
Plasmonic nanohole array biosensor for label-free and real-time analysis of live cell secretion†
Xiaokang
Li
 ,
Maria
Soler
,
Cenk I.
Özdemir
,
Alexander
Belushkin
,
Filiz
Yesilköy
and
Hatice
Altug
*
,
Maria
Soler
,
Cenk I.
Özdemir
,
Alexander
Belushkin
,
Filiz
Yesilköy
and
Hatice
Altug
*
Institute of Bioengineering, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne 1015, Switzerland. E-mail: hatice.altug@epfl.ch
First published on 19th May 2017
Abstract
Cell secretion dynamics plays a central role in physiological and disease processes. Due to its various temporal profiles, it is essential to implement a precise detection scheme for continuous monitoring of secretion in real time. The current fluorescent and colorimetric approaches hinder such applications due to their multiple time-consuming steps, molecular labeling, and especially the ‘snapshot’ endpoint readouts. Here, we develop a nanoplasmonic biosensor for real-time monitoring of live cell cytokine secretion in a label-free configuration. Our nanoplasmonic biosensor is composed of gold nanohole arrays supporting extraordinary optical transmission (EOT), which enables sensitive and high-throughput analysis of biomolecules. The nanobiosensor is integrated with an adjustable microfluidic cell module for the analysis of live cells under well-controlled culture conditions. We achieved an outstanding sensitivity for the detection of vascular endothelial growth factor (VEGF) directly in complex cell media. Significantly, the secretion dynamics from live cancer cells were monitored and quantified for 10 hours while preserving good cell viability. This novel approach of probing cytokine secretion activity is compatible with conventional inverted microscopes found in a common biology laboratory. With its simple optical set-up and label-free detection configuration, we anticipate our nanoplasmonic biosensor to be a powerful tool as a lab-on-chip device to analyze cellular activities for fundamental cell research and biotechnologies.
Introduction
The investigation of cellular functions allows us to better understand physiological and pathological processes and thereby improve the diagnosis and treatment of diseases. Significantly, the extracellular production of biomolecules (e.g. cytokines, chemokines, and proteinases) plays a central role in various cellular activities, such as cell proliferation, differentiation, cancer progression, and immune responses.1,2 Quantifying the amount and real-time dynamics of cytokine secretion from live cells provides vital insights for fundamental research and clinical applications. For instance, cytokines expressed in a tumor microenvironment can function to promote or inhibit tumor development and progression in different stages.2 Profiling the time course of changes in cancer cell secretome or signaling factor kinetics provides a means to discover novel biomarkers for cancer diagnosis and to evaluate anti-cancer therapies.3,4 Unveiling the dynamic response of cancer cell signaling offers further understanding of the disease progression and more information to assess drug efficacy.5,6 Currently, the analysis of cytokine secretion is typically done using fluorescent protein tags and sandwiched enzymatic immunochemistry approaches, e.g. enzyme-linked immunosorbent assay (ELISA) and enzyme-linked immunosorbent spot (ELISpot) assay. Fluorescent tags enable live mapping of intracellular signaling, but the relatively large tag molecules hamper the exocytosis of cytokines. Moreover, labeling a large variety of molecules-of-interest is costly and tedious. Enzymatic immunoassay-based methods can reach a high sensitivity for cytokine detection. However, their low detection limit is achieved through a sandwiched detection scheme, which requires labels and multiple time-consuming washing steps, as well as a large sample quantity. More importantly, these current methodologies only provide an endpoint readout, hence they are greatly limited for deciphering the real-time cell secretion dynamics.Over the past decades, label-free biosensors have emerged as a powerful analytical tool for sensitive and real-time detection of biomolecules (e.g. proteins and nucleic acids) and more complex entities such as bacteria, viruses, and mammalian cells.7–10 Among them, plasmonic biosensors have demonstrated high sensitivity and robustness, presenting promising compatibility for laboratory research and development of point-of-care devices.11–14 For instance, surface plasmon resonance (SPR) biosensors enable direct monitoring of biomolecular interactions in real time and accurate quantification of analytes without the need for labels or amplification steps.15,16 However, conventional SPR biosensor platforms require complex prism-coupling instrumentation, which hampers both the high throughput required for multiplexed analysis and the integration into lab-on-a-chip systems. Nanoplasmonic biosensors featuring nanoscale structures (e.g. nanoapertures, nanoparticles) have recently gained significant attention as they can meet the above needs.17 In particular, nanoplasmonic biosensors consisting of gold nanohole arrays (NHAs) have demonstrated unique capabilities for multiplexed and label-free detection in a compact footprint, which are