
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pre-steady-state kinetic analysis of damage recognition by human single-strand selective monofunctional uracil-DNA glycosylase SMUG1†
Alexandra A.
Kuznetsova
a,
Danila A.
Iakovlev
a,
Inna V.
Misovets
b,
Alexander A.
Ishchenko
c,
Murat K.
Saparbaev
c,
Nikita A.
Kuznetsov
 *ab and
Olga S.
Fedorova
*ab
*ab and
Olga S.
Fedorova
*ab
aInstitute of Chemical Biology and Fundamental Medicine (ICBFM), Siberian Branch of Russian Academy of Sciences, 8 Lavrentyev Ave., Novosibirsk 630090, Russia. E-mail: fedorova@niboch.nsc.ru; nikita.kuznetsov@niboch.nsc.ru; Fax: +7 383-3635153; Fax: +7 383-3635153; Tel: +7 383-3635175 Tel: +7 383-3635174
bDepartment of Natural Sciences, Novosibirsk State University (NSU), 2 Pirogova St., Novosibirsk 630090, Russia
cGroupe “Réparation de l’ADN”, Université Paris-Sud XI, UMR8200 CNRS, Institute Gustave Roussy, Villejuif Cedex F-94805, France
First published on 11th October 2017
Abstract
In all organisms, DNA glycosylases initiate base excision repair pathways resulting in removal of aberrant bases from DNA. Human SMUG1 belongs to the superfamily of uracil-DNA glycosylases catalyzing the hydrolysis of the N-glycosidic bond of uridine and uridine lesions bearing oxidized groups at C5: 5-hydroxymethyluridine (5hmU), 5-formyluridine (5fU), and 5-hydroxyuridine (5hoU). An apurinic/apyrimidinic (AP) site formed as the product of an N-glycosylase reaction is tightly bound to hSMUG1, thus inhibiting the downstream action of AP-endonuclease APE1. The steady-state kinetic parameters (kcat and KM; obtained from the literature) correspond to the enzyme turnover process limited by the release of hSMUG1 from the complex with the AP-site. In the present study, our objective was to carry out a stopped-flow fluorescence analysis of the interaction of hSMUG1 with a DNA substrate containing a dU:dG base pair to follow the pre-steady-state kinetics of conformational changes in both molecules. A comparison of kinetic data obtained by means of Trp and 2-aminopurine fluorescence and Förster resonance energy transfer (FRET) detection allowed us to elucidate the stages of specific and nonspecific DNA binding, to propose the mechanism of damaged base recognition by hSMUG1, and to determine the true rate of the catalytic step. Our results shed light on the kinetic mechanism underlying the initiation of base excision repair by hSMUG1 using the “wedge” strategy for DNA lesion search.
Introduction
Human SMUG1 (hSMUG1; single-strand selective monofunctional uracil-DNA glycosylase) is a monofunctional DNA glycosylase belonging to the highly conserved superfamily of uracil-DNA glycosylases (UDGs) specialized for antimutational uracil removal in the base excision DNA repair pathway (BER).1,2 Initially, hSMUG1 was found to be more selective for single-stranded DNA (ssDNA),3 but later it was found that hSMUG1 has higher affinity for and activity toward uracil-containing double-stranded DNA (dsDNA) substrates.4,5 Uracil is generated in DNA because of spontaneous deamination of cytosine or misincorporation of dUMP instead of dTMP in the course of replication. Aside from uracil, SMUG1 also excises oxidized 5-hydroxymethyluracil (5hmU), 5-formyluracil (5fU), and 5-hydroxyuracil (5hoU) but is inactive toward thymine (5-methyluracil).6,7 The 5hmU base is produced via oxidation of thymine.8 Additionally, 5hmU is formed in epigenetic processes via deamination of 5hmC catalyzed by AID/APOBEC enzymes. This damaged base induces G:C→A:T mutation by insertion of a noncognate nucleotide, dAMP, opposite 5hmU during DNA replication. Its subsequent oxidation produces 5fU, whose ionized form is coupled with incoming dGTP in an antiwobble pair.9,10The superfamily of UDGs is classified into six families by substrate specificity, sequence homology, and structural similarity.11–13 Alignment of major motifs and structural organization of hSMUG1 and UDGs from other structural families revealed that the C-terminal motif of hSMUG1 contains the conserved histidine residue of UNG family 1. On the other hand, the N-terminal catalytic motif of hSMUG1 contains the asparagine that is found in MUG family 2. Therefore, hSMUG1 is a member of structural family 3, which represents the hybrid type of the active site.12
Mutational and structural analysis together with steady-state kinetic studies have revealed that in hSMUG1 Asn85 and His239 catalyze the cleavage of the N-glycosidic bond. Asn163 and Phe98 discriminate pyrimidine bases through π–π stacking with Phe98 and specific hydrogen bonding to the Asn163. The Gly87–Met91 region recognizes the C5 substituent through water-bridged (uracil) or direct (hoU, hmU, and fU) hydrogen bonds.14 In hSMUG1, the 239–249 sequence serves as a “wedge” penetrating the DNA double helix in the region of a specific site.5,14–18 Formation of the catalytic complex leads to the dissociative SN1-like cleavage of the N-glycosidic bond of an everted damaged base placed in the active site.14,19 AP-sites formed as products of the N-glycosylase reaction are tightly bound to hSMUG1 thereby inhibiting their cleavage by AP-endonucleases.15 The same mechanism may be responsible for the catalytic efficiency of hSMUG1 as determined by enzyme turnover. The steady-state kinetic parameters kcat and KM have been identified for dU:dA (0.05–0.07 s−1 and 4.0–11.3 μM),4,15 dU:dG (0.03–0.05 s−1 and 0.5–1.3 μM)4,15 and single stranded U (0.21–0.29 s−1 and 1.7–3.0 μM) oligonucleotide substrates.4,15 For DNA-substrates containing dU:dG base pairs alternatively it was obtained that the values of kcat and KM were equal to 0.014 s−1 and 2.2 nM, respectively.14 Nevertheless, steady-state studies have been unable to characterize the real catalytic power of hSMUG1 and to obtain the values of microscopic rate constants for the separate steps in the reaction pathway from recognition of specific sites to the formation of the final products.
To gain a deeper insight into the damage recognition mechanism, we performed a pre-steady-state kinetic analysis of conformational transitions of hSMUG1 and DNA in the course of the catalytic cycle under conditions when there is no large excess of enzyme or substrate. The conformational dynamics were directly recorded by the stopped-flow technique combined with fluorescence detection. Conformational changes in the protein were monitored by changes in Trp fluorescence intensity. To examine the conformational dynamics of DNA, the fluorescent base 2-aminopurine (aPu) and dye–quencher pair FAM/BHQ1 were incorporated into DNA. A DNA duplex containing the dU:dG base pair served as a “full enzymatic cycle” DNA substrate; this cycle includes DNA binding and N-glycosidic bond cleavage. A DNA duplex containing an uncleavable analog of the natural AP-site (3-hydroxytetrahydrofuran-2-yl)methyl phosphate (F-site) instead of dU was used as a DNA product. The DNA duplex containing dC:dG enabled us to follow the conformational changes in the enzyme and DNA during the formation of the nonspecific complex. A comparison of fluorescence kinetic data obtained by Trp, aPu, and FRET detection allowed us to elucidate the stages of specific and nonspecific DNA binding and to propose the mechanism of damaged-base recognition by hSMUG1.
