
Withaferin A induces cell death and differentiation in multiple myeloma cancer stem cells†
Mark E.
Issa
and
Muriel
Cuendet
*
School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Rue Michel Servet 1, CH-1211 Geneva 4, Switzerland. E-mail: Muriel.Cuendet@unige.ch; Fax: +41 22 379 3399; Tel: +41 22 379 3386
First published on 17th November 2016
Abstract
Multiple myeloma (MM) remains an incurable malignancy despite the development of novel therapeutics. This is believed to be due to a subset of rare chemotherapy-resistant cancer stem cells (CSCs). Differentiation therapy represents one strategy aimed at reducing the stemness of CSCs. The anticancer effect of withaferin A (WFA) was studied in MM-CSCs and RPMI 8226 MM tumoral plasma cells (RPMIs). WFA exhibited growth inhibitory effects in both MM-CSCs and RPMIs, with IC50 values of 649 and 224 nM, respectively. WFA also induced a G2 cell cycle arrest, as well as cell death and apoptosis. Although, WFA did not exhibit a direct anti-migratory effect, a remarkable morphological change was observed in MM-CSCs in response to WFA treatment. Using qPCR gene expression analyses, WFA caused a reduction in stemness markers, and a promotion of differentiation markers in MM-CSCs. These results warrant further investigation of WFA in relevant MM animal models.
Introduction
Despite recent advances in the treatment of multiple myeloma (MM), MM patients are still confronted with an inevitable deadly relapse. This is due to the development of drug resistance that renders virtually all available MM treatments ineffective.1 One hypothesis that explains the almost 100% relapse frequency among MM patients is the existence of MM cancer stem cells (CSCs).2–4 MM-CSCs are equipped with metabolic tools making them highly resistant to conventional cytotoxic therapies, and therefore MM-CSCs are believed to be the cause behind the high mortality rate observed among MM patients.2–4 Novel molecules targeting MM-CSCs may prove an effective prevention strategy to reduce the frequency of relapses in MM.Besides being resistant, CSCs are characterized with high tumorigenicity and the ability to differentiate into heterogeneous tumoral cell populations.5 Importantly, tumoral cells derived from the differentiation of CSCs are not believed to be tumorigenic. Accordingly, the promotion of CSCs differentiation may be an effective strategy to reduce their malignancy.5
Natural products remain a major reservoir of compounds capable of controlling cellular characteristics such as resistance, proliferation and differentiation.6 Importantly, a multitude of compounds inspired from natural products were developed and are currently used in the clinic for the management of cancer. Examples include taxol, vinblastine and all-trans retinoic acid (ATRA).6,7 Withaferin A (WFA) is a steroidal lactone isolated from the leaves of Withania somnifera (Fig. 1A).8W. somnifera, also known as winter cherry, is used in ayurvedic medicine to treat sedation and sexual vitality.8 WFA has been demonstrated to exert an array of biological activities relevant in neurological disorders, inflammation and cancer.8 WFA showed cytotoxic effects in several cancer cell lines, including breast cancer, colorectal cancer, leukemia, lymphoma and MM,8 and dose-dependently inhibited the in vitro mammosphere formation of MCF-7 and SUM-159 breast cancer cells.9In vivo, WFA reduced the number of mammosphere formation and exhibited a trend towards a reduction in mammosphere size,9 suggesting that WFA exhibited some activity against the putative breast CSCs.
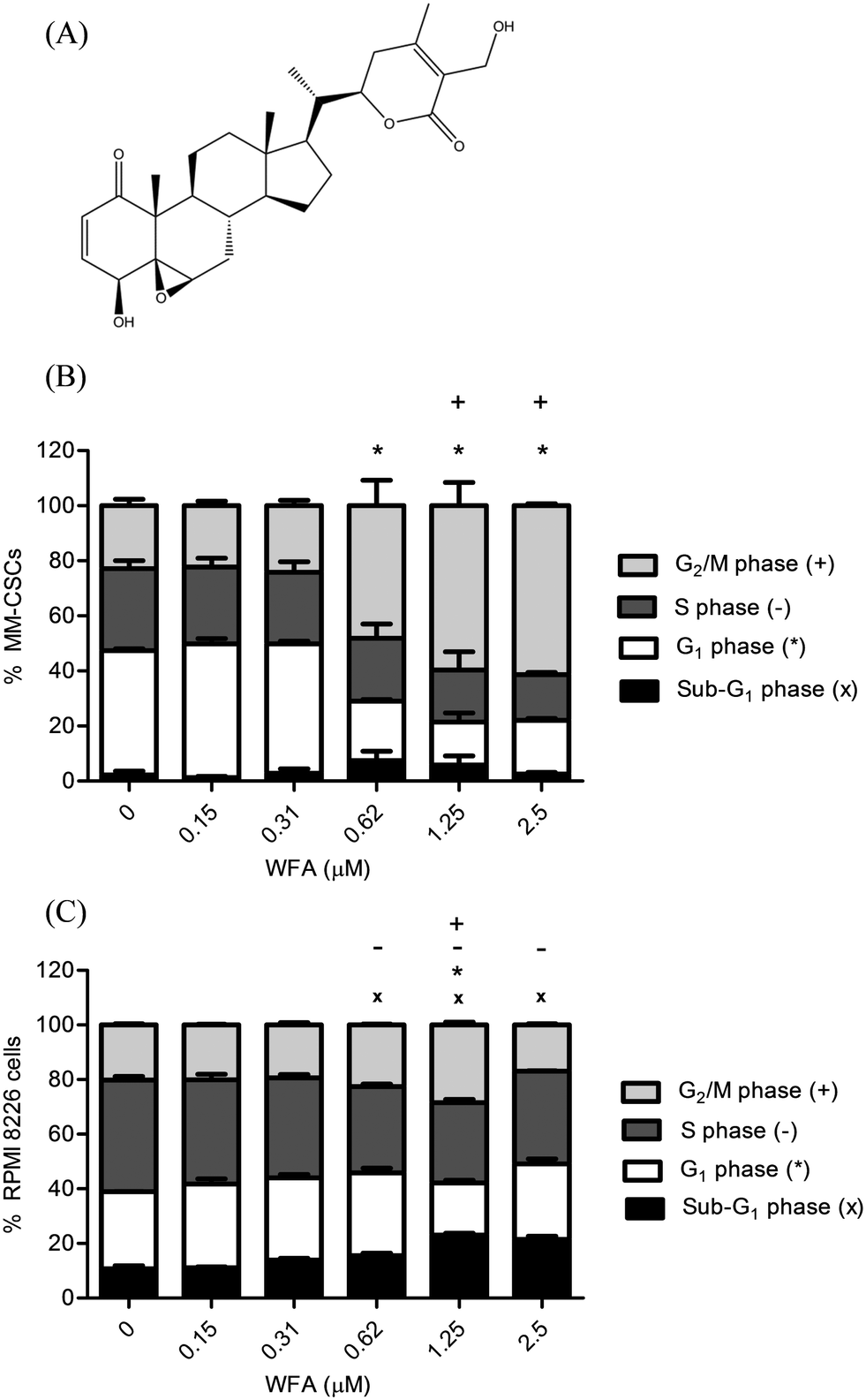 | ||
| Fig. 1 Effect of WFA on MM cell cycle distribution. Chemical structure of WFA (A). MM-CSCs (B) or RPMIs (C) were treated with vehicle control or increasing concentrations of WFA for 24 h. Cells were analyzed for cell cycle distribution as described in Materials and methods. Results represent the means ± SEM of at least 3 independent experiments. Percentage of cells in the sub-G1 phase (x) in G1 phase (*), S phase (−) or G2/M phase (+) significantly different from control values as determined by a one-way ANOVA, followed by Bonferroni multiple comparison test (p < 0.05). | ||
The purpose of this project was to study the effects of WFA on MM-CSCs viability and differentiation. Anticancer endpoints included growth inhibition, cell cycle distribution and cytotoxicity in MM-CSCs. These effects were compared to those in the tumoral plasma RPMI 8226 cell line. The effects of WFA on MM-CSCs migration and differentiation were examined, and a mechanism of action by which WFA induced MM-CSCs differentiation was identified.
