
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA structurally guided dissection-then-evolution strategy for ligand optimization of smoothened receptor†‡
Lintao
Ye
 abef,
Kang
Ding
ceaf,
Fei
Zhao
a,
Xiaoyan
Liu
a,
Yiran
Wu
a,
Yang
Liu
a,
Dongxiang
Xue
abef,
Fang
Zhou
abef,
Xianjun
Zhang
adef,
Raymond C.
Stevens
af,
Fei
Xu
af,
Suwen
Zhao
*af and
Houchao
Tao
*a
abef,
Kang
Ding
ceaf,
Fei
Zhao
a,
Xiaoyan
Liu
a,
Yiran
Wu
a,
Yang
Liu
a,
Dongxiang
Xue
abef,
Fang
Zhou
abef,
Xianjun
Zhang
adef,
Raymond C.
Stevens
af,
Fei
Xu
af,
Suwen
Zhao
*af and
Houchao
Tao
*a
aiHuman Institute, ShanghaiTech University, 2F Building 6, 99 Haike Road, Pudong New District, Shanghai 201210, China. E-mail: zhaosw@shanghaitech.edu.cn; taohch@shanghaitech.edu.cn
bShanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Building 3, Room 426, Shanghai, 201203, China
cKey Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, 200031, China
dInstitute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, 200031, China
eUniversity of Chinese Academy of Sciences, No. 19A, Yuquan Road, Beijing 100049, China
fSchool of Life Science and Technology, ShanghaiTech University, Shanghai 201210, China
First published on 28th April 2017
Abstract
We present herein a novel dissection-then-evolution strategy for ligand optimization. Using the co-crystal structure of the smoothened receptor (SMO) as a guide, we studied the modular contribution of LY2940680 by systematically “silencing” the specific interaction between the individual residue(s) and the fragment in the ligand. Following evolution by focusing on the benzoyl part finally yielded an improved ligand 21.
The smoothened receptor (SMO) is a transmembrane protein categorized as a member of Frizzled G protein-coupled receptors (GPCRs). SMO plays an essential role in the regulation of the evolutionarily conserved and physiologically important Hedgehog (Hh) signaling pathway.1,2 Disruption of this pathway results in disorders of embryonic development and tissue regeneration that are related to birth defects or cancers.3,4 Thus, SMO has been a well-pursued and now validated target for anticancer agent development.5–8 Many SMO ligands have been discovered, including the steroidal alkaloid cyclopamine and other synthetic compounds.9–11 Among these, vismodegib (GDC-0449)12 and sonidegib (LDE225)13 have been approved by the FDA in 2012 and 2015, respectively, for the treatment of basal-cell carcinoma (BCC). Nevertheless, their clinical use has been associated with many severe side reactions,14,15 as well as several instances of mutation-related drug resistance.16–18 In addition, many fundamental questions remain regarding understanding SMO's involvement in those physiological mechanisms.19 Development of a persistent and reliable ligand that would address these issues is highly sought after.
The evolution of SMO ligands can now be guided by the high resolution structures that were obtained recently.20 As part of the achievements from the intense and flourishing research on SMO, several atomic resolution structures of SMO, including those of the transmembrane domain (TMD)21,22 and extracellular domain (ECD),23,24 and the very recent multi-domain structures have emerged.25 These structures clearly defined the molecular contacts of co-crystallized SMO ligands and provided great insights into the design of tools and probes for further biophysical studies. For example, originating from the first co-crystallized SMO ligand LY2940680,21,26 our groups designed a super-stabilizing tool ligand TC114 (Fig. S1, ESI‡) by the introduction of a new functional group. This modification established an extra interaction that restricts motion to hold the receptor's conformation and facilitate crystallization for structural studies of multi-domain SMO.27 This success encouraged us to look for an additional improved ligand with potential applications in pharmacological development and probe design.