Materials and methods
Construction of hSMUG1 expression vector
Total RNA was isolated from HeLa cells according to the instruction of an Oligotex Direct mRNA Kit (Qiagen, Hilden, Germany). The quality of RNA was assessed using the A260/A280 ratio, and the integrity was evaluated on 1% agarose gel. The RNAs were then reverse transcribed into cDNA using a Boehringer Mannheim cDNA synthesis kit (Boehringer Mannheim GmbH, Mannheim, Germany), following the manufacturer's instructions. The PCR DNA fragment, which contained the hSMUG1 gene and NdeI and BamHI restriction sites, was obtained using specific primers GCGGCCTGGTGCCGCGCGGCAGCCATCCCCAGGCTTTCCTGCTGGGG and GCTTGTCGACGGAGCTCGAATTCGGATCTCATTTCAACAGCAGTGGCAGCAGC. The size of the DNA fragment was 864![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) bp. The PCR products were separated on 1.0% agarose/TAE gels, excised and purified using a QIAquick PCR purification kit (Qiagen, Hilden, Germany). Finally, the gene of hSMUG1 was cloned into the pET28c expression vector using NdeI and BamHI restriction sites and sequenced.
bp. The PCR products were separated on 1.0% agarose/TAE gels, excised and purified using a QIAquick PCR purification kit (Qiagen, Hilden, Germany). Finally, the gene of hSMUG1 was cloned into the pET28c expression vector using NdeI and BamHI restriction sites and sequenced.
Protein expression and purification
The full-size hSMUG1 was isolated from Escherichia coli Rosetta 2 cells transformed with plasmid pET28c-hSMUG1. Cells of E. coli Rosetta 2 were grown in Luria-Bertani (LB) medium (1 L) containing 50 μg mL−1 kanamycin at 37 °C to an optical density of 0.6–0.7 at 600 nm. After that, the temperature was lowered to 20 °C, and transcription was induced by addition of isopropyl-β-D-thiogalactopyranoside to 0.2 mM. After the induction, the cells were incubated for 16 h and then centrifuged (12000 rpm, 10 min). A cell suspension was prepared in 30 mL of buffer I (20 mM HEPES-NaOH, pH 7.8) containing 100 mM NaCl and a protease inhibitor cocktail (Complete, Germany). The cells were lysed using a Thermo French Pressure Cell Press. All the subsequent procedures were conducted at 4 °C. The cell lysate was centrifuged (30000 rpm, 40 min), and the supernatant was loaded onto column I (Q-Sepharose Fast Flow, Amersham Biosciences, Sweden) with subsequent washing with buffer solution I (20 mM HEPES-NaOH, pH 7.8) containing 100 mM NaCl. Fractions containing the protein were collected and loaded onto column II (HiTrap-Helating™, Amersham Biosciences, Sweden) in buffer solution II (20 mM HEPES-NaOH, pH 7.8) containing 500 mM NaCl and 20 mM imidazole. Chromatography was run in buffer solution II and a linear gradient of 20 → 500 mM imidazole. The solution's absorbance was detected at a wavelength of 280 nm. The protein purity was determined by gel electrophoresis. Fractions containing the hSMUG1 protein were dialyzed against a buffer (20 mM HEPES-NaOH, pH 7.5, 1 mM EDTA, 1 mM dithiothreitol, 250 mM NaCl, 50% glycerol) and stored at −20 °C. The protein concentration was calculated based on the optical density of the protein solution at 280 nm and a molar extinction coefficient of 28460 M−1 cm−1.20
All the experiments on the enzymatic reaction were conducted in a buffer consisting of 50 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, and 7% glycerol at 25 °C.
Oligodeoxynucleotides (ODNs)
The sequences of the DNA substrates used in this work are presented in Table 1. The oligonucleotides were synthesized by standard phosphoramidite methods on an ASM-700 synthesizer (BIOSSET Ltd, Novosibirsk, Russia) using phosphoramidites purchased from Glen Research (Sterling, VA). The synthetic oligonucleotides were uncoupled from the solid support with ammonium hydroxide according to the manufacturer's protocol. The deprotected oligonucleotides were purified by high-performance liquid chromatography. The purity of ODNs exceeded 98% as estimated by electrophoresis in a 20% denaturing polyacrylamide gel after staining with the Stains-All dye (Sigma-Aldrich). Concentrations of the oligonucleotides were determined by means of A260. The ODN duplexes were prepared by annealing of modified and complementary strands in a 1:1 molar ratio in a buffer consisting of 50 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, and 7% glycerol.
| Shorthand | Sequence |
|---|---|
| a aPu is 2-aminopurine, FAM is 6-carboxyfluorescein, BHQ1 is black hole quencher. | |
| X-substrate, X = U | 5′-GCTCAXGTACAGAGCTG-3′ |
| X-ligand, X = F-site and C | 3′-CGAGTGCATGTCTCGAC-5′ |
| X-aPu-substrate, X = U | 5′-GCTCAX(aPu)TACAGAGCTG-3′ |
| X-aPu-ligand, X = F-site | 3′-CGAGTGCATGTCTCGAC-5′ |
| X-FRET-substrate, X = U | 5′-FAM-GCTCAXGTACAGAGCTG-3′ |
| X-FRET-ligand, X = F-site and C | 3′-CGAGTGCATGTCTCGAC-BHQ1-5′ |
Stopped-flow fluorescence measurements
These measurements with fluorescence detection were carried out mostly as described previously.21,22 In brief, we used a SX.18MV stopped-flow spectrometer (Applied Photophysics Ltd, UK) equipped with a 150 W Xe arc lamp and an optical cell with 2 mm path length. The dead time of the instrument is 1.4 ms. The fluorescence of Trp was excited at λex = 290 nm and monitored at λem > 320 nm as transmitted by the filter WG-320 (Schott, Mainz, Germany). When we used substrates containing aPu residues, fluorescence was excited at a wavelength of 310 nm and detected at wavelengths longer than 370 nm (LG-370 filter, Corion, USA). If a 6-carboxyfluorescein (FAM) fluorophore was present in the ODN duplex (FRET-X-substrates), the wavelength λex = 494 nm was used to excite these residues, and their emission was analyzed at λem > 515 nm (Schott filter OG-515). All the experiments were conducted at 25 °C in the buffer 50 mM Tris–HCl pH 7.5, 50 mM KCl, 1.0 mM EDTA, 1.0 mM DDT, and 7% glycerol (v/v).In a standard procedure, the solution of hSMUG1 was placed in one instrument's syringe and rapidly mixed in the reaction chamber with the substrate from another syringe. The reported concentrations of reactants are those in the reaction chamber after mixing. Typically, each trace shown in the figures is the average of four or more fluorescence traces recorded in individual experiments. In the figures, if necessary for better presentation, the curves were manually moved apart. This procedure does not affect the results of fitting because the background fluorescence is fitted separately for each curve.
