
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and in vitro study of novel borneol derivatives as potent inhibitors of the influenza A virus†‡
A. S.
Sokolova
 *ab,
O. I.
Yarovaya
ab,
M. D.
Semenova
a,
A. A.
Shtro
c,
I. R.
Orshanskaya
c,
V. V.
Zarubaev
c and
N. F.
Salakhutdinov
ab
*ab,
O. I.
Yarovaya
ab,
M. D.
Semenova
a,
A. A.
Shtro
c,
I. R.
Orshanskaya
c,
V. V.
Zarubaev
c and
N. F.
Salakhutdinov
ab
aNovosibirsk Institute of Organic Chemistry, Siberian Branch of the Russian Academy of Sciences, Lavrentjev Avenue 9, 630090 Novosibirsk, Russia. E-mail: asokolova@nioch.nsc.ru
bNovosibirsk State University, Pirogova St. 2, 630090 Novosibirsk, Russia
cLaboratory of Chemotherapy, Influenza Research Institute, Prof. Popova St. 15/17, 197376 St. Petersburg, Russia
First published on 3rd March 2017
Abstract
Herein, we present the design and synthesis of a series of novel heterocyclic derivatives of (−)-borneol and (−)-isoborneol as potent inhibitors of the influenza A virus. All compounds were tested for their toxicity against MDCK cells and for virus-inhibiting activity against the influenza virus A/Puerto Rico/8/34 (H1N1). Compounds 7, 16 and 26 containing a morpholine fragment exhibited the highest efficiency as agents inhibiting the replication of the influenza virus A(H1N1) with selectivity indices of 82, 45 and 65, correspondingly. Derivatives 9 (SI = 23) and 18 (SI = 25) containing a 1-methylpiperazine motif showed moderate antiviral activity. Structure–activity analysis of this new series of borneol derivatives revealed that a 1,7,7-trimethylbicyclo[2.2.1]heptan scaffold is required for the antiviral activity.
Introduction
Influenza A viruses are important pathogens which are capable of causing significant morbidity and mortality in humans. The influenza virus is a negative-sense segmented single-strand RNA-genome virus belonging to the Orthomyxoviridae family. Two main approaches, vaccination and chemotherapy, are used to control influenza in humans. Due to the short life cycle and lack of error-correcting activity of polymerases, influenza viruses are able to rapidly evolve. On the one hand, this results in escape from immune response and antigenic drift that, in turn, makes necessary the annual checking of the efficacy of the vaccine strain composition against circulating viruses. On the other hand, drug-resistant strains of the influenza virus can quickly emerge and spread. In particular, since the mid-1990s, mutation of resistance to adamantane derivatives (amantadine and rimantadine) has emerged, and currently the vast majority of influenza isolates are rimantadine-resistant.1,2 In 2009, a novel triple reassortant influenza A(H1N1)pdm09 virus of swine origin, which was also naturally resistant to adamantanes, emerged and quickly spread worldwide.3 Similarly, oseltamivir-resistant H1N1 viruses appeared in 2007 and spread worldwide with almost 100% drug resistance in 2008/2009 (ref. 4–6) due to mutations in the NA gene.7 Drug resistance substantially restricts the use of antivirals and makes necessary the search for and development of novel virus-inhibiting compounds with alternative targets and mechanisms of activity.Borneol, a bicyclic monoterpenoid alcohol, exists as two enantiomers, the D and L forms. Both the borneol forms occur in the essential oils of numerous medicinal plants, such as valerian (Valeriana officinalis), chamomile (Matricaria chamomilla) and lavender (Lavandula officinalis). Generally, there are two isomers of borneol: borneol and isoborneol, which vary with the location of the hydroxyl group. These substances have a broad spectrum of biological activity. Recently, it has been established that isoborneol shows antiviral activity against herpes simplex virus type 1,7 and both enantiomers of borneol were found to have a highly efficacious positive modulating action at mammalian GABA (γ-aminobutyric acid) inhibitory neurotransmission receptors.8 In addition, borneol shows inhibitory effects on several Gr (−) and Gr (+) pathogenic microorganisms.9
It can be assumed that cage compounds like borneol derivatives containing a 1,7,7-trimethylbicyclo[2.2.1]heptan scaffold have high potential in the synthesis of antiviral agents. Our previous works discovered this scaffold as a promising anti-influenza agent.10–12 The target compound was identified as an inhibitor of the H1N1 influenza virus with a selectivity index (SI) value of 500; this camphor derivative was called camphecene (Fig. 1).13 In contrast to deitiforine that targets the M2 proton channel, camphecene was shown to directly inhibit the acid-induced membrane-disrupting activity of the viral hemagglutinin of influenza A viruses.14 Moreover there are other cage compounds with antiviral activity, for example amino camphor derivatives were identified as a new class of M2 inhibitors with moderate activity.15 Also, compounds based on 1- and 2-adamantylamines exhibited high M2 inhibitor activity.16,17 The ability to inhibit the influenza virus H3N2 was found in amino and imine derivatives with a pinene scaffold;18 compounds containing a pinanamine scaffold with secondary amine and imidazole may increase inhibition of A/M2 channel activity.19
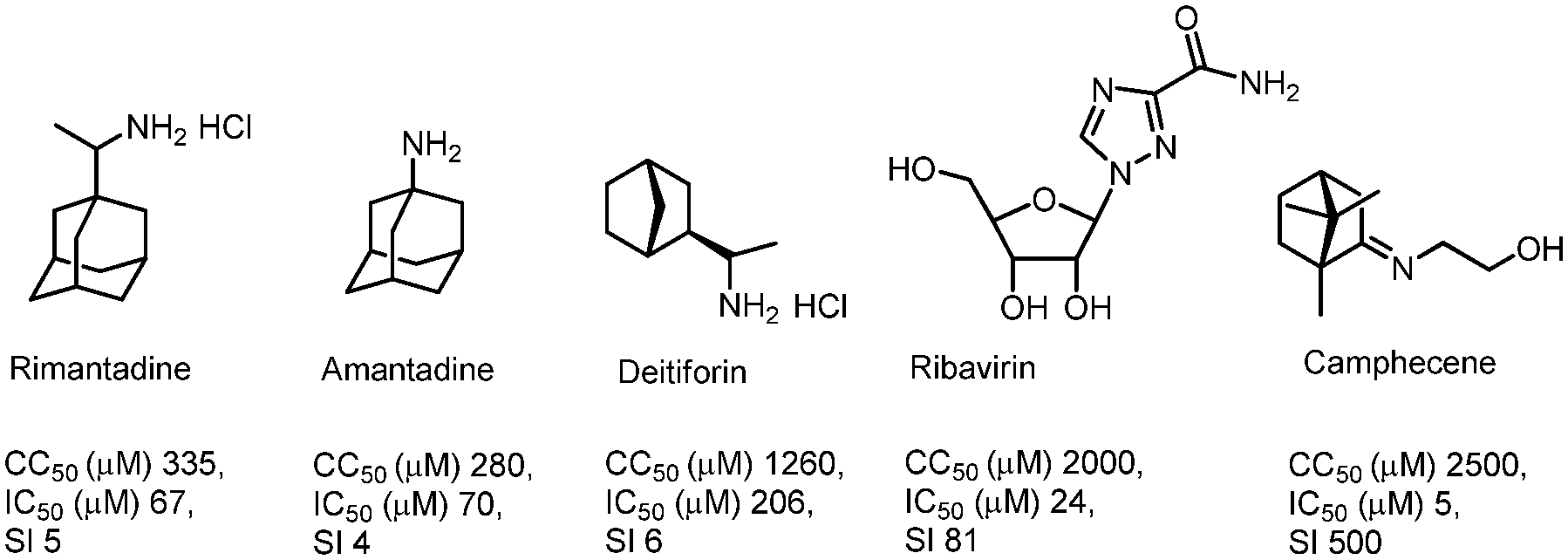 | ||
| Fig. 1 Structural formula and anti-influenza A(H1N1)pdm09 activity of anti-influenza drugs and the camphor derivative (CC50 – the median cytotoxic dose; IC50 – the concentration causing inhibition of 50% of virus activity; SI – the selectivity index). | ||
In view of the above, in the present work, several series of borneol derivatives containing a cage structure based on the 1,7,7-trimethylbicyclo[2.2.1]heptan scaffold were synthesized and tested against the influenza virus. According to the literature data, the general direction in the synthesis of biologically active borneol derivatives includes preparation of different borneol esters. These esters were shown to be inhibitors of cyclooxygenase enzymes20 and the replication of coxsackievirus B3. Also, bornyl ester derivatives showed cytotoxic activity against tumor cell lines.21,22 Therefore in this work the general approach to the synthesis of the target compounds included synthesis of different borneol esters containing various heterocycles. Nitrogen heterocycles are attractive structural units in medicinal chemistry. There are a lot of highly effective drugs with the active ingredient containing nitrogen heterocyclic fragments, such as piperazine, piperidine and morpholine.23 In our search for antiviral agents, we screened several different nitrogen-containing heterocyclic fragments for their ability to inhibit reproduction. Moreover, we studied the influence of the length of the aliphatic linker between the ester group and the heterocyclic scaffold.