Using known SMO structures as a guide, we present herein a dissection-then-evolution strategy for ligand optimization. To this end, we first carried out a comprehensive study on LY2940680, whose interactions between its receptor residues and ligand fragments were revealed at high resolution (Fig. 1A).21 According to the key interactions as indicated, the molecule was dissected into four variable parts (I–IV) and one linker (Fig. 1B). Sequential “silencing”, namely introducing a chemical mutation into LY2940680 to weaken or eliminate a specific ligand–receptor interaction, generated a series of compounds (1–4, Fig. 1B). All of the new compounds were synthesized (Scheme S1, ESI‡) and then tested for activity inhibiting the Hedgehog signaling pathway by a luciferase reporter assay using light II cells, which were NIH-3T3 cells stably transfected with a Gli-responsive firefly luciferase reporter. As clearly indicated (Fig. 1C), compounds with substituents removed on benzoyl (1) or deletion of the pyrazole (4) almost completely abolished the receptor's activity. Meanwhile, the replacement of phthalazine with naphthalene (3) only accounts for a slight reduction of the activity. This may indicate that there is little difference between the electronic interaction of Arg400 with phthalazine and the cation–π interaction with naphthalene. Surprisingly, the replacement of the amide by a tertiary amine in 2 maintained moderate activity with an IC50 of about 70 nM, possibly because of compensation resulting from the missing H-bonding between the carbonyl and Asn219. Additionally, to exclude the potential interruption from the metabolism of these compounds, we checked their stability in cell media (Fig. S3, ESI‡) which indicated that this group of compounds was stable and thus the dissecting effect is only dependent on the structural variation.
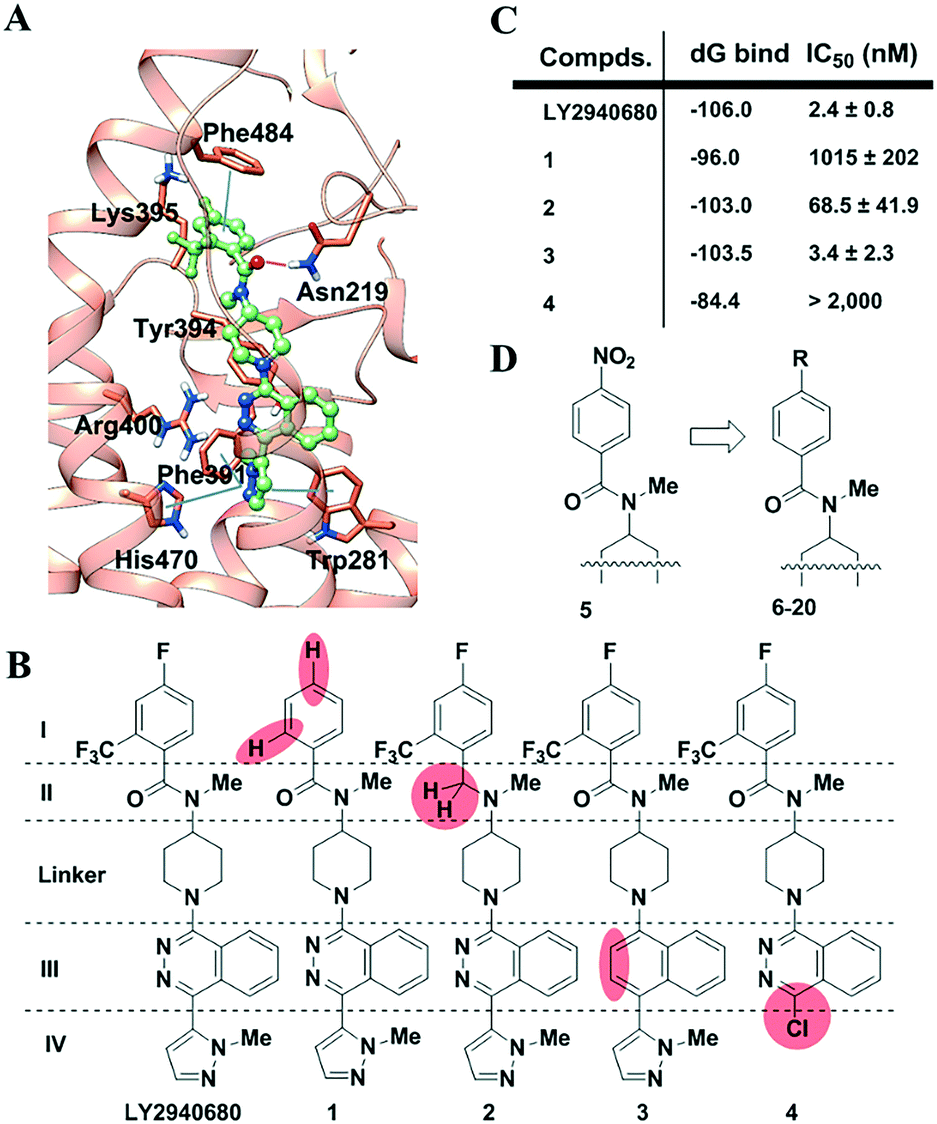 | ||
| Fig. 1 Study of the SMO ligand LY2940680. (A). The key interactions observed in the co-crystal structure of SMO with LY2940680. (B). Design of LY2940680 analogs to analyze the contribution from each part. LY2940680 was dissected into four parts (I–IV) and one linker. Analogs were designed by partially “silencing” the interaction by the removal or substitution of the functional groups, as highlighted in coral in every compound. (C). The relative binding energy was calculated and the potency was evaluated by the Gli assay. IC50 values represent the mean ± SEM of at least three separate experiments carried out in duplicate. All compounds were evaluated following 24 h incubation. (D). New analogs were designed by focusing on the variations in the benzoyl unit. | ||
