
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel bivalent securinine mimetics as topoisomerase I inhibitors†‡
Wen
Hou
 a,
Hui
Lin
a,
Zhen-Ya
Wang
a,
Martin G.
Banwell
b,
Ting
Zeng
a,
Ping-Hua
Sun
a,
Jing
Lin
*a and
Wei-Min
Chen
*a
a,
Hui
Lin
a,
Zhen-Ya
Wang
a,
Martin G.
Banwell
b,
Ting
Zeng
a,
Ping-Hua
Sun
a,
Jing
Lin
*a and
Wei-Min
Chen
*a
aCollege of Pharmacy, Jinan University, Guangzhou 510632, P. R. China. E-mail: linjing_jnu@163.com; twmchen@jnu.edu.cn; Fax: +86 20 8522 4766; Tel: +86 20 8522 1367 Tel: +86 20 8522 4497
bResearch School of Chemistry, Institute of Advanced Studies, Australian National University, Canberra, ACT 2601, Australia
First published on 3rd January 2017
Abstract
A series of novel bivalent securinine mimetics incorporating different linkers between C-15 and C-15′ were synthesized and their topoisomerase I (Topo I) inhibitory activities evaluated. It was thus revealed that mimetic R2 incorporating a rigid m-substituted benzene linker exhibits Topo I inhibitory activity three times that of parent securinine. Comprehensive structure–activity relationship analyses in combination with docking studies were used to rationalize the potent activity of these bivalent mimetics. Mechanistic studies served to confirm the deductions arising from docking studies that the active bivalent mimetics not only inhibited complexation between Topo I and DNA but also stabilized the Topo I–DNA complex itself.
1 Introduction
The topoisomerase I (Topo I) enzymes are involved in the cutting of one strand of double-stranded and supercoiled DNA, relaxing that strand then re-annealing it in specific ways.1 Given its pivotal role in replication, Topo I was recognized as a target for the development of antitumor drugs in the 1980s.2 As a result of studies in the area the natural product camptothecin (CPT)3 was identified as a potent Topo I inhibitor and the semi-synthetic derivatives topotecan4 and irinotecan5 are now used clinically for treating, inter alia, ovarian, colon and lung cancers.In previous studies6 we identified certain securinine