
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitochondria-targeted betulinic and ursolic acid derivatives: synthesis and anticancer activity†
Darya A.
Nedopekina
 a,
Rinat R.
Gubaidullin
a,
Victor N.
Odinokov
a,
Polina V.
Maximchik
b,
Boris
Zhivotovsky
bc,
Yuriy P.
Bel'skii
d,
Veniamin A.
Khazanov
d,
Arina V.
Manuylova
d,
Vladimir
Gogvadze
*bc and
Anna Yu.
Spivak
*a
a,
Rinat R.
Gubaidullin
a,
Victor N.
Odinokov
a,
Polina V.
Maximchik
b,
Boris
Zhivotovsky
bc,
Yuriy P.
Bel'skii
d,
Veniamin A.
Khazanov
d,
Arina V.
Manuylova
d,
Vladimir
Gogvadze
*bc and
Anna Yu.
Spivak
*a
aInstitute of Petrochemistry and Catalysis, Russian Academy of Sciences, 141 prosp. Oktyabrya, Ufa 450075, Russian Federation. E-mail: spivak.ink@gmail.com
bFaculty of Fundamental Medicine, MV Lomonosov Moscow State University, 11999 Moscow, Russia. E-mail: vlad_gogvadze@rambler.ru
cDivision of Toxicology, Institute of Environmental Medicine, Karolinska Institutet, Box 210, 17177 Stockholm, Sweden
dInnovative Pharmacology Research (IPHAR), 79/4 Elizarova, Tomsk 634021, Russian Federation
First published on 13th September 2017
Abstract
A series of new betulinic and ursolic acid conjugates with a lipophilic triphenylphosphonium cation, meant to enhance the bioavailability and mitochondriotropic action of natural triterpenes, have been synthesized. The in vitro experiments on three human cancer cell lines (MCF-7, HCT-116 and TET21N) revealed that all the obtained triphenylphosphonium triterpene acid derivatives not only showed higher cytotoxicity as compared to betulinic acid but were also markedly superior in triggering mitochondria-dependent apoptosis, as assessed using a range of apoptosis markers such as cytochrome c release, stimulation of caspase-3 activity, and cleavage of poly(ADP-ribose) polymerase, which is one of the targets of caspase 3. The IC50 was much lower for all triphenylphosphonium derivatives when compared to betulinic acid. Out of the tested group of conjugates, the most potent toxicity was exhibited by the betulinic acid conjugate 9 (for 9, the IC50 values against MCF-7 and TET21N cells were 0.70 μM and 0.74 μM; for betulinic acid (BA), IC50 > 25 μM against MCF-7 cells).
1. Introduction
Apoptosis is an evolutionarily conserved and genetically regulated process of critical importance for embryonic development and maintenance of tissue homeostasis in the adult organism.1 Apoptosis and cancer are antagonistic processes in cell physiology. Stimulation of apoptosis is responsible for the elimination of potentially harmful or premalignant cells, whereas suppression of apoptotic pathways can lead to uncontrolled cell proliferation and formation of malignancies. Thus, searching for compounds that can specifically stimulate tumor cell death is very important for developing antitumor strategies.2,3Although it appears that distinct pathways leading to cell death are triggered by different signals, they often merge at a common “regulator” of this multistep process – mitochondria. Specifically, permeabilization of the outer mitochondrial membrane (OMM) and the release of certain proteins (including cytochrome c) from the intermembrane space of mitochondria are regarded as key events in apoptosis induction. Once in the cytosol, cytochrome c interacts with its adaptor molecule, Apaf-1, resulting in the recruitment, processing and activation of pro-caspase-9. Active caspase-9, in turn, cleaves and activates pro-caspase-3 and -7; these effector caspases are responsible for the cleavage of cellular proteins leading to biochemical and morphological characteristic features of apoptosis. Other proteins released from the intermembrane space of mitochondria are the Apoptosis Inducing Factor (AIF), Smac/Diablo and Omi. AIF and the serine protease Omi catalyze caspase-independent downstream events in the apoptotic process. Therefore, permeabilization of the outer mitochondrial membrane (OMM) is considered a crucial event during the early phase of the apoptotic process. It should be mentioned that caspase-8 cleaves not only caspase-3 but also Bid, a cytosolic protein and a proapoptotic member of Bcl-2 family proteins.4 Truncated Bid (tBid) oligomerizes another proapoptotic protein, Bax, which causes permeabilization of the OMM and cytochrome c release. The pore-forming capacity of Bax and subsequent release of cytochrome c can be prevented by overexpression of Bcl-2. The overexpression of Bcl-2 and Bcl-XL counteracts OMM permeabilization and contributes to drug resistance in high-risk NB cells.5 Another mode of OMM permeabilization is stimulation of the so-called mitochondrial permeability transition, a Ca2+-dependent process which results in mitochondrial swelling and OMM rupture. Targeting of mitochondria and OMM permeabilization via induction of mitochondrial permeability transition (MPT) might be powerful tool against protective effects of Bcl-2 and Bcl-XL.5–7
Pentacyclic lupane and ursane type triterpenoids (betulin, betulinic and ursolic acids) are among the most abundant terpenoids found in the plant kingdom. These compounds are of interest for pharmacological research, as they exhibit a broad spectrum of biological properties.8–13 Among these properties of triterpenoids, of special interest is their anticancer activity and the ability to trigger the mitochondrial apoptosis pathway in various types of human cancer cells. Thus, betulinic acid is capable of inducing apoptosis in tumor cells of certain types such as melanoma, lung cancer, ovarian cancer and neuroectodermal tumors.14–16 Betulinic acid stimulates apoptosis with the participation of reactive oxygen species (ROS). ROS contribute to permeabilization of the outer mitochondrial membrane (OMM), releasing apoptogenic mitochondrial proteins (cytochrome c, Smac, apoptosis inducing factor (AIF)) from the intermembrane space, and subsequent activation of the caspase cascade.14–18
In vitro, ursolic acid was shown to inhibit the proliferation of various cancer cell types by suppressing the STAT3 activation pathway.19,20 It can also induce apoptosis, autophagy, and cell cycle arrest through various pathways, such as inhibition of DNA replication, stimulation of reactive oxygen species (ROS) production, and affecting the balance between pro- and antiapoptotic proteins.21–23
The special attractiveness of these natural compounds is due to the absence of their cytotoxic action towards normal human cells (fibroblasts or proliferating normal lymphocytes) and negligible systemic toxicity in in vivo experiments, using xenograft models.24–27 However, the relatively low anticancer potential, high hydrophobicity, and poor blood serum solubilization of triterpene acids markedly hamper their advancement as anticancer drug candidates.
Previously, we synthesized conjugates of lupane triterpenoids with a triphenylphosphonium (TPP+) group, which showed cytotoxicity in vitro at low micromolar concentrations and significantly exceeded the antitumor activity of betulinic acid.28,29 In this paper, we discuss the results of investigations into probable mechanisms underlying the cytotoxic effect of triphenylphosphonium cations of triterpenoids. We have synthesized a new series of triphenylphosphonium salts of betulinic and ursolic acids, studied their cytotoxic activity in two human cancer cell lines and their ability to induce programmed cancer cell death, using markers of apoptosis such as activation of caspase-3, PARP-1 cleavage, permeabilization of the outer mitochondrial membrane, release of cytochrome c and inhibition of the mitochondrial respiratory chain.
2. Results and discussion
2.1. Chemistry
The triphenylphosphonium group was linked to a molecule of betulinic or ursolic acid at the C-28 position of the triterpenoid skeleton through the hydrophobic n-butyl or hydrophilic triethylene glycol spacer. In an attempt to enhance the antitumor activity of triphenylphosphonium salts, we linked the 3β-OH group in compounds 6, 10, 14, and 16 with a molecule of dichloroacetate, a known pyruvate dehydrogenase kinase (PDK) inhibitor.30 Inhibition of PDK stimulates the activity of pyruvate dehydrogenase (PDH) facilitating oxidation of pyruvate in mitochondria. Dichloroacetate was shown to reduce high mitochondrial membrane potential in tumor cells and to increase mitochondrial ROS production in malignant but not in normal cells.31 Previously,32 it has been shown that dichloroacetate appended to the C-3 hydroxy group of betulinic acid provided a synergistic cytotoxic effect against a broad spectrum of cancer cells.The betulinic and ursolic acid triphenylphosphonium salts 9 and 15 containing a triethylene glycol bridge were obtained by alkylation of the 28-carboxyl group of the triterpenoids with triethylene glycol dibromide in DMF in the presence of K2CO3 at 50 °C for 3 h. The dibromides were synthesized from triethylene glycol ditosylate33 by treatment with LiBr in boiling acetone for 14 hours. In the synthesis of triterpene acid conjugates 6 and 14, the C(28)-carboxyl group was alkylated with a twofold excess of 1,4-dibromobutane in DMF and acetonitrile in the presence of K2CO3 at 50 °C. The bromides thus obtained were refluxed with a fivefold molar excess of triphenylphosphine in acetonitrile. The phosphonium salts with 3-dichloroacetates 6, 10, 14, and 16 were prepared by esterification of the C(3)-hydroxyl in the triterpenoid skeleton with dichloroacetic acid using the carbodiimide method (Schemes 1 and 2).
