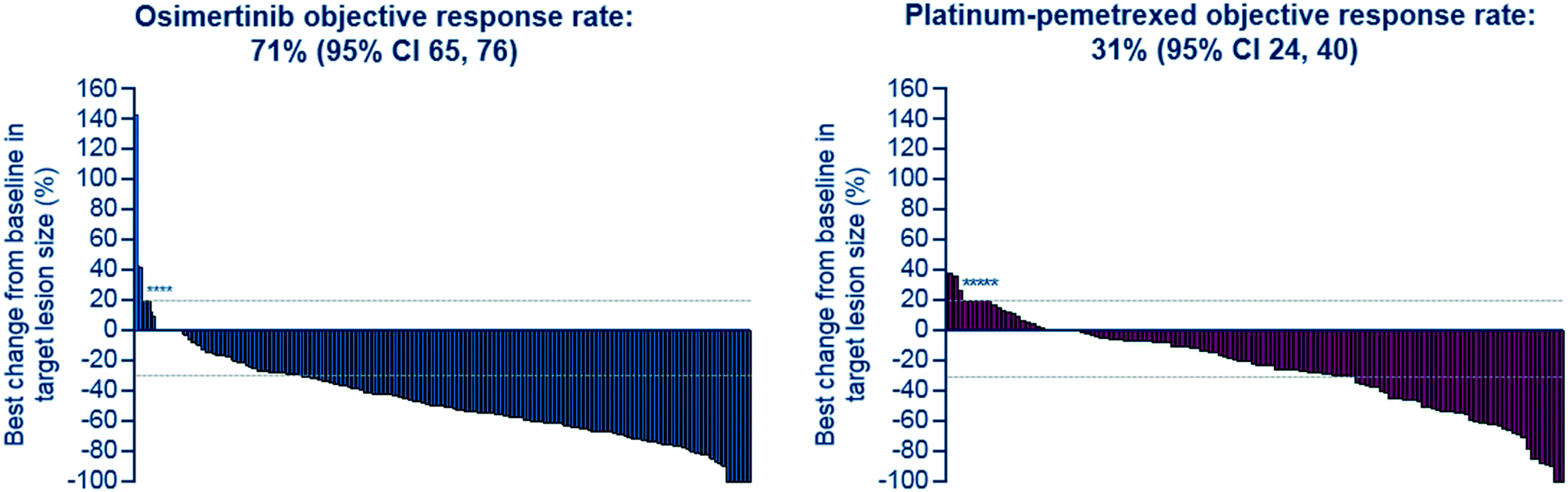
After further medicinal chemistry evolution, exemplar compounds displayed high cellular potency against mutant EGFR accompanied by excellent pharmacokinetic (PK) properties. Subsequent work demonstrated that these compounds could deliver profound regression in mutant EGFR mouse tumour xenograft models (sensitising and T790M resistant mutants) at low oral doses, with a margin to wild-type EGFR. However, as the inhibitor scaffold had originated from an earlier project targeting a methionine gatekeeper kinase (specifically IGF1R), the compounds carried inhibitory activity against both the insulin receptor tyrosine kinase (INSR) and the insulin like growth factor receptor tyrosine kinase (IGF1R). Both of these kinases play a key role in glucose homeostasis and in vivo evaluation of these early compounds showed pronounced hyperglycemic effects in rat. However, the team were subsequently able to establish structure–activity relationships (SAR) that permitted IGF1R and INSR activity to be reduced, without compromising the mutant EGFR potency and wild-type margin required for a clinical candidate. A key compound from this series was shown to be both efficacious in the previously described disease models at a low dose (Fig. 2), as well as being devoid of hyperglycemic effects, and was selected for clinical development as AZD9291, now known as osimertinib. After clinical trials began in March 2013, osimertinib displayed significant anti-tumour activity in NSCLC patients who had become refractory to earlier EGFR inhibitors, with activity being observed in the very first cohort of patients. Subsequent clinical evaluation continued to show clinical responses in patients (Fig. 3), and in November 2015, osimertinib received U.S. FDA approval “for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation-positive non-small cell lung cancer (NSCLC), as detected by an FDA-approved test, who have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy”. Osimertinib is now also approved in regions such as Europe, Korea, Taiwan and Japan and is marketed globally as TAGRISSO™. Other indications for TAGRISSO™ are currently being investigated, including use as a first line therapy as well as a treatment for NSCLC patients suffering from CNS and brain metastases.