Polyacrylamide gel electrophoresis (PAGE) time-course experiments
The reaction mixture contained 5.0 μM U-substrate, 1.0 μM hSMUG1, 50 mM Tris–HCl, pH 7.5, 50 mM KCl, 1.0 mM EDTA, 1.0 mM DDT, and 7% glycerol (v/v). The U-strand was 5′-end labelled using phage T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and [γ-32P]ATP (4500 Ci mol−1) according to the manufacturer's protocol. Labeled oligonucleotide was purified on Sephadex G-25 columns (GE Healthcare, Little Chalfont, UK). The reaction was initiated by adding the enzyme. Aliquots (2 μL) of the reaction mixture were withdrawn and immediately quenched with 2 μL of gel-loading dye containing 7 M urea solution with 0.1% bromophenol blue and 0.1% xylene cyanole. Then, 1.0 μL of 1.0 M NaOH was added with incubation at 56 °C for 15 min to hydrolyze both 5′- and 3′-phosphodiester bonds at AP-sites. Alkali treatment was performed for all samples before gel loading (including zero time point) and resulted in complete splitting of AP-sites generated by hSMUG1. The solution was neutralized with an equivalent amount of hydrochloric acid and loaded on a 20% (w/v) polyacrylamide/7 M urea gel. Conversion of the substrate to the product was analyzed by autoradiography on an Agfa CP-BU X-ray film (Agfa-Geavert, Belgium) and quantified by scanning densitometry in the Gel-Pro Analyzer software, v. 4.0 (Media Cybernetics, USA). The measurement error commonly did not exceed 20%.Global fitting of stopped-flow data
The sets of kinetic curves obtained at different concentrations of the reactants were analyzed in the DynaFit software (BioKin, Pullman, WA)23 as described elsewhere.24–26 This approach is based on fluorescence intensity variation in the course of the reaction owing to the sequential formation and further transformation of the initially formed DNA–enzyme complex and its conformers. The concentrations of each species in the mechanisms are described by a set of differential equations according to the kinetic scheme. The stopped-flow fluorescence traces were directly fitted to the fluorescence intensity (Fc) at any reaction time point (t) as the sum of the background fluorescence (Fb) and fluorescence intensity values of each i-intermediate complex (Fi) formed by the enzyme with DNA.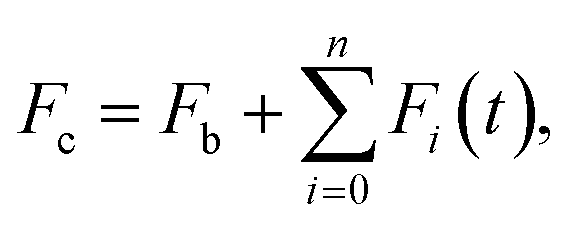 | (1) |
| Fi(t) = fi × [Ei(t)], | (2) |
The software performs numerical integration of a system of ordinary differential equations with subsequent nonlinear least-squares regression analysis. In the fits, we optimized all relevant rate constants for the forward and reverse reactions as well as the specific molar response factors for all the intermediate complexes. During the data processing, the kinetic information was obtained from temporal behavior of the fluorescence intensity, not from the amplitudes of the specific signal contributions. The response factors for different conformers resulting from the fits were not used for determination of the equilibrium constants but rather provided additional information on the fluorescence intensity variations in different complexes and conformers.
Homology modeling and sequence alignment
A model of the structure of hSMUG1 was built by homology modelling in the SWISS-MODEL environment using the structure-based alignment (http://swissmodel.expasy.org).27 Structures of GmeSMUG1 from Geobacter metallireducens (Protein Data Bank codes 5H98 and 5H9I)28 and xSMUG1 from Xenopus laevis (Protein Data Bank codes 1OE4 and 1OE5)5 served as templates. For each model generated based on the selected templates SWISS-MODEL provides the model coordinates along with relevant information to assess the modelling process and to expect the accuracy of the model: the target-template alignment, a step-by-step modelling log, information about the oligomeric state, binding the ligands and cofactors, as well as QMEAN model quality estimation. The “good” models, which are comparable to the experimental structure, reach QMEAN value −0.65.29 An alignment of amino acid sequences of hSMUG1, xSMUG1, and GmeSMUG1 was produced in Clustal Omega.30Results and discussion
Rational approaches to the conformational dynamic analysis
A comparison of pro- and eukaryotic DNA glycosylases that belong to different structural families has revealed that all these enzymes have common global structural features of DNA lesion recognition.31 In general, the mechanism of catalytic-complex formation includes initial DNA binding and formation of a nonspecific complex, DNA bending, damaged base flipping out from the double helix, and its placement into the enzyme's active site.16,21,32–34 Most of the DNA glycosylases insert some amino acids into the resulting void in DNA. Moreover, recent studies suggest that some DNA glycosylases diffusively scan DNA using a “wedge” amino acid residue to identify damaged nucleotides.16–18,32,34–37 The function of the “wedge” amino acid is to sense damaged DNA bases before their eversion into the enzyme's active site. Because mutual conformational changes of the enzyme and DNA play an important role in the damage recognition and in the formation of a catalytic complex, in the present work, we performed a pre-steady-state kinetic analysis of protein–DNA interactions with fluorescent detection to resolve processes of conformational adjustments of DNA glycosylase and DNA. For this purpose, the intrinsic protein fluorescence from tryptophan residues and DNA fluorescence from the incorporated reporters were recorded by the stopped-flow technique.The hSMUG1 molecule contains four Trp residues: Trp62, Trp107, Trp142, and Trp251. Because no structure of hSMUG1 is available, we used the high sequence homology among hSMUG1, xSMUG1, and GmeSMUG1 (Fig. 1A) to obtain a structure of the hSMUG1–DNA complex by homology modelling. It follows from the structures of the xSMUG1–DNA complex5 that the enzyme bound the ends of the DNA duplex. Therefore, these data do not elucidate the lesion recognition state of the enzyme and DNA. Nevertheless crystal-soak experiments with free Ura bases indicate the possible position of the damaged base in the active site of the enzyme. Structural superposition of the noncovalent complex of xSMUG1 with a Ura base (Protein Data Bank code 1OE5)5 and the model structure of hSMUG1 revealed that Trp62, Trp107, and Trp142 are located inside the protein globule and far away (at least 11.9 Å between the N1 atom of uracil and the Cα atom of the Trp142 residue) from the damaged-base-binding pocket of the enzyme (Fig. 1B). Only Trp251 is located in close proximity to the intercalating loop of the enzyme (amino acids 239–249), and therefore this residue most likely can be sensitive to DNA binding.