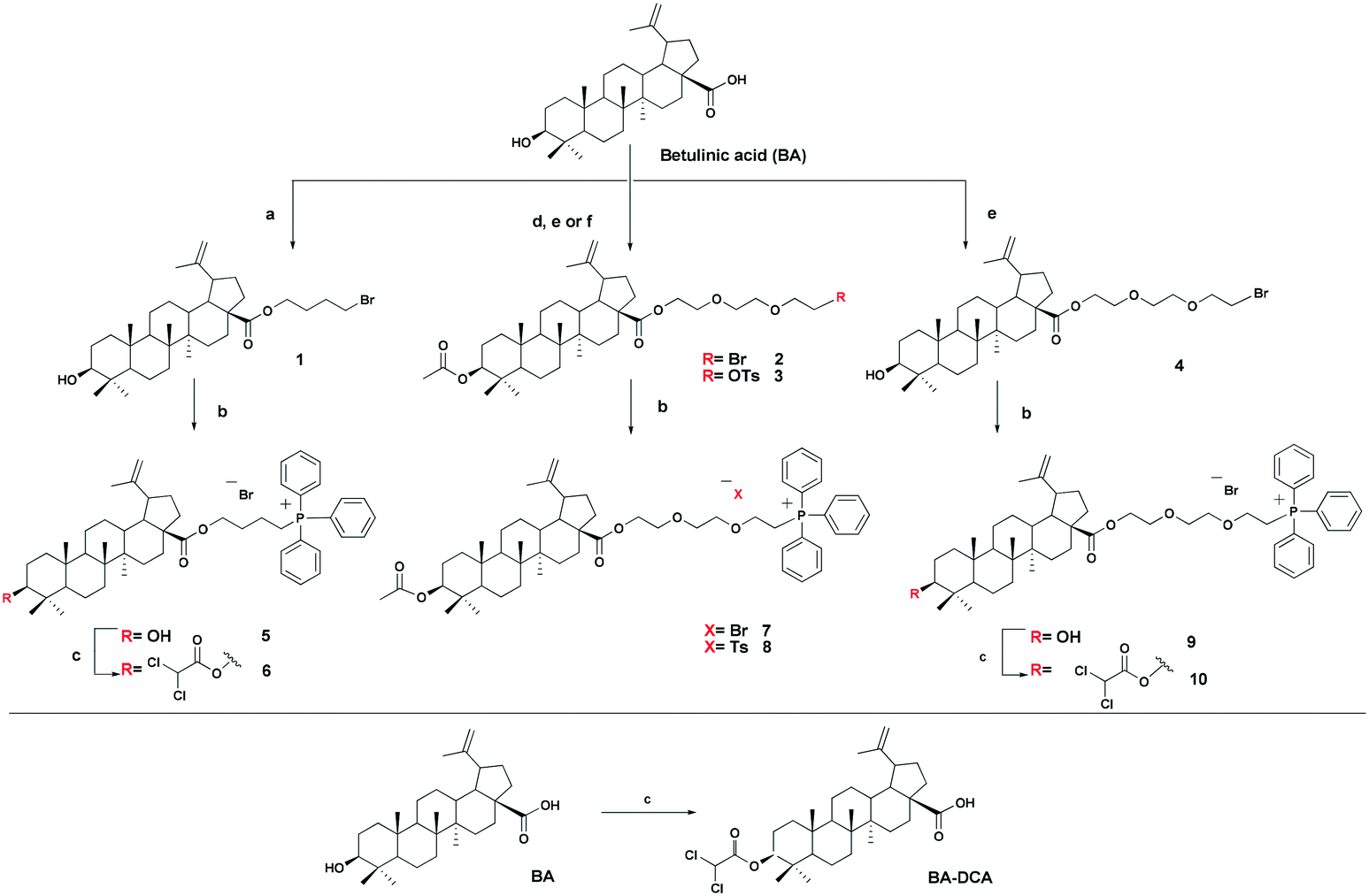 | ||
| Scheme 1 Synthesis of betulinic acid derivatives. Reagent and conditions: a 1,4-dibromobutane, K2CO3, MeCN, DMF, 50 °C, 3h; b PPh3, CH3CN, reflux, Ar, 20–35 h; c Cl2COOH, DMAP, DCC, CH2Cl2, 2–7 h, rt; d AcCI, Py, DMAP, THF, 20 °C; e tri(ethylene glycol) dibromide, K2CO3, DMF, 50 °C, 3 h; f tri(ethylene glycol) ditosylate, K2CO3, MeCN, 70 °C, 7 h. | ||
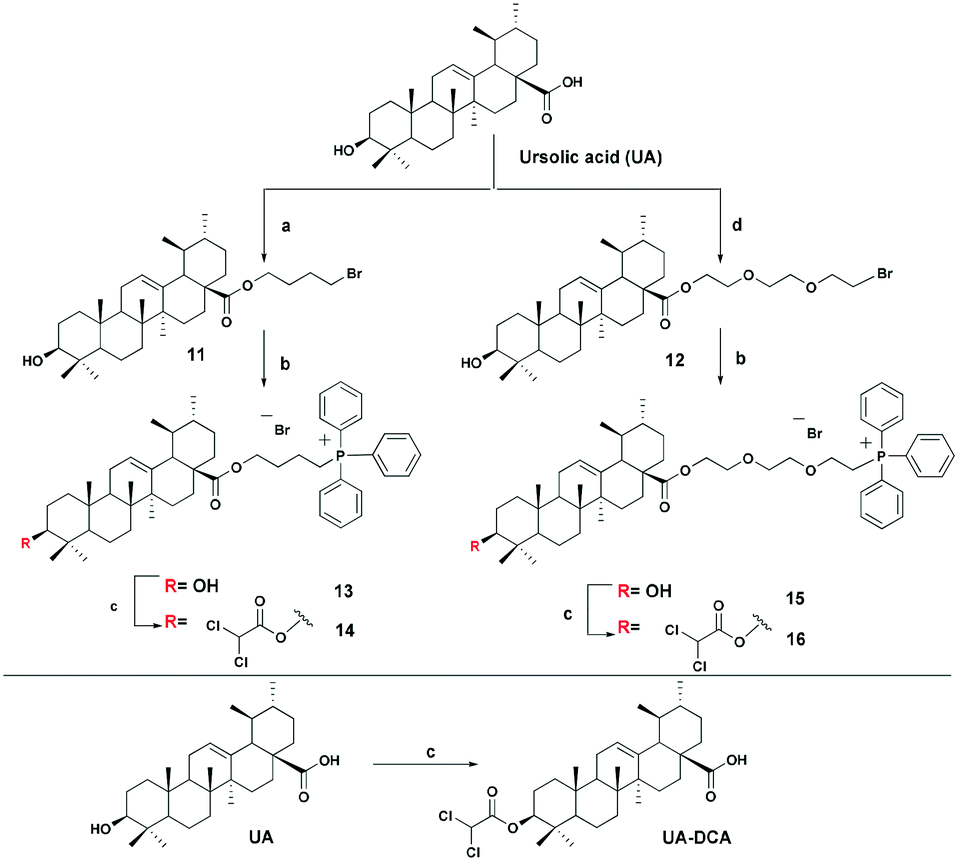 | ||
| Scheme 2 Synthesis of ursolic acid derivatives. Reagents and conditions: a 1,4-dibromobutane, K2CO3, MeCN, DMF, 50 °C, 3 h; b PPh3, CH3CN, reflux, Ar, 20–35 h; c Cl2COOH, DMAP, DCC, CH2Cl2, 2–7 h, rt; d tri(ethylene glycol) dibromide, K2CO3, DMF, 50 °C, 3 h. | ||
2.2. Biological evaluation
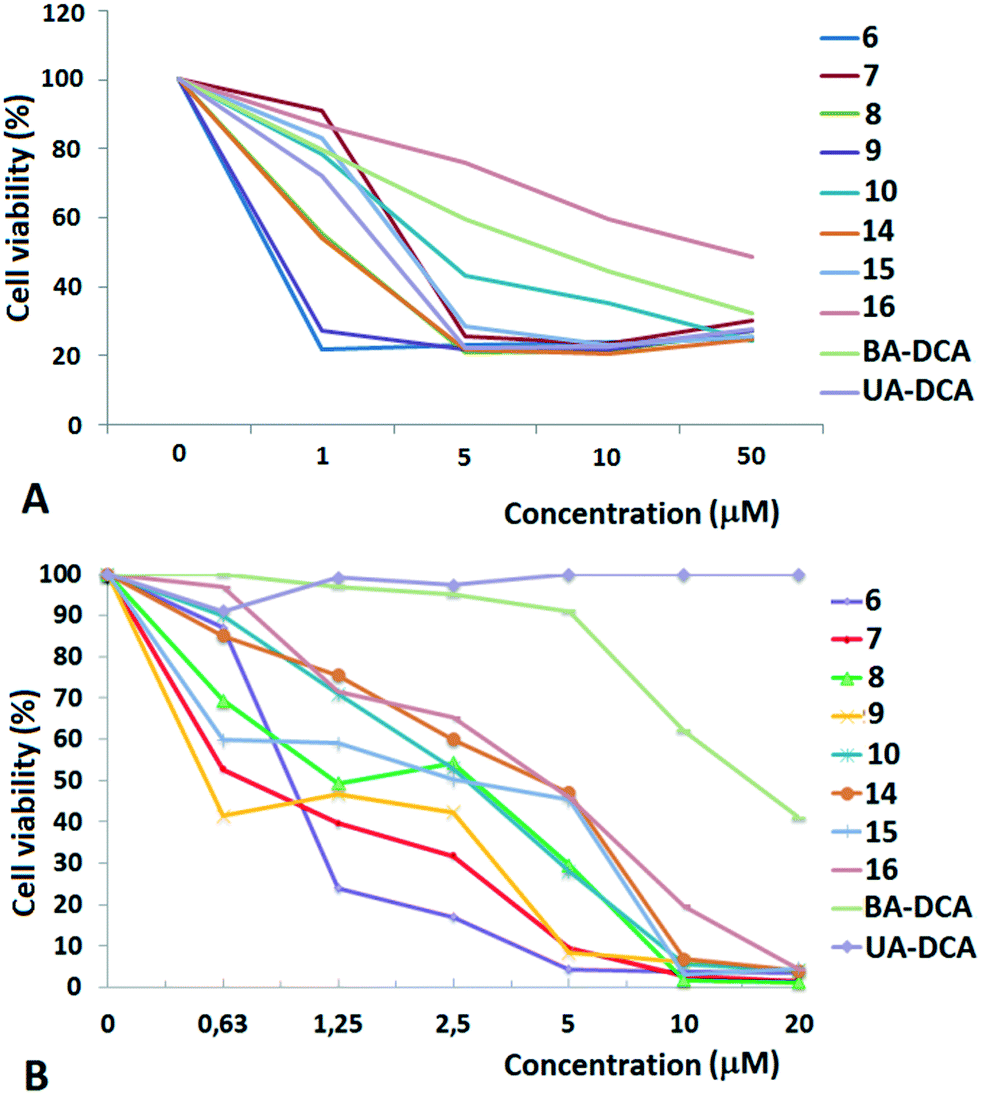 | ||
| Fig. 1 Dose-dependent cytotoxic action of triphenylphosphonium salts of betulinic and ursolic acids on TET21N (neuroblastoma) (A) and MCF-7 (breast adenocarcinoma) (B) cells. The results are expressed as the percentage of viable cells. | ||
| Test compound | TET21N | MCF-7 |
|---|---|---|
| Note: *IC50 (μM) is the half maximal inhibitory concentration for viable cells. Each IC50 (mean ± SE) has been derived from at least three experiments in duplicate. | ||
| 6 | 1.26 ± 0.18 | 0.80 ± 0.08 |
| 7 | 0.98 ± 0.11 | 0.85 ± 0.09 |
| 8 | 1.28 ± 0.18 | 1.51 ± 0.13 |
| 9 | 0.74 ± 0.14 | 0.70 ± 0.11 |
| 10 | 0.95 ± 0.15 | 2.31 ± 0.09 |
| 14 | 4.4 ± 0.34 | 2.76 ± 0.21 |
| 15 | 0.81 ± 0.08 | 1.59 ± 0.11 |
| 16 | 1.18 ± 0.16 | 3.90 ± 0.06 |
| BA-DCA | 4.9 ± 0.2 | 15.89 ± 0.19 |
| UA-DCA | >10 | >20 |
| BA | — | >25 |
High cytotoxic activity against each of the cell lines was found for the betulinic acid conjugate 9 (IC50 values of 0.70–0.74 μM). A considerable activity (<1 μM) was also inherent in the triphenylphosphonium salts of lupane triterpenoids with an acetate or a dichloroacetate function at the triterpenoid's C-3 atom: 6 and 7 against MCF-7 (IC50 values of 0.80 μM and 0.85 μM) and 7, 10 and 15 against TET21N (IC50 values of 0.98 μM, 0.95 μM, and 0.81 μM). Contrary to our expectations, the conjugation of the lupane and ursane triphenylphosphonium salts with dichloroacetate did not bring about a synergistic cytotoxicity. The cytotoxic activity of conjugate 7 against TET21N cells was comparable with that of conjugate 10, while the activity of 9 was even somewhat higher than that of 10 (Table 1). We did not find any noticeable difference between the anticancer activities of n-butyl- and triethylene glycol-bridged conjugates. The triterpenoid skeleton structure had a pronounced effect on the cytotoxicity of phosphonium salts. The antitumor activity of phosphonium lupane triterpenoid derivatives was markedly higher than the activities of the corresponding ursane derivatives, 14, 15 and 16. Betulinic acid and C(3)-dichloroacetyl betulinic and ursolic acid derivatives, BA-DCA and UA-DCA, exhibited markedly lower cytotoxicity than any of the triphenylphosphonium salts. At the concentrations we tested, these compounds were inactive: in the case of TET21N and MCF-7 cells, the IC50 values were 4.91 μM and 15.89 μM for BA-DCA and >10 μM and >20 μM for UA-DCA, respectively.
However, the cytotoxic activity of the newly obtained triphenylphosphonium salts was comparable to their cytotoxic activity against normal mouse splenocytes. Thus the IC50 values were 1.95 μM, 0.66 μM, 1.75 μM and 1.13 μM for compounds 6, 10, 14 and 16, respectively.
Because the antitumor activities of betulinic acid derivatives 6–10 were higher than the activities of the corresponding ursane conjugates 14–16, we have further focused on the ability of BA and its analogues to stimulate cancer cell death.
