
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral disubstituted piperidinyl ureas: a class of dual diacylglycerol lipase-α and ABHD6 inhibitors†‡
Hui
Deng
 a,
Tom
van der Wel
a,
Richard J. B. H. N.
van den Berg
b,
Adrianus M. C. H.
van den Nieuwendijk
b,
Freek J.
Janssen
a,
Marc P.
Baggelaar
a,
Hermen S.
Overkleeft
b and
Mario
van der Stelt
*a
a,
Tom
van der Wel
a,
Richard J. B. H. N.
van den Berg
b,
Adrianus M. C. H.
van den Nieuwendijk
b,
Freek J.
Janssen
a,
Marc P.
Baggelaar
a,
Hermen S.
Overkleeft
b and
Mario
van der Stelt
*a
aDepartment of Molecular Physiology, Leiden Institute of Chemistry, Leiden University, Leiden, The Netherlands. E-mail: m.van.der.stelt@chem.leidenuniv.nl
bDepartment of Bio-organic Synthesis, Leiden Institute of Chemistry, Leiden University, Leiden, The Netherlands
First published on 10th March 2017
Abstract
Inhibitors of diacylglycerol lipases and α,β-hydrolase domain containing protein 6 (ABHD6) are potential leads for the development of therapeutic agents for metabolic and neurodegenerative disorders. Here, we report the enantioselective synthesis and structure activity relationships of triazole ureas featuring chiral, hydroxylated 2-benzylpiperidines as dual inhibitors of DAGLα and ABHD6. The chirality of the carbon bearing the C2 substituent, as well as the position of the hydroxyl (tolerated at C5, but not at C3) has profound influence on the inhibitory activity of both DAGLα and ABHD6, as established using biochemical assays and competitive activity-based protein profiling on mouse brain extracts.
Introduction
Diacylglycerol lipase α and diacylglycerol lipase β (DAGLα and DAGLβ) are intracellular, multi-domain, transmembrane serine hydrolases that employ a Ser-His-Asp catalytic triad to specifically hydrolyse arachidonate-containing diglycerides to form the endocannabinoid 2-arachidonoylglycerol (2-AG) in the brain and peripheral tissues.1,2 Endocannabinoid signalling is involved in various neurophysiological functions, such as learning, memory, pain sensation, adult neurogenesis and regulation of the energy balance.3–5 2-AG is hydrolysed by monoacylglycerol lipase into arachidonic acid, which is a precursor for pro-inflammatory prostaglandins.6–8 Consequently, the development of DAGL inhibitors that perturb 2-AG production is an emerging strategy for potential therapeutic intervention in various human diseases, including metabolic syndrome related diseases and neuroinflammation.9,10Previously, we have reported the discovery of α-ketoheterocycles,11–13 glycinesulfonamides14 and triazole ureas (e.g. DO34 and DH376 (1)),15 as selective DAGL inhibitors (Fig. 1). DH376 and DO34 are brain active DAGL inhibitors that reduce 2-AG levels in a time- and dose-dependent manner in mouse brain. They also reduce lipopolysaccharide-induced pro-inflammatory prostaglandin and cytokine levels in mouse brain, as well as anapyrexia and refeeding in fasted mice.15,30 Of note, most DAGL inhibitors cross-react with α,β-hydrolase domain containing protein 6 (ABHD6), which has a minor role in the hydrolysis of 2-AG,16 degrades bis(monoacylglycero)phosphate,17 and acts as a lysophosphatidyl hydrolase.18 Inhibition of ABHD6 produces neuroprotective, anti-obesity and anti-inflammatory effects in preclinical disease models.19,20 Thus, dual inhibition of DAGLs and ABHD6 may actually be advantageous from a therapeutic point of view.
 | ||
| Fig. 1 Conversion of diacylglycerol (DAG) into 2-arachidonoylglycerol (2-AG) by DAG lipases and chemical structures of their inhibitors DO34, DH376 and (R)-KT109. | ||
The design of DH376 and DO34 was inspired by (R)-KT109 (2a),21,22 the first in vivo active DAGLβ inhibitor. Both compounds are covalent irreversible inhibitors that feature a 2-benzylpiperidine moiety that confers selectivity and activity towards DAGLs and ABHD6. We have reported previously an enantioselective synthesis route for DH376 based on our ample experience with the synthesis of chiral piperidines from easily available starting materials following a strategy that encompasses enzyme-catalysed cyanohydrin synthesis followed by a transamination-reduction-ring-closing metathesis series of events.23–25
Our strategy, as we demonstrated earlier in the synthesis of polyhydroxylated piperidines (termed iminosugars), is especially suited for the construction of chiral, enantiopure 2-alkylpiperidines bearing one or more hydroxyl substituents. We realised that, in this way, piperidinylureas bearing multiple substituents, amongst which solubilizing hydroxyl groups, would be easy to accomplish. To demonstrate the validity of this reasoning, and to extend our panel of putative serine hydrolase inactivators, we set out to make a small library of chiral, disubstituted piperidinylureas. Here, these synthesis efforts as well as the inhibitory potential of the resulting compounds 3, 4a–7a, 4b–7b, 6c, 8 and 9–18, in comparison with lead compounds 2a and 2b against DAGLα and ABHD6 are reported.