Glide XP docking followed by Prime MM-GBSA was used to estimate the relative binding affinities of the compounds mentioned above.28 As shown (Fig. 1C and Table S1, ESI‡), modification of pyridazine leads to the least loss of relative binding energy (ddG = 2.5 kcal mol−1). In comparison, the alteration of other fragments would result in a large loss of the binding energy. For compound 4, the loss of pyrazole leads to a huge loss of ddG (21.6 kcal mol−1), which is mainly from van der Waals energy (ddG VDW = 11.0 kcal mol−1). Similarly, removal of substituents in benzoyl also results in a large loss of ddG (∼10 kcal mol−1). Interestingly, elimination of the carbonyl renders compound 2 with a unique basic tertiary amine which, under physiological conditions, produces a protonated 2-H+ which can still maintain a similar binding free energy to that of the parent compound LY2940680 because the solvation energy (ddG solv = −28.1 kcal mol−1) compensates for the loss of coulomb energy (ddG Coulomb = 28.6 kcal mol−1) of 2-H+.
Overall, in this context, the computational performance showed good agreement with the IC50 values from cell-based functional assays. These results confirmed our hypothesis that modification in part I or IV would be valuable, while that in part II or III would be less sensitive. Furthermore, considering the space limitations of the deeply buried pocket that interacts with part IV, the more outward situated part I fragment seems to be in a better position for modification. It may not be a coincidence that our previous development of TC114 (Fig. S1, ESI‡) in which a nitro group was introduced to the benzoyl yielded successful co-crystallization. However, an aromatic nitro group, like in TC114, was generally avoided by medicinal chemists since its reductive metabolite causes undesired toxicity. Thus, our aim here is to search for a better and more drug-like surrogate for the nitro group (Fig. 1D). To simplify the synthesis and characterization, all the compounds were initially designed without the 6-trifluoromethyl group on the benzoic fragment. For this reason, all the compounds were then compared to that described in ref. 5, a 4-NO2 compound derived from TC114 by removal of the trifluoromethyl group.