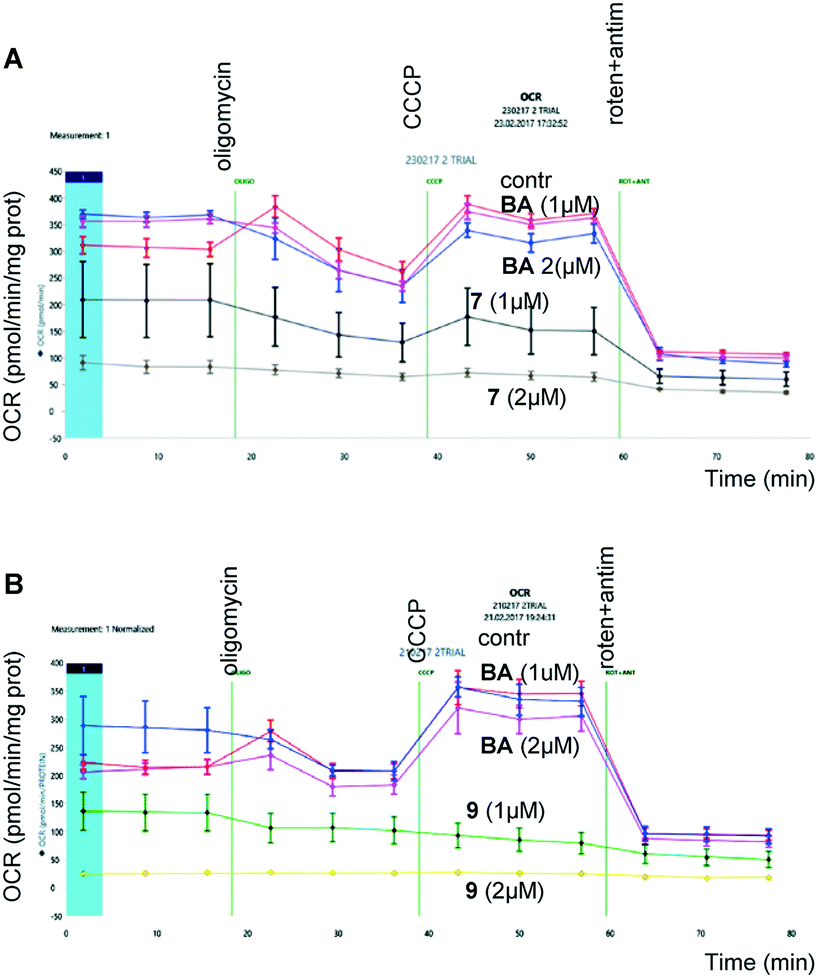 | ||
| Fig. 2 Assessment of oxygen consumption by HCT116 (human colon carcinoma) cancer cells after incubation with betulinic acid and its derivatives, 7 (upper panel) and 9 (lower panel), for 24 hours. | ||
As shown in Fig. 2, the derivatives of BA, 7 (upper panel) and 9 (lower panel), suppressed respiration in a concentration-dependent manner. At the same time, the same concentrations of BA were ineffective. These experiments clearly demonstrate mitochondrial involvement in triggering apoptosis by BA triphenylphosphonium derivatives.
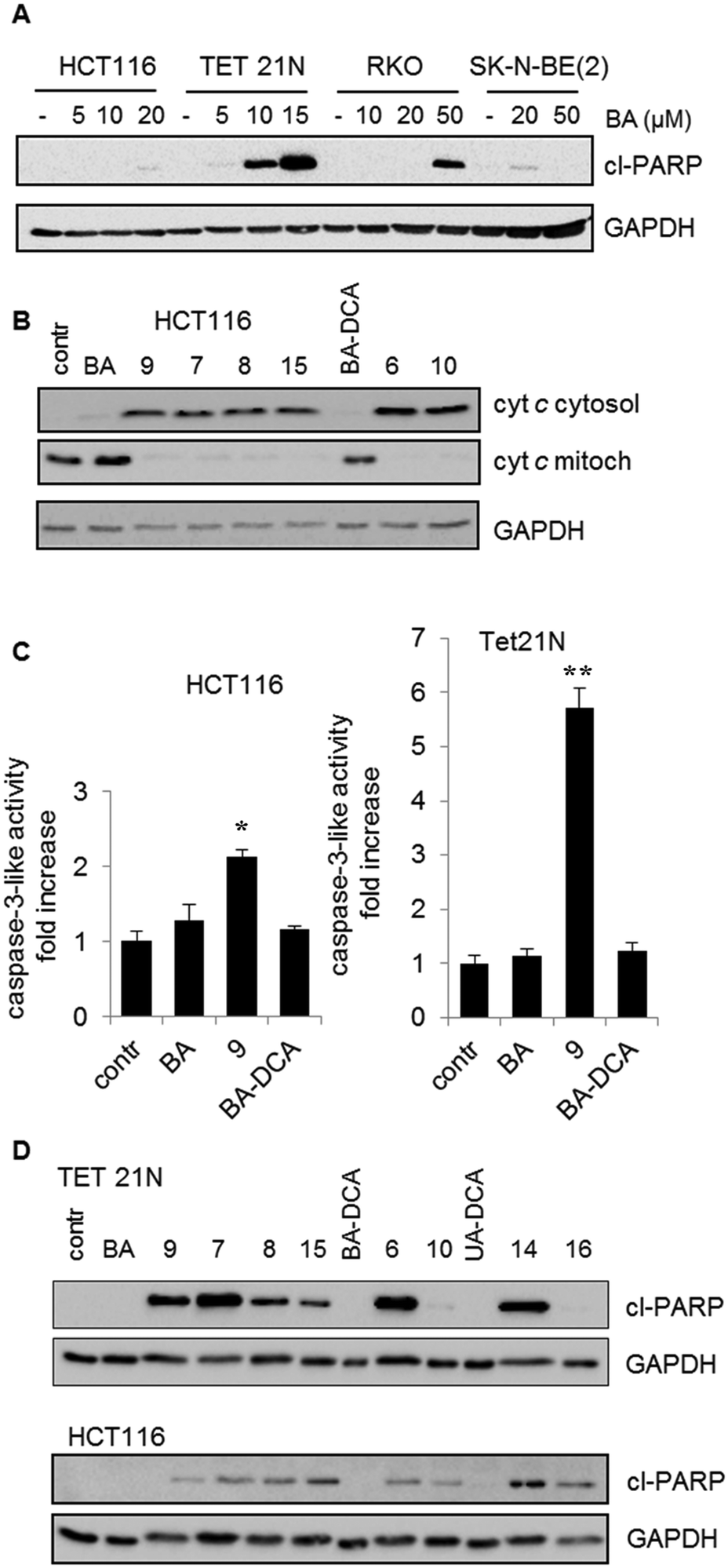 | ||
| Fig. 3 Stimulation of apoptosis (A) by BA in various tumor cell lines: HCT WT (human colon carcinoma cell line and RKO (human rectal carcinoma cell line, TET21N and SK-N-BE (2) (neuroblastoma cell lines); (B) release of cytochrome c from the mitochondria to the cytosol under the action of betulinic acid (BA) and its derivatives with a concentration of 3.5 μM; (C) activation of caspase-3 in HCT and TET21N cells by betulinic acid (BA) and its derivatives, 9 and BA-DCA. The mean values of three experiments ± standard error are given. The statistical significance of the differences was estimated using the Student's t-test, *<0.05, **<0.01; (D) cleavage of PARP by betulinic and ursolic acid derivatives. The assay was based on determining the content of the PARP cleavage product (cl-PARP). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the blot loading control. | ||
As has been mentioned in the introduction, the caspase cascade is triggered by the release of cytochrome c. In order to prove that administration of BA and its analogs results in OMM permeabilization, assessment of cytochrome c release was performed (Fig. 3B). The content of cytochrome c in cytosol and mitochondria was assessed by western blotting after fractionation of HCT116 cells (see 4.2. Biology; 4.2.5.). As can be seen from the results, at 3.5 μM concentration BA failed to trigger the release of cytochrome c, whereas the triphenylphosphonium derivatives of BA were significantly more effective. These experiments clearly demonstrate mitochondrial involvement in triggering apoptosis by BA derivatives.
Next, we analyzed the ability of BA and its derivatives to stimulate caspase-3 activity in HCT116 and TET21 cells. DEVD-AMC was used as a fluorogenic substrate for caspase-3. In agreement with previous data, the results revealed that the triphenylphosphonium salt of betulinic acid 9 considerably surpassed betulinic acid in the caspase-3 activation (Fig. 3C). In addition, the advantage of the derivatives of BA was demonstrated by their ability to cleave PARP (Fig. 3D). The results clearly show that the antitumor activity of the original BA is markedly lower than that of some of its derivatives.
Finally, the antitumor activity of BA and its derivatives was analyzed by the assessment of cells with decreased DNA content. During apoptosis, DNA is cleaved by caspase-activated DNases (CAD), with formation of small fragments. The fragmented 182 bp DNA multimers leak out of the apoptotic cell upon fixation. This results in the appearance of a subpopulation of cells with a reduced DNA content, which can be detected upon staining with propidium iodide. An assessment of the SubG1 population in HCT116 cells is demonstrated in Fig. 4. BA did not stimulate cell death (second quadrant in the second row), whereas its several triphenylphosphonium derivatives, such as 6–9, triggered apoptosis. Thus, the advantage of triphenylphosphonium derivatives of BA in cell death stimulation was confirmed by a number of parameters.
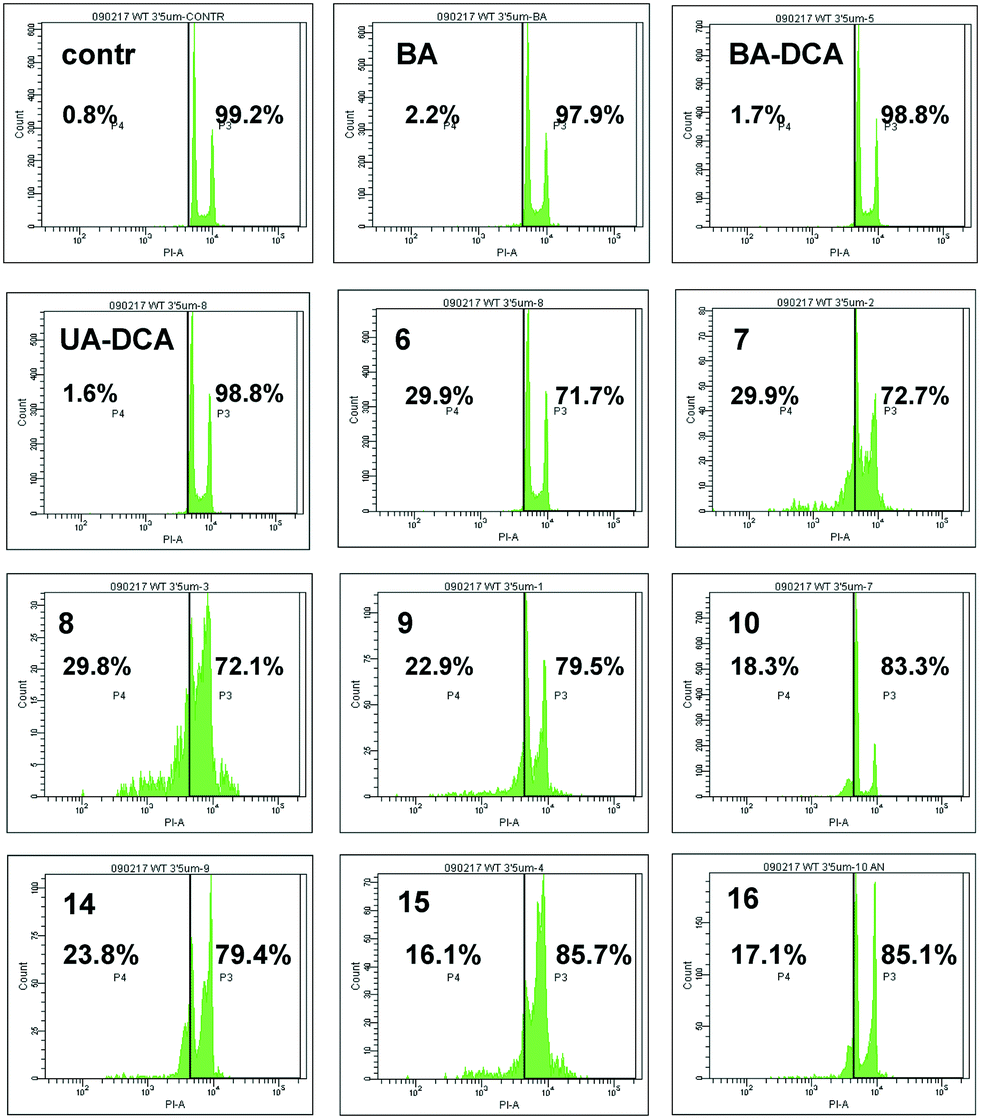 | ||
| Fig. 4 Apoptosis stimulated by betulinic acid (BA) and its derivatives in HCT116 carcinoma cells, shown as the percentage of cells with a reduced amount of DNA (SubG1 population, left part of each quadrant). | ||
3. Conclusions
A range of anticancer agents named mitocans (an abbreviation formed from MITOchondria and CANcer) were shown to cause cell death via targeting mitochondria.35 Thus, one of the mitocans, D-α-tocopherol succinate, a redox-silent analogue of vitamin E (α-TOS), induces multiple changes in tumor cells leading to cell death.36 This compound was shown to selectively kill malignant cells at concentrations which are non-toxic to normal cells. This faculty of α-TOS was explained by its ability to produce ROS (more toxic to malignant cells due to lower antioxidant activity) via interaction with the respiratory chain of mitochondria. Non-malignant cells are protected from α-TOS due to higher levels of esterases which cleave α-TOS with formation of succinate and an antioxidant – α-tocopherol. In the case of BA, the mechanisms might be different, although mitochondrial involvement in cell death execution has been demonstrated by several groups.14–17Recently, conjugation with a lipophilic mitochondriotropic triphenylphosphonium cation (TPP+) has been actively used to enhance the effectiveness and selectivity of mitochondria-targeted anti-cancer agents.37,38 Making a compound positively charged by coupling with TPP+ bearing three positive charges enhances mitochondrial targeting and allows decreasing doses of the drug. The triphenylphosphonium moiety was used to enhance the bioavailability and cytotoxic activity against cancer cells of such compounds as α-tocopheryl succinate,39 curcuminoids,40 alkyl gallates41 and the diterpenoid isosteviol.42 Selective accumulation of lipophilic cationic compounds in mitochondria is associated with the relatively high transmembrane potential of these organelles (Δψ 150–180 mV) in comparison with other organelles and cells. In addition, the transmembrane potential of mitochondria of solid tumor cells is higher than the transmembrane potential of normal cells, which can contribute to the selective cytotoxicity of antitumour substances.