Results and discussion
Chemistry
To systematically investigate the structure–activity relationship of the covalent irreversible inhibitors, we focused our attention first on the modification of 2-alkylpiperidine group, resulting 1,2,3-triazole ureas 3, 4a–7a, 4b–7b, 6c, 8 (Fig. 2, Tables 1 and 2). Next, we explored the influence of electrophilicity of the leaving group (i.e. triazole scaffold) by synthesizing compounds 9–18. The synthesis started with compound 3, as a close homologue of lead compound 2a with a methyloxy moiety inserted into the benzylic position. The synthesis route commenced with O-TBDPS-protected intermediate 19 that was prepared according to previously established procedure.26 Treatment of 19 with hydrogen and 10% Pd/C in MeOH gave hydrogenated intermediate 20, and ensuing desilylation and benzylation of the primary alcohol yielded Boc-protected intermediate 22 (Scheme 1). Removal of the Boc group using 25% (v/v) TFA in DCM gave amine 23 in near quantitative yield. Finally, triphosgene-mediated condensation of 23 with 4-([1,1′-biphenyl]-4-yl)-1H-1,2,3-triazole and isolation of the 1,4-regioisomer by silica gel chromatography provided compound 3 in >95% ee as determined by chiral HPLC.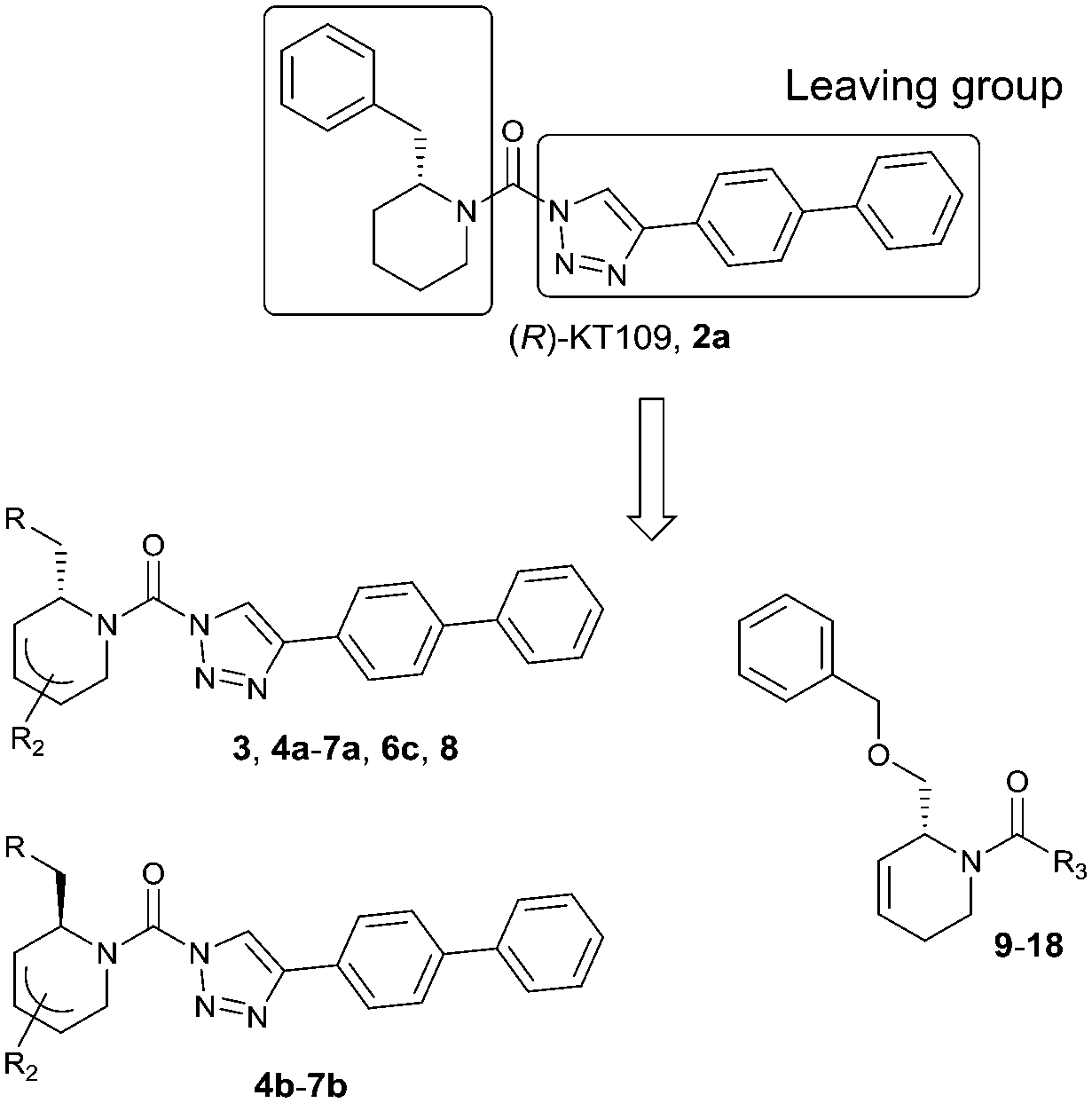 | ||
| Fig. 2 Design of compounds 3, 4a–7a, 4b–7b, 6c, 8 and 9–18 based on lead 2a. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | pIC50 ± SEM (DAGLα) | pIC50 ± SEM (ABHD6) | Entry | R | pIC50 ± SEM (DAGLα) | pIC50 ± SEM (ABHD6) |
| 2a |
|
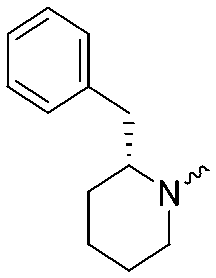
9.1 ± 0.1 | 8.6 ± 0.1 | 2b |
|
7.4 ± 0.1 | 6.2 ± 0.1 |
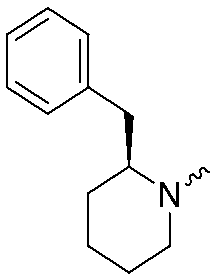
| 3 |
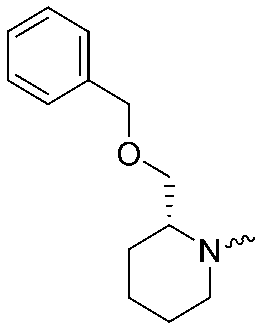
|
7.1 ± 0.1 | 8.5 ± 0.1 | 8 |
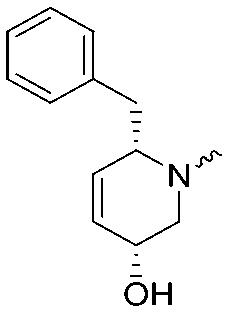
|
7.8 ± 0.1 | 8.5 ± 0.2 |
| 4a |
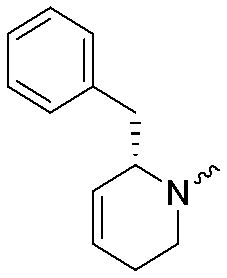
|
9.1 ± 0.1 | 8.6 ± 0.1 | 4b |
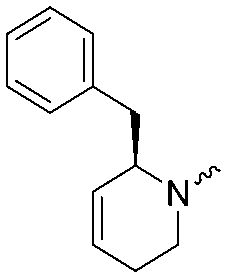
|
7.1 ± 0.1 | 7.6 ± 0.1 |
| 5a |
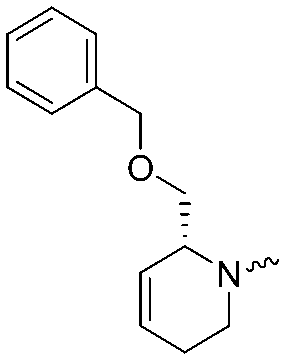
|
7.6 ± 0.1 | 7.9 ± 0.1 | 5b |
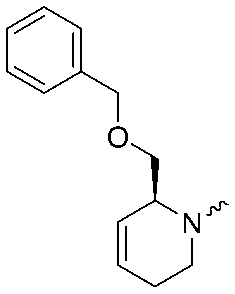
|
5.9 ± 0.2 | 7.0 ± 0.1 |
| 6a |

|
7.6 ± 0.1 | 8.3 ± 0.1 | 6b |

|
<5 | 6.5 ± 0.1 |
| 6c |
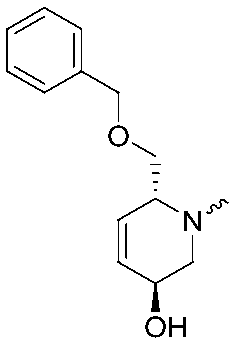
|
7.5 ± 0.2 | 8.0 ± 0.1 | ||||
| 7a |
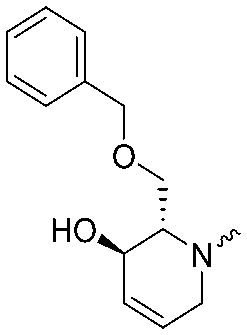
|
<5 | 6.1 ± 0.1 | 7b |
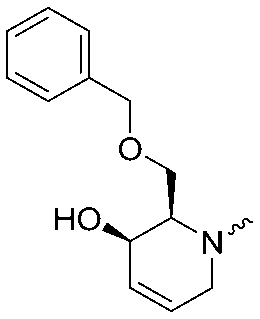
|
<5 | 6.6 ± 0.1 |
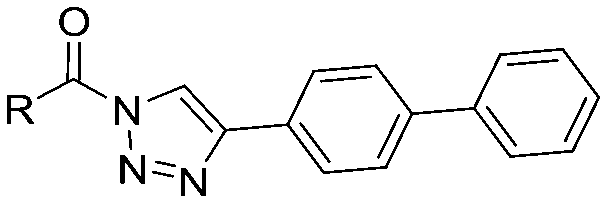
|
|
|||
|---|---|---|---|
| Entry | R | pIC50 ± SEM (DAGLα) | pIC50 ± SEM (ABHD6) |
| 9 |
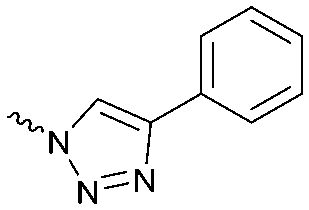
|
6.8 ± 0.1 | 6.8 ± 0.1 |
| 10 |

|
7.8 ± 0.1 | 7.5 ± 0.1 |
| 11 |

|
7.8 ± 0.1 | 7.8 ± 0.1 |
| 12 |

|
7.6 ± 0.1 | 8.2 ± 0.1 |
| 13 |

|
<5 | <5 |
| 14 |
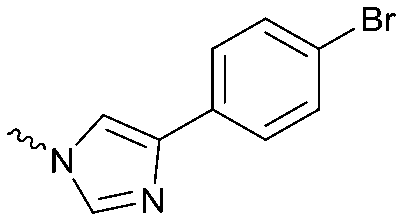
|
<5 | <5 |
| 15 |
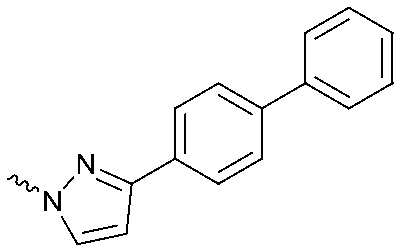
|
<5 | <5 |
| 16 |
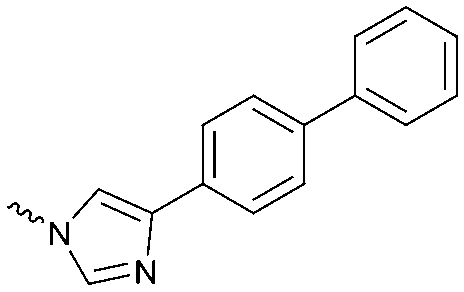
|
<5 | <5 |
| 17 |

|
<5 | <5 |
