
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small molecule piperazinyl-benzimidazole antagonists of the gonadotropin-releasing hormone (GnRH) receptor†
Richard
Fjellaksel
 *abc,
Marc
Boomgaren
c,
Rune
Sundset
ad,
Ira H.
Haraldsen
e,
Jørn H.
Hansen
c and
Patrick J.
Riss
efg
*abc,
Marc
Boomgaren
c,
Rune
Sundset
ad,
Ira H.
Haraldsen
e,
Jørn H.
Hansen
c and
Patrick J.
Riss
efg
aMedical Imaging Group, Department of Clinical Medicine, UiT The Arctic University of Norway, 9037 Tromsø, Norway. E-mail: richard.fjellaksel@uit.no
bDrug Transport and Delivery Group, Department of Pharmacy, UiT The Arctic University of Norway, 9037 Tromsø, Norway
cOrganic Chemistry Group, Department of Chemistry, UiT The Arctic University of Norway, 9037 Tromsø, Norway
dPET imaging center, division of diagnostics, UNN – University Hospital of North-Norway, 9038 Tromsø, Norway
eDepartment of neuropsychiatry and psychosomatic medicine, Oslo University Hospital, Oslo, Norway
fRealomics SFI, Department of Chemistry, University of Oslo, PO BOX 1033, Oslo 0371, Norway
gNorsk Medisinsk Syklotronsenter AS, Postboks 4950 Nydalen, 0424 Oslo, Norway
First published on 14th September 2017
Abstract
In this communication, we report the synthesis and characterization of a library of small molecule antagonists of the human gonadotropin releasing hormone receptor based upon the 2-(4-tert-butylphenyl)-4-piperazinyl-benzimidazole scaffold via Cu-catalysed azide alkyne cycloaddition. Our main purpose was to find a more soluble compound based on the WAY207024 lead with nanomolar potency to inhibit the GnRH receptor. A late stage diversification by the use of click chemistry was, furthermore developed to allow for expansion of the library in future optimisations. All compounds were tested in a functional assay to determine the individual potency of inhibiting stimulation of the receptor by the endogenous agonist GnRH. In conclusion, we found that compound 8a showed improved solubility compared to WAY207024 and nanomolar affinity to GnRH receptor.
Gonadotropin-releasing hormone (GnRH) is a peptide hormone secreted from the hypothalamus into the hypophysial portal bloodstream. Once in circulation, the peptide acts as an endocrine signalling hormone mediating release of follicle stimulating hormone (FSH) and luteinizing hormone (LH) via gonadotropin-releasing hormone receptor (GnRHR) activation in the pituitary gland. LH and FSH directly regulate gender-specific production of androgens, estrogens, progesterone and inhibin in the gonads. The regulatory circuit is closed by the permeability of the blood–brain barrier to steroid hormones such as estrogen and testosterone, which form negative feedback loops. GnRH, LH and FSH are unlikely to permeate the blood–brain barrier, thus impeding any regulatory feedback on hormone balance, which substantiates the impact of testosterone and estrogen on neurochemical correlates of their respective effects on social behaviour, decision-making and ageing. The hypothalamic–pituitary–gonadal (HPG) signalling circuit is key to gender hormone homeostasis, which can lead to substantial implications in cognitive and behavioural traits.1–4
GnRH agonists are well-established pharmaceuticals used at low dose to stimulate hypofunctional GnRH in hypogonadism. At high dose, GnRH agonists deplete GnRH receptor function in the pituitary, limiting LH and FSH secretion and thereby the levels of androgens and estrogens in circulation and suppressing hormone production completely. GnRH receptor agonists play an important role in clinical care, e.g. in the treatment of hormone responsive cancer, reproductive diseases and for behavioral modification of sexual offenders via these mechanisms. However, GnRH agonists cannot address mild GnRH overfunction, which would require attenuation of the signaling circuit, rather than stimulation or depletion. To address this shortcoming, GnRH antagonists have attracted considerable attention in recent years specifically to treat diseases, which require some reduction of GnRH stimulation.5–8
We were interested in the development of a GnRH antagonist of moderate potency to address cognitive and behavioral correlates of GnRH-R attenuation. In our reasoning, a reversibly binding antagonist would reduce gonadal overstimulation by limiting the availability of binding sites by competition with the agonist. Thereby, the central effects of testosterone and estrogen would be buffered, while hormone homeostasis would be preserved. Small molecule antagonists of GnRHR are particularly interesting due to their direct, dose dependent inhibition of GnRH activity, lack of stimulatory side effects on the receptor and superior passive permeability into tissues relative to peptide agonists.