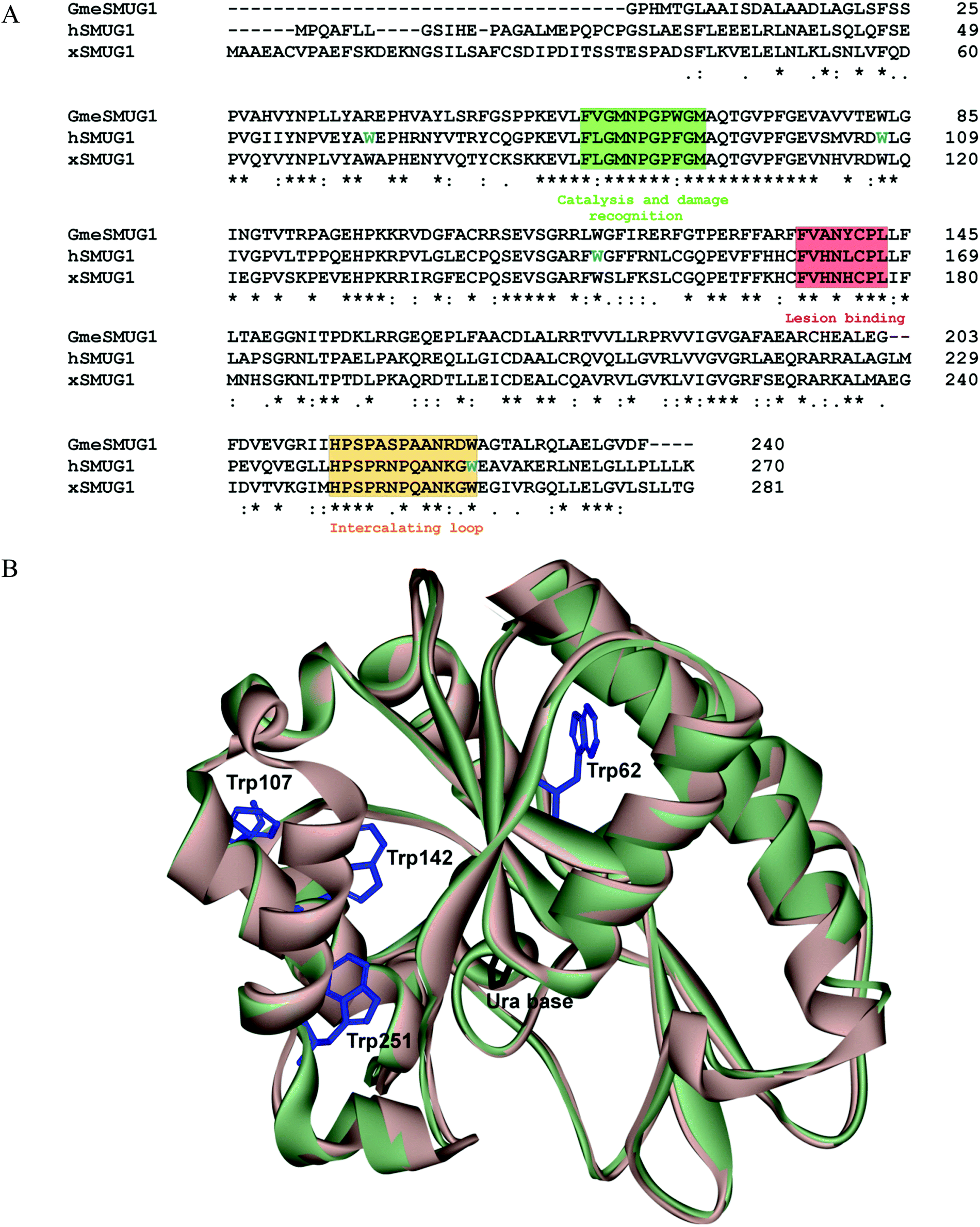 | ||
| Fig. 1 (A) An alignment of sequences of hSMUG1, xSMUG1, and GmeSMUG1. Motifs responsible for lesion binding (red), recognition using the intercalating loop (brown), and catalysis (green) of damaged-base processing are indicated with colors. Trp residues of hSMUG are colored blue. Asterisks indicate identical residues, colons denote conserved residues; dots are residues with at least some physicochemical properties conserved. (B) Structural superposition of the noncovalent complexes of xSMUG1 with a Ura base (Protein Data Bank code 1OE5)5 and model structure of hSMUG1. | ||
To study the conformational transitions in DNA substrates, two approaches were employed. First, the fluorescent analog of DNA base aPu was incorporated on the 3′-side of a damaged nucleotide in DNA substrates as a reporter group of the conformational transitions in the damaged DNA strand near the damaged nucleotide. The aPu residue has a high quantum yield of fluorescence in an aqueous solution, but it is highly quenched when aPu is incorporated into DNA or transferred into a nonpolar environment.38–40 Accordingly, aPu is highly sensitive to the local melting of DNA strands around the damaged nucleotide, to nucleotide flipping out, and to insertion of an enzyme's amino acids into DNA.41–43 Second, Förster resonance energy transfer (FRET) DNA substrates modified at their 5′ termini with the dye–quencher pair FAM/BHQ1 were used for FRET measurements. FRET analysis revealed changes in the distance between the dye and quencher in the processes of DNA helix distortion during the formation of the complex between hSMUG1 and DNA.
Interaction of hSMUG1 with undamaged DNA
The parameters for binding of hSMUG1 to undamaged DNA were obtained by recording the changes of Trp fluorescence and FRET signals using the C-ligand and C-FRET-ligand, respectively. When the C-aPu-ligand was studied, the changes of aPu fluorescence were not observed, indicating the absence of DNA double helix distortion near the aPu residue.In the kinetic curves obtained during the interaction of hSMUG1 with the undamaged C-ligand, a fast single phase of an increase in the Trp fluorescence intensity up to 5 ms is seen (Fig. 2A). When hSMUG1 interacts with the C-FRET-ligand (Fig. 2B), a fast increase in the FRET signal is observed. These data most likely correspond to collisional nonspecific binding of the enzyme to DNA, resulting in the movement of the intercalating loop (239–249) with a change of the Trp251 environment and shielding of FAM fluorescence from the quenching with BHQ1. Both types of data were fitted to a kinetic mechanism consisting of a one-step equilibrium (Scheme 1). The rate constants obtained by the fitting procedure are presented in Table 2.
 | ||
| Fig. 2 Changes in Trp fluorescence intensity (A) and in the FRET signal (B) during the interaction of hSMUG1 with undamaged DNA. Concentrations of the enzyme and C-ligands are indicated in the panels. Jagged traces represent experimental data; smooth curves are the result of the fitting procedure as described in Materials and methods. | ||
 | ||
| Scheme 1 Formation of a nonspecific complex of hSMUG1 with undamaged DNA. | ||
| Constants | Trp | FRET |
|---|---|---|
| K i = ki/k−i, i is the number of a step. | ||
| k 1, M−1 s−1 | (170 ± 60) × 106 | (120 ± 50) × 106 |
| k −1, s−1 | 1260 ± 460 | 850 ± 170 |
| K 1, M−1 | 0.13 × 106 | 0.14 × 106 |
As shown in Table 2, the values of k1 in both cases are in agreement with the typical rate constants of diffusion-controlled DNA–protein bindings.44–48 The rate constant of DNA–protein dissociation was found to be ∼1000 s−1, and a close to analogous value was obtained previously for DNA–glycosylase Fpg. from E. coli (2700 s−1).47 Thus, the human DNA–glycosylase SMUG1 is bound to DNA in a diffusion-controlled process and forms a nonspecific complex, probably by intercalation of the loop (positions 239–249) into the DNA helix.
Interaction of hSMUG1 with the stable analog of a DNA product
N-Glycosylase reaction catalyzed by hSMUG1 results in the formation of an AP-site as a final product. The AP-site is chemically unstable; therefore, in many studies, its stable analog (3-hydroxytetrahydrofuran-2-yl)methyl phosphate (F-site) has been successfully used.49–51 To detect the conformational changes of hSMUG1 during binding of an abasic DNA product we also used the DNA ligand containing an F-site. The interaction of the enzyme with the F-ligand led to a biphasic change in the Trp fluorescence intensity (Fig. 3A). The initial phase of the kinetic curves up to 5 ms represented a small increase in the fluorescence signal. We assumed that this phase corresponds to the movement of the intercalating loop (positions 239–249) in the course of nonspecific-complex formation as shown in the case of a C-ligand. The decrease in the Trp fluorescence intensity in the second phase most likely reflects conformational changes induced by the formation of new specific contacts between the enzyme's active site and an F-ligand. | ||
| Fig. 3 Interaction of hSMUG1 with the stable analog of a DNA product, containing an F-site. Changes in Trp fluorescence intensity (A), aPu fluorescence intensity (B), and FRET signal (C). Concentrations of the enzyme and DNA ligands are indicated in the panels. Jagged traces represent experimental data; smooth curves are the result of the fitting procedure described in Materials and methods. | ||
The nature of these specific interactions was elucidated by analysis of aPu fluorescence because of high sensitivity of an aPu residue to the microenvironment. Mixing of an F-aPu-ligand with hSMUG1 led to a biphasic change in the aPu fluorescence intensity (Fig. 3B) represented by a humped curve. The initial fast increase in the aPu fluorescence intensity up to 5 ms reveals the increase of hydrophilicity of the aPu environment and correlates with the first phase in Trp fluorescence and FRET time courses. In general, these data support the notion about the nature of this phase as the initial DNA binding: movement of the intercalating loop inducing local melting of the DNA helix in the vicinity of the F-site. Our previous data on structurally unrelated DNA glycosylases hOGG152 and Fpg53 have revealed that the fast initial increase in the aPu fluorescence is caused by eversion of the damaged base from the DNA helix. Therefore, we propose that during this time interval, the F-site can also be flipped out from DNA.