| 18 |
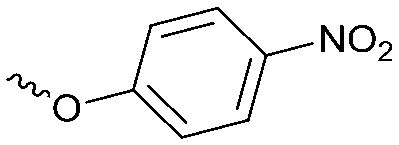
|
<5 | <5 |
 | ||
Scheme 1 Reagents and conditions: (a) 10% Pd/C, H2, MeOH, 95%; (b) TBAF, THF, r.t., 98% (21), 92% (24); (c) BnBr, TBAI, NaH, DMF, 90% (22), 90% (25); (d) 25% TFA, DCM, 84% (26), 85% (28); (e) DIPEA, triphosgene, THF, 0 °C; (f) DIPEA, DMAP, 1,2,3-triazole, THF, 60 °C, 25% (3), 30% (5a), 40% (30) over 2 steps; (g) 1,4-dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (2:1), biphenyl boronic acid, PdCl2 (dppf), 80 °C, 75%; (h) HF-pyridine, THF:pyridine = 1:1 (v/v), 20% over 3 steps (based on 28). :H2O (2:1), biphenyl boronic acid, PdCl2 (dppf), 80 °C, 75%; (h) HF-pyridine, THF:pyridine = 1:1 (v/v), 20% over 3 steps (based on 28). | ||
Following a related sequence of events, we obtained compound 5a (Scheme 1). Compound 5b (the enantiomer of 5a) was synthesized in the same fashion as described for 5a (see ESI,‡ Scheme S1). For the synthesis of compound 6a, we first prepared key intermediate 27 by employing a previously reported method.15,24 Subsequently, removal of the Boc group using 25% (v/v) TFA in DCM generated amine 28 that was directly coupled with 4-([1,1′-biphenyl]-4-yl)-1H-1,2,3-triazole. After silica gel chromatography, 1,4-regioisomer 29 was isolated and ensuing desilylation with HF-pyridine yielded target compound 6a (Scheme 1). In a similar manner, we prepared compounds 4a, 4b, 6b, 6c and 8 with different stereochemistry and substitution pattern on the piperidine ring (see for synthesis details the ESI‡). The synthesis of compound 7b started with piperidene 33 that was prepared according to previously reported method.27,28 Deprotection of 33 with a catalytic amount of p-TsOH yielded diol intermediate 34 that was then regioselectively benzylated using boronic amide as catalyst29 (Scheme 2). After O-silylation and N-Boc deprotection, we obtained free amine 37 that was successfully coupled with triazole using triphosgene. Finally, desylilation (HF-pyridine) gave target compound 7b. Compound 7a (diastereoisomer of 7b) was obtained in the same fashion (see ESI‡).
 | ||
| Scheme 2 Reagents and conditions: (a) cat. p-TsOH, MeOH, 86%; (b) BnBr, K2CO3, KI, MeCN, 60 °C, 89%; (c) TBS-Cl, imidazole, DMF, 95%; (d) 10% TFA, DCM, 0 °C, 69%. (e) DIPEA, triphosgene, THF, 0 °C; (f) DIPEA, DMAP, 1,2,3-triazole, THF, 60 °C; (g) HF-pyridine, THF:pyridine = 1:1 (v/v), 15% over 3 steps. | ||
Compounds 9–17 were prepared by triphosgene-mediated condensation of free amine 26 with the appropriate heterocycle. As an example, heterocycle 17 (Scheme 1) was synthesized by coupling of 26 with 3-bromo-1H-1,2,4-triazole followed by Suzuki coupling with (1,1′-biphenyl)-4-ylboronic acid (Scheme 1). Finally, the para-nitrophenyl carbamate derivative 18 was prepared following a strategy as followed for heterocycle 5a with 4-nitrophenol instead of 4-([1,1′-biphenyl]-4-yl)-1H-1,2,3-triazole (see ESI‡).