derivatives that act as Topo I inhibitors. Since various linked/dimeric forms of other inhibitors, notably benzimidazoles, seem to exhibit better activity than the parent (monomeric) systems,7–11 we were prompted to construct novel, dimeric securinine derivatives for the purpose of examining their Topo I-inhibitory activities. Herein we describe the synthesis, using hetero-Michael addition chemistry,12 of eleven such bivalent securinine “mimetics” and the identification of one displaying three times the inhibitory activity of the parent (monomeric) system. Further, we detail comprehensive structure–activity relationship analyses and docking studies that reveal the origins of this enhanced activity. Mechanistic investigations, including DNA-cleavage studies and electrophoretic mobility shift assays (EMSAs), as well as DNA-insertion assays, are also described that confirm the inferences drawn from the docking studies.
2 Results and discussion
2.1 Chemical synthesis of bivalent securinine mimetics
Inspired by a report11 revealing that certain bisbenzimidazoles were more efficacious than the corresponding monomers as Topo I inhibitors, eleven compounds, namely R1–R6, HR1–HR3, F2 and F3 (Fig. 1), were identified as the most suitable substrates with which to investigate whether or not bivalent securinine mimetics are more active than the analogous monomeric systems. In order to establish any effects that might be exerted by the linker joining the two monomeric units in these bivalent systems, three types of mimetic were designed, namely those incorporating a rigid linker (labeled as the R series), those with a hemi-rigid linker (the HR series) and those with a flexible one (the F series). Additionally, a monomer M2 (Fig. 1) embodying the same linker unit as the most active bivalent mimetic R2 but lacking the second securinine unit was also prepared in order to study whether or not two such units (rather than just the linker itself) was a necessary requirement for enhanced activity.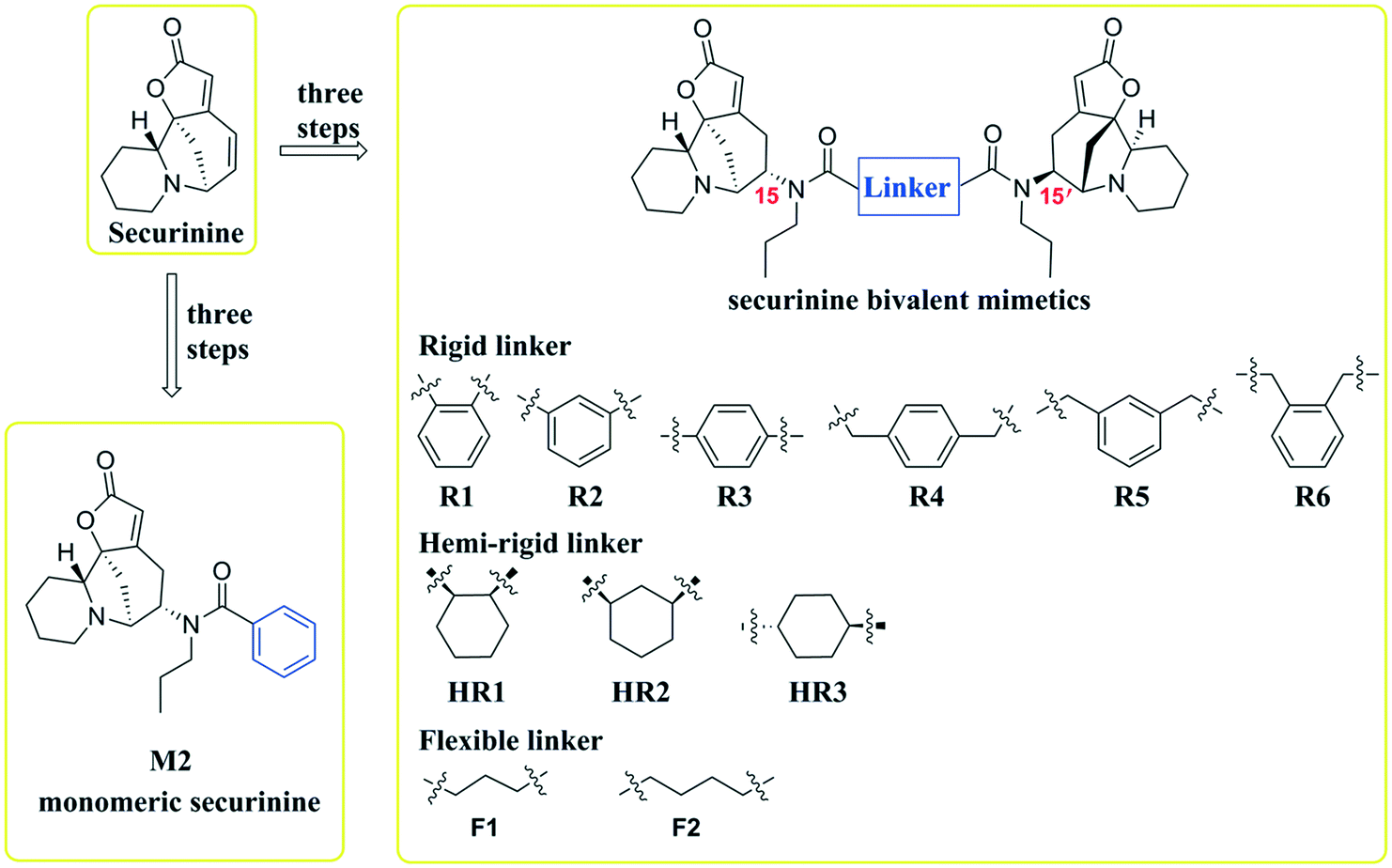 | ||
| Fig. 1 Structures of the target molecules. | ||