Several classes of small molecule antagonists have been developed previously. Pelletier and co-workers reported a 2-(4-tert-butylphenyl)-4-piperazinyl-benzimidazole-scaffold as a small molecule antagonist of the GnRHR with nanomolar inhibition potency, albeit low solubility was cited as a shortcoming by the authors.9,10 Further optimization lead to the discovery of the slightly more soluble WAY 207024 compound as shown in Fig. 1.11
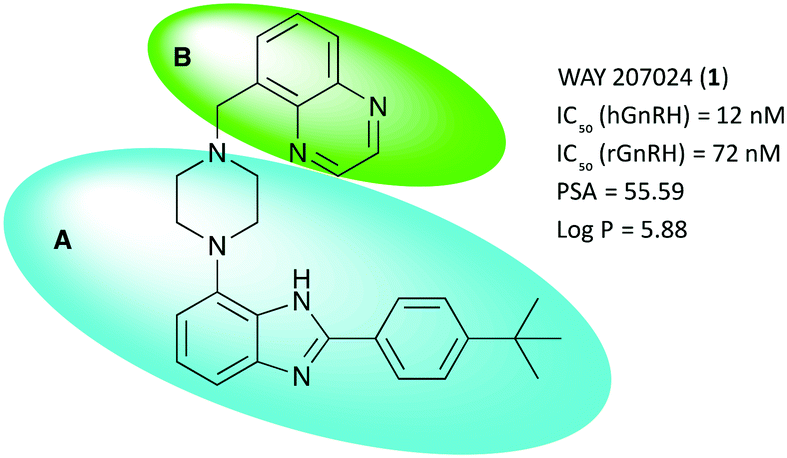 | ||
Fig. 1 Key properties and molecular structure of WAY 207024, the lead compound for GnRH antagonist development. A (blue oval) depicts the scaffold body; B (green oval) depicts the moiety of interest for further structural optimization. IC50 – concentration to inhibit GnRH binding to the receptor by 50%. hGnRH – human GnRH receptor. rGnRH – rat GnRH receptor. PSA – polar surface area in Å2. cLog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P – calculated octanol–water partitioning coefficient. P – calculated octanol–water partitioning coefficient. | ||
Based on these results, we have designed a novel library of potential antagonists addressing the following criteria:
a) A late stage diversification approach based on the 1,3-triazole motive to facilitate straightforward structural modification.
b) A moderate potency permitting competition of the ligand in circulation with endogenous GnRH for GnRHR binding sites, which would make GnRH activation subject to both GnRH concentration and the concentration of the inhibitor in tissue.
c) Improved solubility in aqueous media to improve bioavailability and pharmacokinetic profile of the lead.
Fig. 1 shows the title scaffold and some relevant properties discussed herein. As apparent from the high partitioning coefficient logP, the compound is highly lipophilic, which may hamper solubility in aqueous formulation as well as non-specific binding of the compound in vivo. In previous studies, it was found that the body of the scaffold (blue oval, A) is central to binding selectively the GnRH receptor, whereas structural modifications in the upper section of the lead (green oval, B) with planar, hydrogen bonding functional groups were found to be beneficial for modulating antagonist potency.