Biological evaluation
The potency of 3, 4a–7a, 4b–7b, 6c, 8 and 9–18, as DAGLα inhibitors was established in a colorimetric assay using para-nitrophenylbutyrate as a surrogate substrate and membrane fractions from HEK293T cells overexpressing recombinant human DAGLα. As a reference, we report the biochemical data of (R)-KT109 (2a),30 which we established to be more potent than its enantiomer, (S)-KT109 (2b).30 The same stereochemistry at the C-2 position was preferred for compounds tested (e.g. compare compounds 4a–6avs.4b–6b). A 30–100-fold drop in potency of benzyloxy-containing compounds (3 and 5a) was found. This may suggest that a lipophilic pocket in DAGLα, which accommodates the 2-benzylpiperidine moiety, is restricted in size or, alternatively, that a polar, flexible linker is less preferred. Introduction of polar hydroxyl groups at other positions in the unsaturated piperidines (e.g.4avs.8) also reduced the activity over 20-fold. Of note, introduction of a chiral hydroxyl group at the C-3 position of an unsaturated piperidine ring (compounds 7a and 7b) abolished the activity against DAGLα (pIC50 < 5), whereas a hydroxyl at the C-5 position (compounds 6a and 6c) was allowed. This suggests that the position of the chiral hydroxyl group plays an important role in the binding site of DAGLα. However, a change in conformation of the piperidine ring induced by the double bond can also not be excluded to be responsible for the decrease in potency. Of note, the stereochemistry of the chiral hydroxyl at the C-5 position (6avs.6c) is not important for DAGL activity, which may suggest that this functional group does not make any significant interaction in the binding pocket and may protrude into a solvent exposed region. Compounds 10–12 were equally potent as compound 5a, but 9 showed ∼10-fold less activity. The pyrazoles (13 and 15), imidazoles (14 and 16), 1,2,4-triazole (17) and carbamate (18) were inactive. This is in line with a reduced electrophilicity of their warhead imparted by the heterocycle.15To screen derivatives 3, 4a–7a, 4b–7b, 6c, 8 and 9–18 for ABHD6 inhibitory activity, we employed a real-time, fluorescence-based natural substrate assay with membranes from HEK293T cells expressing recombinant human ABHD6. In general, the inhibitory potency of the compounds followed the same trend as observed for DAGLα inhibition (Tables 1 and 2). To compare the DAGLα and ABHD6 activities of the compounds, we plotted their pIC50 values against both targets (Fig. 3). Most of the compounds were dual DAGLα/ABHD6 inhibitors and a linear relationship (r2 = 0.85) for the potency was observed. Compounds 6b, 7a and 7b were inactive against DAGLα, but still showed inhibition against ABHD6 (pIC50 > 6). Therefore, these compounds could be interesting starting points for the discovery of selective ABHD6 inhibitors.