The concise routes shown in Scheme 1 allowed for the synthesis of all twelve securinine derivatives and the structures of these were established using 1H and 13C NMR spectroscopic as well as low- and high-resolution electrospray ionization (ESI) mass spectrometric techniques.
 | ||
| Scheme 1 Synthetic route used to obtain the targeted securinine analogues. | ||
2.2 Biological studies
 | ||
| Fig. 2 (A) Gel electrophoretic chromatogram arising from Topo I inhibitory of the title securinine derivatives. CPT used a positive control. Supercoiled pBR322 DNA was incubated with Topo I in the presence or absence of compound at 37 °C for 0.5 h. Lane A – pBR322 DNA alone. Lane B – mixture of pBR322 DNA and Topo I. Lane C – mixture of pBR322 DNA, Topo I and 100 μM CPT. Other lanes – mixtures of pBR322 DNA, Topo I and 100 μM of test compound; (B) gray scale value analysis of results shown in (A). | ||
 | ||
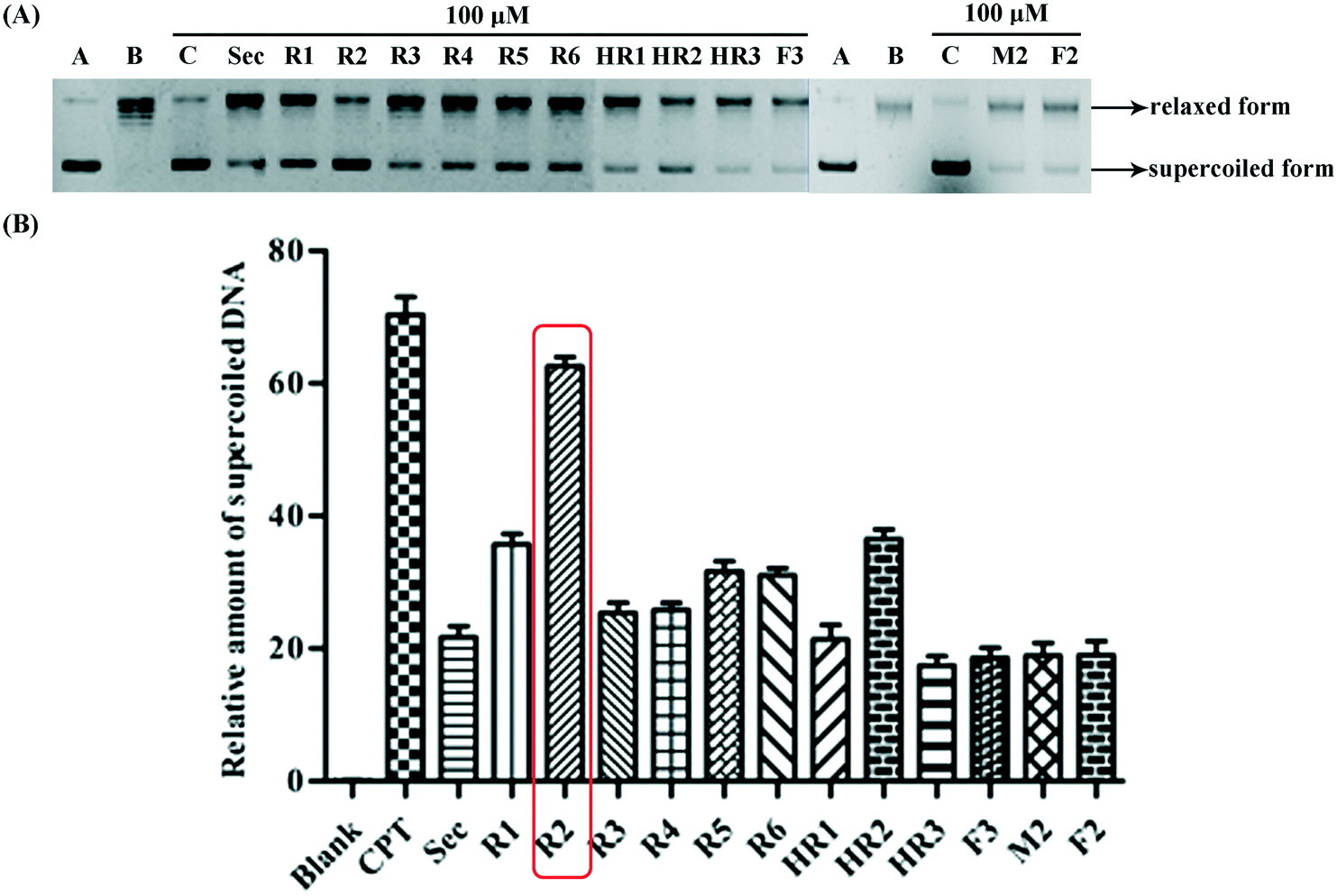
| Fig. 3 Binding model between test compounds and Topo I arising from docking studies. (a) R2 and Topo I (no DNA); (b) R2 and Topo I; (c) HR2 and Topo I; (d) F2 and Topo I; (e) M2 and Topo I; (f) securinine and Topo I. | ||
In order to determine whether or not both securinine units within the bivalent mimetics are necessary for effective binding, the monomeric congener M2 (possessing the same linker as the most active compound R2 but lacking a second securinine residue) and the parent system (viz. securinine) were each subject to docking analysis. As revealed in Fig. 3e and f, neither of these reference substrates bind effectively with the Topo I/DNA complex and thus highlighting the importance of the presence of a bivalent motif.
Overall, then, these docking studies strongly suggest that the excellent Topo I inhibitory activity of the bivalent mimetic R2 arises through a dual inhibitory mechanism involving, (i), binding of it, through π–π stacking, with the covalent Topo I/DNA complex (and thereby stabilizing the same) and, (ii), binding of the second securinine unit within this inhibitor with Topo I (and so inhibiting the normal mode of action of the enzyme).