Experimental
Chemicals and biologicals
WFA was purchased from Enzo Life Sciences (Lausen, Switzerland). Bortezomib (BTZ) and dexamethasone (DEX), MTT, XTT, calcein AM (calcein), and ethidium homodimer (EH) were purchased from Sigma Aldrich (Buchs, Switzerland). Phalloidin and DAPI nuclear stains were purchased from Life Technologies (Zug, Switzerland).Cell culture
Human bone marrow-derived MM-CSCs were obtained from Celprogen (Torrance, CA, USA). MM-CSCs are positive for the putative CSCs markers CD44+, CD166+ and negative for CD138−.10,11 According to Celprogen, MM-CSCs are tumorigenic at <1000 cells.10,11 MM-CSCs were grown in MM-CSCs complete growth medium (Celprogen). Experiments using MM-CSCs were performed at cell passages between 4 to 12. RPMI 8226 cells (RPMIs) were purchased from LGC standards (Middlesex, UK). RPMIs are predominantly CD138+ non-tumorigenic tumoral plasma cells derived from the peripheral blood of a MM patient. RPMIs contain a subpopulation of CD138− cells (2 to 5%) which have been previously demonstrated to be tumorigenic.12 RPMIs were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) (Nuaillé, France), 100 IU mL−1 penicillin and 250 μg mL−1 streptomycin (Life Technologies, Zug, Switzerland). Normal hematopoietic CD34+ stem cells (NSCs) were obtained from Life Technologies. NSCs were maintained in StemPRO-34 medium (Life Technologies), supplemented with Cytokine Mix E (PromoCell). MM-CSCs, RPMIs and NSCs were passaged every 2–3 days, and were maintained in a humidified atmosphere supplemented with 5% CO2 at 37 °C (standard conditions).General experimental procedures
For cell cycle analysis, live/dead cell assay, and Annexin V/Propidium iodide (AV/PI) assays, MM-CSCs were seeded in 6-well plates at a density of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and allowed to adhere for 24 h before WFA treatment at which 300000 cells were present in the well.10 RPMIs and NSCs were seeded at 300000 cells per well in 6-well plates and were treated immediately with increasing concentrations of WFA for 24 h. All flow cytometric analyses were performed using the Attune NxT Acoustic Focusing Flow Cytometer (Applied Biosystems, Life Technologies, Zug, Switzerland).
000 cells per well and allowed to adhere for 24 h before WFA treatment at which 300000 cells were present in the well.10 RPMIs and NSCs were seeded at 300000 cells per well in 6-well plates and were treated immediately with increasing concentrations of WFA for 24 h. All flow cytometric analyses were performed using the Attune NxT Acoustic Focusing Flow Cytometer (Applied Biosystems, Life Technologies, Zug, Switzerland).
MTT/XTT assay
MTT and XTT assays were conducted as previously described.10,13–15 MTT and XTT assays were performed on MM-CSCs and RPMIs, respectively. Briefly, MM-CSCs were plated in 96-well plates at a density of 5000 cells per well and allowed to adhere for 24 h, whereas RPMIs were plated at a density of 15000 cells per well and treated immediately. MM-CSCs and RPMIs were treated with increasing concentrations of WFA (0–10 μM). After 72 h incubation, 20 μL of MTT solution (5 mg mL−1) or 50 μL XTT solution (1 mg mL−1) were added to the cells and incubated for 2 h (MTT assay) or 4 h (XTT assay). The percentage of cell growth was calculated as the absorbance of each test well divided by that of the vehicle control wells and then multiplied by 100. The concentration that caused 50% reduction in cell growth (IC50) was determined from at least five different concentrations using GraphPad Prism 5.0 software.
Cell cycle analysis
Cell cycle analysis in response to WFA was conducted as previously described.10,16 Briefly, floating and adherent cells were collected following WFA treatment, fixed and permeabilized with Fix and Perm reagents (Life Technologies), washed with PBS, adjusted to 106 cells mL−1 in PBS, and then incubated at room temperature for 30 min with the DNA selective stain FxCycle (Life Technologies). Cell cycle analysis was then performed using the Attune flow cytometer according to the manufacturer's instructions.Viability/mortality and apoptosis staining
Viability and apoptosis assays were conducted as previously described.10,17,18 Briefly, MM-CSCs, RPMIs and NSCs were analysed by flow cytometry for viability using the calcein/EH staining, and for apoptosis using the AV/PI staining (Life Technologies). Floating and adherent cells were collected following treatment with WFA, washed with PBS, adjusted to 106 cells mL−1, and then incubated with the corresponding stain at room temperature for 15 min. After the incubation period, cells were analysed using the Attune flow cytometer.Cell migration assay (scratch assay)
Scratch assays were conducted as previously described.10,19 MM-CSCs were plated in 24-well plates at a density of 100000 cells per well. MM-CSCs were incubated for 24 h during which the wells were completely covered with a monolayer of MM-CSCs. A scratch was inflicted in each well using a 10 μL pipette tip and photos were taken shortly afterwards using the Cytation 3 Cell Imaging Reader (Biotek, Winooski, VT, USA). MM-CSCs were then treated with increasing concentrations of WFA for 24 h. Photos of the same spot in each well were then taken to show migration inhibitory capabilities of WFA. A quantification of the inhibitory effect of WFA on cell migration was calculated by dividing the width of the same scratch at 24 h by that at 0 h and multiplied by 100 to denote the percent of unfilled space; the percent recovery was calculated as the percent unfilled space subtracted from 100.