The next phase is characterized by a decrease in the fluorescence intensity within 0.1 s. It is likely that this phase is associated with the insertion of intercalating amino acids into the void in the DNA helix formed by an F-site. The void-filling process leads to transition of the aPu base into a more hydrophobic environment leading to a decrease in the aPu fluorescence intensity within 0.1 s. The kinetic curves obtained for both Trp and aPu fluorescence data were fitted to a two-step equilibrium mechanism (Scheme 2) resulting in calculation of rate and equilibrium constants (Table 3).
 | ||
| Scheme 2 Interaction of hSMUG1 with an F- or F-aPu-ligand. | ||
| Constants | Trp | aPu | FRET |
|---|---|---|---|
| K i = ki/k−i, i is the number of a step. | |||
| k 1, M−1 s−1 | (120 ± 30) × 106 | (130 ± 10) × 106 | (200 ± 50) × 106 |
| k −1, s−1 | 1000 ± 250 | 490 ± 60 | 650 ± 70 |
| K 1, M−1 | 0.12 × 106 | 0.26 × 106 | 0.31 × 106 |
| k 2, s−1 | 60 ± 23 | 25 ± 16 | 7.7 ± 3.5 |
| k −2, s−1 | 24 ± 7 | 55 ± 21 | 18.4 ± 0.9 |
| K 2 | 2.5 ± 1.7 | 0.45 ± 0.46 | 0.42 ± 0.21 |
| K 1 × K2, M−1 | 0.3 × 106 | 0.12 × 106 | 0.13 × 106 |
| k 3, s−1 | 0.13 ± 0.02 | ||
| k −3, s−1 | 0.067 ± 0.004 | ||
| K 3 | 1.9 ± 0.4 | ||
| K 1 × K2 × K3, M−1 | 0.25 × 106 | ||
The time course of a FRET signal obtained during the interaction of hSMUG1 with the F-FRET-ligand has three visible phases: a fast increase followed by a biphasic decrease (Fig. 3C). The fast initial phase (up to 5 ms) represents DNA binding, which likely involves the movement of the intercalating loop of the enzyme and local melting of the DNA duplex, and induces an increase in the FRET signal owing to shielding of the FAM emitter from the BHQ1 quencher. The decrease in the FRET signal at the next steps reflects a decrease in the distance between the FAM and BHQ1 residues, probably, due to DNA bending. This DNA bending proceeds simultaneously with insertion into DNA of the void-filling amino acids, as detected by means of Trp and aPu fluorescence behavior. Moreover, the second decrease phase of the FRET signal proceeding slowly in a time range of 1–100 s reveals an additional conformational adjustment of DNA in complex with the enzyme. FRET kinetic curves were fitted to Scheme 3, which contains three equilibrium steps of the FRET-F-ligand binding by hSMUG1 (Table 3).
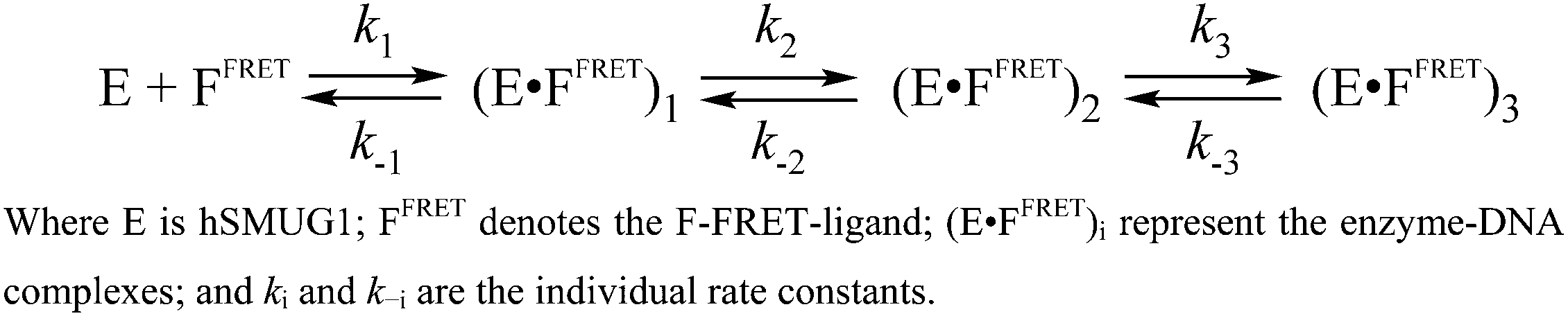 | ||
| Scheme 3 Interaction of hSMUG1 with an F-FRET-ligand. | ||
The rate constants of the formation and dissociation of a primary complex (E·F)1, k1 and k−1, are close (the difference is within the experimental error) and have values similar to those obtained for nonspecific C- and C-FRET-ligands (Table 2). Therefore, the first step is nonspecific binding resulting in local DNA melting and intercalating-loop movement. The second step where Trp and aPu fluorescence intensities as well as the FRET signal decrease can be attributed to the formation of a tight protein–DNA complex, void-filling, and DNA helix bending. The third step observed for the F-FRET-ligand at long periods (>10 s) means a slow process of DNA conformational change in complex with hSMUG1.
The interaction of hSMUG1 with DNA containing uracil
The full hSMUG1 enzymatic cycle was studied with a DNA substrate containing uracil (U-substrate). This process has to proceed through the formation of a catalytically competent complex and subsequent hydrolysis of the N-glycosidic bond of the uridine nucleotide. Trp fluorescence traces corresponding to hSMUG1 interaction with the U-substrate (Fig. 4A) revealed three phases of the process: (1) a slight increase in fluorescence intensity up to 5 ms, (2) a decrease phase up to 0.3 s, and (3) a final increase in the fluorescence intensity over a time range of 0.3–10 s. A comparison of the initial parts (up to 0.2 s) of the kinetic curves obtained for an F-ligand and U-substrate revealed that DNA binding and formation of the catalytic complex lead to similar changes in Trp fluorescence. Therefore, the final increase in Trp fluorescence intensity most likely characterizes the catalytic step of N-glycosidic bond hydrolysis.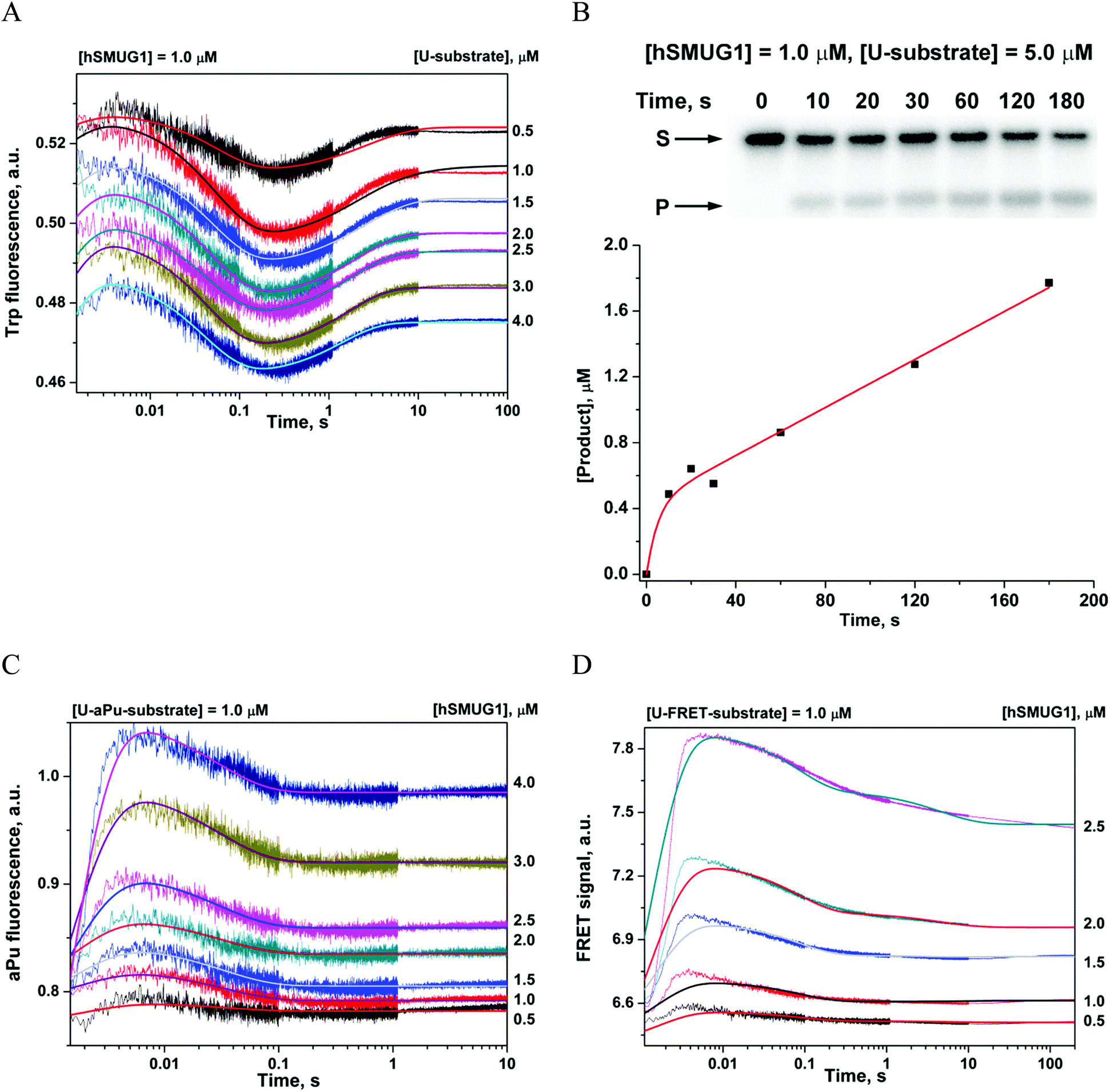 | ||