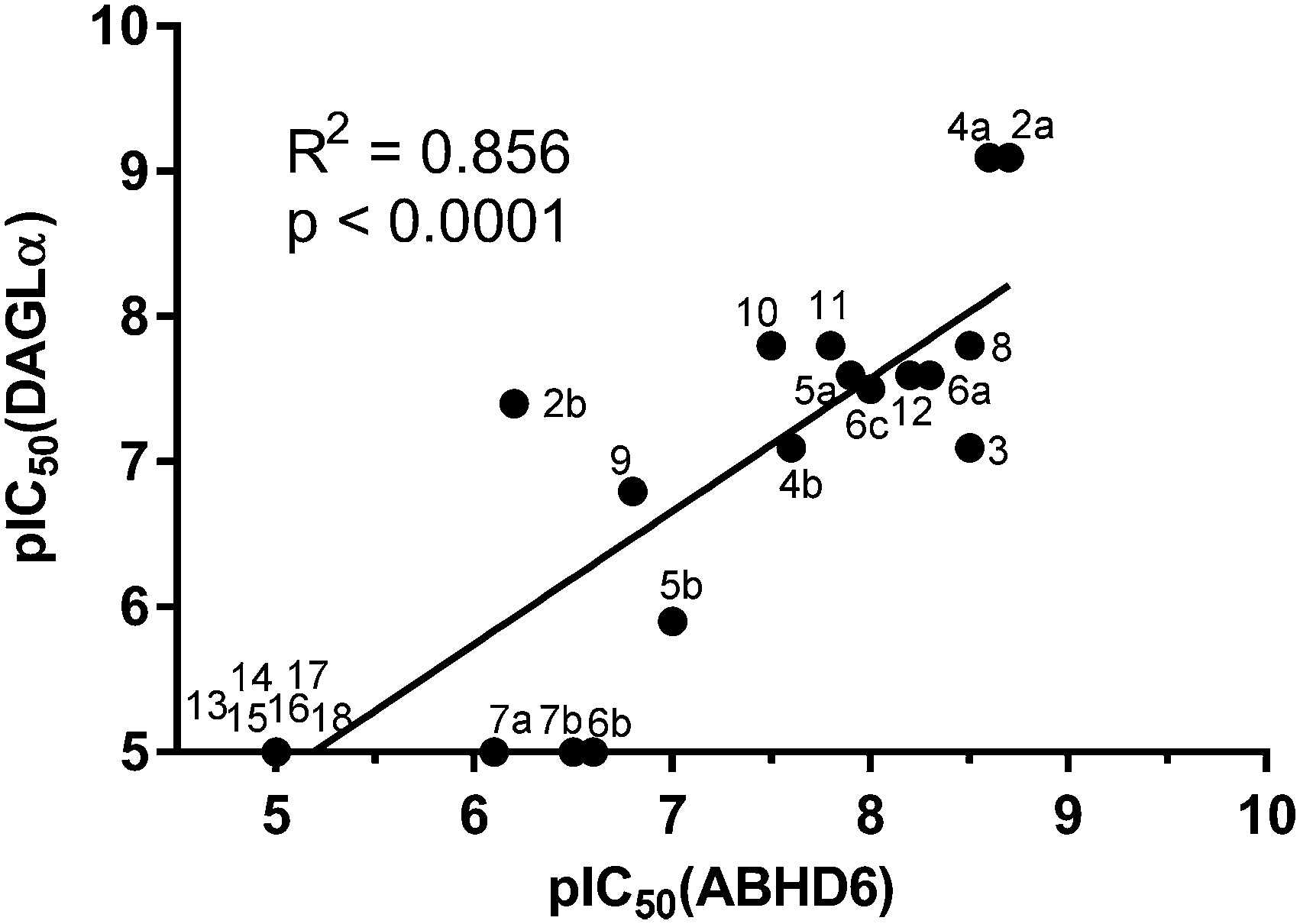 | ||
| Fig. 3 Graphical representation of DAGLα versus ABHD6 inhibition (pIC50) compounds 3, 4a–7a, 4b–7b, 6c, 8 and 9–18. | ||
Finally, to evaluate the selectivity of compounds 3, 4a–7a, 4b–7b, 6c, 8 and 9–18 across a broad panel of serine hydrolases, we applied activity-based protein profiling (ABPP) using mouse brain membrane proteome. Fluorophosphonate (FP)-based probes are routinely used in competitive ABPP experiments to determine the selectivity of serine hydrolase inhibitors.31,32 However, FP-based probes do not label DAGLα. MB064, a Bodipy-tagged tetrahydrolipstatin based β-lactone probe, was therefore previously developed by our group, to detect endogenous DAGLα in brain proteomes.31 Thus, we applied both TAMRA-FP and MB064 to assess the activity and selectivity of our dual DAGLα and ABHD6 inhibitors. In brief, we incubated inhibitors 3, 4a–7a, 4b–7b, 6c, 8 and 9–18 at 10 μM for 30 min with mouse brain membrane homogenates and performed a gel-based ABPP assay using MB064 (0.25 μM, 20 min) or TAMRA-FP (0.5 μM, 20 min). Almost complete blockade of DAGLα and ABHD6 was observed by compounds 3, 4a–6a, 4b, 6c and 8–12, which is consistent with the results of the biochemical assay (Fig. 4a and Table 3). Most compounds showed excellent selectivity over the other serine hydrolases (Fig. 4). Compounds 3, 5a, 6c and 9–12 did, however, reduce the labeling of DDHD2 (Fig. 4a), while compounds 6c, 9 and 10 were non-selective and prevented the labelling of several unknown off-targets (Fig. 4a and b).