Phalloidin cytoskeletal staining
Phalloidin staining was conducted as previously described.20 Briefly, MM-CSCs were seeded in 96-well plates at a density of 5000 cells per well. MM-CSCs were allowed to adhere for a period of 24 h. Increasing concentrations of WFA treatment were applied for 24 h. Phalloidin staining was conducted as per the manufacturer's instructions. Briefly, cells were fixed with 4% paraformalaldehyde for 10 min and perforated with 0.1% triton X-100 for 5 min. They were then incubated with 50 μL of 5 U mL−1 phalloidin for 20 min at room temperature, washed with PBS and incubated with 50 μL of 1 μg mL−1 DAPI for 5 min at room temperature. Photos of MM-CSCs stained with phalloidin/DAPI were taken using the Cytation 3 Cell Imaging Reader. Effects of WFA on MM-CSCs area and circularity were quantified using the Gen5 software provided with the Cytation 3 Cell Imaging Reader.Quantitative real-time PCR analysis of mRNA expression
qPCR analyses were performed as previously described.10,15 Briefly, MM-CSCs were lysed according to the manufacturer's instructions following WFA treatment, and total RNA was collected from cell lysates using the Aurum total RNA Mini Kit (Bio-Rad, Cressier, Switzerland). Reverse transcription was carried out by using 0.5 μg RNA, the “high capacity total RNA to cDNA” Reverse Transcriptase (Life Technologies) and a PCR standard thermal cycler (Applied Biosystems). One μL cDNA was amplified by quantitative PCR using a SYBR Green PCR Kit (Applied Biosystems) and a Step One Plus real-time PCR thermal cycler (Applied Biosystems). Custom primers for GAPDH, 18S, CD44, CD151, CD166, HES1, Kit, MSI2, Notch1, SOX2 and RUNX1 were designed on http://bioinfo.ut.ee/primer3-0.4.0/ and obtained from Life Technologies.21,22 Relative gene expression was calculated by normalizing to the average of two housekeeping genes 18S and GAPDH using the ΔΔCT method.23Statistical analysis
Each experiment was repeated at least three times, and all data were expressed as mean ± SEM. A one-way analysis of variance (ANOVA) was employed for multiple comparisons in experiments with one independent variable; whereas a two-way ANOVA was employed for multiple comparison procedures in experiments with two independent variables. A Bonferroni test was employed for post–hoc analysis of the significant ANOVA. A significant difference in mean values between groups was considered when p ≤ 0.05.Results and discussion
WFA inhibits the growth of MM cells and induces MM cell cycle arrest
Previous results have shown that WFA strongly inhibited the growth of several cancer cell lines, including leukemia, lymphoma and MM.24 In MM, WFA demonstrated promising antiproliferative activity, and induced a dose-dependent cell death in human myeloma cells U226.25,26 However, the effects of WFA on CSCs were not studied. Given the current dogma that CSCs, and not the tumoral cells, are tumorigenic, therefore, it is valuable to address the effects of WFA on CSCs.The growth inhibitory potential of WFA (Fig. 1A) in MM-CSCs was determined. Preliminary MTT assays suggested that a 72 h treatment with WFA inhibited MM-CSCs growth with an IC50 value of 649.7 ± 73.3 nM (Table 1). Because CSCs are believed to be equipped with metabolic tools that render them resistant to conventional therapies,27 the antiproliferative activity of WFA in MM-CSCs was compared to that in RPMIs. WFA inhibited the growth of RPMIs with an IC50 value of 224.0 ± 29.1 nM when using the XTT assay (Table 1). BTZ and DEX were used as positive controls. These results indicated that WFA could inhibit both tumoral cells, which are non-tumorigenic and sensitive to conventional therapies, and CSCs, which are tumorigenic and drug-resistant.
| Compounds | MM-CSCs | RPMIs |
|---|---|---|
| a The antiproliferative activity of WFA and controls was evaluated by MTT/XTT assays as described in Materials and methods. MTT assays were performed with MM-CSCs, whereas XTT assays were performed with RPMIs. Results are the mean of three independent experiments ± SEM. b Positive controls: bortezomib (BTZ) and dexamethasone (DEX). | ||