Indeed, under the conditions of our experiments, conjugation of native triterpenoids, betulinic and ursolic acids with a TPP+ group led to the significant enhancement of cytotoxicity to MCF-7 and TET-21N cancer cells, when compared to BA. The mitochondriotropic analogues of BA were markedly superior to the native triterpenoid in triggering mitochondria-dependent apoptosis, as assessed by a range of apoptosis markers, such as cytochrome c release, stimulation of caspase-3 activity and cleavage of poly(ADP-ribose) polymerase. Thus, structural modifications of BA by conjugation with TPP+ markedly enhance its anticancer properties.
4. Experimental section
4.1. Chemistry
All reagents and solvents were of the purest grade available and generally were used without further treatment. The starting compounds betulinic, ursolic acids and reagents: LiBr, triethylene glycol, p-toluenesulfonyl chloride, PPh3, 1,4-dibromobutane, dimethylaminopyridine (DMAP), dichloroacetic acid, N,N′-dicyclohexylcarbodiimide (DCC) and acetonitrile were purchased from Sigma-Aldrich, Europe. Triethylene glycol ditosylate was prepared by a reported method.33 3-Acetylbetulinic and 3-acetylursolic acids were synthesized from betulinic and ursolic acids according to the typical procedures. IR spectra were recorded on a Specord IR-75 spectrometer for neat samples or for solutions in CHCl3. 1H, 13C, and 31P NMR spectra were recorded on a Bruker Avance-500 spectrometer (1H, 500.13 MHz; 13C, 125.78 MHz) and on a Bruker Avance-400 spectrometer (1H, 400.13 MHz; 13C, 100.62 MHz; 31P, 161.98 MHz) using Me4Si as the internal standard and CDCl3 as the solvent. Mass spectra were recorded on a Bruker-Autoflex III instrument in the MALDI-TOF regime with positive ionization recording using 2,5-dihydroxybenzoic and α-cyano-4-hydroxycinnamic acids as matrices. Optical rotation was measured using a Perkin-Elmer-141 polarimeter. Elemental analysis was performed using a Carlo Erba 1106 analyzer. Sorbfil plates (Sorbpolimer, Krasnodar, Russia) were used for TLC, visualized with anisaldehyde/sulfuric acid. Silica gel L (50–160 lm, KSKG) was used for column chromatography.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1] to afford dibromide. Triethylene glycol dibromide (3 mmol) was added to a stirred suspension of BA or UA (1 mmol) in DMF (5 mL) and K2CO3 (1 mmol), and the mixture was kept at 50 °C for 3 h [TLC monitoring hexane/EtOAc 1:1]. The mixture was then poured into cold H2O (10× volume) and extracted with CHCl3 (2 × 15 mL each). The combined CHCl3 layers were washed with H2O (100 mL) and concentrated. The crude residue was co-evaporated with hexane (100 mL) to remove residual DMF. The residue was chromatographed on silica gel [hexane/EtOAc, 30:1], to obtain a pure compound.
:1] to afford dibromide. Triethylene glycol dibromide (3 mmol) was added to a stirred suspension of BA or UA (1 mmol) in DMF (5 mL) and K2CO3 (1 mmol), and the mixture was kept at 50 °C for 3 h [TLC monitoring hexane/EtOAc 1:1]. The mixture was then poured into cold H2O (10× volume) and extracted with CHCl3 (2 × 15 mL each). The combined CHCl3 layers were washed with H2O (100 mL) and concentrated. The crude residue was co-evaporated with hexane (100 mL) to remove residual DMF. The residue was chromatographed on silica gel [hexane/EtOAc, 30:1], to obtain a pure compound.
8′-Bromo-3′,6′-dioxaoctan-1′-yl-3β-acetoxylup-20(29)-en-28-oate (2). Amorphous powder, 49% yield; [α]21D +12.42° (c 1.21, CHCl3); IR (CHCl3) νmax: 1729 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.84, 0.85, 0.86, 0.92, 0.97 (3H each, all s, H-23–H-27), 0.78–2.28 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.69 (s, 3H, H-30), 2.05 (s, 3H,
O) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.84, 0.85, 0.86, 0.92, 0.97 (3H each, all s, H-23–H-27), 0.78–2.28 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.69 (s, 3H, H-30), 2.05 (s, 3H, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) CO–), 2.96–3.05 (m, 1H, H-19), 3.47 (t, 2H, J = 6 Hz, H-8′), 3.65–3.73 (m, 6H, H-2′, H-4′, H-5′), 3.82 (t, 2H, J = 6 Hz, H-7′), 4.24–4.27 (m, 2H, H-1′), 4.45–4.49 (m, 1H, H-3), 4.61 (s, 1H, H-29), 4.74 (s, 1H, H-29); 13C NMR (100 MHz, CDCl3) δ 14.66 (C-27), 16.01 (C-26), 16.20 (C-25), 16.49 (C-24), 18.18 (C-6), 19.34 (C-30), 20.91 (C-11), 21.33 (CO–), 23.70 (C-12), 25.48 (C-2), 27.94 (C-23), 29.62 (C-15), 30.24 (C-8′), 30.56 (C-16), 32.09 (C-21), 34.27 (C-7), 37.01 (C-22), 37.10 (C-1), 37.80 (C-4), 38.22 (C-13), 38.40 (C-10), 40.72 (C-8), 42.40 (C-14), 46.98 (C-18), 49.40 (C-19), 50.45 (C-9), 55.42 (C-5), 56.54 (C-17), 62.78 (C-1′), 69.38, 70.50, 70.56, 71.27 (C-2′, C-4′, C-5′, C-7′), 80.91 (C-3), 109.65 (C-29), 150.55 (C-20), 170.99 (CH3 O–), 176.00 (C-28); anal. calcd. for C38H61BrO6: C, 65.78; H, 8.86. Found: C, 65.74; H, 8.84.
CO–), 2.96–3.05 (m, 1H, H-19), 3.47 (t, 2H, J = 6 Hz, H-8′), 3.65–3.73 (m, 6H, H-2′, H-4′, H-5′), 3.82 (t, 2H, J = 6 Hz, H-7′), 4.24–4.27 (m, 2H, H-1′), 4.45–4.49 (m, 1H, H-3), 4.61 (s, 1H, H-29), 4.74 (s, 1H, H-29); 13C NMR (100 MHz, CDCl3) δ 14.66 (C-27), 16.01 (C-26), 16.20 (C-25), 16.49 (C-24), 18.18 (C-6), 19.34 (C-30), 20.91 (C-11), 21.33 (CO–), 23.70 (C-12), 25.48 (C-2), 27.94 (C-23), 29.62 (C-15), 30.24 (C-8′), 30.56 (C-16), 32.09 (C-21), 34.27 (C-7), 37.01 (C-22), 37.10 (C-1), 37.80 (C-4), 38.22 (C-13), 38.40 (C-10), 40.72 (C-8), 42.40 (C-14), 46.98 (C-18), 49.40 (C-19), 50.45 (C-9), 55.42 (C-5), 56.54 (C-17), 62.78 (C-1′), 69.38, 70.50, 70.56, 71.27 (C-2′, C-4′, C-5′, C-7′), 80.91 (C-3), 109.65 (C-29), 150.55 (C-20), 170.99 (CH3 O–), 176.00 (C-28); anal. calcd. for C38H61BrO6: C, 65.78; H, 8.86. Found: C, 65.74; H, 8.84.
8′-Bromo-3′,6′-dioxaoctan-1′-yl-3β-hydroxylup-20(29)-en-28-oate (4). White crystals, 61% yield; mp 88–90 °C (EtOH); [α]21D +5° (c 1.02, CHCl3); IR (CHCl3) νmax: 3447 (–OH), 1723 (C
O) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.82, 0.83, 0.93, 0.93, 0.97 (3H each, all s, H-23–H-27), 0.77–2.29 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.69 (s, 3H, H-30), 2.96–3.56 (m, 1H, H-19), 3.14–3.22 (m, 1H, H-3), 3.47–3.48 (m, 2H, H-8′), 3.67–3.72 (m, 6H, H-2′, H-4′, H-5′), 3.81 (t, 2H, J = 6.4 Hz, H-7′), 4.25–4.27 (m, 2H, H-1′), 4.61 (s, 1H, H-29), 4.74 (s, 1H, H-29); 13C NMR (100 MHz, CDCl3) δ 14.91 (C-27), 15.38 (C-26), 16.02 (C-25), 16.14 (C-24), 18.30 (C-6), 19.37 (C-30), 20.90 (C-11), 25.53 (C-12), 27.41 (C-2), 27.99 (C-23), 29.64 (C-21), 30.24 (C-8′), 30.61 (C-15), 32.11 (C-16), 34.35 (C-7), 37.02 (C-22), 37.19 (C-1), 38.25 (C-4), 38.72 (C-13), 38.86 (C-10), 40.71 (C-8), 42.41 (C-14), 46.96 (C-18), 49.41 (C-19), 50.55 (C-9), 55.35 (C-5), 56.55 (C-17), 62.79 (C-1′), 69.38, 70.50, 70.59, 71.27 (C-2′, C-4′, C-5′, C-7′), 78.94 (C-3), 109.59 (C-29), 150.58 (C-20), 176.01 (C-28); anal. calcd. for C36H59BrO5: C, 66.34; H, 9.12. Found: C, 66.32; H, 9.13.