A series of compounds (5–20) were then synthesized (Scheme S2, ESI‡). Following our established procedure in search for stabilizing molecules on GPCRs, the compounds were first screened via a thermal stability assay on purified SMO (Table 1 and Fig. S2, ESI‡). This assay shows the fluorescence (365/460 nm) increase during the thermal denaturation upon reaction of exposed cysteines with a thiol-specific fluorochrome, CPM dye (N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide). The thermal stabilization capability of the test compounds was denoted as the midpoint temperature (Tm) at which 50% of the fluorescence increase was observed. The CPM assay has been widely used in the high throughput screening of tool compounds on membrane proteins.29,30
|
|
||||
|---|---|---|---|---|
| Compds. | R | CPM assay Tm (°C) | MM-GBSA dG bind (kcal mol−1) | Gli-Luc reporterIC50a (nM) |
| a IC50 values represent the mean ± SEM of at least three independent experiments carried out in duplicate. All analogues were evaluated following 24 h incubation. | ||||
| APO | NA | 56.3 | NA | NA |
| 5 | NO2 | 61.0 | −100.6 | 235.6 ± 88.5 |
| 6 | F | 59.1 | −93.6 | 1575 ± 506 |
| 7 | NH2 | 58.2 | −92.3 | 2018 ± 556 |
| 8 | NHCH2CO2H | 56.8 | −88.8 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 9 | CHO | 59.0 | −97.9 | 2891 ± 407 |
| 10 | NMe2 | 58.5 | −96.3 | 1415 ± 338 |
| 11 | SO2Me | 56.3 | −87.3 | >10000 |
| 12 | SO3H | 59.4 | −86.6 | 2682 ± 898 |
| 13 | SO2NH2 | 57.1 | −98.1 | 3242 ± 712 |
| 14 | CO2Me | 60.3 | −100.6 | >10000 |
| 15 | CO2H | 56.8 | −89.5 | >10000 |
| 16 | CONH2 | 59.7 | −97.5 | 3018 ± 223 |
| 17 | B(OH)2 | 59.4 | −101.1 | 870 ± 358 |
| 18 | CH2CHO | 60.2 | −99.0 | >10000 |
| 19 | OC6H5 | 59.6 | −111.8 | 269.7 ± 59.1 |
| 20 | OCH2C6H5 | 58.4 | −113.2 | 1799 ± 443 |

As indicated in Table 1, almost all the compounds were able to stabilize SMO to some extent when compared to the APO sample (Tm = 56.3 °C). Among them, 5 remained the best stabilizer (Tm = 61.0 °C). Surprisingly, little improvement was observed on the negatively charged compounds 8, 12 and 15 which, according to the design with different acidity and alternative linker length, are expected to form an ionic pair with the positively charged ε-amine of Lys395. This may be due to the high desolvation penalty of the highly solvated ligand anion that weakens the contribution from the ionic pair. Simple substituents, like fluorine (6), showed a slight benefit on stabilization (Tm = 59.1 °C). Other neutral compounds without a negatively induced dipole, such as 7 and 10, exhibited even less favorable interactions (Tm ≈ 58 °C), while compounds with possible negative dipoles to mimic the nitro group, like sulfone 11 and ester 14, could indeed promote the expected interaction. The corresponding sulfonamide 13 and amide 16, however, showed less capability to stabilize. This may be due to an attenuated π–π interaction between the ligand and Phe484 by a relatively weaker electron-withdrawing group than the nitro in compound 5. An aldehyde is proposed to form a reversible covalent Schiff base with an amine. However, 9 did not significantly enhance the stability of SMO. 18 with an extended linker is a better ligand in this regard as it may have a higher tendency to form a covalent bond. Boronic acids, though not often, have been found in marketed drugs. Functionally, phenyl boronic acid usually has a pKa of 9–10 that makes it protonated under physiological conditions. It has been reported to behave as the bioisostere of phenol,31 and it was proposed to form a hydrogen bond or a boron–nitrogen bond at times. Thus, it could be an alternative to explore the interaction with Lys395. However, compound 17 did not provide encouraging results (Tm = 59.4 °C). Finally, two phenyl-substituted compounds 19 and 20 exhibited moderately enhanced stabilization on SMO.