We surmised that the 1,3-functionalised triazole moiety would resemble well the planar geometry required for successful structural modification and simultaneously allow for introducing a broad spectrum of structural variations in the last synthetic stage using one robust reaction. Since the 1,3-functionalised triazole is obtained via the Cu-catalysed azide-alkyne cycloaddition reaction, we opted for working with a building block based on A to obtain an alkyne substrate for the reaction.
The overall design strategy in this communication was to generate triazoles from different functionalized azides that would be beneficial for the potency to inhibit the GnRH receptor while improving the aqueous solubility. We incorporated a variety of azides via click chemistry, including sugar moieties, which have been thoroughly demonstrated to effect favourable solubility characteristics and are discussed extensively in the literature.12–15 Furthermore, the reduced solubility of the alkyl and aromatic azides would also counteract our intention to improve aqueous solubility through reducing clogP (and hence, the bioavailability and pharmacokinetically desirable profile).
The desired intermediate (7) was synthesised in 19% overall yield over six steps on a multigram scale. In brief, 2,5-difluoronitrobenzene was converted to phenylene diamine 4 over three steps as described previously.9 Contrary to the published method, we found that the oxidative conversion of 4 into imidazole 6 proceeded smoothly in absence of a transition metal catalyst with air as the oxidant. The observation of 6b as a minor by-product suggests a Cannizarro-like side reaction.16 The route is shown in Scheme 1.