Results and discussion
Chemistry
The borneol derivatives were prepared according to the synthetic route as outlined in Scheme 1. In our work, we use (−)-borneol 1, obtained from natural materials by the method described in a patent.24 Reaction between the (−)-borneol and chlorides, namely, α or β chlorinated acids, led to esters 2 and 3, respectively. By nucleophilic substitution reaction which involved different nitrogen-containing nucleophiles, (1S,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl 2-chloroacetate 2 and (1S,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl 3-chloropropanoate 3 were transformed into target compounds 4–21. The compounds were obtained in good yields ranging from 52% to 91%.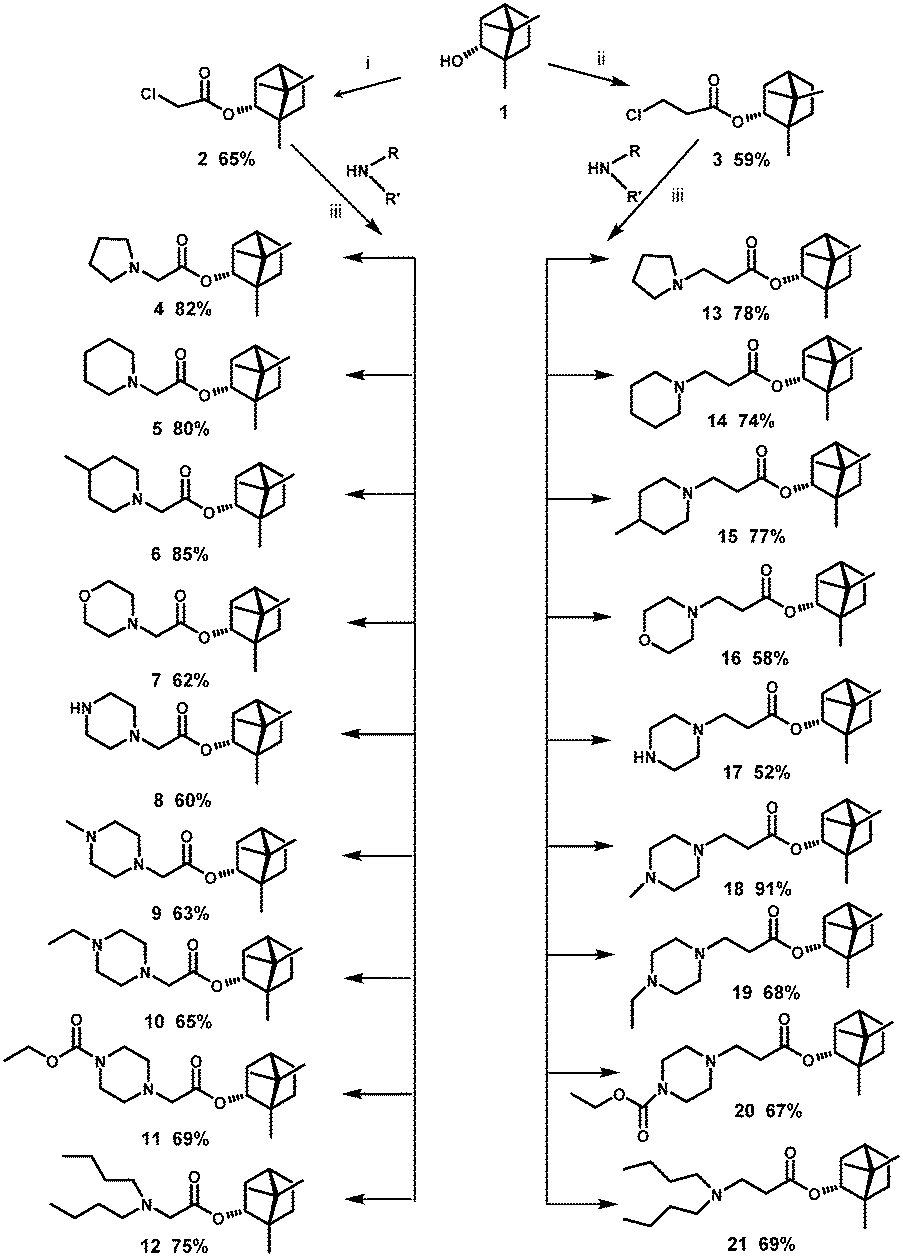 | ||
| Scheme 1 Reagents and conditions: (i) 2-chloroacetyl chloride, Et3N, CH2Cl2 (dry), 25 °C; (ii) 3-chloropropanoyl chloride, Et3N, CH2Cl2 (dry), 25 °C; (iii) Et3N, CH2Cl2 (dry), 25 °C. | ||