promising for miniaturization and integration into lab-on-a-chip devices.18,19 The sub-wavelength NHAs fabricated on optically thick metal films support the extraordinary optical transmission (EOT) phenomenon, which is attributed to the excitation of plasmons by grating coupling of light at normal incidence and hence resulting in a strong field enhancement in the vicinity of the nanoholes.20 The light confinement on the nanometer scale allows the NHAs to be highly sensitive to minute local refractive index changes, such as those induced by the binding of a few biomolecules to the sensor surface. These changes can be readily monitored by tracking the variations of the transmission spectra, providing real-time sensitive detection and quantification of the biomolecules in a label-free manner. Several studies have demonstrated the excellent performance of NHA-based biosensors for the direct detection of proteins, nucleic acids, exosomes, or microorganisms.21–24 Furthermore, NHAs have shown an outstanding multiplexing capability as high-throughput analysis platforms since they can be organized in a highly compact manner. Chang et al. fabricated an array of up to 1.5 million nanohole sensor pixels on a single glass slide with a packing density of 816 sensors per mm2, which is comparable to that of state-of-the-art fluorescence-based microarrays.25 Moreover, as the EOT phenomenon circumvents the need of the prism-coupling mechanism,26 the NHA-based biosensor could be readily fit with a lens-free on-chip imaging set-up, providing an ideal sensing format for miniaturized point-of-care devices.27
Although nanoplasmonic biosensors are highly promising for sensitive biomolecular detection, their application to the incorporation of live cells and the study of cellular functions is still in its infancy. Extension to in situ cell analysis applications requires the biosensor to support a highly robust and stable microenvironment for long-term cell characterization, which can range from several hours to days. So far, cell secretion analysis with nanoplasmonic biosensors has been limited either to protein quantification in isolated blood, serum samples and culture supernatants or to the monitoring of immediate cell responses covering only a few hours (1–3 h).28–31 The integration of cell culture modules is thereby necessary to control and preserve cell viability and facilitate long-term analysis of cellular activities under appropriate conditions. Furthermore, the device needs to operate in complex solutions (e.g. cell culture media) to maintain high cell functionality. This implies that the sensor should be able to discriminate the target proteins from other fouling agents, ensuring high detection selectivity. And finally, it is important to mention that the secreted cytokines or signaling factors are usually small biomolecules (<30 kDa) produced at low concentrations. Therefore it demands a highly sensitive performance of the biosensor to detect the molecules in a direct manner, without the need for labels or amplification steps.
In this work, we introduce a nanoplasmonic biosensor platform integrated with a microfluidic live cell culture for real-time monitoring of cellular secretion directly in complex culture media. The platform consists of a highly versatile microfluidic cell culture module combined with a readily implementable NHA-based biosensor. The sensing principle of our plasmonic sensor enables high-resolution spectroscopic imaging of multiple sensor arrays simultaneously by utilizing normal broadband illumination and simple bright-field transmission optics. This operation scheme provides direct compatibility of our platform with a conventional inverted microscope, which is usually available in a general biology laboratory. We demonstrated the excellent performance of our biosensor by analyzing the real-time secretion of vascular endothelial growth factor (VEGF) from live cancer cells. This cytokine plays a key role in various processes in developing cancers, such as angiogenesis, progression, and metastasis.32 Thus, the label-free monitoring of VEGF secretion could be of genuine interest for cancer research and the development of anti-cancer therapies. We performed stable real-time monitoring of cytokine secretion over 10 hours by employing a comparable number of cells to that adopted in conventional methods. Cell viability was not only well preserved throughout the measurements but we could also recover the cells after the experiment for further studies. The nanoplasmonic biosensor demonstrated successful functionality with complex cell culture media by achieving an outstanding sensitivity of 145 pg mL−1 (∼5 pM) for VEGF detection. We obtained equivalent results from independent cell measurements, and the detection outcome was validated with standard ELISA experiments, confirming the good reproducibility and accuracy of the biosensor. Similarly, our NHA-based biosensor platform could be extended to the analysis of other cell types (e.g. stem cells) and also for the real-time monitoring of cytokine secretion in a multiplexed configuration. This platform may provide new analysis tools that offer significant insights into cell biology and facilitate the development of more efficient therapies.