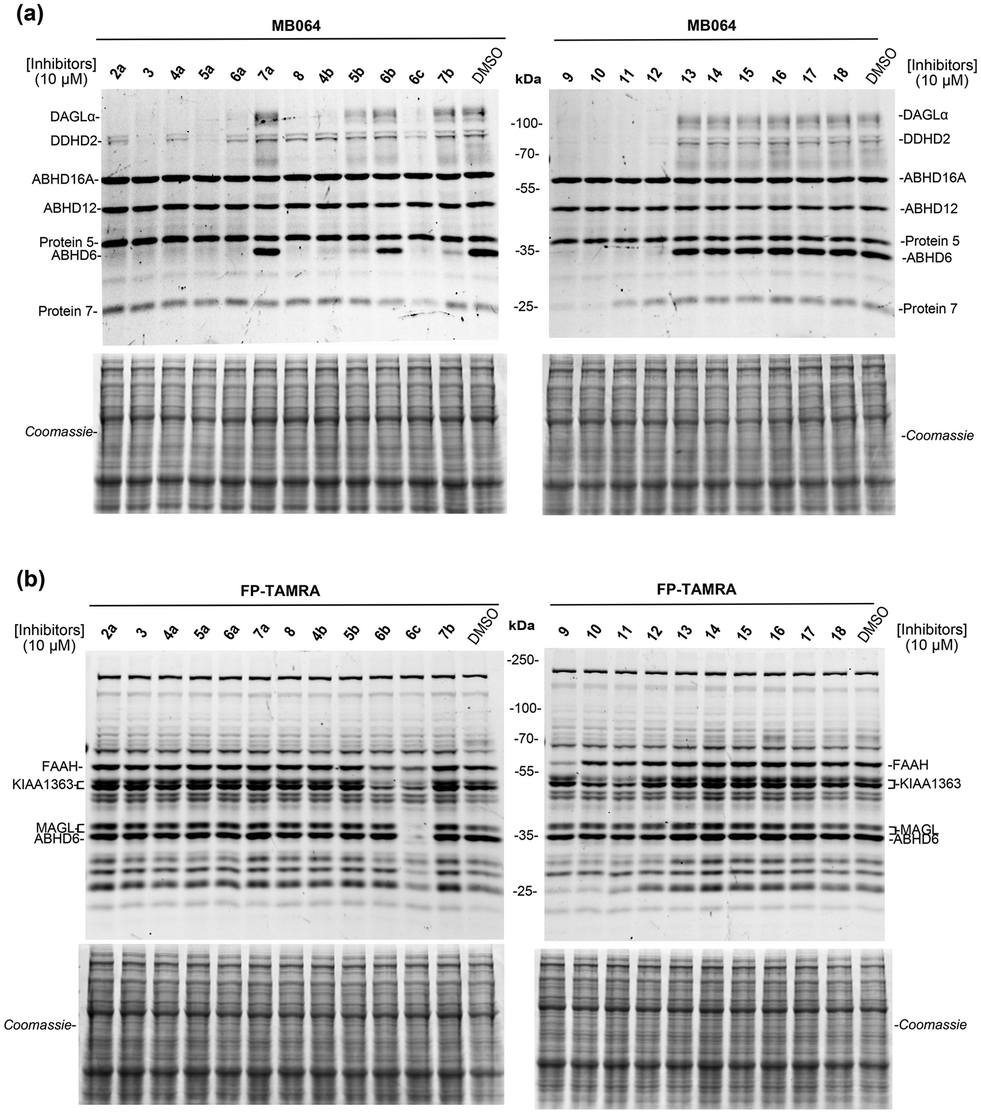 | ||
| Fig. 4 (a and b) Selectivity profile of compounds 2a, 3, 4a–7a, 4b–7b, 6c, 8 and 9–18 (10 μM, 30 min) across mouse brain membrane serine hydrolases as determined by competitive ABPP using two broad-spectrum probes MB064 (0.25 μM, 20 min) (a) and FP-TAMRA (0.5 μM, 20 min) (b). Coomassie staining gel were used as a loading control. | ||
| Entry | Inhibition (%) | Entry | Inhibition (%) | ||
|---|---|---|---|---|---|
| DAGLα | ABHD6 | DAGLα | ABHD6 | ||
| 2a | 99 ± 0 | 95 ± 1 | 7b | 18 ± 11 | 83 ± 2 |
| 3 | 96 ± 1 | 96 ± 0 | 9 | 89 ± 5 | 89 ± 2 |
| 4a | 99 ± 0 | 96 ± 1 | 10 | 95 ± 2 | 93 ± 1 |
| 5a | 94 ± 2 | 96 ± 0 | 11 | 93 ± 3 | 94 ± 1 |
| 6a | 87 ± 5 | 95 ± 2 | 12 | 93 ± 3 | 86 ± 2 |
| 7a | 6 ± 13 | 18 ± 9 | 13 | −3 ± 5 | −3 ± 4 |
| 8 | 92 ± 2 | 96 ± 2 | 14 | −9 ± 15 | −15 ± 15 |
| 4b | 83 ± 5 | 90 ± 3 | 15 | −14 ± 18 | −3 ± 19 |
| 5b | 30 ± 12 | 84 ± 5 | 16 | −13 ± 14 | −28 ± 15 |
| 6b | 5 ± 14 | 50 ± 7 | 17 | 0 ± 12 | −16 ± 9 |
| 6c | 85 ± 2 | 95 ± 2 | 18 | −11 ± 12 | −12 ± 9 |
Conclusions
We reported the enantioselective synthesis and structure–activity relationship studies of chiral, disubstituted piperidinylureas as dual inhibitors of DAGLα and ABHD6. The SAR studies revealed the stereochemistry of the C-2 substitution on the piperidine ring plays an important role. Incorporation of a hydroxyl group at the C-5 position on piperidine ring maintained the activity against DAGLα and ABHD6, whereas a hydroxyl at the C-3 position completely abolished all DAGLα activity. Competitive activity-based protein profiling confirmed the activity of the inhibitors against endogenous DAGLα and ABHD6 and revealed differences in the selectivity profile against other serine hydrolases.Experimental section
Synthesis of compound
The synthesis and characterization of all final compounds 3, 4a–7a, 4b–7b, 6c, 8, 9–18 and intermediates are described in the ESI.‡Biological assays
:1 mixture of polyethyleneimine (60 μg) and plasmid DNA (20 μg) in 2 mL serum free medium. The medium was refreshed after 24 hours, and after 72 h the cells were harvested by suspending them in 20 mL medium. The suspension was centrifuged for 10 min at 1000 rpm, and the supernatant was removed. The cell pellet was stored at −80 °C until use.