 | ||
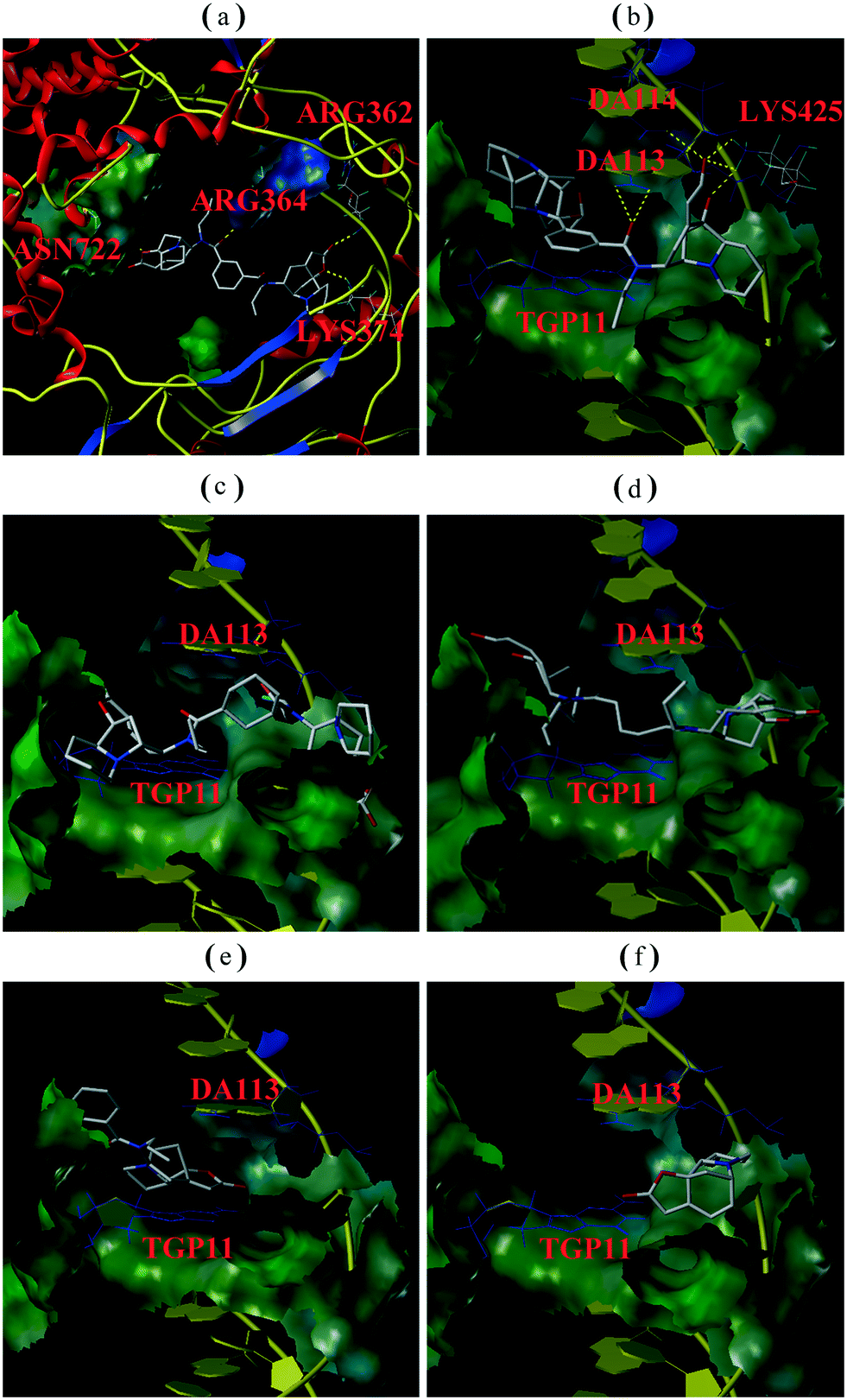
| Fig. 4 (A) Gel electrophoretic chromatogram arising from EMSA of compound R2. CPT used as a negative control. Lane A – pBR322 DNA only. Lane B – mixture of pBR322 DNA and Topo I. Line C – mixture of pBR322 DNA, Topo I and 50 or 100 μM CPT. Other lanes – mixture of pBR322 DNA, Topo I and 50, 100, 200, or 400 μM R2; (B) gray scale value analysis of results shown in (A). | ||
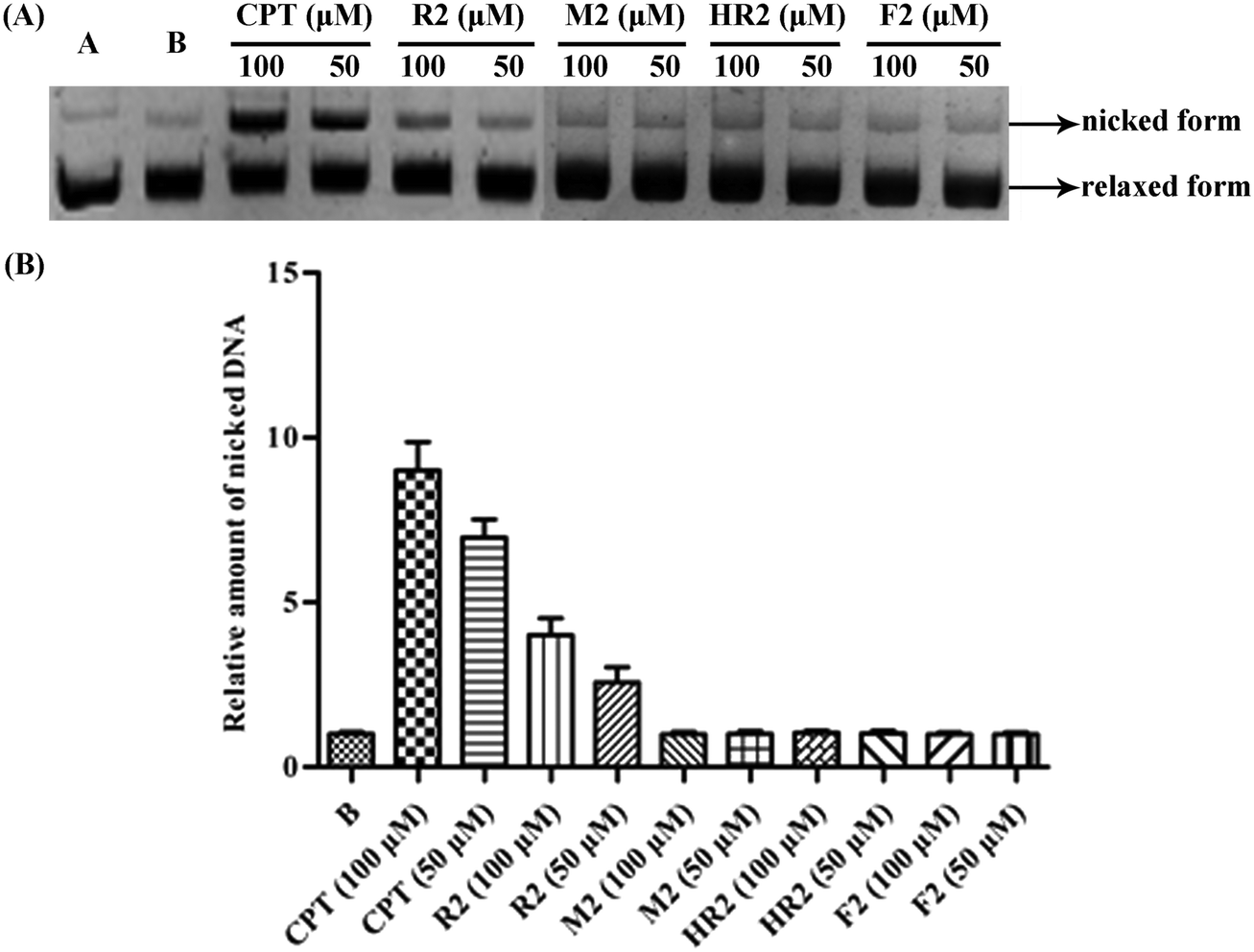 | ||
| Fig. 5 (A) Gel electrophoretic chromatogram arising from Topo I-mediated assay of compounds R2, M2, HR2, and F2. CPT used as a positive control. Lane A – pBR322 DNA only. Lane B – mixture of pBR322 DNA and Topo I. Other lanes – mixture of pBR322 DNA, Topo I and 50, 100 μM of test compound; (B) gray scale value analysis of A. | ||
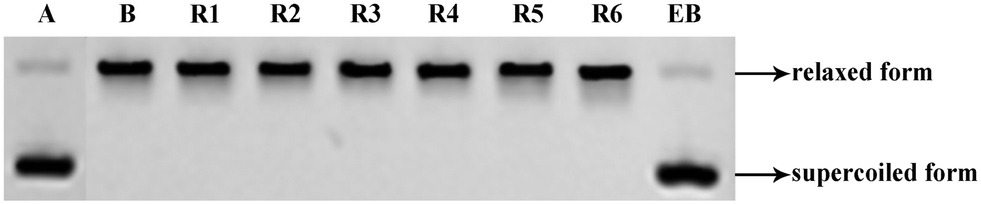 | ||
| Fig. 6 DNA-insertion assay of the R series compounds. EB used as a positive control. Supercoiled pBR322 DNA was incubated at 37 °C for 0.5 h with Topo I in the presence or absence of EB, and with the test compounds. Lane A – pBR322 DNA alone. Lane B – mixture of pBR322 DNA and Topo I. Lane EB – mixture of pBR322 DNA, Topo I and 25 μM EB. Other lanes – mixture of pBR322 DNA, Topo I and 100 μM of test compound. | ||
3 Conclusions
In this study, a series of novel securinine bivalent mimetics and one monomer were designed and synthesized for evaluation as Topo I inhibitors. Compound R2 was found to display about three times the Topo I inhibitory activity of securinine itself and so demonstrating that the development of bivalent securinine mimetics is a novel and feasible strategy for constructing effective inhibitors of this crucial enzyme. Furthermore, through the combined application of docking studies and mechanistically relevant assays, the two-fold binding mode and consequent dual effects of this potent and structurally novel19 inhibitor (viz.R2) have been revealed. The structure–activity relationship data and docking studies reported here also provide a wealth of information for the further development of bivalent mimetics as potentially valuable new Topo I inhibitors.4 Experimental