Materials and methods
Reagents and chemicals
PEGylated alkanethiol compounds HS-C6EG4OH (TH 001-m6.n4-0.2) and HS-C11EG4OCH2COOH (TH 003-m11.n4-0.2) were purchased from Prochimia Surfaces (Sopot, Poland). Crosslinking reagents N-(3-imethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, E1769), sulfo-N-hydroxysulfosuccinimide (sulfo-NHS, 56485) and bis(sulfosuccinimidyl) suberate sodium salt (BS3, S5799) as well as Streptavidin (85878) and calcium ionophore A23187 (C7522) were purchased from Sigma-Aldrich (Buchs, Switzerland). Phosphate saline buffer (PBS, 10010015), Dulbecco's Modified Eagle Medium (DMEM, 42430025), penicillin/streptomycin (PS, 15140130), fetal bovine serum (FBS, 10270106) and TrypLETMExpress (12605036) for the HeLa cell culture were purchased from Thermo Fisher Scientific (Reinach, Switzerland). The human VEGF ELISA kit (ab100662) was obtained from Abcam (Cambridge, UK). All the other chemicals were purchased from the chem shop in EPFL unless otherwise indicated.Fabrication of the NHA biosensor chip
The NHA chip was fabricated based on a deep-UV lithography method that was previously described (Fig. S1a†).27 Briefly, the NHAs were fabricated on a 500 μm-thick silicon wafer substrate that was coated with a 100 nm-thick silicon nitride film on both sides. The nanoholes were first patterned by UV lithography with a stepper (ASML S500/300 DUV). Then the photoresist was developed and etched through the top layer of nitride by SF6/Ar plasma to generate the nanoholes. Oxygen plasma further removed remaining residues. The nitride layer on the backside of the wafer was removed by similar approaches, and the wafer was subsequently immersed in KOH solution to create freestanding nitride membranes. Finally, the Ti adhesion layer (5 nm) and Au sensor layer (120 nm) were evaporated providing the gold NHAs for biosensing without the need for a lift-off process. Each NHA sensor was patterned with a dimension of 100 × 100 μm, and with an edge-to-edge separation of 800 μm, where the nanohole has a diameter of 200 nm and a period of 600 nm (Fig. S1b†).Fabrication of microfluidic devices
Microfluidic devices were fabricated with polydimethylsiloxane (PDMS, SYLGARD® 184 SILICONE ELASTOMER KIT) by a soft lithography technique. The master mold was prepared on a 4′′ Si wafer where the microchannel structures were directly engraved on 50 μm-thick photoresist layer (mr-DWL_40) using a maskless laser aligner (Heidelberg MLA150, Germany). Before the PDMS preparation, a silanization process with trimethylchlorosilane (TMCS, 92360, Sigma) under vacuum for 20 min was performed to passivate the mold surface to aid the release of PDMS from the mold. Then, the PDMS prepolymer was prepared by mixing the elastomer and the curing agent in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio and degassed for 20 min in a desiccator. Following the degassing, the prepolymer was gently poured onto the passivated Si mold and cured at 80 °C for 4 hours to form the microchannels used in the optofluidic detection module and cured overnight for the cell module. Finally, the molded PDMS layer was released from the wafer and inlets and outlets were punched.
:1 weight ratio and degassed for 20 min in a desiccator. Following the degassing, the prepolymer was gently poured onto the passivated Si mold and cured at 80 °C for 4 hours to form the microchannels used in the optofluidic detection module and cured overnight for the cell module. Finally, the molded PDMS layer was released from the wafer and inlets and outlets were punched.
To assemble the microfluidic cell module, the PDMS slab and the glass slide were treated with an oxygen plasma cleaner, followed by immediate contact that results in irreversible bonding. The microfluidic cell chamber was designed to have winding channels (Fig. 1a) of 500 μm wide. The total length of the channel is adjustable according to the number of winding units. The chamber used in this study provides a surface area of ∼1 cm2 for cell growth. For the optofluidic detection module, the sensor microarrays were optically aligned with the corresponding sensor channels embedded in the PDMS slab on an upright microscope. Each sensor channel measures 60 μm high, 500 μm wide and 5 mm long, resulting in a volume of ∼150 nL (Fig. S2†).