8′-Bromo-3′,6′-dioxaoctan-1′-yl-3β-hydroxyurs-12-en-28-oate (12). White crystals; 52% yield; mp 52–54 °C (EtOH); [α]21D +49.92° (c 0.66, CHCl3); IR (CHCl3) νmax: 3448 (–OH), 1722 (C
O) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.75, 0.77, 0.91, 0.98, 1.07 (3H each, all s, H-23–H-27), 0.84 (d, 3H, J = 6 Hz, H-30), 0.93 (d, 3H, J = 6 Hz, H-29), 0.70–2.02 (m, 22H, CH, CH2 in pentacyclic skeleton), 2.23 (d, 1H, J = 11.6 Hz, H-18), 3.18–3.22 (m, 1H, H-3), 3.46 (t, 2H, J = 6.4 Hz, H-8′), 3.65–3.67 (m, 6H, H-2′, H-4′, H-5′), 3.81 (t, 2H, J = 6.4 Hz, H-7′), 4.15 (t, 2H, J = 4.8 Hz, H-1′), 5.24 (br s, 1H, H-12); 13C NMR (100 MHz, CDCl3) δ 15.48 (C-25), 15.66 (C-24), 17.02 (C-26), 17.09 (C-29), 18.31 (C-6), 21.19 (C-30), 23.31 (C-11), 23.54 (C-27), 24.17 (C-16), 27.21 (C-2), 27.98 (C-15), 28.15 (C-23), 30.24 (C-8′), 30.66 (C-21), 33.04 (C-7), 36.61 (C-22), 36.95 (C-10), 38.63 (C-1), 38.74 (C-20), 38.83 (C-19), 39.04 (C-4), 39.54 (C-8), 42.03 (C-14), 47.53 (C-9), 48.05 (C-17), 52.82 (C-18), 55.21 (C-5), 63.22 (C-1′), 69.23, 70.52, 70.57, 71.26 (C-2′, H-4′, C-5′, H-7′), 78.94 (C-3), 125.59 (C-12), 138.07 (C-13), 177.42 (C-28); anal. calcd. for C36H59BrO5: C, 66.34; H, 9.12. Found: C, 66.38; H, 9.11.
8′-Tosyl-3′,6′-dioxaoctan-1′-yl-3β-acetoxylup-20(29)-en-28-oate (3). Prepared by a reported method.42 Triethylene glycol ditosylate (0.73 g, 1.6 mmol) was added to a stirred warm solution (50 °C) of betulinic acid (0.40 g, 0.8 mmol) in dry acetonitrile (12 mL). The reaction mixture was cooled to room temperature, and then K2CO3 (0.062, 2 mmol) was added. The resulting mixture was heated at 70 °C for 7 h [TLC monitoring hexane/EtOAc 2
:1], the precipitate was filtered off and acetonitrile was removed under reduced pressure. The residue was chromatographed on silica gel [hexane/EtOAc, 10:1 → 5:1], to obtain a pure compound. White crystals; 60% yield; mp 38–40 °C (EtOH); [α]17.5D +96° (c 0.49, CHCl3); IR (CHCl3) νmax: 1727 (CO) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.84, 0.85, 0.86, 0.91, 0.96 (3H each, all s, 3H, H-23–H-27), 0.78–2.26 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.69 (s, 3H, H-30), 2.05 (s, 3H, ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) CO–), 2.46 (s, 3H, OTs-), 2.99–3.03 (m, 1H, H-19), 3.59 (s, 4H, H-4′, H-5′), 3.66, 3.70 (2H each, both t, J = 4.8 Hz, H-2′, H-7′), 4.16, 4.22 (2H each, both t, J = 4.8 Hz, H-1′, H-8′), 4.15–4.24 (m, 1H, H-3), 4.60 (s, 1H, H-29), 4.72 (s, 1H, H-29) 7.34, 7.80 (2H each, both d, J = 8 Hz, Ph); 13C NMR (100 MHz, CDCl3) δ 14.98 (C-27), 16.11 (C-26), 16.19 (C-25), 16.49 (C-24), 18.17 (C-6), 19.33 (C-30), 20.90 (C-11), 21.32 (CO–), 21.65 (OTs-), 23.69 (C-12), 25.47 (C-2), 27.94 (C-23), 29.60 (C-15), 30.56 (C-16), 32.07 (C-21), 34.26 (C-7), 36.98 (C-22), 37.09 (C-1), 37.79 (C-4), 38.21 (C-13), 38.39 (C-10), 40.71 (C-8), 42.39 (C-14), 46.98 (C-18), 49.38 (C-19), 50.44 (C-9), 55.42 (C-5), 56.53 (C-17), 62.72 (C-1′), 68.76, (C-8′), 69.18, 69.32, 70.45, 70.78 (C-2′, C-4′, C-5′, C-7′), 80.91 (C-3), 109.64 (C-29), 127.97, 129.82, 133.15, 145.20, (OTs-
CO–), 2.46 (s, 3H, OTs-), 2.99–3.03 (m, 1H, H-19), 3.59 (s, 4H, H-4′, H-5′), 3.66, 3.70 (2H each, both t, J = 4.8 Hz, H-2′, H-7′), 4.16, 4.22 (2H each, both t, J = 4.8 Hz, H-1′, H-8′), 4.15–4.24 (m, 1H, H-3), 4.60 (s, 1H, H-29), 4.72 (s, 1H, H-29) 7.34, 7.80 (2H each, both d, J = 8 Hz, Ph); 13C NMR (100 MHz, CDCl3) δ 14.98 (C-27), 16.11 (C-26), 16.19 (C-25), 16.49 (C-24), 18.17 (C-6), 19.33 (C-30), 20.90 (C-11), 21.32 (CO–), 21.65 (OTs-), 23.69 (C-12), 25.47 (C-2), 27.94 (C-23), 29.60 (C-15), 30.56 (C-16), 32.07 (C-21), 34.26 (C-7), 36.98 (C-22), 37.09 (C-1), 37.79 (C-4), 38.21 (C-13), 38.39 (C-10), 40.71 (C-8), 42.39 (C-14), 46.98 (C-18), 49.38 (C-19), 50.44 (C-9), 55.42 (C-5), 56.53 (C-17), 62.72 (C-1′), 68.76, (C-8′), 69.18, 69.32, 70.45, 70.78 (C-2′, C-4′, C-5′, C-7′), 80.91 (C-3), 109.64 (C-29), 127.97, 129.82, 133.15, 145.20, (OTs-![[P with combining low line]](https://www.rsc.org/images/entities/i_char_0050_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/i_char_0068_0332.gif) ), 150.42 (C-20), 170.99 (CH3O–), 176.02 (C-28); anal. calcd. for C45H68O8S: C, 68.84; H, 8.73. Found: C, 68.86; H, 8.71.
:1]. The mixture was then poured into cold H2O (10× volume) and extracted with CHCl3 (2 × 15 mL each). The combined CHCl3 layers were washed with H2O (100 mL) and concentrated. The crude residue was co-evaporated with H2O (100 mL) to remove residual DMF. The residue was chromatographed on silica gel [hexane/EtOAc, 30:1 → 1:1] to obtain a pure compound.
), 150.42 (C-20), 170.99 (CH3O–), 176.02 (C-28); anal. calcd. for C45H68O8S: C, 68.84; H, 8.73. Found: C, 68.86; H, 8.71.
:1]. The mixture was then poured into cold H2O (10× volume) and extracted with CHCl3 (2 × 15 mL each). The combined CHCl3 layers were washed with H2O (100 mL) and concentrated. The crude residue was co-evaporated with H2O (100 mL) to remove residual DMF. The residue was chromatographed on silica gel [hexane/EtOAc, 30:1 → 1:1] to obtain a pure compound.
4-Bromobutyl-3β-hydroxylup-20(29)-en-28-oate (1). White crystals; 75% yield; mp 78–80 °C (EtOH); [α]21D +27° (c 0.95, CHCl3); IR (CHCl3) νmax: 3448 (–OH), 1726 (C
O) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.77, 0.83, 0.90, 0.92, 0.97 (3H each, all s, H-23–H-27), 0.68–2.26 (m, 24H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.70 (s, 3H, H-30), 2.99–3.04 (m, 1H, H-19), 3.17–3.21 (m, 1H, H-3), 3.46 (t, 2H, J = 7 Hz, H-4′), 4.08–4.18 (m, 2H, H-1′), 4.61 (s, 1H, H-29), 4.75 (s, 1H, H-29); 13C NMR (100 MHz, CDCl3) δ 14.71 (C-27), 15.37 (C-26), 16.04 (C-25), 16.14 (C-24), 18.30 (C-6), 19.38 (C-30), 20.91 (C-11), 25.54 (C-12), 27.42 (C-2), 27.50 (C-3′), 27.99 (C-23), 29.50 (C-2′), 29.67 (C-21), 30.64 (C-15), 32.18 (C-16), 33.02 (C-4′), 34.35 (C-7), 37.05 (C-22), 37.20 (C-1), 38.32 (C-4), 38.73 (C-13), 38.86 (C-10), 40.72 (C-8), 42.42 (C-14), 46.01 (C-18), 49.42 (C-19), 50.56 (C-9), 55.36 (C-5), 56.57 (C-17), 62.84 (C-1′), 78.96 (C-3), 109.64 (C-29), 150.51 (C-20), 176.09 (C-28); anal. calcd. for C34H55O3Br: C, 69.02; H, 9.37. Found: C, 69.12; H, 9.31.
4-Bromobutyl-3β-hydroxyurs-12-en-28-oate (11). White crystals; 69% yield; mp 66–68 °C (EtOH); [α]20D +53.44° (c 0.36, CHCl3); IR (CHCl3) νmax: 3448 (–OH), 1719 (C=O) cm−1; 1H NMR (400 MHz, CDCl3) δ 0.76, 0.79, 0.93, 1.00, 1.09 (3H each, all s, H-23–H-27), 0.88, 0.96 (3H each, both d, J = 6 Hz, H-29, H-30); 0.64–2.05 (m, 22H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′); 2.24 (d, 1H, J = 11 Hz, H-18), 3.23 (dd, 1H, J = 10.8, 4.8 Hz, H-3), 3.43 (t, 2H, J = 7 Hz, H-4′), 4.03 (m, 2H, H-1′), 5.26 (br s, 1H, H-12); 13C NMR (100 MHz, CDCl3) δ 15.50 (C-25), 15.63 (C-24), 17.02 (C-26), 17.16 (C-29), 18.32 (C-6), 21.18 (C-30), 23.30 (C-11), 23.56 (C-27), 24.23 (C-16), 27.23 (C-2), 27.33 (C-3′), 27.97 (C-15), 28.15 (C-23), 29.52 (C-2′), 30.67 (C-21), 33.05 (C-7), 33.10 (C-4′), 36.77 (C-22), 36.98 (C-10), 38.62 (C-1), 38.75 (C-20), 38.88 (C-19), 39.09 (C-4), 39.56 (C-8), 42.07 (C-14), 48.13 (C-9), 48.13 (C-17), 52.88 (C-18), 55.22 (C-5), 63.17 (C-1′), 79.03 (C-3), 125.62 (C-12), 138.23 (C-13), 177.44 (C-28). Anal. calcd. for C34H55O3Br: C, 69.02; H, 9.37. Found: C, 69.11; H, 9.34.
:1]. The solution was cooled and the solvent was evaporated under vacuum. The solid product obtained was washed with hot hexane (2 × 10 mL), dissolved in EtOAc (2–4 mL), and diluted with hexane (12 ml). The precipitate was filtered to obtain the title product.
4-(Triphenylphosphonio)butyl-3β-hydroxylup-20(29)-en-28-oate bromide (5). White crystals; 79% yield; mp 150–152 °C (EtOH); [α]21D −0.38° (c 1.85, CHCl3); IR (CHCl3) νmax: 3446 (–OH), 1718 (C=O); 31P NMR (161 MHz, CDCl3) δ 24.49; 1H NMR (400 MHz, CDCl3) δ 0.73, 0.77, 0.79, 0.90, 0.94 (3H each, all s, H-23–H-27), 0.63–2.12 (m, 24H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.63 (s, 3H, H-30), 2.87–2.89 (m, 1H, H-19), 3.15–3.18 (m, 1H, H-3), 3.96–4.13 (m, 4H, H-4′, H-1′), 4.56 (s, 1H, H-29), 4.66 (s, 1H, H-29), 7.69–7.86 (m, 15H, Ph); 13C NMR (100 MHz, CDCl3) δ 14.65 (C-27), 15.41 (C-26), 15.96 (C-25), 16.13 (C-24), 18.27 (C-6), 19.27 (C-30), 19.42 (d, J = 3 Hz, C-3′), 20.81 (C-11), 22.30 (d, J = 50 Hz, 4′), 25.41 (C-12), 27.37 (C-2), 27.99 (C-23), 29.28 (d, J = 17 Hz, 2′), 29.57 (C-21), 30.52 (C-15), 32.00 (C-16), 34.29 (C-7), 36.91 (C-22), 37.14 (C-1), 38.13 (C-4), 38.68 (C-13), 38.83 (C-10), 40.61 (C-8), 42.31 (C-14), 46.91 (C-18), 49.30 (C-19), 50.46 (C-9), 55.28 (C-5), 56.42 (C-17), 62.75 (C-1′), 78.81 (C-3), 109.70 (C-29), 117.85 (d, J = 85 Hz, Ph), 130.50 (d, J = 13 Hz, Ph), 133.70 (d, J = 10 Hz, Ph), 135.03 (br s, Ph), 150.37 (C-20), 175.91 (C-28); MS: m/z [M−Br]−, found 773.49. [C52H70BrO3P]+ requires 852.42.