To further elucidate the structure activity relationships (SARs), we performed MM-GBSA calculations and measured IC50s via a Gli-luciferase reporter assay. As indicated (Table 1 and Fig. S6, ESI‡), the relative binding free energy and Tm values correlate well, in a linear manner with only a few exceptions. Compounds that deviated from the correlation include the highly acidic phenyl sulfonic acid 12 and phenyl attached 19 and 20. For the latter two compounds, calculations provided an extremely low binding free energy (dG bind < −110). The major difference in the formula came from VDW and lipophilicity (Table S1, ESI‡) when compared to those with nitro embedded 5. The low Tm value reflected that the loading of such a large rigid and hydrophobic group is not favorable for stabilizing SMO in micelles. Nevertheless, the correlations indicated that the calculation method can be a rough alternative for the prediction of a ligand's stabilizing effect.
On the other hand, the Gli-luciferase reporter assay showed a slight consistency with the CPM assay or MM-GBSA calculation. Roughly, compounds with lower binding energy behave relatively well with a few exceptions. 14 has a high Tm value and a low binding energy but exhibited no activity in the Gli assay. This may be ascribed to its in vivo hydrolysis to acid 15 which, in general in this assay, lost activity like 8. Overall, the nitro compound 5 remained the best with an IC50 of 235 nM. Excitingly, 19 ranked second with a close IC50 value, though not as potent as reflected in the MM-GBSA calculation. Other molecules were generally much less effective, in the micro molar range of IC50 values in this assay.
We then further modified 19 to 21 by adding a trifluoromethyl group on the benzoyl unit (Fig. 2A). As observed in the transformation of 5 and TC114,27 this evolution dramatically enhanced the activity in the Gli assay. Trifluoromethyl groups play multiple roles in pharmaceutical development. This strong electron-withdrawing group likely enhanced the π–π interaction with Phe484. In comparison with TC114, 21 could be a promising drug candidate for further evolution since 21 maintained comparable potency when the nitro group was replaced with a pharmacologically safer group. We carefully determined the docking of compound 21 in our recent full-length structure of SMO (Fig. 2B).27 This compound retained all the important interactions with SMO as in the TC114 co-crystallized structure. In particular, 21 establishes two additional key interactions, both involved with the appended benzene ring. One is the cation–π interaction between Lys395 and the benzene,32 and the other is the π–π interaction with Phe484. By comparison, such interactions were not observed in other compounds. For example, a closely resembling compound S1 (Fig. S4, ESI‡), modified from 20 by one more carbon extension in the linker, showed a dramatic decrease in efficacy in the Gli luciferase assay. In general, these extra interactions may promote ligand potency on SMO and more importantly, hold promise in applications against other disease-related SMO mutations. It should also be noted that the newly introduced benzene ring added an excellent knob for the derivation of probes for other biophysical studies. In fact, in our attempts to design tool compounds in our on-going projects, we found that derivatives based on other analogs, as shown in Table 1, would have to pay a price with a decreasing efficacy.
 | ||
| Fig. 2 Compound 21 exhibited enhanced potency. (A). Evolution of 19 by the addition of a trifluoromethyl group on the benzoyl unit generated compound 21 with IC50 of 1.0 nM. (B). Docking study of 21 in the two-domain SMO showed that all the original key interactions were retained. Besides, the embedded benzene ring at the outer end of a molecule of 21 established two additional interactions with Lys395 and Phe484, respectively. | ||
In conclusion, a more drug-like and better probe-oriented ligand 21 was developed for SMO via a structurally guided dissection-then-evolution strategy. We confirmed that the introduction of a phenyl ether into LY2940680 can strengthen its binding and stabilize the SMO. This combined computation and chemistry strategy could be employed on other scaffolds or against other targets for future projects, including imaging, structural exploration and drug discovery.