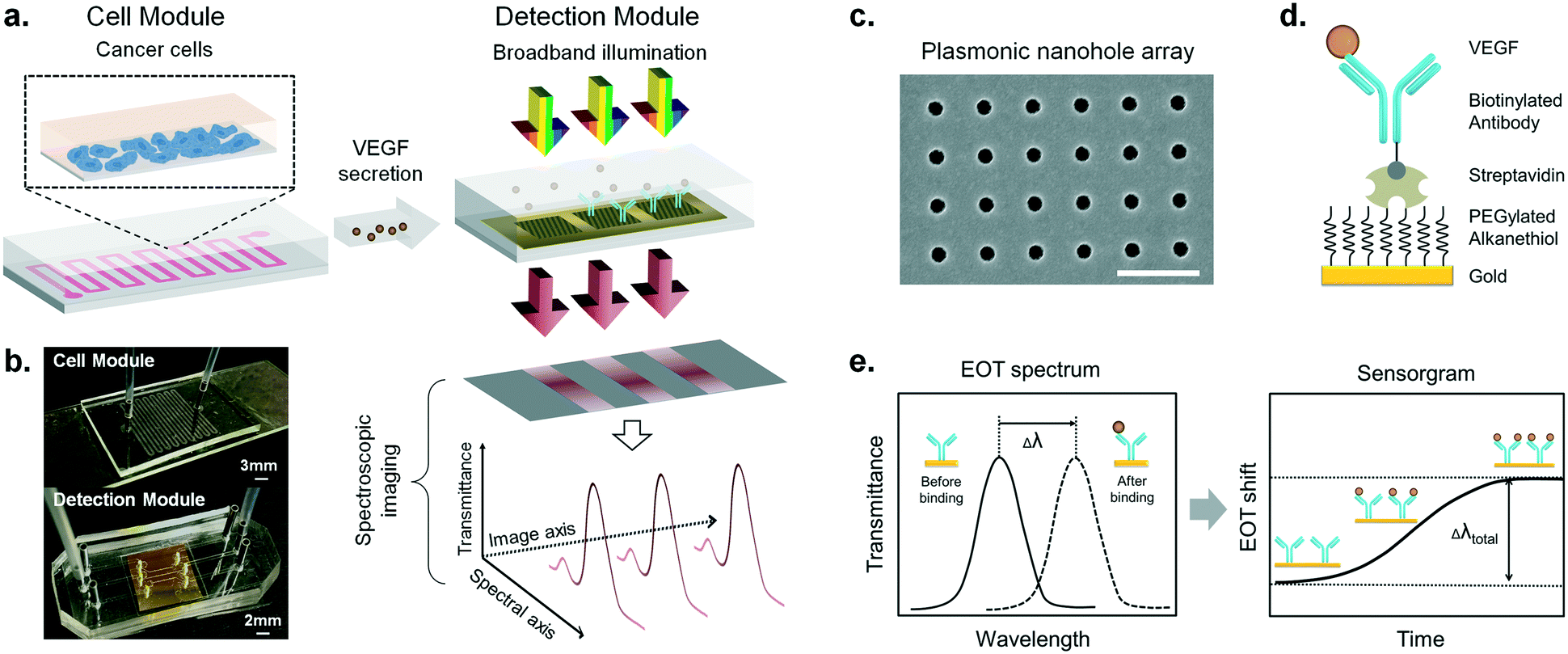 | ||
| Fig. 1 Design, principle and experimental configuration of the microfluidic-integrated biosensor for real-time cytokine secretion analysis. (a) The biosensor system consists of a microfluidic cell module and an optical detection module. Cancer cells grow in a zigzag single-channel PDMS unit, and the secreted cytokines are directly delivered to the adjacent detection module. The detection module illustrates the three in-line nanohole arrays in one microfluidic channel. Two of the arrays are functionalized with specific anti-VEGF antibodies (blue) and the third one without antibodies serves as a negative control sensor. A beam of collimated broadband light illuminates the microarrays at normal incidence. The transmitted light is coupled to a spectrometer and dispersed along one dimension to form a spectroscopic image on a CCD camera. One axis of this image corresponds to the positions of the three sensors, and the other one corresponds to their spectral response. Using this readout scheme, the EOT spectra of all the three sensors are simultaneously recorded for biosensing analysis. (b) Pictures of the cell culture module bonded to a gelatin-coated glass slide with inlet and outlet connections (top), and the detection module consisting of the plasmonic chip embedded in a microfluidic system with three independent channels (bottom). (c) SEM image of the nanohole structures with a hole diameter of 200 nm and a periodicity of 600 nm. Scale bar: 1 μm. (d) The illustration shows the surface chemistry utilized for specific VEGF detection, where the antibody is immobilized by the robust streptavidin–biotin interaction. Streptavidin is covalently bonded to the activated carboxylic groups on the PEGylated alkanethiol molecules sitting on the gold surface. (e) Sensing principle of the real-time plasmonic detection. The EOT spectrum from each nanohole array shows a characteristic resonance peak (solid line). Biomolecular binding on the sensor surface induces an EOT spectral shift of the peak (dashed line). As molecular binding accumulates, the spectral displacements of the resonance peak are then plotted on the sensorgram (EOT shift vs. time) to reveal the real-time binding dynamics. | ||
Biosensor surface functionalization
The NHA plasmonic chip was first cleaned by consecutive washes in acetone, ethanol, and Milli-Q water, and then further treated with a UV-ozone cleaner (Bioforce Nanosciences) for 20 min. The biofunctionalization procedure is illustrated in Fig. S3a.† The cleaned chip was incubated using a mixture of PEGylated alkanethiols ([–COOH]:[–OH] = 1:5, [–SH] total = 1 μM) in absolute ethanol overnight at room temperature. The carboxylic groups on the chip surface were then activated by incubating with 100 μL cross-linking solution containing 200 mM EDC and 50 mM sulfo-NHS in 0.1 M MES buffer (pH = 4.5) for 15 min at room temperature. Subsequently, the activated chip was incubated with 100 μg mL−1 streptavidin solution diluted in PBS for 1 h at room temperature. Biotinylated anti-VEGF was spotted onto the streptavidin-coated NHA chip using a piezoelectric microdispensing system, sciFLEXARRAYER S3 (Scienion, Germany). Due to the minute amount of the liquid droplet during spotting, the antibodies were diluted in PBS buffer solution supplemented with 2% glycerol to prevent liquid evaporation. Meanwhile, the spotting device is equipped with an airtight hood where high humidity (>65%) was supplied and with a cooling unit to maintain the target stage at low temperature (e.g. 10–15 °C). The correlation between the droplet number and the resulting spot size is shown in Fig. S3b.† To fully cover the sensor surface, the droplet size was tuned to reach ∼180 pL. The chip was incubated at 4 °C overnight following the spotting to allow stable immobilization of biotinylated antibodies on the streptavidin layer. The chip was subsequently blocked with complete cell culture media prior to VEGF detection.