8′-(Triphenylphosphonio-3′,6′-dioxaoctan-1′-yl)-3β-acetoxylup-20(29)-en-28-oate bromide (7). White crystals; 70% yield; mp 108–110 °C (EtOH); [α]18D +9.6° (c 0.50, CHCl3); IR (CHCl3) νmax: 1727 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 25.50; 1H NMR (500 MHz, CDCl3) δ 0.81, 0.82, 0.83, 0.88, 0.94 (3H each, all s, H-23–H-27), 0.76–2.25 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.67 (s, 3H, H-30), 2.02 (s, 3H, CO–), 3.28–3.29 (m, 1H, H-19), 3.33–3.40 (m, 6H, H-4′, H-5′, H-7′), 3.89–3.96 (m, 2H, H-8′), 4.07–4.19 (m, 4H, H-1′, H-2′), 4.43–4.47 (m, 1H, H-3), 4.59 (s, 1H, H-29), 4.70 (s, 1H, H-29), 7.64–7.83 (m, 15H, Ph); 13C NMR (126 MHz, CDCl3) δ 14.64 (C-27), 16.02 (C-26), 16.20 (C-25), 16.48 (C-24), 18.16 (C-6), 19.26 (C-30), 20.89 (C-11), 21.31 (CO–), 23.66 (C-12), 25.42 (C-8′, J = 52 Hz), 25.43 (C-2), 27.92 (C-23), 29.59 (C-15), 30.51 (C-16), 32.03 (C-21), 34.26 (C-7), 36.98 (C-22), 37.08 (C-1), 37.77 (C-4), 38.19 (C-13), 38.36 (C-10), 40.70 (C-8), 42.38 (C-14), 46.98 (C-18), 49.33 (C-19), 50.41 (C-9), 55.39 (C-5), 56.51 (C-17), 62.50 (C-1′), 64.06 (C-7′, J = 7 Hz), 69.01, 69.85, 70.24 (C-2′, C-4′, C-5′), 80.88 (C-3), 109.75 (C-29), 118.91 (Ph, J = 86 Hz), 130.07 (Ph, J = 12.7 Hz), 134.04 (Ph, J = 10.2 Hz), 134.65 (Ph, J = 2.6 Hz), 150.40 (C-20), 171.01 (CH3O–), 175.88 (C-28); MS: m/z [M−Br]−, found 875.33. [C56H76BrO6P]+ requires 954.46.
8′-(Triphenylphosphonio-3′,6′-dioxaoctan-1′-yl)-3β-acetoxylup-20(29)-en-28-oate tosyl (8). White crystals; 91% yield; mp 59–61 °C (EtOH); [α]17.5D +7.5° (c 0.39, CHCl3); IR (CHCl3) νmax: 1727 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 25.57; 1H NMR (500 MHz, CDCl3) δ 0.80, 0.82, 0.83, 0.89, 0.95 (3H each, all s, H-23–H-27), 0.77–2.25 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.68 (s, 3H, H-30), 2.03 (s, 3H, CH3CO–), 2.30 (s, 3H, OTs-CH3), 2.96–2.99 (m, 1H, H-19), 3.26–3.38 (m, 6H, H-4′, H-5′, H-7′), 3.84–4.07 (m, 6H, H-1′, H-2′, H-8′), 4.44–4.48 (m, 1H, H-3), 4.60 (s, 1H, H-29), 4.71 (s, 1H, H-29), 7.05 (d, 2H, J = 2 Hz, Ph), 7.62–7.80 (m, 15H, Ph and 2H, OTs-![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) ); 13C NMR (126 MHz, CDCl3) δ 14.65 (C-27), 16.02 (C-26), 16.21 (C-25), 16.49 (C-24), 18.17 (C-6), 19.28 (C-30), 20.90 (C-11), 21.29 (CH3CO–), 21.32 (OTs-CH3), 23.67 (C-12), 24.43 (C-8′, J = 54 Hz), 25.45 (C-2), 27.93 (C-23), 29.60 (C-15), 30.53 (C-16), 32.04 (C-21), 34.27 (C-7), 36.99 (C-22), 37.09 (C-1), 37.78 (C-4), 38.20 (C-13), 38.38 (C-10), 40.70 (C-8), 42.39 (C-14), 46.99 (C-18), 49.34 (C-19), 50.42 (C-9), 55.40 (C-5), 56.51 (C-17), 62.53 (C-1′), 63.06 (C-7′, J = 7.4 Hz), 68.97, 69.84, 70.08 (C-2′, C-4′, C-5′), 80.88 (C-3), 109.75 (C-29), 119.11 (Ph, J = 86 Hz), 130.00 (Ph, J = 12.7 Hz), 134.00 (Ph, J = 10.2 Hz), 134.53 (Ph, J = 2.6 Hz), 126.11, 128.32, 138.54, 144.31, (OTs-Ph), 150.41 (C-20), 171.01 (CH3CO–), 175.89 (C-28); MS: m/z [M − TsO]−, found 875.32. [C63H83O8PS]+ requires 1030.55.
); 13C NMR (126 MHz, CDCl3) δ 14.65 (C-27), 16.02 (C-26), 16.21 (C-25), 16.49 (C-24), 18.17 (C-6), 19.28 (C-30), 20.90 (C-11), 21.29 (CH3CO–), 21.32 (OTs-CH3), 23.67 (C-12), 24.43 (C-8′, J = 54 Hz), 25.45 (C-2), 27.93 (C-23), 29.60 (C-15), 30.53 (C-16), 32.04 (C-21), 34.27 (C-7), 36.99 (C-22), 37.09 (C-1), 37.78 (C-4), 38.20 (C-13), 38.38 (C-10), 40.70 (C-8), 42.39 (C-14), 46.99 (C-18), 49.34 (C-19), 50.42 (C-9), 55.40 (C-5), 56.51 (C-17), 62.53 (C-1′), 63.06 (C-7′, J = 7.4 Hz), 68.97, 69.84, 70.08 (C-2′, C-4′, C-5′), 80.88 (C-3), 109.75 (C-29), 119.11 (Ph, J = 86 Hz), 130.00 (Ph, J = 12.7 Hz), 134.00 (Ph, J = 10.2 Hz), 134.53 (Ph, J = 2.6 Hz), 126.11, 128.32, 138.54, 144.31, (OTs-Ph), 150.41 (C-20), 171.01 (CH3CO–), 175.89 (C-28); MS: m/z [M − TsO]−, found 875.32. [C63H83O8PS]+ requires 1030.55.
9′-(Triphenylphosphonio-3′,6′-dioxaoctan-1′-yl)-3β-hydroxylup-20(29)-en-28-oate bromide (9). White crystals; 69% yield; mp 95–97 °C (EtOH); [α]18D +3.32° (c 0.62, CHCl3); IR (CHCl3) νmax: 3370 (−OH), 1722 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 25.45; 1H NMR (500 MHz, CDCl3) δ 0.72, 0.77, 0.86, 0.93, 0.94 (3H each, all s, H-23–H-27), 0.64–2.19 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.65 (s, 3H, H-30), 2.93–2.95 (m, 1H, H-19), 3.15–3.17 (m, 1H, H-3), 3.15–3.39 (m, 6H, H-4′, H-5′, H-7′), 3.88–3.93 (m, 2H, H-8′), 4.06–4.14 (m, 4H, H-1′, H-2′), 4.57 (s, 1H, H-29), 4.69 (s, 1H, H-29), 7.64–7.83 (m, 15H, Ph); 13C NMR (125 MHz, CDCl3) δ 14.68 (C-27), 15.42 (C-26), 16.01 (C-25), 16.14 (C-24), 18.27 (C-6), 19.28 (C-30), 20.87 (C-11), 25.41 (C-8′, J = 52 Hz), 25.46 (C-12), 27.34 (C-2), 27.99 (C-23), 29.59 (C-21), 30.52 (C-15), 32.04 (C-16), 34.32 (C-7), 36.97 (C-22), 37.15 (C-1), 38.21 (C-4), 38.68 (C-13), 38.82 (C-10), 40.67 (C-8), 42.38 (C-14), 46.96 (C-18), 49.33 (C-19), 50.47 (C-9), 55.29 (C-5), 56.51 (C-17), 62.51 (C-1′), 64.06 (C-7′, J = 7 Hz), 69.01, 69.84, 70.24 (C-2′, C-4′, C-5′), 78.80 (C-3), 109.70 (C-29), 118.82 (Ph, J = 87 Hz), 130.07 (Ph, J = 12.7 Hz), 134.03 (Ph, J = 10.2 Hz), 134.69 (Ph, J = 2.6 Hz), 150.42 (C-20), 175.88 (C-28); MS: m/z [M−Br]−, found 833.32. [C54H74BrO5P]+ requires 912.45.
4-(Triphenylphosphonio)butyl-3β-hydroxyurs-12-en-28-oate bromide (13). White crystals; 85% yield; mp 166–168 °C (EtOH); [α]20D +3.02° (c 0.50, CHCl3); IR (CHCl3) νmax: 3367 (–OH), 1716 (C
O); 31P NMR (161 MHz, CDCl3) δ 24.48; 1H NMR (400 MHz, CDCl3) δ 0.64, 0.77, 0.87, 0.98, 1.03 (3H each, all s, H-23–H-27), 0.82, 0.93 (3H each, both d, J = 6 Hz, H-29, H-30), 0.67–2.10 (m, 22H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 2.08 (d, 1H, J = 10.4 Hz, H-18), 3.20 (dd, 1H, J = 10.4, 4.4 Hz, H-3), 3.94–4.08 (m, 4H, H-4′, H-1′), 5.07 (br s, 1H, H-12), 7.68–7.80 (m, 15H, Ph); 13C NMR (100 MHz, CDCl3) δ 15.47 (C-25), 15.67 (C-24), 17.01 (C-26), 17.02 (C-29), 18.29 (C-6), 19.43 (d, J = 3 Hz, C-3′), 21.16 (C-30), 22.30 (d, J = 50 Hz, 4′), 23.27 (C-11), 23.52 (C-27), 24.10 (C-16), 27.18 (C-2), 27.93 (C-15), 28.15 (C-23), 29.37 (d, J = 17 Hz, 2′), 30.59 (C-21), 32.95 (C-7), 36.66 (C-22), 36.91 (C-10), 38.60 (C-1), 38.72 (C-20), 38.84 (C-19), 38.95 (C-4), 39.44 (C-8), 41.96 (C-14), 47.45 (C-9), 48.00 (C-17), 52.82 (C-18), 55.15 (C-5), 62.85 (C-1′), 78.91 (C-3), 118.14 (d, J = 86 Hz, Ph), 125.46 (C-12), 130.51 (d, J = 13 Hz, Ph), 133.64 (d, J = 9 Hz, Ph), 135.03 (br s, Ph), 138.09 (C-13), 177.44 (C-28); MS: m/z [M−Br]−, found 773.49. [C52H70BrO3P]+ requires 852.42.