4.1 Chemistry
(5S,6S,11aS,11bS)-5-(Propylamino)-5,6,9,10,11,11a-hexahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-2(4H)-one (KA). The title compound was prepared using a modification of a previously reported procedure.21 Thus, a magnetically stirred solution of securinine (1.90 g, 9.0 mmol, 1.0 equiv.) in dichloromethane (DCM, 30 mL) maintained at ambient temperatures was treated with K3PO4 (9.5 mg, 0.045 mmol, 0.05 equiv.) then n-propylamine (1.59 g, 27.0 mmol, 3.0 molar equiv.). After 8 h the now milk-white mixture was concentrated under reduced pressure and the residue diluted with water (100 mL) then extracted with DCM (3 × 50 mL). The combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure. The residue thus obtained was subject to flash chromatography (silica gel, 200
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v DCM/MeOH) to give, after concentration of the appropriate fractions, compound KA21 (1.20 g, 49%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ 5.57 (d, J = 2.1 Hz, 1H), 3.22–3.16 (m, 1H), 3.01 (dd, J = 6.3 and 4.2 Hz, 1H), 2.92–2.75 (m, 4H), 2.58–2.40 (m, 4H), 1.89–1.79 (m, 2H), 1.59–1.25 (m, 8H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 174.5, 173.2, 110.9, 91.4, 63.0, 59.8, 59.6, 49.7, 48.8, 32.3, 30.4, 25.7, 23.8, 23.3, 21.5, 11.9. These data were essentially identical, in all respects, with those reported21 previously.
:1 v/v DCM/MeOH) to give, after concentration of the appropriate fractions, compound KA21 (1.20 g, 49%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ 5.57 (d, J = 2.1 Hz, 1H), 3.22–3.16 (m, 1H), 3.01 (dd, J = 6.3 and 4.2 Hz, 1H), 2.92–2.75 (m, 4H), 2.58–2.40 (m, 4H), 1.89–1.79 (m, 2H), 1.59–1.25 (m, 8H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 174.5, 173.2, 110.9, 91.4, 63.0, 59.8, 59.6, 49.7, 48.8, 32.3, 30.4, 25.7, 23.8, 23.3, 21.5, 11.9. These data were essentially identical, in all respects, with those reported21 previously.