To calibrate the optical responses to different concentrations of VEGF, the VEGF standards were diluted in PBS to achieve gradient concentrations and injected into the optofluidic chip at a flow rate of 20 μL min−1. Each concentration was measured in triplicate. The selectivity and specificity of the biosensor were confirmed by introducing a nonspecific cytokine (interleukin-2) as a negative control, and no signal was observed (Fig. S3b†). In addition, the calibration curve was also obtained by spiking different concentrations of VEGF into the cell medium, and no significant differences were observed when compared to PBS (Fig. S3c†). This excludes possible matrix interferences. The chip can be regenerated by injecting 20 mM NaOH solution post each injection. The limit of detection (LOD) of the biosensor is defined as LOD = 3 × σ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) , where σ is the standard deviation of the background optical signal.
, where σ is the standard deviation of the background optical signal.
Cell culture
HeLa cells were cultivated in high glucose Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 100 units per mL penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified 5% CO2 incubator. The cells were subcultured routinely, harvested and counted based on a general protocol. Before cell seeding in the microfluidic culture chamber, the cells were rinsed with PBS and detached with TrypLE™Express. 105 cells were injected into the culture chamber and incubated overnight at 37 °C before the measurement.Sandwiched ELISA assay
To analyze the stimulatory impact of calcium ionophore A23187, cells were cultured in a 48-well plate and incubated overnight before being assayed. The following day, the cells were rinsed two times with PBS followed by the addition of 100 μL fresh medium supplemented with 1–100 μM A23187 to each well. Each condition was measured in duplicate wells. Supernatants were collected after 24 h incubation. To validate the dynamics of VEGF secretion detected by the NHA biosensor, cells were cultured in the microfluidic chamber with 10 μM A23187 in flow and the supernatants were harvested every hour. The VEGF components were analyzed by sandwiched ELISA assay according to the manufacturer's manual.VEGF immunofluorescence staining and imaging
Immunofluorescence staining assay was performed to analyze the VEGF production in HeLa cells. Cells were first cultured on gelatin (G6144, Sigma)-coated coverslips overnight. The cells were rinsed with PBS three times and fixed with ice-cold methanol at −20 °C for 10 min, followed by another PBS rinsing. Then, the cells were blocked with 5% w/v bovine serum albumin (37525, Sigma) and incubated with 5 μg mL−1 monoclonal mouse anti-human VEGF (ab1316, Abcam) overnight at 4 °C. This was followed by 1 h incubation with a 1:500 Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (ab150113, Abcam) at room temperature. Fluoroshield mounting medium with DAPI (ab104139, Abcam) was used to stain the cell nuclei while preserving fluorescence. The stained sample was finally imaged with Nikon Ti-U inverted microscope.
Spectral measurement set-up and data processing
To facilitate the EOT measurement, a spectrometer (Shamrock 303i, Andor) and a deep-cooled CCD camera (iKon-M, Andor) were installed onto an inverted microscope (Nikon Ti-U). A broadband tungsten-halogen lamp illuminated the NHA chip at normal incidence. The EOT signals were collected using a 4× objective (CFI Plan Fluor, NA = 0.13) lens and entered the spectrometer through a 400 μm-wide slit opening. Andor Solis software was employed to analyze the spectra. Image mode in the software was used to capture spectra from multiple NHAs sensors simultaneously in one microchannel. The spectral images were obtained with 0.5 s exposure, and a grating of 600 lines per mm was used to improve the pixel resolution. The spectra were extracted from each spectral image by a custom MATLAB image processing script. The resonance peak position was defined by calculating the centroid of the peak within a fixed wavelength window (40% maximum intensity) and plotted in real time. Sensorgrams show the centroid shifting versus time of the 3 independent sensor arrays simultaneously. To quantify the spectral shift and interpolate the calibration curve, curve fitting of the sensorgrams was performed with nonlinear regression using GraphPad Prism 7.Results and discussion