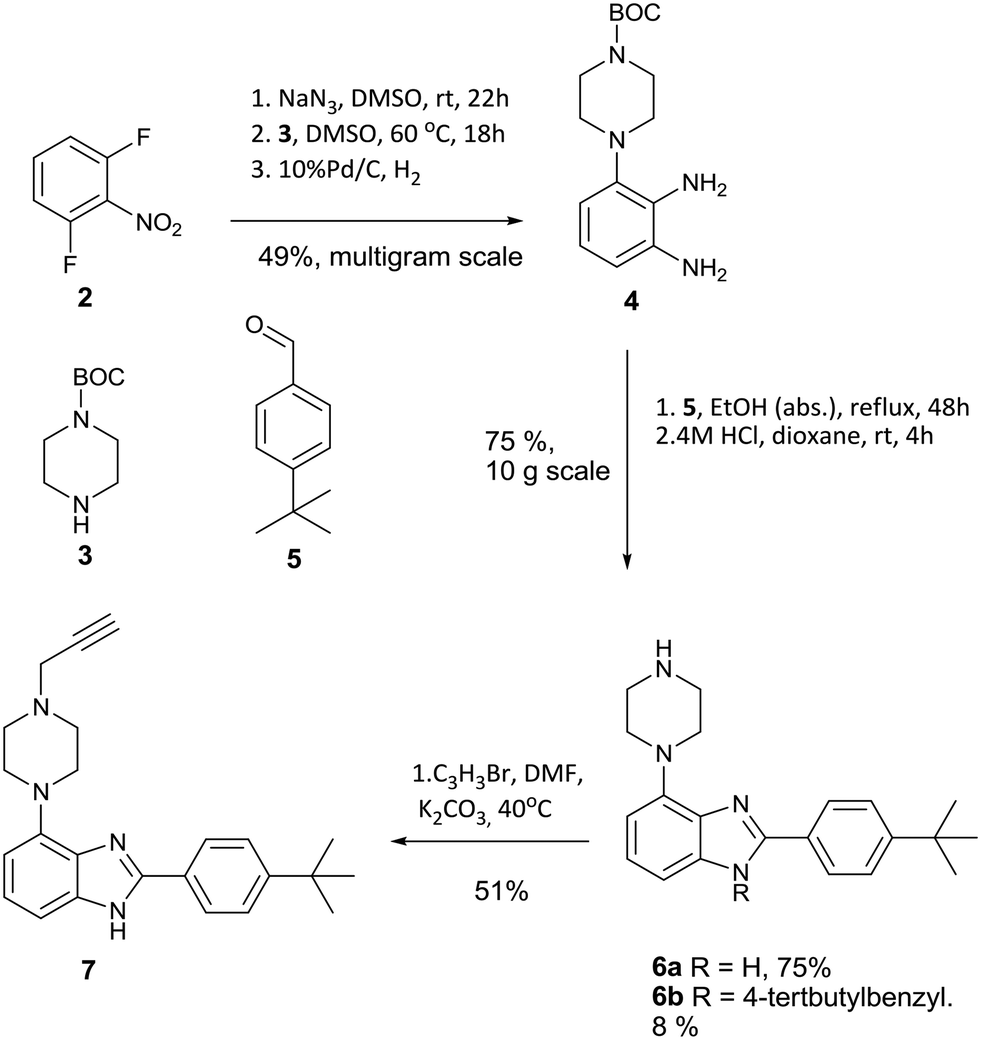 | ||
| Scheme 1 Synthetic route to alkyne intermediate 7. | ||
A further modification was made relative to literature to facilitate cleavage of the BOC protecting group. Substitution of the TFA used originally with HCl in dioxane gave a colourless solid instead of an oily product, which benefits the scalability of this route. Notably, compound 4 is fully converted into heterocyclic products, although the competing formation of 6b in about 8% yield limits the overall yield of 6a to 75% over two steps. Intermediate 7 was synthesized by alkylation with propargyl bromide in dry DMF using potassium carbonate as base in 51% yield to furnish the desired substrate 7 for azide alkyne cycloaddition.
With a robust and high yielding route to gram amounts of 7 in hand, further diversification using the copper-catalysed azide-alkyne cycloaddition (CuAAC) reaction with a range of commercially available azide substrates became possible. See Scheme 2 for an overview.
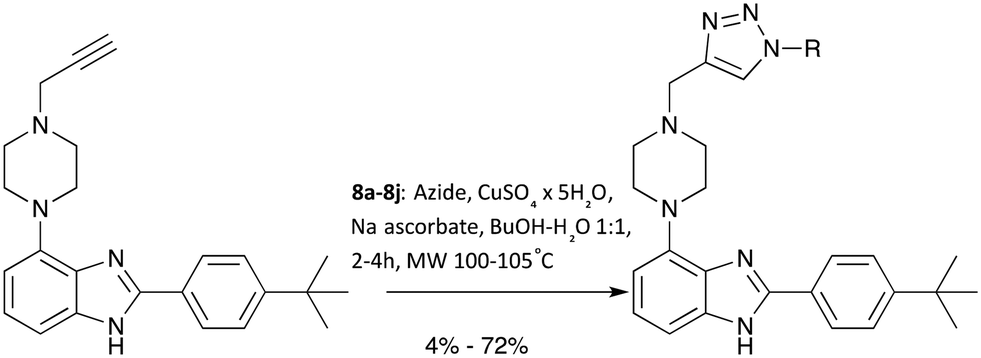 | ||
| Scheme 2 Diversification of the scaffold body using CuAAC. | ||
Since products 8a–8j were obtained in more than sufficient yields for biological testing following a published protocol, no attempts to optimise the reaction were made at this point.