N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N-propylbenzamide (M2). Benzoic acid (10 mmol) was added to thionyl chloride (5 mL, 25 mmol) and the resulting solution heated, with magnetic stirring, under reflux for 5 h while being maintained under an atmosphere of nitrogen. The cooled reaction mixture was concentrated under reduced pressure and a portion of the benzoyl chloride (0.3 mmol, 0.6 equiv.) thus obtained added, dropwise over 0.33 h, to a magnetically stirred solution of KA (138 mg, 0.5 mmol, 1.0 equiv.) in anhydrous DCM (10 mL) containing di-iso-propylethylamine (DIPEA, 387 mg, 1.5 mmol, 3.0 equiv.) maintained at −20 °C. After 1 h the reaction mixture was warmed to ca. 22 °C, stirred at this temperature for 16 h then treated with aqueous NaHCO3 so as to achieve pH ≥ 8. The ensuing mixture was concentrated under reduced pressure and the residue extracted with DCM (3 × 20 mL). The combined organic phases were dried (MgSO4), filtered and concentrated under reduced pressure and the residue subjected to flash chromatography (silica gel, 100
:1 v/v DCM:MeOH) to give, after concentration of the appropriate fractions, compound M2 (61 mg, 32%) as a brown solid. 1H NMR (300 MHz, CDCl3) δ 7.40–7.34 (m, 3H), 7.32–7.26 (m, 2H), 5.67 (s, 1H), 4.24 (t, J = 15.5 Hz, 1H), 3.27 (dd, J = 15.5 and 7.5 Hz, 1H), 3.15 (m, 1H), 2.90 (m, 5H), 2.59 (dd, J = 11.0 and 5.8 Hz, 1H), 1.92–1.72 (m, 2H), 1.63–1.44 (m, 3H), 1.42–1.21 (m, 5H), 0.70 (t, J = 6.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.8, 172.5, 165.1, 137.3, 129.4, 128.6, 126.2, 110.3, 90.9, 63.0, 60.0, 57.6, 48.3, 33.6, 29.7, 27.3, 26.4, 24.2, 24.0, 22.1, 11.2; MS (ESI, +ve) m/z 381 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z (M + H)+ calcd for C23H29N2O3 381.2173, found 381.2174.
:1 v/v DCM:MeOH) to give the relevant diamide. The spectral data sets acquired on each of these are given below.
N 1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N2-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N2-dipropylphthalamide (R1). N 1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N2-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N2-dipropylphthalamide (R1) was obtained as a yellow solid. Yield 33%. 1H NMR (300 MHz, CDCl3) δ 7.40 (broad s, 2H), 7.26 (broad s, 2H), 5.67 (broad s, 2H), 4.00–2.00 (m, 15H), 1.98–1.08 (m, 20H), 0.99–0.79 (m, 4H), 0.64 (broad s, 5H); 13C NMR (75 MHz, CDCl3) δ 173.8, 172.7, 171.4, 138.2, 126.5, 110.2, 90.8, 62.6, 59.9, 57.8, 53.7, 48.4, 33.6, 29.6, 27.3, 26.3, 24.1, 22.1, 11.1; MS (ESI, +ve) m/z 683 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C40H51N4O6 683.3803, found 683.3806.
N 1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N3-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N3-dipropylisophthalamide (R2) was obtained a white solid. Yield 46%. 1H NMR (300 MHz, CDCl3) δ 7.50–7.20 (m, 4H), 5.68 (s, 2H), 4.20 (broad s, 2H), 3.50–2.70 (m, 14H), 2.61 (m, 2H), 1.90–1.76 (m, 4H), 1.59–1.16 (m, 16H), 0.70 (broad s, 6H); 13C NMR (75 MHz, CDCl3) δ (major rotamer) 173.7, 172.7, 171.3, 137.8, 128.7, 126.9, 124.2, 110.2, 90.8, 62.8, 59.9, 57.8, 48.3, 33.6, 29.7, 27.3, 26.3, 24.1, 22.0, 14.0, 11.2; MS (ESI, +ve) m/z 683 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C40H51N4O6 683.3803, found 683.3810.
N 1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N4-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N4-dipropylterephthalamide (R3). N 1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N4-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N4-dipropylterephthalamide (R3) was obtained as a yellow solid. Yield 33%. 1H NMR (300 MHz, CDCl3) δ 7.31 (broad s Hz, 4H), 5.65 (s, 2H), 4.19 (broad s, 2H), 3.50–2.65 (m, 14H), 2.58 (m, 2H), 1.80 (m, 4H), 1.65–1.00 (m, 16H), 0.65 (broad s, 6H); 13C NMR (75 MHz, CDCl3) δ 173.8, 172.7, 171.4, 138.2, 126.4, 110.1, 90.8, 62.6, 59.9, 57.8, 53.6, 48.3, 33.6, 29.6, 27.2, 26.3, 24.1, 22.0, 11.1; MS (ESI, +ve) m/z 683 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C40H51N4O6 683.3803, found 683.3800.
N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-2-(4-(2-oxo-2-(((5S,6S,11aR,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)(propyl)amino)ethyl)phenyl)-N-propylacetamide (R4). N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-2-(4-(2-oxo-2-(((5S,6S,11aR,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)(propyl)amino)ethyl)phenyl)-N-propylacetamide (R4) was obtained as a white solid. Yield 36%. 1H NMR (300 MHz, CDCl3) δ 7.23 (m, 1H), 7.05 (m, 3H), 5.64 (s, 2H), 4.28 (broadened s, 2H), 3.79–3.39 (m, 6H), 3.30–3.10 (m, 4H), 3.05–2.76 (m, 10H), 2.48 (broadened s, 2H), 1.92–1.15 (m, 18H), 0.87 (broadened s, 6H); 13C NMR (75 MHz, CDCl3) δ (mixture of rotamers) 174.4, 172.8, 171.3, 135.5, 129.3, 129.0, 127.4, 110.0, 91.1, 61.6, 60.0, 57.4, 48.4, 41.1, 33.4, 26.8, 26.4, 25.3, 24.1, 22.1, 11.3; MS (ESI, +ve) m/z 711 [(M + H)+, 97.2%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C42H55N4O6 711.4116, found 711.4126.