Biosensor system assembly and working principle
Our integrated biosensor is composed of two complementary and adjustable microfluidic modules, namely, the cell culture module and the optical detection module (Fig. 1a). For the microfluidic cell module, we fabricated a single zigzag channel in a polydimethylsiloxane (PDMS) slab that was permanently bonded to a glass slide (Fig. 1b). The growth area could be easily adjusted to provide an appropriate number of cells by simply changing the length and width of the channel. The cell module also includes inlet and outlet tubing connections for introducing fresh culture media and chemical stimuli and directly delivering the secreted proteins to the adjacent detection module. The optical detection module consists of a nanoplasmonic biosensor based on gold NHAs, also integrated with a PDMS microfluidic system (Fig. 1b). The nanoholes (200 nm diameter, 600 nm period) were fabricated by deep-UV lithography on freestanding SiNx membranes, and each NHA was defined to have an area of 100 × 100 μm2 (Fig. 1c and S1†). In this module, the microfluidic system includes three parallel microchannels, and each channel covers three in-line NHAs with a volume of 150 nL. The complete biosensor system including the two modules has approximate dimensions of 2.5 × 6 cm2. Taking advantage of the multiplexing capability of our biosensor, we functionalized two of the NHAs with anti-VEGF antibodies, leaving the third one as a negative control. For our experiments, we employed one microfluidic channel at a time, but it is worth mentioning that we could also incorporate multiple cell culture modules for different channels, enabling the simultaneous analysis of distinct types of cells. Antibodies specific to VEGF were selectively immobilized on the gold nanohole arrays as biorecognition molecules to capture the analytes (Fig. 1d). In this way, the VEGF secreted by the cancer cells can be directly captured by the specific antibodies without the need for sample pretreatment or purification steps. For real-time analysis, we placed the biosensor on the inverted microscope, and a collimated broadband light source illuminated the three in-line NHAs at normal incidence (Fig. 1a). The transmitted light is collected simultaneously by a 4× objective lens and entered the imaging spectrometer through a 400 μm-wide input slit. The three sensors are simultaneously imaged along the vertical slit of the spectrometer. The spectral content of each sensor is dispersed by a medium grating (600 lines per mm) and recorded on a 1024 × 1024 pixel CCD with a pixel size of 13 × 13 μm2. In this optical read-out scheme, the recorded two-dimensional CCD data contains a spatially resolved 1-D spectral image: one axis of the image corresponds to the positions of three sensors, and the other axis corresponds to their spectral response (Fig. 1a). Using this readout scheme, the EOT spectrum from each sensor unit is simultaneously acquired for biosensing analysis (Fig. 2a). The spectral coverage and resolution are controlled by the choice of the grating. The number of spectrally resolvable sensors is determined by the total magnification of the system, as well as the size and positioning of the NHA sensors on the chip. For instance, with one sensor unit measuring 100 × 100 μm2 and being separated by 50 μm in between, we could image 20 in-line sensors with a 4× objective lens simultaneously. In our case, the spectral content of each NHA sensor is