| WFA | 649.7 ± 73.3 | 224.0 ± 29.1 |
| BTZb | 8.4 ± 0.3 | 3.6 ± 0.5 |
| DEXb | >100000 |
7.8 ± 0.7 |
The effect of WFA on cell cycle distribution in MM-CSCs (Fig. 1B) and RPMIs (Fig. 1C) was analyzed using the DNA specific FxCycle violet stain. Results showed that the proportion of MM-CSCs in sub-G1 phase tended to increase but without statistical significance. However, the proportion of MM-CSCs in the G1 phase significantly decreased from 45.1% with the vehicle control to 21.5%, 15.5% and 19.5% at 0.62, 1.25, and 2.5 μM WFA, respectively. The amount of MM-CSCs in the S phase did not appear to change significantly. The amount of MM-CSCs in the G2/M phase significantly increased from 22.9% in the vehicle control to 59.6% and 61.4% at 1.25 and 2.5 μM WFA, respectively.
Cell cycle analyses in RPMIs showed that the proportion of cells in the sub-G1 phase significantly increased in a dose dependent manner from 10.8% in the vehicle control to 15.6%, 23.1% and 21.5% after 0.62, 1.25 and 2.5 μM WFA treatment, respectively. The proportion of RPMIs in the G1 phase significantly decreased from 28.2% with the vehicle control to 19.1% at 1.25 μM WFA. The amount of MM-CSCs in the S phase significantly decreased from 40.9% in the vehicle control to 31.7%, 29.4% and 34.0% at 0.62, 1.25, and 2.5 μM WFA, respectively. Finally, the amount of RPMIs in the G2/M phase significantly increased from 20.2% with the vehicle control to 28.4% at 1.25 μM WFA.
These results are in alignment with those found in other hematological cancer lines. Previous studies showed that increasing doses of WFA caused a significant decrease in G1 and S phases, and a significant increase in the G2/M phase in lymphoma cancer lines.24 Also, WFA inhibited the growth of leukemic cell lines by stopping cell cycle progression in the G2/M phase in leukemic cancer cells.28 The fact that both MM-CSCs and RPMIs displayed a G2/M cell cycle arrest in response to WFA treatment suggested that these results are not only consistent with other hematological cancer lines but also between tumorigenic and non-tumorigenic cells.
WFA induces MM cells death and apoptosis
Previous results revealed that WFA induced a dose-dependent cell death in several cancer lines, including the neuroblastoma Be(2)-c, SMS-KCNR, SH-SY5Y, LAN-5 and IMR-32, and the MM cell line U266.25 Interestingly, WFA demonstrated cytotoxic activity against the chemotherapy-resistant MM cell line MM1.RL.25 As cytotoxic effects could not be concluded from MTT/XTT proliferation assays, the effect of WFA on MM cell viability and mortality were pursued. Flow cytometric analyses of calcein/EH staining were performed in MM-CSCs and RPMIs in response to WFA treatment. A 24 h treatment resulted in a dose-dependent reduction of viable MM-CSCs (Fig. 2A) and RPMIs (Fig. 2B), as indicated by the significant decrease in calcein+/EH− cells. MM-CSCs treated with vehicle control displayed a viability (calcein+/EH−) of 95.7%, which significantly decreased to 64.6% starting at 2.5 μM, and reached 12.1% at 10 μM WFA. Similarly, the percentage of dying (calcein+/EH+) and dead (calcein−/EH+) MM-CSCs increased significantly from 3.6% and 0.7% with the vehicle control to 31.5% and 56.4% at 10 μM WFA, respectively. In comparison, RPMIs appeared to be slightly more prone to the cytotoxic effects of WFA. RPMIs treated with vehicle control displayed a percentage of viable cells of 67.7%, which significantly decreased to 32.1% and 8.2% at 1.25 and 10 μM WFA, respectively. The percentage of dead cells was of 21.4% in the vehicle control, and significantly increased starting at 1.25 μM to reach 87.0% at 10 μM WFA. Taken together, these results suggested that WFA was not only cytostatic, but also cytotoxic to the resistant MM-CSCs and to the sensitive tumoral plasma RPMIs.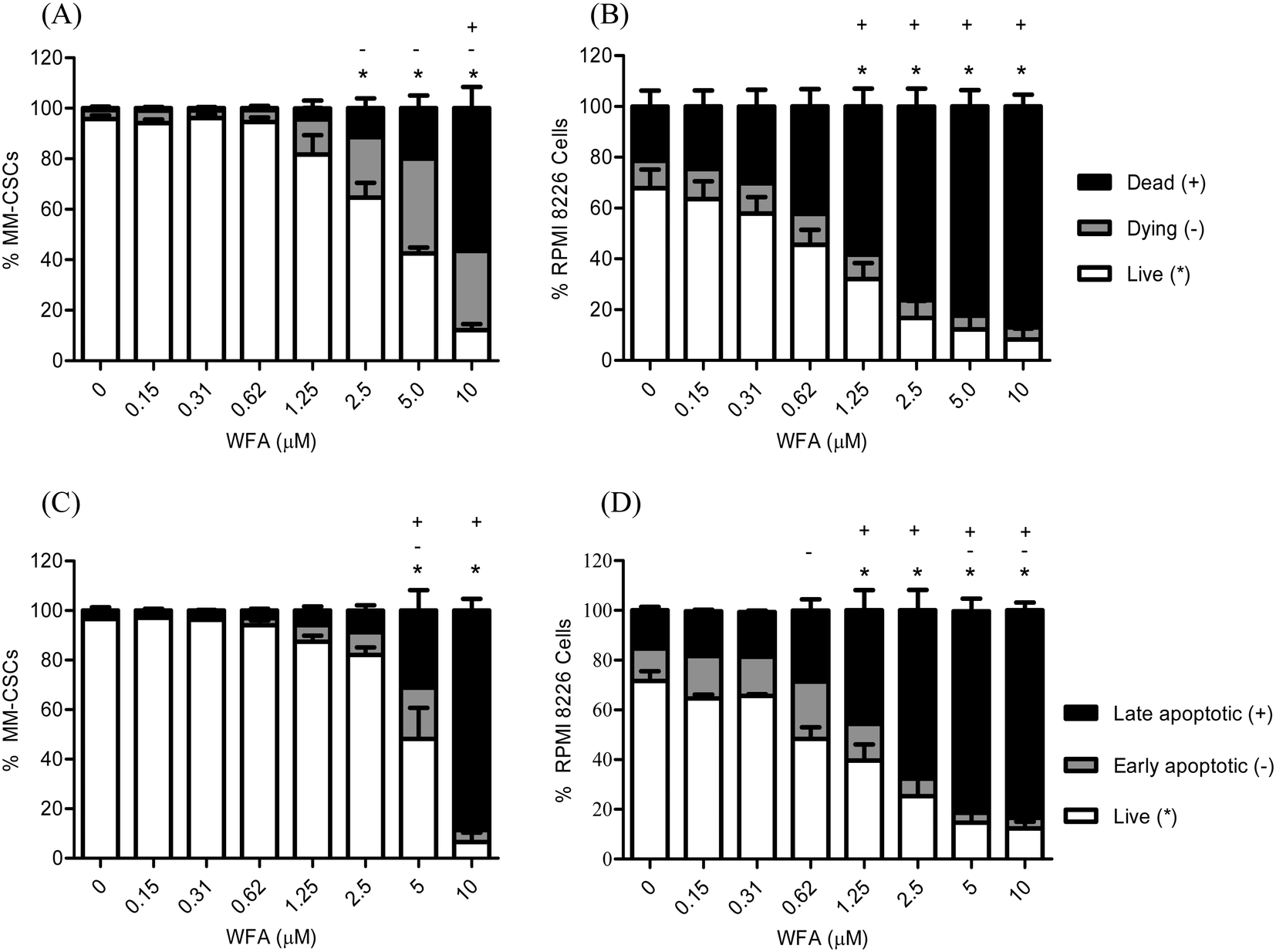 | ||