2,2′-(1,3-phenylene)bis(N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N-propylacetamide) (R5). 2,2′-(1,3-phenylene)bis(N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N-propylacetamide) (R5) was obtained as a yellow solid. Yield 32%. 1H NMR (300 MHz, CDCl3) δ 7.17 (s, 4H), 5.65 (s, 2H), 4.34 (m, 2H), 3.75–3.41 (m, 6H), 3.33–3.09 (m, 4H), 3.02–2.77 (m, 10H), 2.53 (m, 2H), 1.76–1.22 (m, 18H), 0.88 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 174.4, 172.8, 171.5, 133.7, 129.1, 110.1, 91.1, 61.6, 60.0, 57.3, 53.5, 48.4, 40.8, 33.4, 26.8, 26.3, 25.4, 24.2, 22.1, 11.2 (signals due to two carbons obscured or overlapping); MS (ESI, +ve) m/z 711 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C42H55N4O6 711.4116, found 711.4126.
N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-2-(2-(2-oxo-2-(((5S,6S,11aR,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)(propyl)amino)ethyl)phenyl)-N-propylacetamide (R6). N-((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-2-(2-(2-oxo-2-(((5S,6S,11aR,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)(propyl)amino)ethyl)phenyl)-N-propylacetamide (R6) was obtained as a white solid. Yield 43%. 1H NMR (300 MHz, CDCl3) δ 7.24–7.07 (m, 4H), 5.71 (m, 2H), 4.38 (m, 2H), 3.75–2.70 (m, 20H), 2.56 (m, 2H), 1.89–1.26 (m, 18H), 0.90 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 174.1, 172.9, 171.4, 134.5, 129.5, 127.5, 110.2, 91.0, 61.5, 60.1, 57.2, 53.5, 48.4, 38.9, 33.4, 26.7, 26.4, 25.3, 24.1, 22.1, 11.2; MS (ESI, +ve) m/z 711 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C42H55N4O6 711.4116, found 711.4126.
(1R,2S)-N1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N2-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N2-dipropylcyclohexane-1,2-dicarboxamide(HR1). (1R,2S)-N1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N2-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N2-dipropylcyclohexane-1,2-dicarboxamide(HR1) was obtained as a white solid. Yield 21%. 1H NMR (300 MHz, CDCl3) δ 5.56 (m, 2H), 4.30 (m, 2H), 3.75–2.35 (m, 18H), 2.30–1.00 (m, 28H), 0.89–0.80 (m, 6H); 13C NMR (75 MHz, CDCl3) δ (mixture of rotamers) 175.3, 175.2, 174.6, 173.2, 172.8, 110.2, 109.9, 91.4, 91.1, 60.2, 59.9, 56.6, 48.5, 48.3, 41.9, 33.3, 30.1, 29.1, 27.0, 26.6, 26.5, 26.1, 24.3, 24.2, 23.3, 23.1, 22.2, 14.1, 11.3, 11.2, 11.1; MS (ESI, +ve) m/z 689 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C40H57N4O6 689.4273, found 689.4274.
(1R,3S)-N1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N3-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N3-dipropylcyclohexane-1,3-dicarboxamide(HR2). (1R,3S)-N1-((5S,6S,11aR,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N3-((5S,6S,11aS,11bS)-2-oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N3-dipropylcyclohexane-1,3-dicarboxamide(HR2) was obtained as a yellow oil. Yield 26%. 1H NMR (300 MHz, CDCl3) δ 5.65 (broad s, 2H), 4.33 (m, 2H), 3.41–2.60 (m, 14H), 2.54–2.23 (m, 4H), 1.95–1.10 (m, 28H), 0.92–0.73 (m, 6H); 13C NMR (75 MHz, CDCl3) δ (mixture of rotamers) 175.6, 175.5, 174.5, 174.4, 172.7, 172.7, 110.1, 93.5, 91.1, 91.0, 61.5, 61.4, 59.9(4), 59.8(9), 56.7, 56.6, 53.5, 48.3(7), 48.3(5), 48.3(3), 47.5(7), 47.5(6), 41.2, 40.7, 33.2, 29.6, 26.4, 26.2(4), 26.2(1), 22.1(7), 22.1(0), 11.2(4), 11.1(5); MS (ESI, +ve) m/z 689 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C40H57N4O6 689.4273, found 689.4274.
(1S,4S)-N1,N4-bis((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N4-dipropylcyclohexane-1,4-dicarboxamide (HR3). (1S,4S)-N1,N4-bis((5S,6S,11aS,11bS)-2-Oxo-2,4,5,6,9,10,11,11a-octahydro-8H-6,11b-methanofuro[2,3-c]pyrido[1,2-a]azepin-5-yl)-N1,N4-dipropylcyclohexane-1,4-dicarboxamide (HR3) was obtained as a white solid. Yield 8%. 1H NMR (300 MHz, CDCl3) δ 5.65 (m, 2H), 4.28–4.05 (m, 2H), 3.35–2.25 (m, 20H), 1.90–1.05 (m, 26H), 0.76 (m, 6H); 13C NMR (75 MHz, CDCl3) δ (mixture of rotamers) 176.2, 174.4, 172.7, 110.0, 91.0, 61.7, 60.0, 57.1, 53.5, 48.3, 48.0, 40.6, 33.2, 29.4, 28.1, 26.8, 26.4, 25.9, 24.1, 22.1, 11.1; MS (ESI, +ve) m/z 689 [(M + H)+, 100%]; HRMS (ESI, +ve) m/z [M + H]+ calcd for C40H57N4O6 689.4273, found 689.4274.
4.2 Modeling and biological evaluation
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81273362).References
- J. C. Wang, J. Mol. Biol., 1971, 55, 523–533 CrossRef CAS PubMed.