| Fig. 4 Interaction of hSMUG1 with DNA containing uracil. Changes in Trp fluorescence intensity (A). Accumulation of a product formed in the reaction of N-glycosidic bond cleavage, as determined by PAGE (B). Changes in aPu fluorescence intensity (C), and FRET signal (D). Concentrations of the enzyme and each DNA substrate are indicated in the panels. Jagged traces represent experimental data; smooth curves were constructed by the fitting procedure as described in Materials and methods. | ||
The minimal kinetic mechanism describing these fluorescence changes consists of two reversible binding steps and one irreversible step of catalysis (Scheme 4). Given that hSMUG1 binds tightly to product AP-sites, we hypothesized that the dissociation of the enzyme–product complex does not occur during the registration time (10 s), and product P stays tightly associated with the enzyme in this period. The rate and equilibrium constants obtained by Trp fluorescence trace fitting are shown in Table 4.
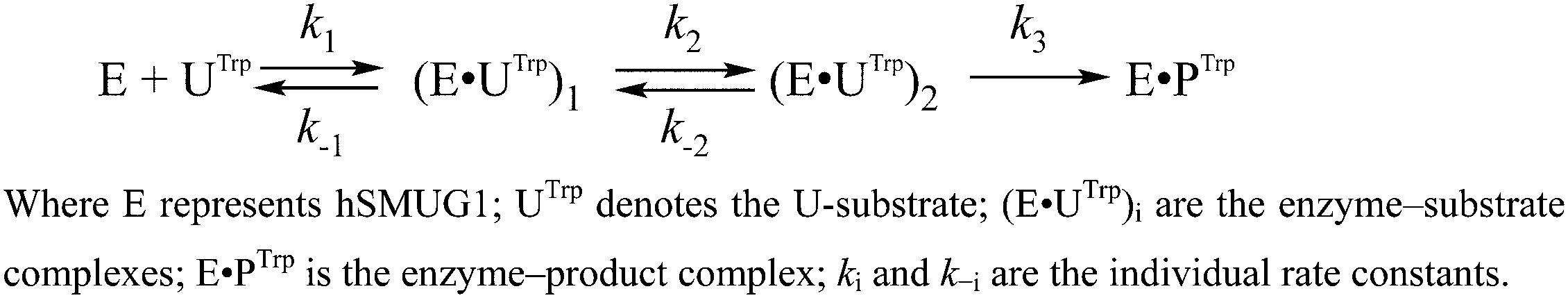 | ||
| Scheme 4 Interaction of hSMUG1 with a U-substrate. | ||
| Constants | Trp | aPu | FRET |
|---|---|---|---|
| K i = ki/k−i, i is the number of a step. | |||
| k 1, M−1 s−1 | (115 ± 20) × 106 | (130 ± 60) × 106 | (140 ± 20) × 106 |
| k −1, s−1 | 760 ± 90 | 240 ± 30 | 410 ± 20 |
| K 1, M−1 | 0.15 × 106 | 0.54 × 106 | 0.34 × 106 |
| k 2, s−1 | 30 ± 8 | 15 ± 8 | 3.6 ± 0.8 |
| k −2, s−1 | 11.5 ± 0.8 | 25 ± 6 | 9.7 ± 0.2 |
| K 2 | 2.6 | 0.6 | 0.37 |
| K 1 × K2, M−1 | 0.3 × 106 | 0.32 × 106 | 0.13 × 106 |
| k 3, s−1 | 1.7 ± 0.2 | 0.6 ± 0.1 | |
| k −3, s−1 | 0.11 ± 0.02 | ||
| K 3 | 5.4 | ||
| K 1 × K2 × K3, M−1 | 0.68 × 106 | ||
PAGE analysis of the reaction product formation (Fig. 4B) showed that in the initial region of the kinetic curves (up to 10 s), a burst is observed. This type of curve indicates the presence of a rate-limiting step after the catalytic reaction. Indeed, hSMUG1 binds tightly to product AP-sites, and steady-state rate constant kcat, which was found to be 0.014–0.05 s−1 as determined elsewhere,4,14,15 is much less than rate constant k3, which characterizes the catalytic step in Scheme 4. Therefore, we believe that changes in Trp fluorescence allow us to determine the “true” value of the catalytic rate constant.
The shape of the kinetic curves recorded by means of aPu fluorescence (Fig. 4C) and FRET (Fig. 4D) for U-aPu and U-FRET substrates, respectively, was similar to the shapes obtained for DNA containing an F-site. The similarity of kinetic curves obtained for cleavable (U-substrates) and uncleavable (F-ligands) DNAs supports the idea that catalytic reaction of N-glycosidic bond hydrolysis does not require additional conformational changes of DNA. The minimal kinetic schemes describing the observed changes in aPu fluorescence intensity (Scheme 5) and in the FRET signal (Scheme 6) were identical to those proposed for DNA containing an F-site (Table 4). It should be noted that the initial increase phase of the FRET signal is not well fitted to the kinetic scheme due to the fact that the registered process is very fast. Indeed, the dead time of the device is 1.4 ms, while the end of the growth phase occurs up to 4 ms. Therefore, this phase on the kinetic curves reflects only the end of the ongoing process. Nevertheless, the proposed mechanism allowed estimation of the values of the rate constants of this phase.
 | ||
| Scheme 5 Interaction of hSMUG1 with a U-aPu-substrate. | ||
 | ||
| Scheme 6 Interaction of hSMUG1 with a U-FRET-substrate. | ||
It should be noted that hSMUG1 removes uracil from both ssDNA and dsDNA. Control experiments with ssDNA were performed to verify the possibility of the influence of enzyme binding to ssDNA on the kinetics of interaction with dsDNA (ESI,† Fig. S1). The obtained data support the conclusion that interaction of hSMUG1 with ssDNA cannot be detected using Trp and aPu fluorescence. In the first case complex formation between hSMUG1 and ssU oligonucleotides does not provide enough quenching of Trp fluorescence by nucleic bases, probably, due to different modes of interaction with ssDNA and dsDNA. In the case of ssUaPu oligonucleotides, the aPu base is located in the aqueous polar environment and has a high level of fluorescence intensity, which does not allow the process of Ura base eversion to be registered as in the case of a double stranded U-aPu/G-substrate. Control experiments with the FAM-U/G duplex without BHQ1 were also carried out. As shown in the ESI,† Fig. S1, changes of FAM fluorescence without the BHQ1 quencher are barely noticeable in comparison with the case of the presence of the quencher.