The authors (FX, RCS, SZ, and HT) are thankful to Shanghai Municipal Government, ShanghaiTech University for financial support, the Shanghai PuJiang Talent Program (16PJ1407300; SZ) and the Natural Science Foundation of Shanghai (16ZR1448500; SZ). We thank Y. Zhao for providing the NIH3T3 cell line for compound evaluation assays. We thank J. Yang and W. Tan for performing the cytotoxicity assay. We also thank Z. J. Liu, G. Song and J. Cheng for their helpful discussion and assistance in manuscript preparation.
Notes and references
- Y. Zhao, C. Tong and J. Jiang, Nature, 2007, 450, 252 CrossRef CAS PubMed.
- P. W. Ingham and A. P. McMahon, Genes Dev., 2001, 15, 3059 CrossRef CAS PubMed.
- M. P. di Magliano and M. Hebrok, Nat. Rev. Cancer, 2003, 3, 903 CrossRef CAS PubMed.
- J. Jiang and C.-C. Hui, Dev. Cell, 2008, 15, 801 CrossRef CAS PubMed.
- E. Dolgin, Nat. Med., 2011, 17, 523 CrossRef CAS PubMed.
- C. A. Maschinot, J. R. Pace and M. K. Hadden, Curr. Med. Chem., 2015, 22, 4033 CrossRef CAS PubMed.
- G. Liu, J. Yang, J. Wang, X. Liu, W. Huang, J. Li, W. Tan and A. Zhang, J. Med. Chem., 2016, 59, 11050 CrossRef CAS PubMed.
- H. J. Sharpe, W. Wang, R. N. Hannoush and F. J. de Sauvage, Nat. Chem. Biol., 2015, 11, 246 CrossRef CAS PubMed.
- J. K. Chen, J. Taipale, M. K. Cooper and P. A. Beachy, Genes Dev., 2002, 16, 2743 CrossRef CAS PubMed.
- J. K. Chen, J. Taipale, K. E. Young, T. Maiti and P. A. Beachy, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14071 CrossRef CAS PubMed.
- H. Tao, Q. Jin, D.-I. Koo, X. Liao, N. P. Englund, Y. Wang, A. Ramamurthy, P. G. Schultz, M. Dorsch, J. Kelleher and X. Wu, Chem. Biol., 2011, 18, 432 CrossRef CAS PubMed.
- A. Dlugosz, S. Agrawal and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2012, 11, 437 CrossRef CAS PubMed.
- S. Pan, X. Wu, J. Jiang, W. Gao, Y. Wan, D. Cheng, D. Han, J. Liu, N. P. Englund, Y. Wang, S. Peukert, K. Miller-Moslin, J. Yuan, R. Guo, M. Matsumoto, A. Vattay, Y. Jiang, J. Tsao, F. Sun, A. C. Pferdekamper, S. Dodd, T. Tuntland, W. Maniara, J. F. Kelleher, III, Y.-m. Yao, M. Warmuth, J. Williams and M. Dorsch, ACS Med. Chem. Lett., 2010, 1, 130 CrossRef CAS PubMed.
- S. E. Gould, J. A. Low, J. C. Marsters, Jr., K. Robarge, L. L. Rubin, F. J. de Sauvage, D. P. Sutherlin, H. Wong and R. L. Yauch, Expert Opin. Drug Discovery, 2014, 9, 969 CrossRef CAS PubMed.
- E. Pak and R. A. Segal, Dev. Cell, 2016, 38, 333 CrossRef CAS PubMed.
- G. J. P. Dijkgraaf, B. Alicke, L. Weinmann, T. Januario, K. West, Z. Modrusan, D. Burdick, R. Goldsmith, K. Robarge, D. Sutherlin, S. J. Scales, S. E. Gould, R. L. Yauch and F. J. de Sauvage, Cancer Res., 2011, 71, 435 CrossRef CAS PubMed.