| Fig. 2 Effect of WFA on MM cell viability. Cells were treated with vehicle control or increasing concentrations of WFA for 24 h, and then stained with calcein AM/ethidium homodimer for cell viability/mortality (A and B), or annexin V/propidium iodide for apoptosis (C and D). Results represent the means ± SEM of at least 3 independent experiments. Percentage of live (*), dying (−) and dead (+) cells; or live (*), early apoptotic (−) and late apoptotic (+) cells significantly different from control values as determined by a one-way ANOVA, followed by Bonferroni multiple comparison test (p < 0.05). | ||
Because WFA induced cell death in MM cells, the effects of WFA on apoptosis was measured in both MM-CSCs and RPMIs. Flow cytometric detection of AV/PI staining was used, and a significant induction of apoptosis was observed in both cell lines (Fig. 2C and D). The proportion of viable cells (AV−/PI−) was significantly lower starting at 5 μM WFA in MM-CSCs, and starting at 1.25 μM WFA in RPMIs. However, the proportion of late apoptotic AV+/PI+ MM-CSCs was significantly higher starting at 5 μM WFA in MM-CSCs, and at 1.25 μM WFA in RPMIs. These results indicate that WFA induced apoptosis in both MM-CSCs and RPMIs, but required slightly higher concentrations in the CSCs than in the tumoral plasma cells.
WFA is less toxic towards normal hematopoietic stem cells than towards cancer stem cells
Due to the extensive metabolic and epigenetic profiles that CSCs and normal hematopoietic stem cells (NSCs) share, it is therefore doomed challenging to target CSCs but leave NSCs intact.11,29–31 The importance of CSCs-selective targeting comes from the fact that NSCs are extremely important to replenish the various dying/damaged tissues in the body. The attrition of NSCs may have severe adverse effects; thus, the discovery of compounds that selectively target CSCs, while NSCs remain reasonably healthy, is highly desirable. For these reasons, the cytotoxic effects of WFA were also examined on NSCs to determine if WFA exhibited any selectivity towards MM-CSCs (Fig. 3A). Results showed that a 24 h WFA treatment did not cause a significant decrease in NSCs viability at doses lower than 5 μM. In addition, the percentage of dying and dead NSCs was only significant at 10 μM WFA compared to no treatment (Fig. 3A). A direct comparison of viable NSCs to viable MM-CSCs was made (Fig. 3B). The results showed that at 5 μM WFA, the proportion of NSCs was higher than that of MM-CSCs, although not statistically significant. However, the proportion of viable MM-CSCs was significantly lower than that of NSCs when cells were treated with 10 μM WFA for 24 h. These results indicated that WFA may exhibit a degree of selectivity towards MM-CSCs. Although WFA did not demonstrate much selectivity towards NSCs, the difference seen at 10 μM remains a valuable starting point. The discovery of the responsible targets that mediate the death of CSCs in response to WFA represents valuable information as some medicinal chemistry work could be pursued to improve the selectivity of WFA towards CSCs over NSCs.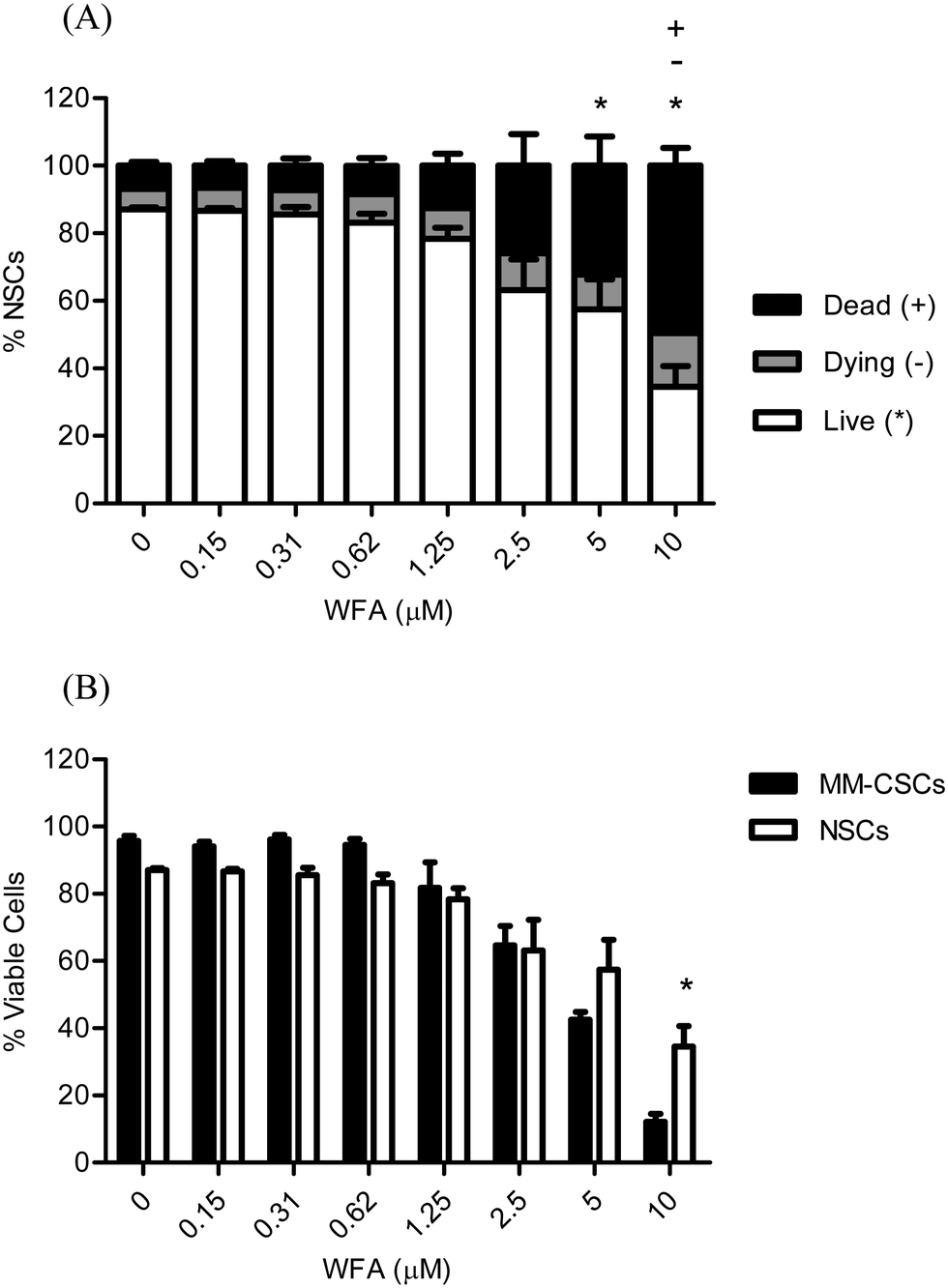 | ||
| Fig. 3 Effect of WFA on NSCs viability. NSCs were treated with vehicle control or increasing concentrations of WFA for 24 h, and then stained with calcein AM/ethidium homodimer for cell viability/mortality (A). The percentage of live cells in response to WFA treatment of NSCs and MM-CSCs were compared using a two-way ANOVA (B). Results represent the means ± SEM of at least 3 independent experiments. Percentage of live (*), dying (−) and dead (+) cells significantly different from control values as determined by a one-way ANOVA, followed by Bonferroni multiple comparison test (p < 0.05). | ||
WFA does not appear to inhibit MM-CSCs migration