- W. E. Ross, Biochem. Pharmacol., 1985, 34, 4191–4195 CrossRef CAS PubMed.
- Y.-H. Hsiang, R. Hertzberg, S. Hecht and L. F. Liu, J. Biol. Chem., 1985, 260, 14873–14878 CAS.
- J. K. Roberts, A. V. Birg, T. Lin, V. M. Daryani, J. C. Panetta, A. Broniscer, G. W. Robinson, A. J. Gajjar and C. F. Stewart, Drug Metab. Dispos., 2016, 44, 1116–1122 CrossRef CAS PubMed.
- Q. Hu, Q. Wang, H. Zhu, Y. Yao and Q. Song, J. Cancer Res. Ther., 2016, 12, 881–887 CrossRef PubMed.
- W. Hou, Z.-Y. Wang, C.-K. Peng, J. Lin, X. Liu, Y.-Q. Chang, J. Xu, R.-W. Jiang, H. Lin, P.-H. Sun and W.-M. Chen, Eur. J. Med. Chem., 2016, 122, 149–163 CrossRef CAS PubMed.
- R. Gaur, A. S. Pathania, F. A. Malik, R. S. Bhakuni and R. K. Verma, Eur. J. Med. Chem., 2016, 122, 232–246 CrossRef CAS PubMed.
- N. Dunlap, T. L. J. Salyard, A. L. Pathiranage, J. Stubblefield, S. L. Pitts, R. E. Ashley and N. Osheroff, Bioorg. Med. Chem. Lett., 2014, 24, 5627–5629 CrossRef CAS PubMed.
- M. Venkatraj, K. K. Ariën, J. Heeres, J. Joossens, J. Messagie, J. Michael, P. V. D. Veken, G. Vanham, P. J. Lewi and K. Augustyns, Bioorg. Med. Chem. Lett., 2012, 22, 7174–7178 CrossRef CAS PubMed.
- N. Dunlap, T. L. J. Salyard, A. L. Pathiranage, J. Stubblefield, S. L. Pitts, R. E. Ashley and N. Osheroff, Bioorg. Med. Chem. Lett., 2014, 24, 5627–5629 CrossRef CAS PubMed.
- O. Y. Susova, A. A. Ivanov, S. S. M. Ruiz, E. A. Lesovaya, A. V. Gromyko, S. A. Streltsov and A. L. Zhuze, Biochemistry, 2010, 75, 695–701 CAS.
- Z. H. Wang, Z. J. Wu, D. F. Yue, Y. You, X. Y. Xu, X. M. Zhang and W. C. Yuan, Org. Biomol. Chem., 2016, 14, 6568–6576 CAS.
- V. A. D'yakonov, L. U. Dzhemileva, A. A. Makarov, A. R. Mulukova, D. S. Baev, E. K. Khusnutdinova, T. G. Tolstikova and U. M. Dzhemilev, Bioorg. Med. Chem. Lett., 2015, 25, 2405–2408 CrossRef PubMed.
- V. Vadwai and S. Devaraj, Int. J. Bioinf. Res. Appl., 2009, 5, 603–615 CrossRef CAS PubMed.
- N. Wu, X.-W. Wu, K. Agama, Y. Pommier, J. Du, D. Li, L.-Q. Gu, Z.-S. Huang and L.-K. An, Biochemistry, 2010, 49, 10131–10136 CrossRef CAS PubMed.
- D. K. Trask, J. A. DiDonato and M. T. Muller, EMBO J., 1984, 3, 671–676 CAS.
- I. L. Gromova, E. Kjeldsen, J. Q. Svejstrup, J. Alsner, K. Christiansen and O. Westergaard, Nucleic Acids Res., 1993, 21, 593–600 CrossRef CAS PubMed.
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS PubMed.
- A. Singh, K. Nepali, N. Kaur, G. Singh, S. Sharma and P. Sharma, Recent Pat. Anticancer Drug Discov., 2016, 1033–1056 Search PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- G. Tang, X. Liu, N. Ma, X. Huang, Z.-L. Wu, W. Zhang, Y. Wang, B.-X. Zhao, Z.-Y. Wang, W.-C. Ye, L. Shi and W.-M. Chen, ACS Chem. Neurosci., 2016, 7, 1442–1451 CrossRef CAS PubMed.
- A. Kumar and U. Bora, Interdiscip. Sci.: Comput. Life Sci., 2014, 6, 285–291 CrossRef CAS PubMed.
- M.-J. Wang, Y.-Q. Liu, L.-C. Chang, C.-Y. Wang, Y.-L. Zhao, X.-B. Zhao, K. Qian, X. Nan, L. Yang, X.-M. Yang, H.-Y. Hung, J.-S. Yang, D.-H. Kuo, M. Goto, S. L. Morris-Natschke, S.-L. Pan, C.-M. Teng, S.-C. Kuo, T.-S. Wu, Y.-C. Wu and K.-H. Lee, J. Med. Chem., 2014, 57, 6008–6018 CrossRef CAS PubMed.
- B.-L. Yao, Y. Mai, S.-B. Chen, H. Xie, P.-F. Yao, T. Ou, J. Tan, H. Wang, D. Li, S.-L. Huang, L.-Q. Gu and Z.-S. Huang, Eur. J. Med. Chem., 2015, 92, 540–553 CrossRef CAS PubMed.
- D. E. Pulleyblank and A. R. Morgan, J. Mol. Biol., 1975, 91, 1–13 CrossRef CAS PubMed.
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00563b |
| This journal is © The Royal Society of Chemistry 2017 |