Following synthesis and characterisation, inhibition of receptor activation by LHRH/GnRH was measured in division arrested cell lines from Multispan Inc. stably expressing functional hGnRH receptors. As described in the literature, the solubility of 1 and some of its derivatives is rather poor, which lead to a cut-off at 5 × 103 nM for the maximum concentration in the cell culture assay. Compounds insoluble in DMSO at 10 mM were dropped from testing. Nonetheless, concentration dependent competition studies were performed in vitro in presence of 5 nM LnRH antagonist with the remainder of derivatives. Compounds showing a pIC50 > 5 were tested again over a wide range of concentrations (logarithmic, 0.1–103 nM). A one-site competitive inhibition model was found to work best for fitting of the data curves. To evaluate the obtained inhibition potency values and to validate the assay, we included the well-described, commercially available GnRH antagonists T98475 (8k) and AG04557 (8l) as independent, positive controls and the metabolically stable peptide agonist buserelin as negative control. The lead compound WAY207024 (1) was included as a reference in the cell assay. All IC50 values obtained for each test compound in the concentration dependent screening were converted into Ki values using the equation of Cheng and Prusoff.15
As illustrated in Fig. 2, compounds derived from 1 using CuAAC were found to exhibit competitive antagonism against the endogeneous agonist GnRH. While alkyl piperazines 7a and 7b show only moderate potency, 21 and 43-fold lower than 1, respectively, their clogP values are lower than that of 1, which results in higher solubility (>100 nM). In line with our working hypothesis, the introduction of a planar triazole-core has a positive effect on potency. The 2-fluoroethyl triazole 8a is equally potent as 1, Interestingly the thermodynamic solubility assay showed that 8a is 1.5 times more soluble than 1, 3 μg ml−1 at pH 7.4 (phosphate buffered saline) and 2.098 mg mL−1 at pH 1.2 (simulated gastric fluid) which was indicated by the lower clogP-value. The introduction of an N-BOC-Ala functionalised triazole 8b further improves polarity, albeit at the cost of a drop of one order of magnitude in potency. While these results may indicate that the triazole moiety is a useful pharmacophore, its vicinity was found to be much less tolerant to the introduction of pyranose moieties (8c–f), which was attempted to improve solubility. While these sugar derivatives lead to a major improvement of polarity as indicated by clogP values between 2 and 5, their potency to inhibit GnRH binding dropped out of the desired range. Masking the polyol as a tetraacetate (8c) helped with retaining potency, which