One important characteristic of CSCs is their ability to migrate and consequently cause metastasis; therefore compounds capable of inhibiting this CSCs characteristic would be valuable.32 The effect of a 24 h WFA treatment on the migration of MM-CSCs was determined using the scratch, aka wound healing assay.19 0.62 and 1.25 μM WFA did not appear to inhibit MM-CSCs migration. However, at 2.5 μM, MM-CSCs migration was halted (Fig. 4), and at 5 and 10 μM treatment, the cells started to die and lift off the plate (data not shown). The lack of dose–response, combined with the halt in migration at only one dose, renders it difficult to state that WFA inhibits MM-CSCs migration. Further studies are needed to characterize the nature of the observed single dose inhibition of MM-CSCs migration. However, one interesting observation was that at 1.25 μM, clear morphological changes were observed in MM-CSCs. MM-CSCs were larger and their circularity was increased. For this reason, the effects of WFA on MM-CSCs morphology and differentiation were pursued.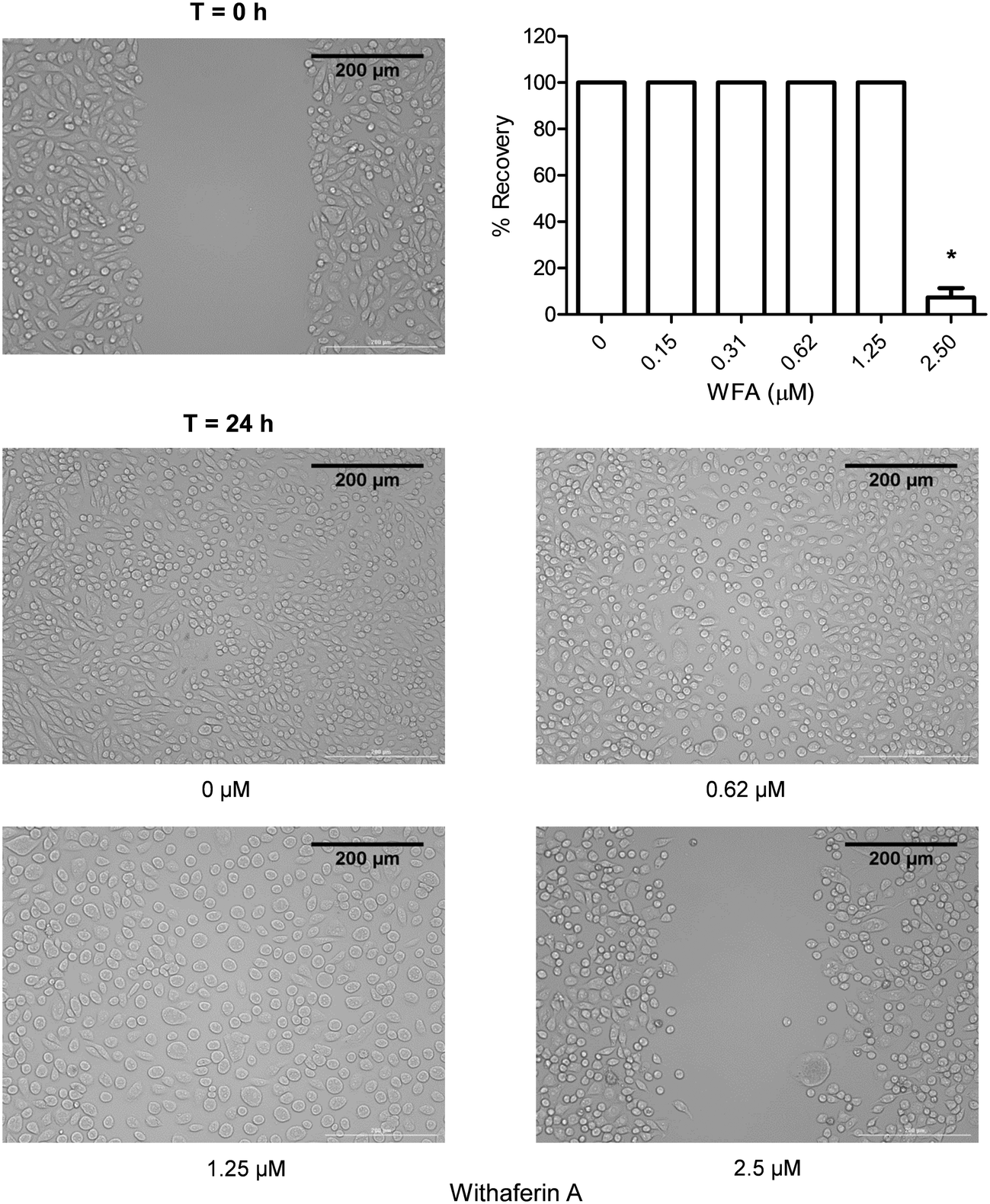 | ||
| Fig. 4 Effect of WFA on MM-CSCs migration. Confluent MM-CSCs were scratched and treated with increasing concentrations of WFA for 24 h. Results are representative of 3 independent experiments (scale bar = 200 μm). *Anti-migratory effect significantly different from vehicle control values as determined by a one-way ANOVA, followed by Bonferroni multiple comparison test (p < 0.05). | ||
WFA alters MM-CSCs morphology
To provide a more visible effect of the impact of WFA on MM-CSCs morphology, a phalloidin cytoskeletal staining was conducted. Phalloidin staining confirmed the effect of WFA observed in the scratch assay. A 24 h WFA treatment caused clear alterations in MM-CSCs morphology (Fig. 5). At vehicle control, MM-CSCs appeared polygonal with clear angles reminiscent of a fibroblast-like morphology. Starting at 0.62 μM WFA treatment, MM-CSCs appeared to start enlarging in size, became more circular, and displayed an epithelial/lymphoblastic-like morphology. At 1.25 μM WFA treatment, an apparent morphological difference was observed. MM-CSCs displayed larger cytoplasmic surface area, more spherical shape and polygonally-shaped MM-CSCs were no longer observed. In addition, a quantitative evaluation indicated that MM-CSCs area and circularity significantly increased in response to 1.25 μM WFA. These morphological differences pointed out that WFA may induce the differentiation of MM-CSCs.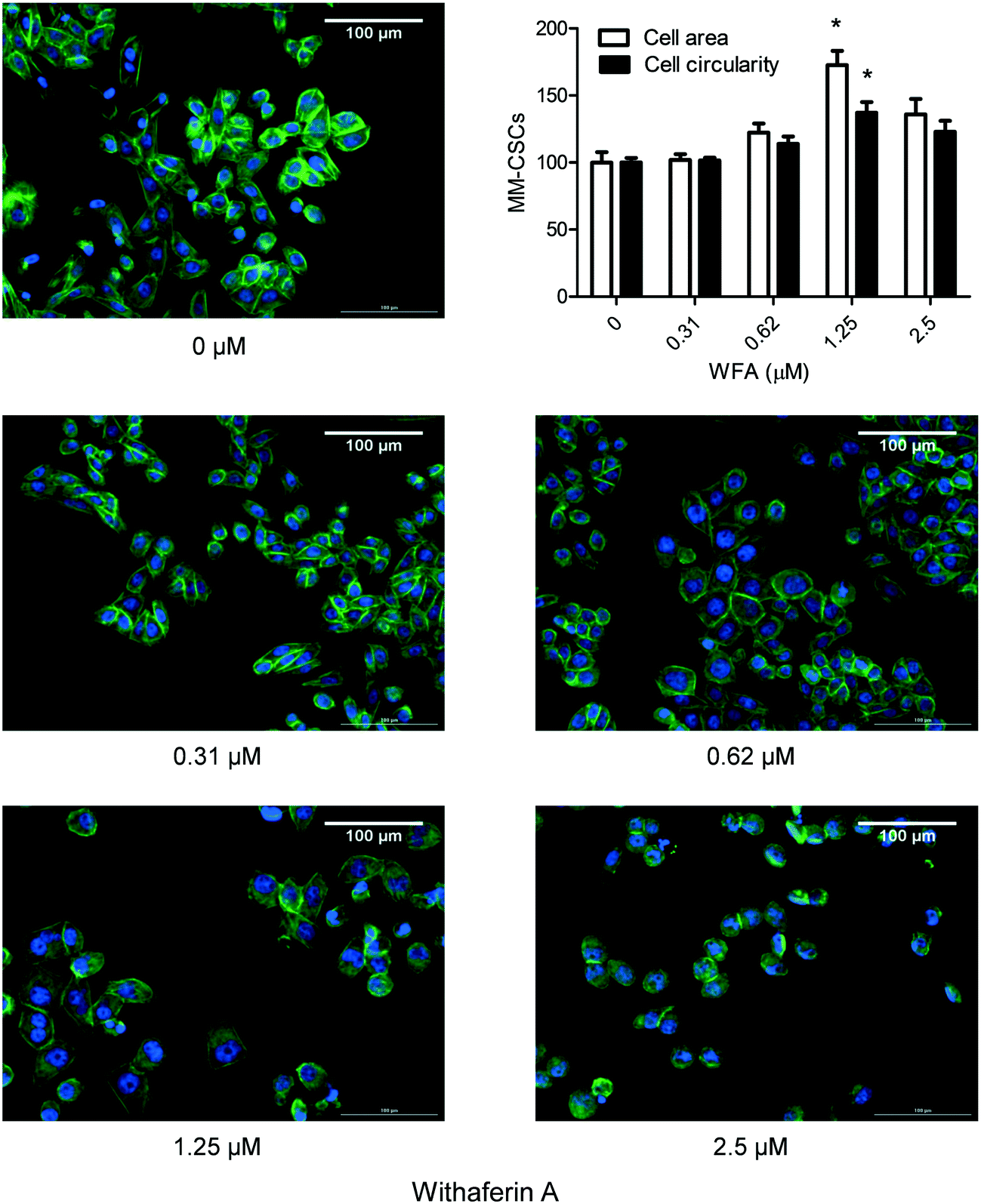 | ||
| Fig. 5 Effect of WFA on MM-CSCs morphology. MM-CSCs were treated with vehicle control or increasing concentrations of WFA for 24 h. Phalloidin staining was performed as described in Materials and methods. Results are representative of 3 independent experiments (scale bar = 100 μm). The effect of WFA on MM-CSCs area and circularity is displayed. Results represent the means ± SEM of at least 3 independent experiments. *Significantly different from vehicle control values as determined by a one-way ANOVA, followed by Bonferroni multiple comparison test (p < 0.05). | ||
WFA alters the expression of genes involved in stemness and cell differentiation
Having observed alterations in MM-CSCs morphology, the effect of WFA on the expression of genes implicated in stem cell maintenance, cell fate and differentiation were measured (Fig. 6). The expression levels of the putative CSCs markers, MSI2 (musahi 2), CD166, CD151, SOX2, CD44 and Kit were evaluated in MM-CSCs after a 24 h treatment with WFA. These markers have been previously used to isolate CSCs and are currently believed to mimic “stemness”.11,33–39MSI2, CD166, CD151 and SOX2 were downregulated in response to WFA treatment, whereas CD44 and Kit were upregulated. The downregulation of MSI2, CD166, CD151 and SOX2 indicated that WFA might reduce the stemness of MM-CSCs. As for CD44 and Kit, although significant upregulation was observed, it has been previously shown that CD44 and Kit play roles not only in the maintenance of stem cells but also in the regulation of their differentiation. The mRNA levels of CD44 and Kit increased strongly with the differentiation of natural killer (NK) cells.40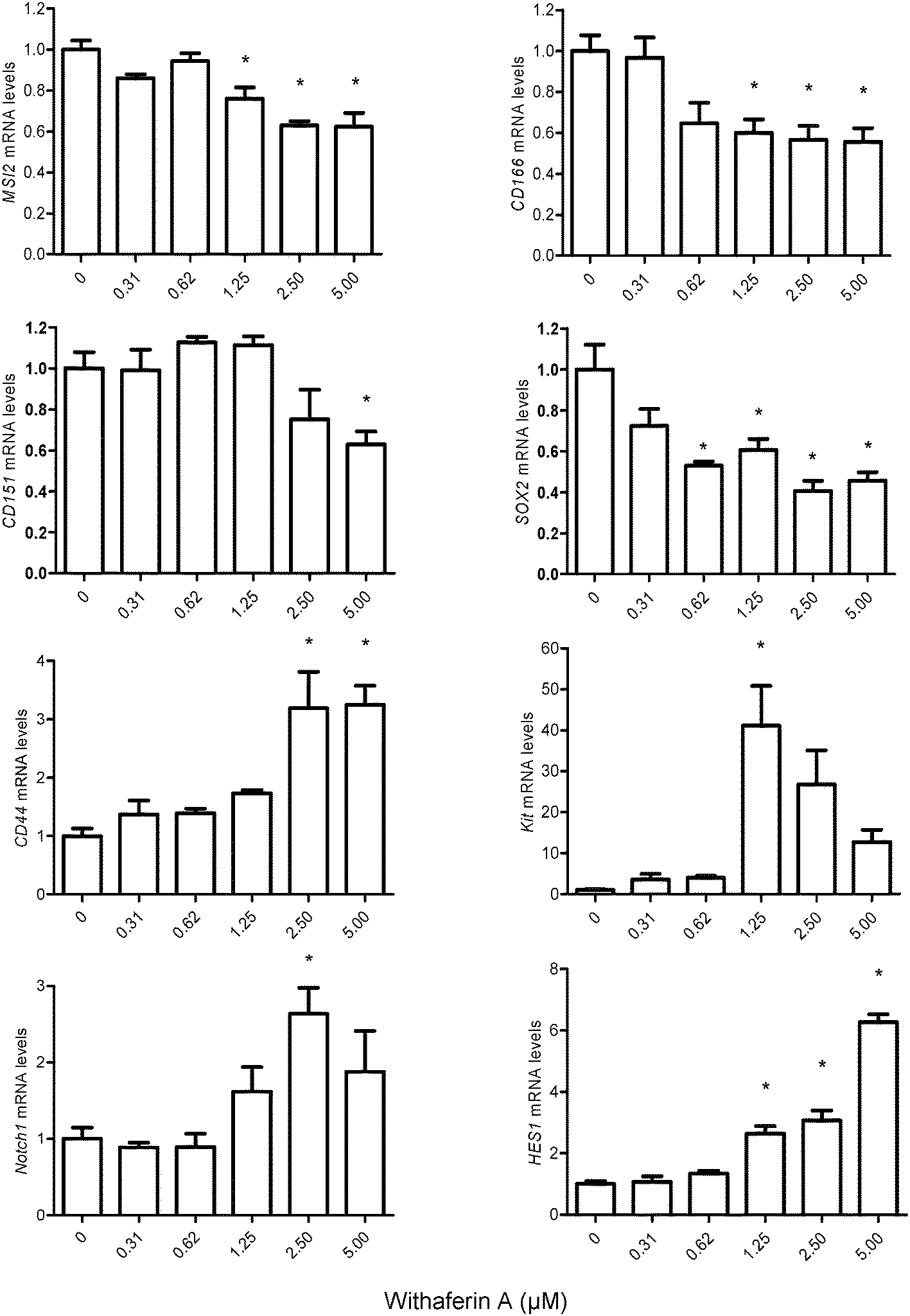 | ||