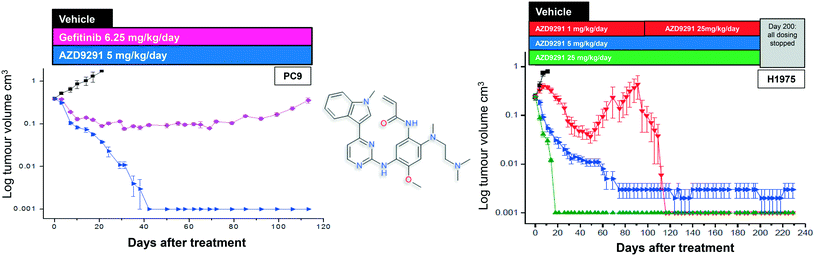 |
| | Fig. 2 Chemical structure of osimertinib with in vivo xenograft activity and in the sensitising (PC-9, left) and T790M resistant models (H1975, right).3 Figure adapted from Cross et al. (2014) Cancer Discovery, 4, 1046–1061. | |
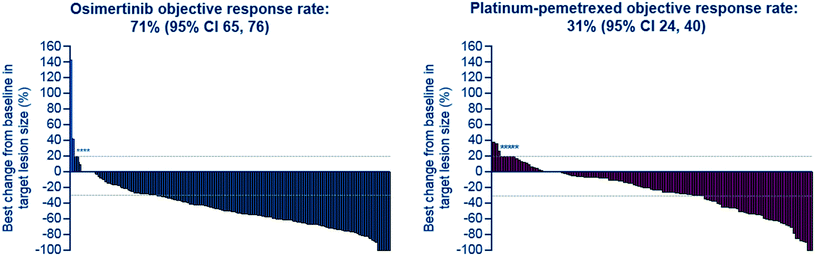 |
| | Fig. 3 AURA3: tumour response by investigator assessment. Best percentage change in target lesion size is the maximum reduction from baseline or the minimum increase. *Represents imputed values: if it is known that the patient has died, has new lesions or progression of assessments, best change will be imputed as 20%.4 | |
TAGRISSO™ was discovered as a result of an extremely rapid drug discovery and development programme, with contributions from many scientists and offers new treatment options for patients suffering from NSCLC. As well as acknowledging the contribution of all those involved in the discovery and development of this molecule within AstraZeneca, the authors would like to express their thanks and appreciation to all of our collaborators, clinicians and above all the patients involved in the clinical trials and their families.
References
- R. A. Ward, M. J. Anderton, S. Ashton, P. A. Bethel, M. Box, S. Butterworth, N. Colclough, C. G. Chorley, C. Chuaqui, D. A. Cross, L. A. Dakin, J. E. Debreczeni, C. Eberlein, M. R. Finlay, G. B. Hill, M. Grist, T. C. Klinowska, C. Lane, S. Martin, J. P. Orme, P. Smith, F. Wang and M. J. Waring, Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR), J. Med. Chem., 2013, 56, 7025–7048 CrossRef CAS PubMed.
- M. R. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. McFarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor, J. Med. Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- D. A. Cross, S. E. Ashton, S. Ghiorghiu, C. Eberlein, C. A. Nebhan, P. J. Spitzler, J. P. Orme, M. R. Finlay, R. A. Ward, M. J. Mellor, G. Hughes, A. Rahi, V. N. Jacobs, M. Red Brewer, E. Ichihara, J. Sun, H. Jin, P. Ballard, K. Al-Kadhimi, R. Rowlinson, T. Klinowska, G. H. Richmond, M. Cantarini, D. W. Kim, M. R. Ranson and W. Pao, AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer, Cancer Discovery, 2014, 4, 1046–1061 CrossRef CAS PubMed.
- T. S. Mok, Y.-L. Wu, M.-J. Ahn, M. C. Garassino, H. R. Kim, S. S. Ramalingam, F. A. Shepherd, Y. He, H. Akamatsu, W. S. M. E. Theelen, C. K. Lee, M. Sebastian, A. Templeton, H. Mann, M. Marotti, S. Ghiorghiu and V. A. Papadimitrakopoulou, N. Engl. J. Med., 2017, 376, 629–640 CrossRef PubMed.
Footnote |
| † The authors declare no competing interests. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.