recorded from ∼30 pixels, and the entire spectral coverage ranges from 830 nm to 890 nm. In aqueous solution, the EOT resonance peak of the NHA sensor is located at approximately 850 nm, which is well accommodated within the spectroscopic imaging range. The capture of VEGF on the sensor induces a change of refractive index proximate to the surface and thus results in a peak shift of the EOT spectrum. By tracing the spectral shifts continuously, we are able to resolve the sensorgram (i.e. binding curve) of the VEGF binding kinetics, and thus monitor the dynamics of cell secretion (Fig. 1e). We implemented a custom MATLAB script based on the centroid algorithm to track the spectral shifts.33 This data processing method calculates the center of mass of the resonance peak and plots the corresponding wavelength as a function of time. Unlike the conventional determination of the maximum value of the peak, this centroid algorithm can extraordinarily minimize the systematic noise and increase the signal-to-noise ratio, therefore reaching lower limits of detection.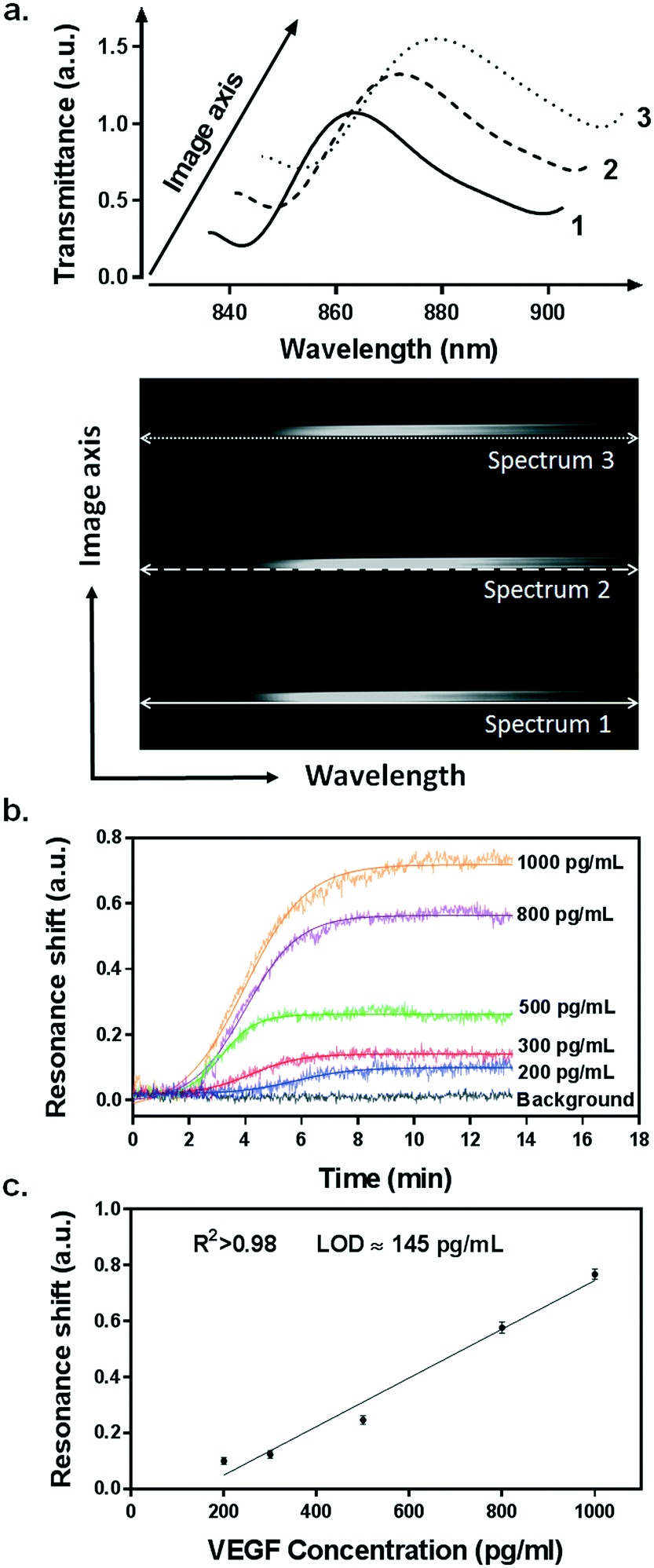 | ||
| Fig. 2 Spectroscopic imaging of the NHAs and evaluation of the plasmonic biosensor sensitivity for direct detection of VEGF. (a) The spectral image recorded by the spectrometer and CCD camera shows the spectral information from the three in-line NHA sensors simultaneously (bottom). Each spectrum is then extracted from the image, showing the EOT resonance peaks for detection analysis (top). (b) The sensorgrams of the direct detection of VEGF ranging from 200–1000 pg mL−1. (c) Standard calibration curve for VEGF detection. Data plots represent the mean and standard deviation of each concentration measured in triplicate. Plots are fitted to a linear regression model (R2 > 0.98), and the limit of detection (LOD) is calculated as the concentration corresponding to 3 times the standard deviation of the blank signal. | ||
Surface functionalization and biosensor calibration for VEGF detection
Before measuring the cell secretion, we characterized the performance of our biosensor for VEGF detection. In affinity-based biosensors, it is essential to establish a robust and stable surface chemistry strategy that ensures sensitive, selective and reproducible detection of the analyte of interest. This is particularly important for live cell secretion analysis since we monitor the cellular activity for extended periods of time and we need to detect low concentrations of proteins (usually in the range of pg mL−1). We employed the well-known biotin/streptavidin interaction for the biofunctionalization of the NHA sensors (Fig. 1d and S3a†). This strategy enables highly stable immobilization of biotinylated antibodies due to the high affinity between the streptavidin and the biotin molecules (kD ≈ 10−14 mol L−1). Furthermore, since the biotin tag is conjugated to the Fc domain of the antibodies (i.e. non-antigen binding domain), the antibodies are immobilized in an oriented manner, leading their antigen binding sites (Fab domains) entirely available for antigen capture.First of all, we created an anti-fouling layer on the sensor surface that prevents binding of nonspecific molecules. We took advantage of polyethylene glycol (PEG) derived compounds, which have demonstrated high resistance to protein adsorption due to their hydrophilic properties.34 In particular, we created a mixed self-assembled monolayer (SAM) consisting of two kinds of PEGylated alkanethiols carrying a reactive carboxylic functional group (–COOH) and a non-reactive hydroxyl group (–OH), respectively. The latter was used as a lateral spacer to minimize steric hindrance issues during protein attachment. The –COOH functional groups were activated by the general carbodiimide chemistry (i.e. EDC/NHS activation) and then a layer of streptavidin was covalently bonded. Finally, we employed the state-of-the-art piezoelectric micro spotting technique (Scienion, Germany) to locally deposit the biotinylated anti-VEGF antibodies on two of the NHA sensors (spatial precision <5 μm, volume <1 nL). This allowed us to define the third sensor in the same channel as a reference to compensate for the possible false positive signal produced by nonspecific molecular binding or systemic perturbations. Once the plasmonic microarray was functionalized, it was assembled with microfluidics and connected to a syringe pump and a manual injection valve for the characterization measurements.