| Fig. 6 Effect of WFA on the mRNA level of genes involved in stem cell regulation. MM-CSCs were treated with vehicle control or increasing concentrations of WFA for 24 h. Relative mRNA expression of MSI2, CD166, CD151, SOX2, CD44, Kit, Notch1, and HES1 in MM-CSCs after WFA treatment is shown. Each bar represents the mean ± SEM of 3 independent experiments. *Values significantly different from vehicle control, as measured by one-way ANOVA followed by Bonferroni multiple comparison test (p < 0.05). | ||
Notch signaling has been demonstrated to regulate cell-fate decisions, and favours the differentiation of NK cells over B-cells from a common progenitor.41–44 The mRNA level of genes involved in the Notch pathway,45–47 including Notch1, HES1 and RUNX1 (data not shown) were significantly upregulated in response to WFA. As these genes are Notch targets, the results indicated that Notch signaling pathway might be activated in response to WFA treatment. Taken together, the downregulation of CSC markers, the upregulation of specific NK cell differentiation markers and that of Notch targets in response to WFA treatment, indicated that WFA might promote the differentiation of MM-CSCs towards a lymphoid lineage.
Overall, these results suggest that WFA may be promoting the differentiation of MM-CSCs towards a NK cell lineage as the observed gene expression alterations are reminiscent of NK cell differentiation. Although terminal differentiation of a NK cell lineage from MM-CSCs is not yet observed, further studies are required to elucidate the stage of differentiation WFA is capable of inducing in MM-CSCs. In addition, stemness reduction has been argued to be one important strategy to combat the tumorigenic aspect of CSCs.48 Thus, the reduction of CSCs stemness markers observed in MM-CSCs in response to WFA may suggest that WFA is capable of reducing self-renewal of MM-CSCs.
Conclusions
The induction of CSCs differentiation has been proposed as a therapy for the management of cancer. Differentiation therapy has proven highly plausible and beneficial with the use of all-trans retinoic acid to treat promyelocytic leukemia. Thus, the proof of concept exists, and the current thought is to apply it to CSCs. CSCs have been demonstrated to exhibit high self-renewal capabilities. Therefore, diminishing these capabilities would theoretically render CSCs less malignant. In this study, WFA was demonstrated to induce cell death in MM-CSCs, and at relatively low WFA doses was capable of inducing MM-CSCs differentiation as demonstrated by the clear morphological changes and alterations in gene expression levels reminiscent of hematopoietic stem cell differentiation. The ability of WFA to induce both MM-CSCs death and differentiation, two major processes in the progression of cancer, renders it a molecule valuable for further investigation in relevant MM in vivo models.Acknowledgements
The authors would like to thank Ms. Hieu Hien Tran for providing assistance in data collection. This work is part of the European COST Action CM1106: “Chemical Approaches to Targeting Drug Resistance in Cancer Stem Cells”. The authors are grateful to Dr Nadine Martinet for creating the compound library.References
- S. V. Rajkumar, Nat. Rev. Clin. Oncol., 2011, 8, 479–491 CrossRef CAS PubMed.
- G. W. Basak and E. Carrier, Biol. Blood Marrow Transplant., 2010, 16, 587–594 CrossRef PubMed.
- R. Hajek, S. A. Okubote and H. Svachova, Br. J. Haematol., 2013, 163, 551–564 CrossRef PubMed.
- J. Kellner, B. Liu, Y. Kang and Z. Li, J. Hematol. Oncol., 2013, 6, 91 CrossRef PubMed.
- D. Zieker, S. Buhler, Z. Ustundag, I. Konigsrainer, S. Manncke, K. Bajaeifer, J. Vollmer, F. Fend, H. Northoff, A. Konigsrainer and J. Glatzle, Langenbecks Arch. Chir., 2013, 398, 603–608 CrossRef PubMed.
- S. Mondal, S. Bandyopadhyay, M. K. Ghosh, S. Mukhopadhyay, S. Roy and C. Mandal, Anti-Cancer Agents Med. Chem., 2012, 12, 49–75 CrossRef CAS PubMed.
- M. Herreros-Villanueva, T. K. Er and L. Bujanda, Pancreas, 2015, 44, 918–924 CrossRef CAS PubMed.
- W. Vanden Berghe, L. Sabbe, M. Kaileh, G. Haegeman and K. Heyninck, Biochem. Pharmacol., 2012, 84, 1282–1291 CrossRef CAS PubMed.
- S. H. Kim and S. V. Singh, Cancer Prev. Res., 2014, 7, 738–747 CrossRef CAS PubMed.