8′-(Triphenylphosphonio-3′,6′-dioxaoctan-1′-yl)-3β-hydroxyurs-12-en-28-oate bromide (15). White crystals; 69% yield; mp 81–83 °C (EtOH); [α]18D +28.61° (c 0.57, CHCl3); IR (CHCl3) νmax: 3151 (−OH), 1722 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 25.46; 1H NMR (500 MHz, CDCl3) δ 0.71, 0.75, 0.88, 0.96, 1.06 (3H each, all s, H-23–H-27), 0, 83 (d, 3H, J = 6 Hz, H-30), 0.93 (d, 3H, J = 6 Hz, H-29), 0.69–2.01 (m, 22H, CH, CH2 in pentacyclic skeleton), 2.19 (d, 1H, J = 11.5 Hz, H-18), 3.19–3.21 (m, 1H, H-3), 3.27–3.36 (m, 6H, H-4′, H-5′, H-7′), 3.90–3.98 (m, 4H, H-2′, H-8′), 4.13–4.15 (m, 2H, H-1′), 5.24 (br s, 1H, H-12), 7.64–7.84 (m, 15H, Ph); 13C NMR (125 MHz, CDCl3) δ 15.48 (C-25), 15.69 (C-24), 17.02 (C-26), 17.12 (C-29), 18.29 (C-6), 21.15 (C-30), 23.29 (C-11), 23.50 (C-27), 24.18 (C-16), 25.42 (C-8′, J = 52 Hz), 27.18 (C-2), 27.96 (C-15), 28.15 (C-23), 30.61 (C-21), 33.03 (C-7), 36.66 (C-22), 36.93 (C-10), 38.59 (C-1), 38.72 (C-20), 38.85 (C-19), 39.01 (C-4), 39.52 (C-8), 42.03 (C-14), 47.46 (C-9), 48.07 (C-17), 52.86 (C-18), 55.15 (C-5), 62.92 (C-1′), 64.06 (C-7′, J = 7 Hz), 68.86, 69.87, 70.27 (C-2′, C-4′, C-5′), 78.86 (C-3), 118.81 (Ph, J = 87 Hz), 125.57 (C-12), 130.08 (Ph, J = 13 Hz), 134.07 (Ph, J = 10 Hz), 134.66 (Ph, J = 3 Hz), 138.06 (C-13), 177.38 (C-28); MS: m/z [M−Br]−, found 833.32 [C54H74BrO5P]+ requires 912.45.
:1], the precipitate was filtered off and CH2Cl2 was removed under reduced pressure. The residue was chromatographed on silica gel [CHCl3/MeOH, 50:1 → 5:1], to obtain a pure compound.
4′-(Triphenylphosphonio)butyl-3β-dichloroacetoxylup-20(29)-en-28-oate bromide (6). White crystals; 70% yield; mp 123–125 °C (EtOH); [α]17.5D +4.3° (c 0.53, CHCl3); IR (CHCl3) νmax: 1722 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 24.56; 1H NMR (500 MHz, CDCl3) δ 0.80, 0.81, 0.86, 0.87, 0.90 (3H each, all s, H-23–H-27), 0.75–2.11 (m, 24H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 1.63 (s, 3H, H-30), 2.86–2.88 (m, 1H, H-19), 3.90–4.13 (m, 4H, H-4′, H-1′), 4.54–4.55 (m, 2H, H-29, H-3), 4.66 (s, 1H, H-29), 5.92 (s, 1H, CCl2CO–), 7.68–7.85 (m, 15H, Ph); 13C NMR (100 MHz, CDCl3) δ 14.61 (C-27), 15.95 (C-26), 16.14 (C-25), 16.34 (C-24), 18.03 (C-6), 19.23 (C-30), 19.50 (d, J = 3 Hz, C-3′), 20.85 (C-11), 22.30 (d, J = 50 Hz, 4′), 23.19 (C-12), 25.32 (C-2), 27.81 (C-23), 29.34 (d, J = 17 Hz, 2′), 29.54 (C-21), 30.49 (C-15), 31.97 (C-16), 34.15 (C-7), 36.90 (C-22), 37.04 (C-1), 38.05 (C-4), 38.20 (C-13), 38.21 (C-10), 40.62 (C-8), 42.32 (C-14), 46.90 (C-18), 49.26 (C-19), 50.36 (C-9), 55.33 (C-5), 56.39 (C-17), 62.72 (C-1′), 64.81 (HCl2CO–), 84.93 (C-3), 109.77 (C-29), 118.15 (d, J = 85 Hz, Ph), 130.53 (d, J = 12.4 Hz, Ph), 133.66 (d, J = 10 Hz, Ph), 135.07 (br s, Ph), 150.31 (C-20), 164.29 (CHCl2O–), 175.88 (C-28); MS: m/z [M−Br]−, found 883.51. [C54H70BrCl2O4P]+ requires 962.36.
8′-(Triphenylphosphonio-3′,6′-dioxaoctan-1′-yl)-3β-dichloroacetoxylup-20(29)-en-28-oate bromide (10). White crystals; 82% yield; mp 98–100 °C (EtOH); [α]17.5D +8.4° (c 0.49, CHCl3); IR (CHCl3) νmax: 1722 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 25.49; 1H NMR (500 MHz, CDCl3) δ 0.83, 0.86, 0.87, 0.88, 0.94 (3H each, all s, H-23–H-27), 0.77–2.21 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.66 (s, 3H, H-30), 2.93–2.97 (m, 1H, H-19), 3.27–3.39 (m, 6H, H-4′, H-5′, H-7′), 3.90–4.15 (m, 6H, H-2′, H-8′, H-1′), 4.54–4.57 (m, 2H, H-3, H-29), 4.69 (s, 1H, H-29), 5.91 (s, 1H, CCl2CO–), 7.64–7.80 (m, 15H, Ph); 13C NMR (125 MHz, CDCl3) δ 14.64 (C-27), 15.99 (C-26), 16.16 (C-25), 16.33 (C-24), 18.05 (C-6), 19.26 (C-30), 20.91 (C-11), 23.19 (C-12), 25.25 (C-8′, J = 52 Hz), 27.38 (C-2), 27.80 (C-23), 29.56 (C-21), 30.49 (C-15), 32.00 (C-16), 34.19 (C-7), 36.96 (C-22), 37.06 (C-1), 38.15 (C-4), 38.21 (C-13), 38.21 (C-10), 40.68 (C-8), 42.39 (C-14), 46.96 (C-18), 49.30 (C-19), 50.37 (C-9), 55.33 (C-5), 56.48 (C-17), 62.51 (C-1′), 64.03 (C-7′, J = 7 Hz), 64.81 (CHCl2CO–), 69.01, 69.84, 70.23 (C-2′, C-4′, C-5′), 84.94 (C-3), 109.76 (C-29), 118.82 (Ph, J = 87 Hz), 130.00 (Ph, J = 12.7 Hz), 134.03 (Ph, J = 10.2 Hz), 134.67 (Ph, J = 2.6 Hz), 164.28 (CHCl2CO–), 150.37 (C-20), 175.86 (C-28); MS: m/z [M−Br]−, found 943.59. [C56H74BrCl2O6P]+ requires 1022.38.
4′-(Triphenylphosphonio)butyl-3β-dichloroacetoxyurs-12-en-28-oate bromide (14). White crystals; 71% yield; mp 138–140 °C (EtOH); [α]17.5D +24.7° (c 0.63, CHCl3); IR (CHCl3) νmax: 1721 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 24.52; 1H NMR (400 MHz, CDCl3) δ 0.65, 0.80, 0.90, 0.91, 1.03 (3H each, all s, H(23)–H(27)), 0.82, 0.92 (3H each, both d, J = 6 Hz, H-29, H-30), 0.67–2.10 (m, 22H, CH, CH2 in pentacyclic skeleton, 4H, H-2′, H-3′), 2.08 (d, 1H, J = 10.4 Hz, H-18), 3.96–4.07 (m, 4H, H-4′, H-1′), 4.58–4.61 (m, 1H, H-3), 5.09 (br s, 1H, H-12), 5.94 (s, 1H, CCl2CO–), 7.69–7.87 (m, 15H, Ph); 13C NMR (100 MHz, CDCl3) δ 15.49 (C-25), 16.60 (C-24), 17.00 (C-26), 17.04 (C-29), 18.07 (C-6), 19.36 (d, J = 3 Hz, C-3′), 21.16 (C-30), 22.36 (d, J = 50 Hz, 4′), 23.06 (C-2), 23.26 (C-11), 23.50 (C-27), 24.09 (C-16), 27.95 (C-15), 27.96 (C-23), 29.17 (d, J = 17 Hz, 2′), 30.57 (C-21), 32.82 (C-7), 36.64 (C-22), 36.80 (C-10), 38.10 (C-1), 38.11 (C-20), 38.84 (C-19), 38.94 (C-4), 39.45 (C-8), 41.96 (C-14), 47.38 (C-9), 47.97 (C-17), 52.81 (C-18), 55.21 (C-5), 62.80 (C-1′), 64.80 (HCl2CO–), 84.91 (C-3), 118.65 (d, J = 85.5 Hz, Ph), 125.26 (C-12), 130.12 (d, J = 12.4 Hz, Ph), 133.10 (d, J = 9.9 Hz, Ph), 134.98 (br s, Ph), 138.15 (C-13), 164.32 (CHCl2O–), 177.40 (C-28); MS: m/z [M−Br]−, found 883.54. [C54H70BrCl2O4P]+ requires 962.36.
8′-(Triphenylphosphonio-3′,6′-dioxaoctan-1′-yl)-3β-dichloroacetoxyurs-12-en-28-oate bromide (16). White crystals; 87% yield; mp 96–98 °C (EtOH); [α]17.5D +24.3° (c 0.61, CHCl3); IR (CHCl3) νmax: 1722 (C
O) cm−1; 31P NMR (161 MHz, CDCl3) δ 25.52; 1H NMR (500 MHz, CDCl3) δ 0.72, 0.75, 0.88, 0.96, 1.06 (3H each, all s, H-23–H-27), 0.83 (d, 3H, J = 6 Hz, H-30), 0.93 (d, 3H, J = 6 Hz, H-29), 0.70–2.13 (m, 22H, CH, CH2 in pentacyclic skeleton), 2.19 (d, 1H, J = 11.5 Hz, H-18), 3.27–3.37 (m, 6H, H-4′, H-5′, H-7′), 3.90–3.98 (m, 4H, H-2′, H-8′), 4.14–4.15 (m, 2H, H-1′), 4.58–4.59 (m, 1H, H-3), 5.21 (br s, 1H, H-12), 5.93 (s, 1H, CCl2CO–), 7.64–7.84 (m, 15H, Ph); 13C NMR (125 MHz, CDCl3) δ 15.51 (C-25), 16.60 (C-24), 17.03 (C-26), 17.09 (C-29), 18.08 (C-6), 21.14 (C-30), 23.05 (C-2), 23.29 (C-11), 23.47 (C-27), 24.15 (C-16), 25.42 (C-6′, J = 52 Hz), 27.95 (C-15), 27.94 (C-23), 30.59 (C-21), 32.90 (C-7), 36.64 (C-22), 36.81 (C-10), 38.11 (C-1), 38.10 (C-20), 38.84 (C-19), 39.00 (C-4), 39.52 (C-8), 42.04 (C-14), 47.38 (C-9), 48.06 (C-17), 52.84 (C-18), 55.21 (C-5), 62.92 (C-1′), 64.06 (C-5′, J = 7 Hz), 64.80 (HCl2CO–), 68.85, 69.86, 70.26 (C-2′–C-4′), 84.89 (C-3), 118.70 (d, J = 69 Hz, Ph), 125.33 (C-12), 129.10 (d, J = 10 Hz, Ph), 133.08 (d, J = 8 Hz, Ph), 134.67 (br s, Ph), 138.14 (C-13), 164.28 (CHCl2O–), 177.35 (C-28); MS: m/z [M−Br]−, found 943.59. [C56H74BrCl2O6P]+ requires 1022.38.
3β-Dichloroacetoxyurs-12-en-28-oic acid (UA-DCA). White crystals; 71% yield; mp 258–260 °C (EtOH); [α]17.5D +75.5° (c 0.48, CHCl3); IR (CHCl3) νmax: 1748 (C
O) cm−1; 1H NMR (500 MHz, CDCl3) δ 0.79, 0.92, 0.95, 0.99, 1.09 (3H each, all s, H-23–H-27), 0.88 (d, 3H, J = 6 Hz, H-30), 0.97 (d, 3H, J = 6.5 Hz; H-29), 1.27–2.03 (m, 22H, CH, CH2 in pentacyclic skeleton), 2.20 (d, 1H, J = 11 Hz, H-18), 4.62–4.65 (m, 1H, H-3), 5.25 (t, 1H, J = 3.5 Hz, H-12), 5.96 (s, 1H, CCl2CO–); 13C NMR (125 MHz, CDCl3) δ 15.51 (C-25), 16.56 (C-24), 17.01 (C-26), 17.09 (C-29), 18.05 (C-6), 21.19 (C-30), 23.10 (C-2), 23.27 (C-11), 23.62 (C-27), 23.99 (C-16), 27.95 (C-15), 27.96 (C-23), 30.58 (C-21), 32.77 (C-7), 36.71 (C-22), 36.90 (C-10), 38.10 (C-1), 38.15 (C-20), 38.82 (C-19), 39.00 (C-4), 39.48 (C-8), 41.88 (C-14), 47.43 (C-9), 47.97 (C-17), 52.45 (C-18), 55.25 (C-5), 64.82 (HCl2CO–), 84.94 (C-3), 125.58 (C-12), 137.99 (C-13), 164.32 (CHCl2O–), 184.31 (C-28); anal. calcd. for C32H48Cl2O4: C, 67.71; H, 8.52. Found: C, 67.69; H, 8.49.