It is well recognised that the stereochemistry of molecules is often crucial in deriving a SAR. The next step in borneol-based SAR analysis was using as the starting materials (−)-isoborneol 22 with different stereochemistries of the hydroxyl group. Compounds 7 and 16 with a morpholine fragment showed the highest antiviral activity, so we synthesised analogues 23 and 24 based on (−)-isoborneol (Scheme 2).
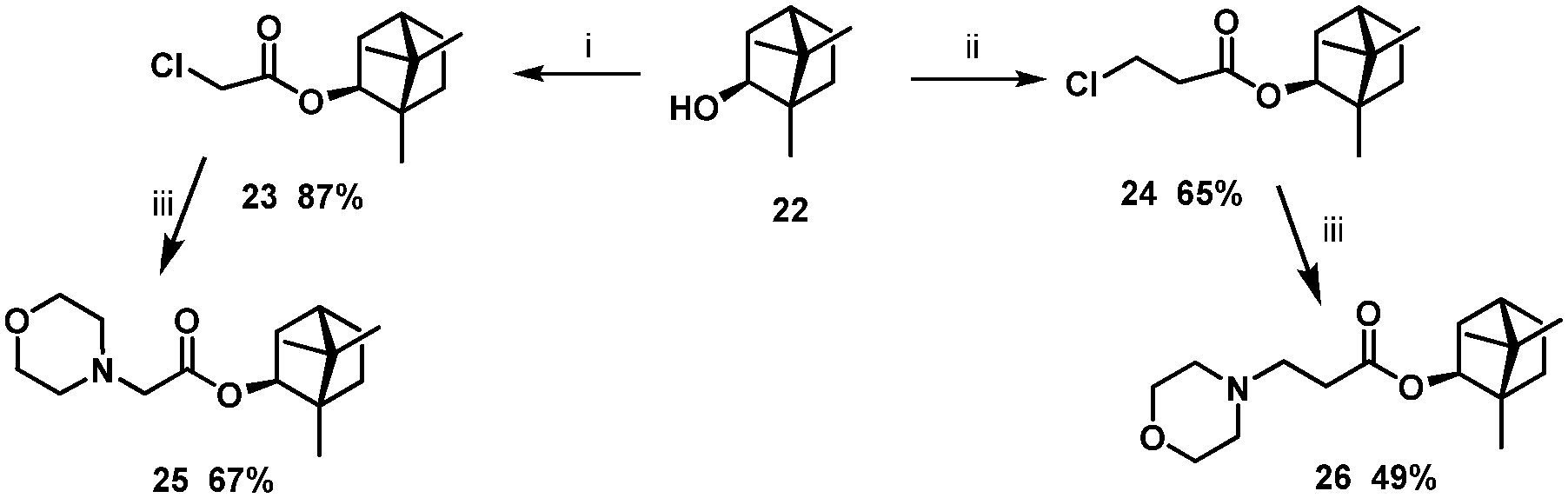 | ||
| Scheme 2 Reagents and conditions: (i) 2-chloroacetyl chloride, Et3N, CH2Cl2 (dry), 25 °C; (ii) 3-chloropropanoyl chloride, Et3N, CH2Cl2 (dry), 50 °C; (iii) Et3N, CH2Cl2 (dry), 25 °C. | ||
To establish the key structural requirements for antiviral activity, we synthesized derivatives 27 (ref. 25) and 28 (ref. 26) without the 1,7,7-trimethylbicyclo[2.2.1]heptan fragment (Fig. 2). As the nitrogen-containing nucleophile, we chose morpholine because borneol derivatives 7 and 16 with the morpholine fragment showed considerable antiviral activity.