does not bode well for making use of the solubility improvement by glycosylation. However, when varying the azide component to a synthetic glycoside to obtain 8g, a viable compromise between solubility and inhibition potency was obtained. Derivative 8g was measured to be soluble at pH 1.2, 1.2 mg ml−1 and <1 μg ml−1 at pH 7.4. With a potency of 38 nM, barely threefold lower than that of 1, this hit may create an opening for further exploration using a library of diverse analogues of 8g. Products 8h–j, obtained from aromatic and heteroaromatic azides and 7, did not lead to increased solubility or affinities compared to WAY207024.
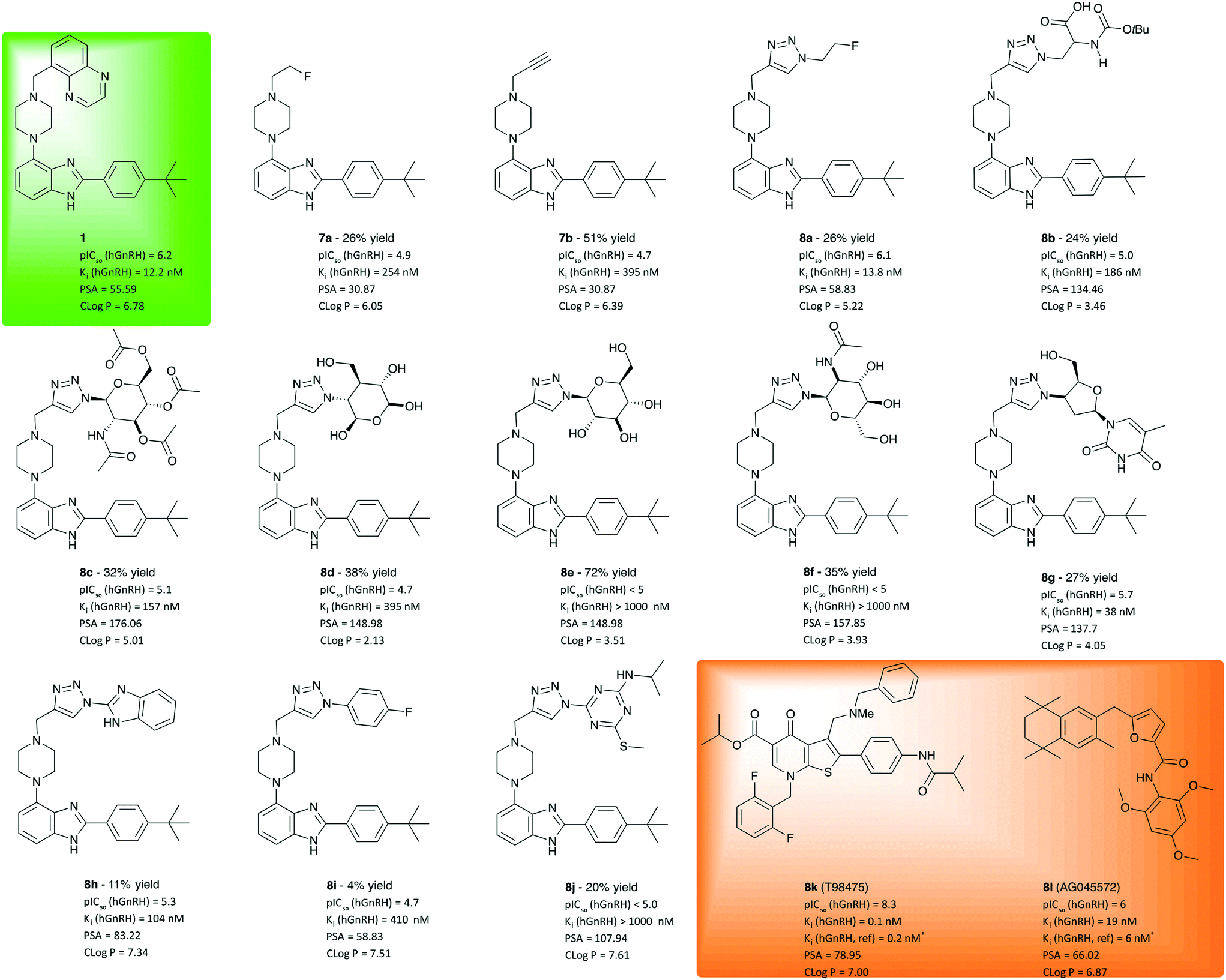 | ||
| Fig. 2 Investigated molecular entities, isolated yields, inhibition potency, inhibition constant and properties computed for evaluation. For comparison, test results of lead 1 (green box) and positive controls 8k and 8l should be considered. aReference value from literature.17,18 IC50 values are given as the geometric mean of two experiments, see ESI† for details. Ki was calculated from mean value using the Cheng–Prusoff equation (Ki = IC50/(1 + [L]/Kd)).19cReplicated twice with broader range of concentrations. dBased on single experiment. | ||
In conclusion, 18 analogues were synthesized using an approach of late-stage diversification via CuAAC and evaluated for potency to inhibit hGnRHR activation. While molecular diversity can easily be introduced into the scaffold body via this route few compounds with hGnRH inhibition potency in the desired range were identified indicating limited tolerance for structural modification in the vicinity of the triazole. The pyranose derivatives did not give the desired inhibition to the GnRH receptors neither did the less soluble compounds 8h and 8i. Nonetheless, compounds 8a and 8g emerge as highly promising candidates for further investigation in behavioural animal models as a couple of GnRH modulating antagonists.
Conflicts of interest
The author declare no competing interests.Acknowledgements
RF and RS acknowledge the Northern Norway Regional Health Authority for funding [project number: SFP1196-14]. PJR gratefully acknowledges the research council of Norway and the University of Oslo (realomics SFI). JHH gratefully acknowledges the Department of Chemistry at UiT, The Arctic University of Norway, for funding parts of this research project.Notes and references
- S. Meethal, M. Smith, R. Bowen and C. Atwood, Endocrine, 2005, 26, 317–325 CrossRef CAS PubMed.
- V. A. Tobin, R. P. Millar and B. J. Canny, Endocrinology, 1997, 138, 3314–3319 CrossRef CAS PubMed.
- G. Zhang, J. Li, S. Purkayastha, Y. Tang, H. Zhang, Y. Yin, B. Li, G. Liu and D. Cai, Nature, 2013, 497, 211–216 CrossRef CAS PubMed.