3β-Dichloroacetoxylup-20(29)-en-28-oic acid (BA-DCA). White crystals; 65% yield; mp 267–269 °C (EtOH), lit.32 mp 269 °C; [α]17.5D +17.2° (c 0.58, CHCl3); IR (CHCl3) νmax: 1742 (C
O) cm−1, lit. 1745 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.89, 0.91, 0.92, 0.96, 1.00 (3H each, all s, H-23–H-27), 0.83–2.32 (m, 24H, CH, CH2 in pentacyclic skeleton), 1.72 (s, 3H, H-30), 3.00–3.06 (m, 1H, H-19), 4.60–4.64 (m, 2H, H-29, H-3), 4.76 (s, 1H, H-29), 5.96 (s, 1H, CCl2CO–); 13C NMR (125 MHz, CDCl3) δ 14.68 (C-27), 16.03 (C-26), 16.15 (C-25), 16.33 (C-24), 18.06 (C-6), 19.36 (C-30), 20.88 (C-11), 23.24 (C-2), 25.40 (C-12), 27.85 (C-23), 29.69 (C-21), 30.57 (C-15), 32.15 (C-16), 34.19 (C-7), 37.06 (C-22), 37.12 (C-1), 38.26 (C-4), 38.27 (C-13), 38.42 (C-10), 40.70 (C-8), 42.45 (C-14), 46.96 (C-18), 49.25 (C-19), 50.36 (C-9), 55.37 (C-5), 56.44 (C-17), 64.83 (HCl2CO–), 84.97 (C-3), 109.79 (C-29), 150.34 (C-20), 164.33 (CHCl2O–), 182.76 (C-28); anal. calcd. for C32H48Cl2O4: C, 67.71; H, 8.52. Found: C, 67.73; H, 8.54.
4.2. Biology (materials and methods)
000 rpm for 20 min at 4 °C to separate the insoluble material, followed by the determination of protein concentration using the BSA assay (Pierce). Equal amounts of protein from each sample were mixed with Laemmle's loading buffer, boiled for 5 min and subjected to SDS-PAGE. Membranes were blocked for 1 h with 5% non-fat milk in PBS at room temperature and subsequently probed with the primary antibody of interest. Blots were revealed either using the ECL Western Blotting Substrate (Promega, USA) or SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, USA).
000 cells per well were grown in 96-well plates (Seahorse Bioscience, Billerica, MA), allowed to adhere overnight, and treated with BA or its derivatives for 24 h. The cells were then washed with assay medium (for Mitotest: unbuffered DMEM supplemented with 5 mM glucose, 2 mM glutamine, pH 7.4) before incubation with assay medium (0.175 mL) for 1 h at 37 °C in a CO2-free incubator. The following states of respiration were evaluated: basal respiration, proton leak after oligomycin injection, spare respiratory capacity after injection of the uncoupler CCCP (carbonyl cyanide 3-chlorophenylhydrazone), and non-mitochondrial respiration after injection of rotenone and antimycin A. The data were normalized to the total protein in each well.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was performed under financial support from the Russian Science Foundation (Grant 16-13-10051). A part of this work (Analysis of mitochondrial respiration and Analysis of antitumor activity of BA derivatives, Fig. 2–4) was supported by a grant from the Russian Science Foundation (14-25-00056).Notes and references
- R. C. Taylor, S. P. Cullen and S. J. Martin, Mol. Cell. Biol., 2008, 9, 231–241 CAS.
- B. Zhivotovsky and S. Orrenius, Exp. Cell Res., 2010, 1374–1383 CrossRef CAS PubMed.
- K. Viktorsson, R. Lewensohn and B. Zhivotovsky, Adv. Cancer Res., 2005, 94, 143–196 CrossRef CAS PubMed.
- C. Kantari and H. Walczak, Biochim. Biophys. Acta, 2011, 1813, 558–563 CrossRef CAS PubMed.
- V. Gogvadze, B. Zhivotovsky and S. Orrenius, Mol. Aspects Med., 2010, 31, 60–74 CrossRef CAS PubMed.
- S. Vyas, E. Zaganjor and M. C. Haigis, Cell, 2016, 166, 555–566 CrossRef CAS PubMed.
- V. Gogvadze, S. Orrenius and B. Zhivotovsky, Semin. Cancer Biol., 2009, 19, 57–66 CrossRef CAS PubMed.
- R. H. Cichewicz and S. A. Kouzi, Med. Res. Rev., 2004, 24, 90–114 CrossRef CAS PubMed.
- J. Sarek, M. Kvasnica, M. Vlk, M. Urban, P. Dzubak and M. Hajduch, in Research on Melanoma: A Glimpse into Current Directions and Future Trends, ed. M. Murph, In Tech, Rijeka, Croatia – European Union, 2011, vol. 7, p. 125 Search PubMed.
- R. Mukherjee, V. Kumar, S. K. Srivastava, S. K. Agarwal and A. C. Burman, Anti-Cancer Agents Med. Chem., 2006, 6, 271–679 CrossRef CAS PubMed.
- M. Ali-Seyed, I. Jantan, K. Vijayaraghavan and S. N. A. Bukhari, Chem. Biol. Drug Des., 2016, 87, 517 CAS.
- R. Csuk, Expert Opin. Ther. Pat., 2014, 24, 1–11 CrossRef PubMed.
- D.-M. Zhang, H.-G. Xu, L. Wang, Y.-J. Li, P.-H. Sun, X.-M. Wu, G.-J. Wang, W.-M. Chen and W.-C. Ye, Med. Res. Rev., 2015, 35, 1127–1155 CrossRef CAS PubMed.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- X. Wang, X. Lu, R. Zhu, K. Zhang, S. Li, Z. Chen and L. Li, Neurochem. Res., 2017, 42, 1130–1140 CrossRef CAS PubMed.
- Y. M. Tan, R. Yu and J. M. Pezzuto, Clin. Cancer Res., 2003, 9, 2866–2875 CAS.
- S. Fulda and G. Kroemer, Drug Discovery Today, 2009, 14, 885–890 CrossRef CAS PubMed.
- S. Fulda and G. Kroemer, Antioxid. Redox Signaling, 2011, 15, 2937–2949 CrossRef CAS PubMed.
- S. Shishodia, S. Majumdar, S. Banerjee and B. B. Aggarwal, Clin. Cancer Res., 2003, 63, 4375–4383 CAS.
- A. K. Pathak, M. Bhutani, A. S. Nair, K. S. Ahn, A. Chakraborty, H. Kadara, S. Guha, G. Sethi and B. B. Aggarwal, Clin. Cancer Res., 2007, 5, 943–955 CAS.
- H. Chen, Y. Gao, A. Wang, X. Zhou, Y. Zheng and J. Zhou, Eur. J. Med. Chem., 2015, 92, 648–655 CrossRef CAS PubMed.
- V. H. Villar, O. Vögler, F. Barceló, J. M. Broto, J. M. Serra, V. R. Gutiérrez and R. Alemany, PLoS One, 2016, 11, e0155946 Search PubMed.
- M. K. Shanmugam, X. Dai, A. P. Kumar, B. K. H. Tan, G. Sethi and A. Bishayee, Biochem. Pharmacol., 2013, 85, 1579–1587 CrossRef CAS PubMed.
- A. A. Damle, Y. P. Pawar and A. A. Narkar, Indian J. Exp. Biol., 2013, 51, 485–491 CAS.
- V. Zuco, R. Supino, S. C. Righetti, L. Cleris, E. Marchesi, C. Gambacorti-Passerini and F. Formelli, Cancer Lett., 2002, 175, 17–25 CrossRef CAS PubMed.
- F. B. Mullauer, L. Bloois, J. B. Daalhuisen, M. S. T. Brink, G. Storm, J. P. Medema, R. M. Schiffelers and J. H. Kessler, Anti-Cancer Drugs, 2011, 22, 223–233 CrossRef CAS PubMed.
- F. B. Mullauer, J. H. Kessler and J. P. Medema, Anti-Cancer Drugs, 2010, 21, 215–227 CrossRef CAS PubMed.
- A. Yu. Spivak, D. A. Nedopekina, E. R. Shakurova, R. R. Khalitova, R. R. Gubaidullin, V. N. Odinokov, U. M. Dzhemilev, Y. P. Bel'skii, N. V. Bel'skaya, S. A. Stankevich, E. V. Korotkaya and V. A. Khazanov, Russ. Chem. Bull., 2013, 62, 188–198 CrossRef CAS.
- A. Yu. Spivak, D. A. Nedopekina, R. R. Khalitova, R. R. Gubaidullin, V. N. Odinokov, Y. P. Bel'skii, N. V. Bel'skaya and V. A. Khazanov, Med. Chem. Res., 2017, 26, 518–531 CrossRef CAS.
- S. Kankotia and P. W. Stacpoole, Biochim. Biophys. Acta, 2014, 1846, 617–629 CAS.
- W. Zhang, S.-L. Zhang, X. Hu and K. Y. Tam, Int. J. Biol. Sci., 2015, 11, 1390–1400 CrossRef CAS PubMed.
- S. Saha, M. Ghosh and S. K. Dutta, Sci. Rep., 2015, 5, 7762 CrossRef CAS PubMed.
- M. Ouchi, Y. Inoue, T. Kanzaki and T. Hakushi, J. Org. Chem., 1984, 49, 1408–1412 CrossRef CAS.
- P. J. Duriez and G. M. Shah, Biochem. Cell Biol., 1997, 75, 337–349 CrossRef CAS PubMed.
- J. Neuzil, L. F. Dong, J. Rohlena, J. Truksa and S. J. Ralph, Mitochondrion, 2013, 13, 199–208 CrossRef CAS PubMed.
- J. Neuzil, X. F. Wang, L. F. Dong, P. Low and S. J. Ralph, FEBS Lett., 2006, 580, 5125–5129 CrossRef CAS PubMed.
- J. S. Modica-Napolitano and V. Weissig, Int. J. Mol. Sci., 2015, 16, 17394–17421 CrossRef PubMed.
- J. S. Modica-Napolitano, M. Kulawiec and K. K. Singh, Curr. Mol. Med., 2007, 7, 121–131 CrossRef CAS PubMed.
- J. Kovarova, M. Bajzikova, M. Vondrusova, J. Stursa, J. Goodwin, M. Nguyen, R. Zobalova, E. A. Pesdar, J. Truksa, M. Tomasetti, L.-F. Dong and J. Neuzil, Redox Rep., 2014, 19, 16–25 CrossRef CAS PubMed.
- J. A. Jara, V. Castro-Castillo, J. Saavedra-Olavarría, L. Peredo, M. Pavanni, F. Jana, M. E. Letelier, E. Parra, M. I. Becker, A. Morello, U. Kemmerling, J. D. Maya and J. Ferreira, J. Med. Chem., 2014, 57, 2440–2454 CrossRef CAS PubMed.
- C. A. Reddy, V. Somepalli, T. Golakoti, A. K. Kanugula, S. Karnewar, K. Rajendiran, N. Vasagiri, S. Prabhakar, P. Kuppusamy, S. Kotamraju and V. K. Kutala, PLoS One, 2014, 9, e89351 Search PubMed.
- I. Yu. Strobykina, M. G. Belenok, M. N. Semenova, V. V. Semenov, V. M. Babaev, I. K. Rizvanov, V. F. Mironov and V. E. Kataev, J. Nat. Prod., 2015, 78, 1300–1308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00248c |
| This journal is © The Royal Society of Chemistry 2017 |