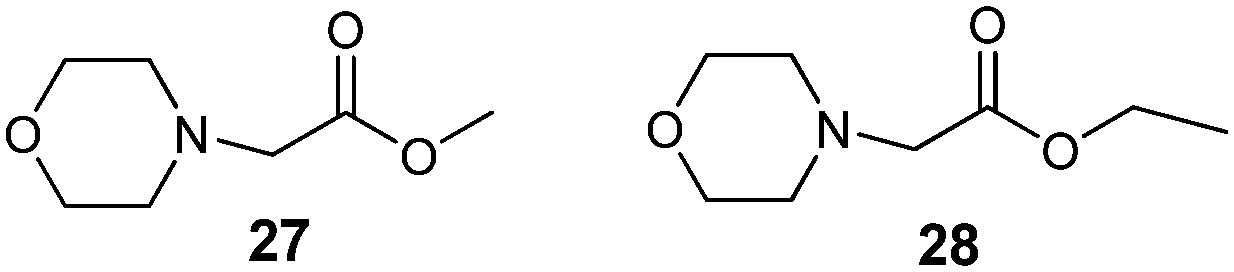 | ||
| Fig. 2 Compounds without the 1,7,7-trimethylbicyclo[2.2.1]heptan fragment. | ||
The structures of the new, synthesized compounds were confirmed by means of the NMR spectra (1H, 13C NMR) and HR-MS and IR spectra. The 1H NMR spectra of compounds 13–21 and 26 revealed upfield signals of the methylene protons of N–CH2 at δ 2.6–2.8 ppm in contrast to the protons of the starting compound 3 at δ 3.74 ppm. Also, in the 1H NMR spectra of compounds 4–12 and 25 an upfield singlet at δ 3.1 ppm was observed in comparison with the singlet of compound 2 that was revealed at δ 4.03 ppm.
Study of antiviral activity
The obtained compounds 4–21, 25, and 26 were studied as potential antiviral agents (Table 1).| Compound | CC50, μM | IC50, μM | SI | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pa Pa |
|---|---|---|---|---|
|
a logP, the octanol–water partitioning coefficient, was calculated using ACDLabs/ChemSketch 12.01 (http://www.acdlabs.com).