A comparison of the rate constants obtained for DNA duplexes containing a Ura base or F-site (Tables 3 and 4) revealed that the formation of the first enzyme–substrate complex proceeds at a similar rate constant k1. By contrast, association constant K1 is slightly higher for uracil-containing DNA duplexes, thus indicating tighter binding with a specific substrate. Therefore, these data suggest that the initial DNA binding proceeds more efficiently in the case of a DNA duplex containing a Ura base. The second detected step, which consists of insertion of void-filling amino acids into DNA, has rate constant k2 that is twofold lower for DNA duplexes containing a Ura base in comparison with an F-site. These data allow us to suppose that the void-filling process depends on the nature of the damaged nucleotide, and the formation of additional contacts with the Ura base in the active site of the enzyme led to a decrease in the rate of catalytic-complex formation. The product accumulation curve detected by PAGE analysis revealed the burst phase already in the first time point up to 10 s. Although time resolution of the burst phase does not allow at what time a burst occurs before 10 s to be determined, a comparison of PAGE analysis with the Trp fluorescence data revealed increase of fluorescence also up to 10 s. Therefore a suggestion was made that the phase of the increase in Trp fluorescence reflects a catalytic process and allows a true value of the rate constant of the catalytic reaction to be calculated.
Conclusions
Combining the fluorescence data characterizing conformational transitions of hSMUG1 and DNA in the course of the catalytic cycle and PAGE analysis of product accumulation allowed us to propose a stepwise mechanism of specific DNA binding and catalysis (Fig. 5). Our kinetic characterization has determined the order of structural rearrangements in the enzyme and DNA substrate in the mechanism of action of DNA glycosylase hSMUG1. From the standpoint of the overall kinetic mechanism (Fig. 5), the first two equilibrium steps coincide with each other for all three types of fluorescence detection, implying synchronous conformational changes both in the enzyme and in the DNA substrate. At the first equilibrium binding step, hSMUG1 nonspecifically interacts with DNA, thereby causing movement of the intercalating loop of the enzyme, local DNA melting near the specific site, and probably, damage eversion from DNA. In recent years, it was reported that DNA glycosylases with different structures and specificity have common structural features of DNA lesion recognition including insertion of “wedge” amino acids into DNA.17,18,35,54–56 Therefore, the movement of the intercalating loop is probably associated with the “wedge” strategy of hSMUG1 for DNA lesion search. At the second step, the intercalating loop of SMUG1 is completely inserted into the void in DNA. It should be noted that k2 is twofold higher for DNA duplexes containing an F-site than for duplexes containing a Ura base if we compare on average the data obtained by three types of detection. This finding indicates that the rate of interaction of amino acids of the intercalating loop with the damaged nucleotide is dependent on the nature of the damage. Isomerization of the enzyme–substrate complex at this step produces a catalytically active conformation. The catalytic reaction proceeds at the third step of the interaction. The “true” value of the rate constant of the catalytic reaction was determined by Trp fluorescence changes, which is two orders higher than previously published values of kcat.14,15 Rate-limiting dissociation of the enzyme–product complex is clearly detectable by PAGE as a slow phase of product accumulation. Moreover, the third step detected by FRET analysis also reflects fine DNA tuning in the course of the product release because of a good agreement between the rate constant of this step and the value of steady-state kcat.14,15 | ||
| Fig. 5 A summary kinetic scheme of the steps detected by different fluorescent reporters in the course of interaction of hSMUG1 with DNA. Step 1 (detected by Trp, aPu fluorescence, and FRET analysis) is characterized by initial DNA binding involving a movement of the intercalating loop of the enzyme, DNA melting in the vicinity of the specific site, and possibly Ura base eversion from DNA; step 2 (detected by Trp, aPu fluorescence, and FRET analysis) consists of insertion of void-filling amino acids into DNA; step 3 (detected by Trp fluorescence and PAGE) is hydrolysis of the N-glycosidic bond; step 4 (detected by FRET and PAGE) represents a slow process of product release accompanied by a DNA conformational change. | ||
Abbreviations
| SMUG1 | Human single-stranded selective monofunctional uracil-DNA glycosylase |
| AP-site | Apurinic/apyrimidinic site |
| F-site | (3-Hydroxytetrahydrofuran-2-yl)methyl phosphate |
| ODN | Oligodeoxyribonucleotide |
| PAGE | Polyacrylamide gel electrophoresis |
| BER | Base excision repair |
| FRET | Förster resonance energy transfer. |
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was supported partially by a Russian Government-funded project (No. VI.57.1.2, 0309-2016-0001), SB RAS Comprehensive Scientific Program (No. II.2Π/VI.57-6, 0309-2015-0025), and grants from the Russian Foundation of Basic Research (numbers 16-04-00037 and 15-04-00467). The part of the work with Trp, aPu, and FRET detection combined with stopped-flow kinetics was specifically funded by Russian Science Foundation grant 16-14-10038.References
- R. J. Boorstein, A. Cummings, Jr., D. R. Marenstein, M. K. Chan, Y. Ma, T. A. Neubert, S. M. Brown and G. W. Teebor, J. Biol. Chem., 2001, 276, 41991–41997 CrossRef CAS PubMed.
- T. Lindahl, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3649–3653 CrossRef CAS.
- K. A. Haushalter, P. T. Stukenberg, M. W. Kirschner and G. L. Verdine, Curr. Biol., 1999, 9, 174–185 CrossRef CAS PubMed.
- B. Kavli, O. Sundheim, M. Akbari, M. Otterlei, H. Nilsen, F. Skorpen, P. A. Aas, L. Hagen, H. E. Krokan and G. Slupphaug, J. Biol. Chem., 2002, 277, 39926–39936 CrossRef CAS PubMed.
- J. E. A. Wibley, T. R. Waters, K. Haushalter, G. L. Verdine and L. H. Pearl, Mol. Cell, 2003, 11, 1647–1659 CrossRef CAS PubMed.
- A. Masaoka, M. Matsubara, R. Hasegawa, T. Tanaka, S. Kurisu, H. Terato, Y. Ohyama, N. Karino, A. Matsuda and H. Ide, Biochemistry, 2003, 42, 5003–5012 CrossRef CAS PubMed.
- S. Bjelland and E. Seeberg, Mutat. Res., 2003, 531, 37–80 CrossRef CAS PubMed.
- H. Nilsen, K. A. Haushalter, P. Robins, D. E. Barnes, G. L. Verdine and T. Lindahl, EMBO J., 2001, 20, 4278–4286 CrossRef CAS PubMed.
- H. Terato, A. Masaoka, M. Kobayashi, S. Fukushima, Y. Ohyama, M. Yoshida and H. Ide, J. Biol. Chem., 1999, 274, 25144–25150 CrossRef CAS PubMed.
- A. Masaoka, H. Terato, M. Kobayashi, Y. Ohyama and H. Ide, J. Biol. Chem., 2001, 276, 16501–16510 CrossRef CAS PubMed.
- H. E. Krokan, F. Drablos and G. Slupphaug, Oncogene, 2002, 21, 8935–8948 CrossRef CAS PubMed.
- N. Schormann, R. Ricciardi and D. Chattopadhyay, Protein Sci., 2014, 23, 1667–1685 CrossRef CAS PubMed.
- L. H. Pearl, Mutat. Res., 2000, 460, 165–181 CAS.
- M. Matsubara, T. Tanaka, H. Terato, E. Ohmae, S. Izumi, K. Katayanagi and H. Ide, Nucleic Acids Res., 2004, 32, 5291–5302 CrossRef CAS PubMed.