- M. E. Issa, S. Berndt, G. Carpentier, J. M. Pezzuto and M. Cuendet, Cancer Biol. Ther., 2016, 17, 966–975 CrossRef CAS PubMed.
- J. P. Medema, Nat. Cell Biol., 2013, 15, 338–344 CrossRef CAS PubMed.
- W. Matsui, C. A. Huff, Q. Wang, M. T. Malehorn, J. Barber, Y. Tanhehco, B. D. Smith, C. I. Civin and R. J. Jones, Blood, 2004, 103, 2332–2336 CrossRef CAS PubMed.
- N. W. Roehm, G. H. Rodgers, S. M. Hatfield and A. L. Glasebrook, J. Immunol. Methods, 1991, 142, 257–265 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. E. Issa, S. R. Hall, S. N. Dupuis, C. L. Graham, D. L. Jakeman and K. B. Goralski, Anti-Cancer Drugs, 2014, 25, 255–269 CrossRef CAS PubMed.
- J. M. Klco, D. H. Spencer, T. L. Lamprecht, S. M. Sarkaria, T. Wylie, V. Magrini, J. Hundal, J. Walker, N. Varghese, P. Erdmann-Gilmore, C. F. Lichti, M. R. Meyer, R. R. Townsend, R. K. Wilson, E. R. Mardis and T. J. Ley, Blood, 2013, 121, 1633–1643 CrossRef CAS PubMed.
- M. Li, X. Liu, X. Sun, Z. Wang, W. Guo, F. Hu, H. Yao, X. Cao, J. Jin, P. G. Wang, J. Shen and Z. Li, Inflammation, 2013, 36, 888–896 CrossRef CAS PubMed.
- V. Thayanithy, C. Park, A. L. Sarver, R. V. Kartha, D. M. Korpela, A. J. Graef, C. J. Steer, J. F. Modiano and S. Subramanian, PLoS One, 2012, 7, e43720 CAS.
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS PubMed.
- D. Baddeley, D. Crossman, S. Rossberger, J. E. Cheyne, J. M. Montgomery, I. D. Jayasinghe, C. Cremer, M. B. Cannell and C. Soeller, PLoS One, 2011, 6, e20645 CAS.
- T. Koressaar and M. Remm, Bioinformatics, 2007, 23, 1289–1291 CrossRef CAS PubMed.
- A. Untergasser, I. Cutcutache, T. Koressaar, J. Ye, B. C. Faircloth, M. Remm and S. G. Rozen, Nucleic Acids Res., 2012, 40, e115 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- M. K. McKenna, B. W. Gachuki, S. S. Alhakeem, K. N. Oben, V. M. Rangnekar, R. C. Gupta and S. Bondada, Cancer Biol. Ther., 2015, 16, 1088–1098 CrossRef CAS PubMed.
- L. P. Yco, G. Mocz, J. Opoku-Ansah and A. S. Bachmann, Biochem. Insights, 2014, 7, 1–13 Search PubMed.
- N. Malara, D. Foca, F. Casadonte, M. F. Sesto, L. Macrina, L. Santoro, M. Scaramuzzino, R. Terracciano and R. Savino, Cell Cycle, 2008, 7, 3235–3245 CrossRef CAS PubMed.
- A. Singh and J. Settleman, Oncogene, 2010, 29, 4741–4751 CrossRef CAS PubMed.
- S. Okamoto, T. Tsujioka, S. I. Suemori, J. I. Kida, T. Kondo, Y. Tohyama and K. Tohyama, Cancer Sci., 2016, 107, 1302–1314 CrossRef CAS PubMed.
- L. V. Nguyen, R. Vanner, P. Dirks and C. J. Eaves, Nat. Rev. Cancer, 2012, 12, 133–143 CAS.
- M. Shackleton, Semin. Cancer Biol., 2010, 20, 85–92 CrossRef CAS PubMed.
- P. Munoz, M. S. Iliou and M. Esteller, Mol. Oncol., 2012, 6, 620–636 CrossRef CAS PubMed.
- M. Ratajczak, M. Tarnowski, M. Staniszewska, T. Sroczynski and B. Banach, Minerva Med., 2010, 101, 179–191 CAS.
- C. Margaritescu, D. Pirici, I. Cherciu, A. Barbalan, T. Cartana and A. Saftoiu, J. Gastrointestin. Liver Dis., 2014, 23, 161–170 Search PubMed.
- J. Tilghman, P. Schiapparelli, B. Lal, M. Ying, A. Quinones-Hinojosa, S. Xia and J. Laterra, Neoplasia, 2016, 18, 185–198 CrossRef CAS PubMed.
- T. Fang, H. Lv, F. Wu, C. Wang, T. Li, G. Lv, L. Tang, L. Guo, S. Tang, D. Cao, M. Wu, W. Yang and H. Wang, Cancer Lett., 2016, 384, 50–59 CrossRef PubMed.
- I. V. Lundberg, S. Edin, V. Eklof, A. Oberg, R. Palmqvist and M. L. Wikberg, BMC Cancer, 2016, 16, 471 CrossRef PubMed.
- J. Jiao, A. Hindoyan, S. Wang, L. M. Tran, A. S. Goldstein, D. Lawson, D. Chen, Y. Li, C. Guo, B. Zhang, L. Fazli, M. Gleave, O. N. Witte, I. P. Garraway and H. Wu, PLoS One, 2012, 7, e42564 CAS.
- J. Taggart, T. C. Ho, E. Amin, H. Xu, T. S. Barlowe, A. R. Perez, B. H. Durham, P. Tivnan, R. Okabe, A. Chow, L. Vu, S. M. Park, C. Prieto, C. Famulare, M. Patel, C. J. Lengner, A. Verma, G. Roboz, M. Guzman, V. M. Klimek, O. Abdel-Wahab, C. Leslie, S. D. Nimer and M. G. Kharas, Nat. Commun., 2016, 7, 10739 CrossRef CAS PubMed.
- V. K. Rajasekhar, L. Studer, W. Gerald, N. D. Socci and H. I. Scher, Nat. Commun., 2011, 2, 162 CrossRef PubMed.
- A. Poli, T. Michel, M. Theresine, E. Andres, F. Hentges and J. Zimmer, Immunology, 2009, 126, 458–465 CrossRef CAS PubMed.
- A. Bigas, A. Robert-Moreno and L. Espinosa, Int. J. Dev. Biol., 2010, 54, 1175–1188 CrossRef CAS PubMed.
- K. Aoyama, C. Delaney, B. Varnum-Finney, A. D. Kohn, R. T. Moon and I. D. Bernstein, Stem Cells, 2007, 25, 2488–2497 CrossRef CAS PubMed.
- S. Stier, T. Cheng, D. Dombkowski, N. Carlesso and D. T. Scadden, Blood, 2002, 99, 2369–2378 CrossRef CAS PubMed.
- R. C. Beck, M. Padival, D. Yeh, J. Ralston, K. R. Cooke and J. B. Lowe, Biol. Blood Marrow Transplant., 2009, 15, 1026–1037 CrossRef CAS PubMed.
- M. Katoh and M. Katoh, Int. J. Oncol., 2007, 31, 461–466 CAS.
- J. T. Park, I. M. Shih and T. L. Wang, Cancer Res., 2008, 68, 8852–8860 CrossRef CAS PubMed.
- D. D. Dudley, H. C. Wang and X. H. Sun, PLoS One, 2009, 4, e6678 Search PubMed.
- V. Plaks, N. Kong and Z. Werb, Cell Stem Cell, 2015, 16, 225–238 CrossRef CAS PubMed.
Footnote |
| † The authors declare no competing interests. |
| This journal is © The Royal Society of Chemistry 2017 |