All values are presented as the mean ± SD from at least three independent experiments.
|
||||
| 1 | >2142.9 | 155.8 ± 18.3 | >14 | 2.71 |
| 4 | 301.9 ± 21.4 | 34.0 ± 4.2 | 9 | 4.29 |
| 5 | 137.6 ± 9.9 | 15.1 ± 1.0 | 9 | 4.85 |
| 6 | 93.9 ± 7.3 | 10.2 ± 1.4 | 9 | 5.34 |
| 7 | 580.1 ± 41.1 | 7.1 ± 0.8 | 82 | 3.53 |
| 8 | 102.9 ± 8.2 | 7.1 ± 0.5 | 14 | 3.68 |
| 9 | 100.0 ± 7.3 | 4.4 ± 0.5 | 23 | 3.26 |
| 10 | 51.6 ± 4.1 | 3.2 ± 0.4 | 16 | 3.79 |
| 11 | >852.0 | 683.1 ± 62.8 | 1 | 4.11 |
| 12 | 69.3 ± 4.9 | >34.1 | >2 | 6.78 |
| 13 | 124.7 ± 10.1 | 14.3 ± 1.6 | 9 | 4.45 |
| 14 | 69.6 ± 5.2 | 7.8 ± 0.8 | 9 | 5.02 |
| 15 | 56.4 ± 3.8 | 15.3 ± 2.0 | 4 | 5.51 |
| 16 | 552.5 ± 31.1 | 12.2 ± 1.6 | 45 | 3.36 |
| 17 | 12.6 ± 1.0 | 6.8 ± 0.9 | 2 | 3.51 |
| 18 | 48.1 ± 3.3 | 1.9 ± 0.4 | 25 | 3.09 |
| 19 | 104.7 ± 8.7 | 8.4 ± 0.9 | 12 | 3.62 |
| 20 | 149.5 ± 11.7 | >98.4 | 2 | 3.94 |
| 21 | 41.8 ± 2.2 | 7.1 ± 0.9 | 6 | 6.94 |
| 22 | >974 | >974 | 1 | 2.71 |
| 25 | >533.8 | 73.7 ± 8.5 | 7 | 3.53 |
| 26 | 508.5 ± 36.1 | 7.8 ± 1.1 | 65 | 3.36 |
| 27 | >1886.8 | >1886.8 | 1 | 0.11 |
| 28 | >1734.1 | 982.7 ± 75.3 | >2 | 0.64 |
| Rimantadine | 335.2 ± 26.8 | 67.0 ± 4.9 | 5 | |
| Amantadine | 284.1 ± 21.4 | 64.2 ± 4.7 | 4 | |
| Deitiforine | 1266.2 ± 81.5 | 208.6 ± 15.4 | 6 | |
| Ribavirin | >2000.0 | 24.6 ± 3.1 | >81.0 | |
| Oseltamivir | 160.3 ± 11.4 | 0.2 ± 0.03 | 781 | |
Adamantane- and norbornane-based derivatives were used as reference compounds due to their close similarity to the compounds under investigation in having rigid cage fragments in their structures. To study the structure–activity relationship, we examined changes of the aliphatic linker and heterocyclic fragment in terms of their effect on toxicity and virus-inhibiting activity. In general, compounds with longer linkers (C2) were slightly more toxic compared to their C1 analogues. The piperazine derivatives 9 and 18 exhibited high antiviral activity with IC50 values of 4.4 ± 0.5 and 1.9 ± 0.4 respectively. Together, with potent activities, these compounds showed high toxicity which results in a relatively low therapeutic index. It was found that the morpholine substituent resulted in a strong reduction of toxicity compared to piperazine derivatives. This led to higher SIs, although the IC50 values of morpholine and piperazine derivatives did not differ (compounds 7 and 8, 16 and 17). Acylation of one of the nitrogen atoms of the piperazine moiety led to strongly decreased toxicity of the compound although its antiviral properties did not increase. The derivatives containing 1-methylpiperazine and 1-ethylpiperazine motifs showed moderate antiviral activity.
It was also demonstrated that in the case of a short linker, the stereochemistry of the compound does not affect the cytotoxicity of the compound but is of critical significance for anti-viral activity. Indeed, compounds 7 and 25 are based on two optical isomers of borneol, and their values of IC50 differ tenfold (7.1 and 73.7, correspondingly). This, however, was not important for two other isomers, 16 and 26, with a longer linker and which have similar values of both toxicity and activity and, therefore, similar SIs (45 and 65, respectively). This phenomenon should be further studied, in particular using computer simulation of ligand–target interaction.