- H. S. Pettersen, O. Sundheim, K. M. Gilljam, G. Slupphaug, H. E. Krokan and B. Kavli, Nucleic Acids Res., 2007, 35, 3879–3892 CrossRef CAS PubMed.
- N. A. Kuznetsov, C. Bergonzo, A. J. Campbell, H. Li, G. V. Mechetin, C. de los Santos, A. P. Grollman, O. S. Fedorova, D. O. Zharkov and C. Simmerling, Nucleic Acids Res., 2015, 43, 272–281 CrossRef CAS PubMed.
- S. R. Nelson, A. R. Dunn, S. D. Kathe, D. M. Warshaw and S. S. Wallace, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2091–2099 CrossRef CAS PubMed.
- A. J. Lee and S. S. Wallace, Free Radical Biol. Med., 2017, 107, 170–178 CrossRef CAS PubMed.
- R. M. Werner and J. T. Stivers, Biochemistry, 2000, 39, 14054–14064 CrossRef CAS PubMed.
- S. C. Gill and P. H. von Hippel, Anal. Biochem., 1989, 182, 319–326 CrossRef CAS PubMed.
- D. A. Yakovlev, A. A. Kuznetsova, O. S. Fedorova and N. A. Kuznetsov, Acta Naturae, 2017, 9, 88–98 CAS.
- A. A. Kuznetsova, N. A. Kuznetsov, Y. N. Vorobjev, N. P. Barthes, B. Y. Michel, A. Burger and O. S. Fedorova, PloS One, 2014, 9, e100007 Search PubMed.
- P. Kuzmic, Anal. Biochem., 1996, 237, 260–273 CrossRef CAS PubMed.
- N. A. Kuznetsov, Y. N. Vorobjev, L. N. Krasnoperov and O. S. Fedorova, Nucleic Acids Res., 2012, 40, 7384–7392 CrossRef CAS PubMed.
- N. A. Kuznetsov, V. V. Koval, D. O. Zharkov and O. S. Fedorova, DNA Repair, 2012, 11, 884–891 CrossRef CAS PubMed.
- V. V. Koval, N. A. Kuznetsov, A. A. Ishchenko, M. K. Saparbaev and O. S. Fedorova, Mutat. Res., 2010, 685, 3–10 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–258 CrossRef CAS PubMed.
- Z. Zhang, J. Shen, Y. Yang, J. Li, W. Cao and W. Xie, ACS Chem. Biol., 2016, 11, 1729–1736 CrossRef CAS PubMed.
- P. Benkert, M. Biasini and T. Schwede, Bioinformatics, 2011, 27, 343–350 CrossRef CAS PubMed.
- F. Sievers, A. Wilm, D. Dineen, T. J. Gibson, K. Karplus, W. Li, R. Lopez, H. McWilliam, M. Remmert, J. Soding, J. D. Thompson and D. G. Higgins, Mol. Syst. Biol., 2011, 7, 539 CrossRef PubMed.
- S. C. Brooks, S. Adhikary, E. H. Rubinson and B. F. Eichman, Biochim. Biophys. Acta, 2013, 1834, 247–271 CrossRef CAS PubMed.
- O. A. Kladova, A. A. Kuznetsova, O. S. Fedorova and N. A. Kuznetsov, Genes, 2017, 8, 1–13 CrossRef PubMed.
- N. A. Kuznetsov, A. S. Kiryutin, A. A. Kuznetsova, M. S. Panov, M. O. Barsukova, A. V. Yurkovskaya and O. S. Fedorova, J. Biomol. Struct. Dyn., 2017, 35, 950–967 CAS.
- A. A. Kuznetsova, N. A. Kuznetsov, A. A. Ishchenko, M. K. Saparbaev and O. S. Fedorova, Biochim. Biophys. Acta, 2014, 1840, 387–395 CrossRef CAS PubMed.
- A. J. Lee and S. S. Wallace, Radiat. Phys. Chem., 2016, 128, 126–133 CrossRef CAS PubMed.
- N. A. Kuznetsov, A. A. Kuznetsova, Y. N. Vorobjev, L. N. Krasnoperov and O. S. Fedorova, PloS One, 2014, 9, e98495 Search PubMed.
- N. A. Kuznetsov, O. A. Kladova, A. A. Kuznetsova, A. A. Ishchenko, M. K. Saparbaev, D. O. Zharkov and O. S. Fedorova, J. Biol. Chem., 2015, 290, 14338–14349 CrossRef CAS PubMed.
- E. L. Rachofsky, R. Osman and J. B. A. Ross, Biochemistry, 2001, 40, 946–956 CrossRef CAS PubMed.
- J. M. Jean and K. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 37–41 CrossRef CAS.
- J. M. Jean and K. B. Hall, Biochemistry, 2002, 41, 13152–13161 CrossRef CAS PubMed.
- L. C. Sowers, Y. Boulard and G. V. Fazakerley, Biochemistry, 2000, 39, 7613–7620 CrossRef CAS PubMed.
- C. A. Dunlap and M. D. Tsai, Biochemistry, 2002, 41, 11226–11235 CrossRef CAS PubMed.
- J. T. Stivers, K. W. Pankiewicz and K. A. Watanabe, Biochemistry, 1999, 38, 952–963 CrossRef CAS PubMed.
- O. G. Berg and P. H. Vonhippel, Annu. Rev. Biophys. Biophys. Chem., 1985, 14, 131–160 CrossRef CAS PubMed.
- M. Slutsky and L. A. Mirny, Biophys. J., 2004, 87, 4021–4035 CrossRef CAS PubMed.
- S. E. Halford, Biochem. Soc. Trans., 2009, 37, 343–348 CrossRef CAS PubMed.
- V. V. Koval, N. A. Kuznetsov, D. O. Zharkov, A. A. Ishchenko, K. T. Douglas, G. A. Nevinsky and O. S. Fedorova, Nucleic Acids Res., 2004, 32, 926–935 CrossRef CAS PubMed.
- N. A. Kuznetsov, V. V. Koval, D. O. Zharkov, G. A. Nevinsky, K. T. Douglas and O. S. Fedorova, Nucleic Acids Res., 2005, 33, 3919–3931 CrossRef CAS PubMed.
- A. A. Kuznetsova, N. A. Kuznetsov, A. A. Ishchenko, M. K. Saparbaev and O. S. Fedorova, Biochim. Biophys. Acta, 2014, 1840, 3042–3051 CrossRef CAS PubMed.
- N. A. Kuznetsov, V. V. Koval and O. S. Fedorova, Biochemistry, 2011, 76, 118–130 CAS.
- O. S. Fedorova, N. A. Kuznetsov, V. V. Koval and D. G. Knorre, Biochemistry, 2010, 75, 1225–1239 CAS.
- N. A. Kuznetsov, V. V. Koval, G. A. Nevinsky, K. T. Douglas, D. O. Zharkov and O. S. Fedorova, J. Biol. Chem., 2007, 282, 1029–1038 CrossRef CAS PubMed.
- N. A. Kuznetsov, V. V. Koval, D. O. Zharkov, Y. N. Vorobiev, G. A. Nevinsky, K. T. Douglas and O. S. Fedorova, Biochemistry, 2007, 46, 424–435 CrossRef CAS PubMed.
- A. R. Dunn, N. M. Kad, S. R. Nelson, D. M. Warshaw and S. S. Wallace, Nucleic Acids Res., 2011, 39, 7487–7498 CrossRef CAS PubMed.
- A. Prakash, S. Doublie and S. S. Wallace, Prog. Mol. Biol. Transl. Sci., 2012, 110, 71–91 CAS.
- A. J. Lee, D. M. Warshaw and S. S. Wallace, DNA Repair, 2014, 20, 23–31 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mb00457e |
| This journal is © The Royal Society of Chemistry 2017 |