- C. Eisenegger, M. Naef, R. Snozzi, M. Heinrichs and E. Fehr, Nature, 2010, 463, 356–359 CrossRef CAS PubMed.
- H. van Poppel and S. Nilsson, Urology, 2008, 71, 1001–1006 CrossRef PubMed.
- R. L. Gustofson, J. H. Segars and F. W. Larsen, Hum. Reprod., 2006, 21, 2830–2837 CrossRef CAS PubMed.
- J. B. Engel and A. V. Schally, Nat. Clin. Pract. Endocrinol. Metab., 2007, 3, 157–167 CrossRef CAS PubMed.
- P. Briken, A. Hill and W. Berner, J. Clin. Psychiatry, 2003, 64, 890–897 CrossRef CAS PubMed.
- J. C. Pelletier, M. Chengalvala, J. Cottom, I. Feingold, L. Garrick, D. Green, D. Hauze, C. Huselton, J. Jetter, W. Kao, G. S. Kopf, J. T. T. Lundquist, C. Mann, J. Mehlmann, J. Rogers, L. Shanno and J. Wrobel, Bioorg. Med. Chem., 2008, 16, 6617–6640 CrossRef CAS PubMed.
- D. B. Hauze, M. V. Chengalvala, J. E. Cottom, I. B. Feingold, L. Garrick, D. M. Green, C. Huselton, W. Kao, K. Kees, J. T. Lundquist Iv, C. W. Mann, J. F. Mehlmann, J. F. Rogers, L. Shanno, J. Wrobel and J. C. Pelletier, Bioorg. Med. Chem. Lett., 2009, 19, 1986–1990 CrossRef CAS PubMed.
- J. C. Pelletier, M. V. Chengalvala, J. E. Cottom, I. B. Feingold, D. M. Green, D. B. Hauze, C. A. Huselton, J. W. Jetter, G. S. Kopf, J. T. Lundquist, R. L. Magolda, C. W. Mann, J. F. Mehlmann, J. F. Rogers, L. K. Shanno, W. R. Adams, C. O. Tio and J. E. Wrobel, J. Med. Chem., 2009, 52, 2148–2152 CrossRef CAS PubMed.
- R. D. Egleton and T. P. Davis, NeuroRx, 2005, 2, 44–53 CrossRef PubMed.
- A. Banerjee, S. Maschauer, H. Hübner, P. Gmeiner and O. Prante, Bioorg. Med. Chem. Lett., 2013, 23, 6079–6082 CrossRef CAS PubMed.
- R. Haubner, B. Kuhnast, C. Mang, W. A. Weber, H. Kessler, H.-J. Wester and M. Schwaiger, Bioconjugate Chem., 2004, 15, 61–69 CrossRef CAS PubMed.
- M. Schottelius, F. Rau, J. C. Reubi, M. Schwaiger and H.-J. Wester, Bioconjugate Chem., 2005, 16, 429–437 CrossRef CAS PubMed.
- R. Chebolu, D. N. Kommi, D. Kumar, N. Bollineni and A. K. Chakraborti, J. Org. Chem., 2012, 77, 10158–10167 CrossRef CAS PubMed.
- E. A. Iatsimirskaia, M. L. Gregory, K. L. Anderes, R. Castillo, K. E. Milgram, D. R. Luthin, V. P. Pathak, L. C. Christie, H. Vazir, M. B. Anderson and J. M. May, Pharm. Res., 2002, 19, 202–208 CrossRef CAS.
- N. Cho, M. Harada, T. Imaeda, T. Imada, H. Matsumoto, Y. Hayase, S. Sasaki, S. Furuya, N. Suzuki, S. Okubo, K. Ogi, S. Endo, H. Onda and M. Fujino, J. Med. Chem., 1998, 41, 4190–4195 CrossRef CAS PubMed.
- Y.-C. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical characterisation of all compounds. See DOI: 10.1039/c7md00320j |
| This journal is © The Royal Society of Chemistry 2017 |