Also, the structure–activity analysis of this new series of borneol derivatives revealed that the 1,7,7-trimethylbicyclo[2.2.1]heptan scaffold is required for the antiviral activity, as compounds 27 and 28 lacking this cage fragment did not possess virus-inhibiting activity. Among all the compounds tested, the highest activity was found in substances 7, 16 and 26, combining a morpholine fragment and 1,7,7-trimethylbicyclo[2.2.1]heptan (SI 82, 45, and 65 correspondingly).
Lipophilicity is a physicochemical property of principal importance in drug discovery and development.27 The quantitative descriptor of lipophilicity, the partition coefficient P, is defined as the ratio of the concentrations of a neutral compound in organic and aqueous phases under equilibrium conditions. To correlate the antiviral activity of the present series of compounds with their lipophilicity, logP values were calculated using ACDLabs/ChemSketch 12.01. All the new, examined compounds have the optimum lipophilicity range with a logP value of 3.09–3.79.
Conclusions
In summary, we presented a novel series of borneol derivatives containing different heterocyclic fragments. All of the compounds were investigated against the influenza virus A/Puerto Rico/8/34 (H1N1) in MDCK cells, and the structure–activity relationship was studied. Among these novel derivatives, compounds 7, 16 and 26 with 1,7,7-trimethylbicyclo[2.2.1]heptan and morpholine fragments were found to possess the highest efficacy in virus inhibition. Structure–activity analysis showed that an increase of linker length leads to enhancement of the toxicity. The results obtained suggest that a bulky lipophilic unit such as the 1,7,7-trimethylbicyclo[2.2.1]heptan scaffold was necessary for activity.Acknowledgements
This work has been supported by Russian Scientific Foundation grant 15-13-00017.References
- R. A. Bright, D. K. Shay, B. Shu, N. J. Cox and A. I. Klimov, JAMA, J. Am. Med. Assoc., 2006, 295, 891 CrossRef CAS PubMed.
- V. M. Deyde, X. Xu, R. A. Bright, M. Shaw, C. B. Smith, Y. Zhang, Y. Shu, L. V. Gubareva, N. J. Cox and A. I. Klimov, J. Infect. Dis., 2007, 196, 249 CrossRef CAS PubMed.
- F. S. Dawood, S. Jain, L. Finelli, M. W. Shaw, S. Lindstrom, R. J. Garten, L. V. Gubareva, X. Xu, C. B. Bridges and T. M. Uyeki, N. Engl. J. Med., 2009, 360, 2605 CrossRef PubMed.
- CDC, http://www.cdc.gov/flu/weekly/weeklyarchives2008-2009/weekly15.htm, 2009.
- N. J. Dharan, L. V. Gubareva, J. J. Meyer, M. Okomo-Adhiambo, R. C. McClinton, S. A. Marshall, K. St. George, S. Epperson, L. Brammer, A. I. Klimov, J. S. Bresee and A. M. Fry, JAMA, J. Am. Med. Assoc., 2009, 301, 1034 CrossRef CAS PubMed.
- A. Moscona, N. Engl. J. Med., 2009, 360, 953 CrossRef CAS PubMed.
- M. Armaka, E. Papanikolaou, A. Sivropoulou and M. Arsenakis, Antiviral Res., 1999, 43, 79 CrossRef CAS PubMed.
- R. E. Granger, E. L. Campbell and G. A. R. Johnston, Biochem. Pharmacol., 2005, 69, 1101 CrossRef CAS PubMed.
- N. Tabanca, N. Kirimer, B. Demirci, F. Demirci and K. H. Baser, J. Agric. Food Chem., 2001, 49, 4300 CrossRef CAS PubMed.
- A. S. Sokolova, O. I. Yarovaya, A. V. Shernyukov, M. A. Pokrovsky, A. G. Pokrovsky, V. A. Lavrinenko, V. V. Zarubaev, T. S. Tretiak, O. I. Kiselev, A. B. Beklemishev and N. F. Salakhutdinov, Bioorg. Med. Chem., 2013, 21, 6690 CrossRef CAS PubMed.