Cell pellets were thawed on ice and suspended in lysis buffer A (20 mM HEPES, 2 mM DTT, 0.25 M sucrose, 1 mM MgCl2, 25 U mL−1 Benzonase). The suspension was homogenized by polytrone (3 × 7 s) and incubated for 30 min on ice. The suspension was subjected to ultracentrifugation (93.000 × g, 30 min, 4 °C, Beckman Coulter, Type Ti70 rotor) to yield the cytosolic fraction in the supernatant and the membrane fraction as a pellet. The pellet was resuspended in lysis buffer B (20 mM HEPES, 2 mM DTT). The protein concentration was determined with Quick Start Bradford reagent (BioRad) or Qubit™ fluorometric quantitation (Life Technologies). The protein fractions were diluted to a total protein concentration of 1 mg mL−1 and stored in small aliquots at −80 °C until use.
000 × g, 45 min, 4 °C, Beckman Coulter, Type Ti70 rotor) to yield the cytosolic fraction in the supernatant and the membrane fraction as a pellet. The pellet was resuspended in storage buffer B (20 mM HEPES pH 7.2, 2 mM DTT). The total protein concentration was determined with Quick Start Bradford reagent (Bio-Rad) or Qubit™ fluorometric quantitation (Life Technologies). Membranes and supernatant were flash frozen in liquid nitrogen and stored in aliquots at −80 °C until use.
Acknowledgements
This work was supported by grants from the Chinese Scholarship Council (to H. D.); the Netherlands Research Council – Chemical Sciences ECHO grant (to M. v. d. S.); and an ECHO-STIP Grant (M. v. d. S.).Notes and references
- T. Bisogno, F. Howell, G. Williams, A. Minassi, M. G. Cascio, A. Ligresti, I. Matias, A. Schiano-Moriello, P. Paul, E. J. Williams, U. Gangadharan, C. Hobbs, V. Di Marzo and P. Doherty, J. Cell Biol., 2003, 163, 463–468 CrossRef CAS PubMed.
- M. Reisenberg, P. K. Singh, G. Williams and P. Doherty, Philos. Trans. R. Soc., B, 2012, 367, 3264–3275 CrossRef CAS PubMed.
- I. Katona and T. F. Freund, Annu. Rev. Neurosci., 2012, 35, 529–558 CrossRef CAS PubMed.
- Y. Gao, D. V. Vasilyev, M. B. Goncalves, F. V. Howell, C. Hobbs, M. Reisenberg, R. Shen, M. Y. Zhang, B. W. Strassle, P. Lu, L. Mark, M. J. Piesla, K. Deng, E. V. Kouranova, R. H. Ring, G. T. Whiteside, B. Bates, F. S. Walsh, G. Williams, M. N. Pangalos, T. A. Samad and P. Doherty, J. Neurosci., 2010, 30, 2017–2024 CrossRef CAS PubMed.
- A. Tanimura, M. Yamazaki, Y. Hashimotodani, M. Uchigashima, S. Kawata, M. Abe, Y. Kita, K. Hashimoto, T. Shimizu, M. Watanabe, K. Sakimura and M. Kano, Neuron, 2010, 65, 320–327 CrossRef CAS PubMed.
- D. K. Nomura, B. E. Morrison, J. L. Blankman, J. Z. Long, S. G. Kinsey, M. C. G. Marcondes, A. M. Ward, Y. K. Hahn, A. H. Lichtman, B. Conti and B. F. Cravatt, Science, 2011, 334, 809–813 CrossRef CAS PubMed.
- R. A. Kohnz and D. K. Nomura, Chem. Soc. Rev., 2014, 43, 6859–6869 RSC.
- C. A. Rouzer and L. J. Marnett, Chem. Rev., 2011, 111, 5899–5921 CrossRef CAS PubMed.
- R. A. Kohnz and D. K. Nomura, Chem. Soc. Rev., 2014, 43, 6859–6869 RSC.
- G. G. Muccioli, Drug Discovery Today, 2010, 15, 474–483 CrossRef CAS PubMed.
- M. P. Baggelaar, F. J. Janssen, A. C. M. van Esbroeck, H. den Dulk, M. Allara, S. Hoogendoorn, R. McGuire, B. I. Florea, N. Meeuwenoord, H. van den Elst, G. A. van der Marel, J. Brouwer, V. Di Marzo, H. S. Overkleeft and M. van der Stelt, Angew. Chem., Int. Ed., 2013, 52, 12081–12085 CrossRef CAS PubMed.
- M. P. Baggelaar, P. J. P. Chameau, V. Kantae, J. Hummel, K. L. Hsu, F. Janssen, T. van der Wel, M. Soethoudt, H. Deng, H. den Dulk, M. Allara, B. I. Florea, V. Di Marzo, W. J. Wadman, C. G. Kruse, H. S. Overkleeft, T. Hankemeier, T. R. Werkman, B. F. Cravatt and M. van der Stelt, J. Am. Chem. Soc., 2015, 137, 8851–8857 CrossRef CAS PubMed.