- H. J. Sharpe, G. Pau, G. J. Dijkgraaf, N. Basset-Seguin, Z. Modrusan, T. Januario, V. Tsui, A. B. Durham, A. A. Dlugosz, P. M. Haverty, R. Bourgon, J. Y. Tang, K. Y. Sarin, L. Dirix, D. C. Fisher, C. M. Rudin, H. Sofen, M. R. Migden, R. L. Yauch and F. J. de Sauvage, Cancer Cell, 2015, 27, 327 CrossRef CAS PubMed.
- S. X. Atwood, K. Y. Sarin, R. J. Whitson, J. R. Li, G. Kim, M. Rezaee, M. S. Ally, J. Kim, C. Yao, A. L. S. Chang, A. E. Oro and J. Y. Tang, Cancer Cell, 2015, 27, 342 CrossRef CAS PubMed.
- A. M. Arensdorf, S. Marada and S. K. Ogden, Trends Pharmacol. Sci., 2016, 37, 62 CrossRef CAS PubMed.
- J. Shonberg, R. C. Kling, P. Gmeiner and S. Loeber, Bioorg. Med. Chem., 2015, 23, 3880 CrossRef CAS PubMed.
- C. Wang, H. Wu, V. Katritch, G. W. Han, X.-P. Huang, W. Liu, F. Y. Siu, B. L. Roth, V. Cherezov and R. C. Stevens, Nature, 2013, 497, 338 CrossRef CAS PubMed.
- C. Wang, H. Wu, T. Evron, E. Vardy, G. W. Han, X.-P. Huang, S. J. Hufeisen, T. J. Mangano, D. J. Urban, V. Katritch, V. Cherezov, M. G. Caron, B. L. Roth and R. C. Stevens, Nat. Commun., 2014, 5, 4355 CAS.
- R. Rana, C. E. Carroll, H.-J. Lee, J. Bao, S. Marada, C. R. R. Grace, C. D. Guibao, S. K. Ogden and J. J. Zheng, Nat. Commun., 2013, 4, 3965 Search PubMed.
- P. Huang, D. Nedelcu, M. Watanabe, C. Jao, Y. Kim, J. Liu and A. Salic, Cell, 2016, 166, 1176 CrossRef CAS PubMed.
- E. F. X. Byrne, R. Sircar, P. S. Miller, G. Hedger, G. Luchetti, S. Nachtergaele, M. D. Tully, L. Mydock-McGrane, D. F. Covey, R. P. Rambo, M. S. P. Sansom, S. Newstead, R. Rohatgi and C. Siebold, Nature, 2016, 535, 517 CrossRef CAS PubMed.
- M. H. Bender, P. A. Hipskind, A. R. Capen, M. Cockman, K. M. Credille, H. Gao, J. A. Bastian, J. M. Clay, K. L. Lobb, D. J. Sall, M. L. Thompson, T. Wilson, G. N. Wishart and B. K. R. Patel, Cancer Res., 2011, 71, 2819 Search PubMed.
- X. Zhang, F. Zhao, Y. Wu, J. Yang, G. W. Han, S. Zhao, A. Ishchenko, L. Ye, X. Lin, K. Ding, V. Dharmarajan, P. R. Griffin, C. Gati, G. Nelson, M. S. Hunter, M. A. Hanson, V. Cherezov, R. C. Stevens, W. Tan, H. Tao and F. Xu, Nat. Commun., 2017 DOI:10.1038/ncomms15383.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2011, 79, 2794 CrossRef CAS PubMed.
- X. Zhang, R. C. Stevens and F. Xu, Trends Biochem. Sci., 2015, 40, 79 CrossRef PubMed.
- A. I. Alexandrov, M. Mileni, E. Y. T. Chien, M. A. Hanson and R. C. Stevens, Structure, 2008, 16, 351 CrossRef CAS PubMed.
- Y. Kong, J. Grembecka, M. C. Edler, E. Hamel, S. L. Mooberry, M. Sabat, J. Rieger and M. L. Brown, Chem. Biol., 2005, 12, 1007 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459 CrossRef CAS.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Materials, synthetic protocols, computational process and additional data. See DOI: 10.1039/c7md00104e |
| This journal is © The Royal Society of Chemistry 2017 |