- A. S. Sokolova, O. I. Yarovaya, D. V. Korchagina, V. V. Zarubaev, T. S. Tretiak, P. M. Anfimov, O. I. Kiselev and N. F. Salakhutdinov, Bioorg. Med. Chem., 2014, 22, 2141 CrossRef CAS PubMed.
- A. S. Sokolova, O. I. Yarovaya, D. S. Baev, A. V. Shernyukov, A. A. Shtro, V. V. Zarubaev and N. F. Salakhutdinov, Eur. J. Med. Chem., 2016, 127, 661–670 CrossRef PubMed.
- A. S. Sokolova, O. I. Yarovaya, A. V. Shernyukov, Yu. V. Gatilov, Yu. V. Razumova, V. V. Zarubaev, T. S. Tretiak, A. G. Pokrovsky, O. I. Kiselev and N. F. Salakhutdinov, Eur. J. Med. Chem., 2015, 105, 263 CrossRef CAS PubMed.
- V. V. Zarubaev, A. V. Garshinina, T. S. Tretiak, V. A. Fedorova, A. A. Shtro, A. S. Sokolova, O. I. Yarovaya and N. F. Salakhutdinov, Antiviral Res., 2015, 120, 126 CrossRef CAS PubMed.
- X. Zhao, Z. W. Zhang, W. Cui, S. Chen, Y. Zhou, J. Dong, Y. Jie, J. Wan, Y. Xu and W. Hu, Med. Chem. Commun., 2015, 6, 727 RSC.
- F. Li, Ch. Ma, W. F. DeGrado and J. Wang, J. Med. Chem., 2016, 59, 1207 CrossRef CAS PubMed.
- A. Kolocouris, Ph. Spearpoint, S. R. Martin, A. J. Hay, M. López-Querol, F. X. Sureda, E. Padalko, J. Neyts and E. De Clercq, Bioorg. Med. Chem. Lett., 2008, 18, 6156 CrossRef CAS PubMed.
- X. Zhao, C. Li, S. Zeng and W. Hu, Eur. J. Med. Chem., 2011, 46, 52 CrossRef CAS PubMed.
- X. Zhao, Y. Jie, M. R. Rosenberg, J. Wan, S. Zeng, W. Cui, Y. Xiao, Z. Li, Z. Tu, M. G. Casarotto and W. Hu, Antiviral Res., 2012, 96, 91 CrossRef CAS PubMed.
- L. Jiufeng, N. Kefeng, Z. Yang, W. Dongzhi and R. Yuhong, Lett. Drug Des. Discovery, 2015, 12(1), 72 Search PubMed.
- R. Schobert, B. Biersack, A. Dietrich, A. Grotemeier, T. Muller, B. Kalinowski, S. Knauer, W. Voigt and R. Paschke, J. Med. Chem., 2007, 50(6), 1288 CrossRef CAS PubMed.
- H. Z. Chen, Y. B. Chen, Y. P. Lv, F. Zeng, J. Zhang, Y. L. Zhou, H. B. Li, L. F. Chen, B. J. Zhou, J. R. Gao and C. N. Xia, Bioorg. Med. Chem. Lett., 2014, 24, 4367 CrossRef CAS PubMed.
- M. Asif, Lett. Drug Des. Discovery, 2015, 1, 5 Search PubMed.
- T. P. Kukina, E. V. Malykhin, S. A. Popov and A. M. Chibiriaev, Patent RU, 2464035CAN157:586524, 2012 Search PubMed.
- V. K. Tammara, M. M. Narurkar, A. M. Crider and M. A. Khan, J. Pharm. Sci., 1994, 83, 644 CrossRef CAS PubMed.
- M. Di Braccio, G. Grossi, S. Alfei, V. Ballabeni, M. Tognolini, L. Flammini, C. Giorgio, S. Bertoni and E. Barocelli, Eur. J. Med. Chem., 2014, 86, 394 CrossRef CAS PubMed.
- E. Rutkowska, K. Pajak and K. Jozwiak, Acta Pol. Pharm., 2013, 70(1), 3 CAS.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00657d |
| This journal is © The Royal Society of Chemistry 2017 |