- F. J. Janssen, M. P. Baggelaar, J. J. A. Hummel, H. S. Overkleeft, B. F. Cravatt, D. L. Boger and M. van der Stelt, J. Med. Chem., 2015, 58, 9742–9753 CrossRef CAS PubMed.
- F. J. Janssen, H. Deng, M. P. Baggelaar, M. Allara, T. van der Wel, H. den Dulk, A. Ligresti, A. C. M. van Esbroeck, R. McGuire, V. Di Marzo, H. S. Overkleeft and M. van der Stelt, J. Med. Chem., 2014, 57, 6610–6622 CrossRef CAS PubMed.
- D. Ogasawara, H. Deng, A. Viader, M. P. Baggelaar, A. Breman, H. den Dulk, A. M. van den Nieuwendijk, M. Soethoudt, T. van der Wel, J. Zhou, H. S. Overkleeft, M. Sanchez-Alavez, S. Mo, W. Nguyen, B. Conti, X. Liu, Y. Chen, Q. S. Liu, B. F. Cravatt and M. van der Stelt, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 26–33 CrossRef CAS PubMed.
- W. R. Marrs, J. L. Blankman, E. A. Horne, A. Thomazeau, Y. H. Lin, J. Coy, A. L. Bodor, G. G. Muccioli, S. S. J. Hu, G. Woodruff, S. Fung, M. Lafourcade, J. P. Alexander, J. Z. Long, W. W. Li, C. Xu, T. Moller, K. Mackie, O. J. Manzoni, B. F. Cravatt and N. Stella, Nat. Neurosci., 2010, 13, 951–967 CrossRef CAS PubMed.
- M. A. Pribasnig, I. Mrak, G. F. Grabner, U. Taschler, O. Knittelfelder, B. Scherz, T. O. Eichmann, C. Heier, L. Grumet, J. Kowaliuk, M. Romauch, S. Holler, F. Anderl, H. Wolinski, A. Lass, R. Breinbauer, G. Marsche, J. M. Brown and R. Zimmermann, J. Biol. Chem., 2015, 290, 29869–29881 CrossRef CAS PubMed.
- G. Thomas, J. L. Betters, C. C. Lord, A. L. Brown, S. Marshall, D. Ferguson, J. Sawyer, M. A. Davis, J. T. Melchior, L. C. Blume, A. C. Howlett, P. T. Ivanova, S. B. Milne, D. S. Myers, I. Mrak, V. Leber, C. Heier, U. Taschler, J. L. Blankman, B. F. Cravatt, R. G. Lee, R. M. Crooke, M. J. Graham, R. Zimmermann, H. A. Brown and J. M. Brown, Cell Rep., 2013, 5, 508–520 CrossRef CAS PubMed.
- A. Fisette, S. Tobin, L. Decarie-Spain, K. Bouyakdan, M. L. Peyot, S. R. Madiraju, M. Prentki, S. Fulton and T. Alquier, Cell Rep., 2016, 17, 1217–1226 CrossRef CAS PubMed.
- F. Tchantchou and Y. M. Zhang, J. Neurotrauma, 2013, 30, 565–579 CrossRef PubMed.
- K. L. Hsu, K. Tsuboi, A. Adibekian, H. Pugh, K. Masuda and B. F. Cravatt, Nat. Chem. Biol., 2012, 8, 999–1007 CrossRef CAS PubMed.
- K. L. Hsu, K. Tsuboi, L. R. Whitby, A. E. Speers, H. Pugh, J. Inloes and B. F. Cravatt, J. Med. Chem., 2013, 56, 8257–8269 CrossRef CAS PubMed.
- A. M. van den Nieuwendijk, M. Ruben, S. E. Engelsma, M. D. Risseeuw, R. J. van den Berg, R. G. Boot, J. M. Aerts, J. Brussee, G. A. van der Marel and H. S. Overkleeft, Org. Lett., 2010, 12, 3957–3959 CrossRef CAS PubMed.
- A. M. C. H. van den Nieuwendijk, R. J. B. H. N. van den Berg, M. Ruben, M. D. Witte, J. Brussee, R. G. Boot, G. A. van der Marel, J. M. F. G. Aerts and H. S. Overkleeft, Eur. J. Org. Chem., 2012, 3437–3446 CrossRef CAS.
- J. B. Jiang, M. Artola, T. J. M. Beenakker, S. P. Schroder, R. Petracca, C. de Boer, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codee and H. S. Overkleeft, Eur. J. Org. Chem., 2016, 3671–3678 CrossRef CAS.
- Y. Banba, C. Abe, H. Nemoto, A. Kato, I. Adachi and H. Takahata, Tetrahedron: Asymmetry, 2001, 12, 817–819 CrossRef CAS.
- P. Garner, J. M. Park and E. Malecki, J. Org. Chem., 1988, 53, 4395–4398 CrossRef CAS.
- H. Takahata, Y. Banba, H. Ouchi and H. Nemoto, Org. Lett., 2003, 5, 2527–2529 Search PubMed.
- D. Lee, C. L. Williamson, L. N. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260–8267 CrossRef CAS PubMed.
- H. Deng, S. Kooijman, A. M. van den Nieuwendijk, D. Ogasawara, T. van der Wel, F. van Dalen, M. P. Baggelaar, F. J. Janssen, R. J. B. H. N. van den Berg, H. den Dulk, B. F. Cravatt, H. S. Overkleeft, P. C. Rensen and M. van der Stelt, J. Med. Chem., 2017, 60, 428–440 CrossRef CAS PubMed.
- H. S. Hoover, J. L. Blankman, S. Niessen and B. F. Cravatt, Bioorg. Med. Chem. Lett., 2008, 18, 5838–5841 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- T. van der Wel, F. J. Janssen, M. P. Baggelaar, H. Deng, H. den Dulk, H. S. Overkleeft and M. van der Stelt, J. Lipid Res., 2015, 56, 927–935 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7md00029d |
| This journal is © The Royal Society of Chemistry 2017 |