We utilized a standard calibration curve to evaluate the sensitivity of our plasmonic biosensor for VEGF detection. Samples spiked with various concentrations of recombinant VEGF, ranging from 200 pg mL−1 to 1 ng mL−1, were introduced and flowed over the functionalized microarrays. The binding of VEGF to the antibodies was clearly detected by resonance peak shifting (Δλ) in a concentration-dependent manner (Fig. 2b). The values obtained from each sample were plotted and fitted to a linear regression model (Fig. 2c). We determined the limit of detection (LOD) of our biosensor, which is the lowest analyte concentration that the biosensor can reliably detect. It is defined as the concentration with a signal corresponding to three times the standard deviation of the blank.35 We achieved an outstanding LOD of 145 pg mL−1 (5.37 pM) for VEGF, which overlaps the physiological concentration range reported in other related biological studies.36,37
Probing the optimal condition for VEGF secretion from HeLa cells
In parallel with the biosensor characterization, we determined the stimulation scheme for VEGF secretion of HeLa cells. The endogenous production of VEGF was first confirmed by immunofluorescence assay (Fig. 3a). Currently, there is no uniform protocol to stimulate the VEGF secretion from the cells. A calcium ionophore, namely A23187, has been utilized as a popular stimulus for VEGF secretion in various studies. It can increase the intracellular Ca2+ levels rapidly and allow divalent ions to cross plasma membranes. Various reports have demonstrated a significant increase in VEGF secretion from different cells stimulated by the ionophore, albeit reporting a broad range of concentrations from hundreds of nanomolars to micromolars.36,38 Therefore, we tested a variety of cell culture conditions to stimulate the exocytosis of VEGF from HeLa cells. Cells were grown in multi-well plates and incubated overnight with different concentrations of A23187, ranging from 1 to 100 μM. Supernatants were collected and analyzed by sandwiched VEGF ELISA assay. As shown in Fig. 3b, the VEGF secretion showed a biphasic profile depending on the concentration of A23187. A marked increase in VEGF production was achieved with 1 and 10 μM of A23187, which was more than two times higher than the control group (i.e. Ctrl, w/o stimulus). Whereas, the stimuli exhibited significant cytotoxicity with concentrations greater than 100 μM. Cellular viability was significantly reduced by exhibiting apoptotic cellular morphology under optical microscopy (data not shown), along with scarce secretion of VEGF. Therefore, to facilitate maximum VEGF secretion and avoid cytotoxicity induced by the ionophore, the concentration of A23187 was selected to be 10 μM for the following studies.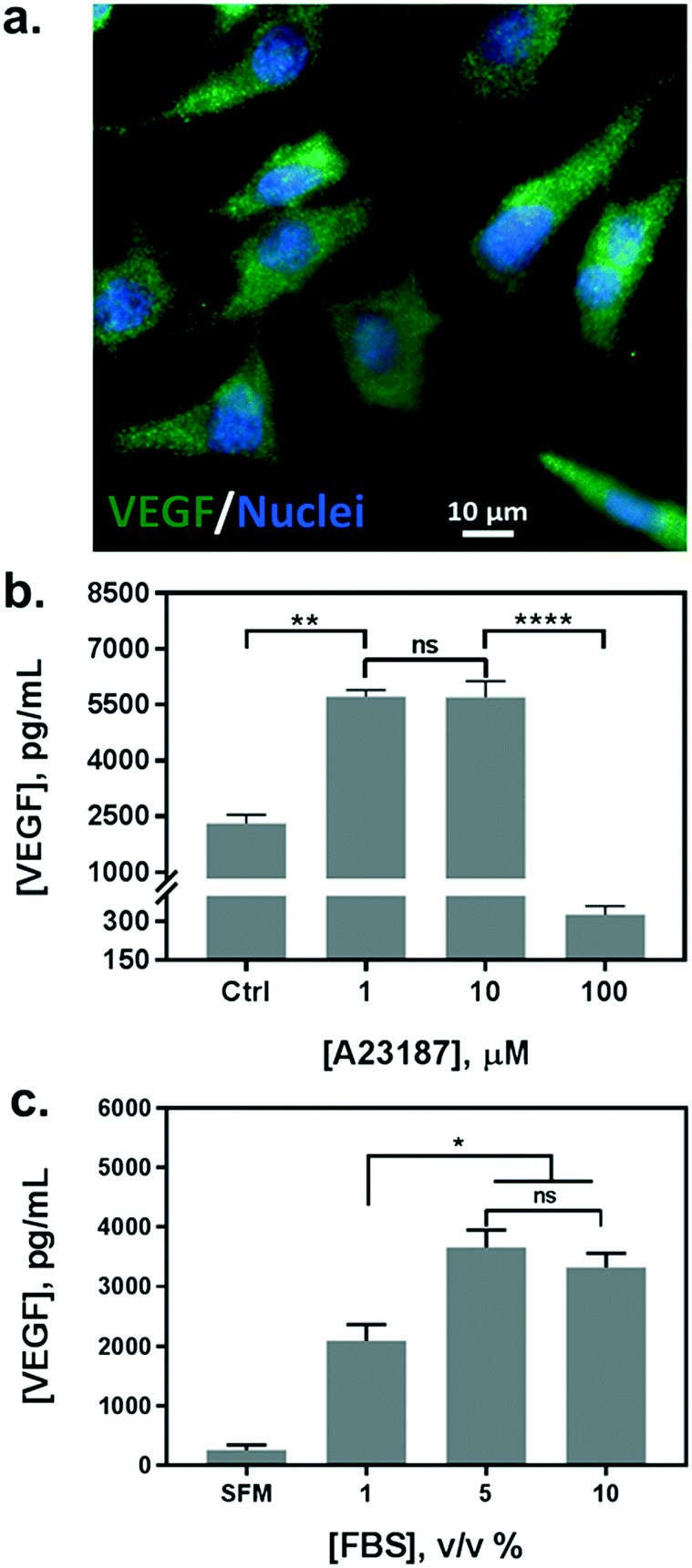 | ||
| Fig. 3 Assessment and optimization of cell culture conditions for VEGF secretion from HeLa cells. (a) Immunofluorescence assay for the confirmation of endogenous production of VEGF in HeLa cells. VEGF was stained with Alexa Fluor 488 (green) while cell nuclei were highlighted by DAPI (blue). (b) VEGF concentration measured in cell culture supernatants after administration of calcium ionophore at different levels ([A23187] = 0 as Ctrl; p < 0.005: **; p < 0.0001: ****; ns: non-significant; n = 2) (c) VEGF concentration measured in cell culture supernatants in the presence of different serum concentrations ([FBS] = 0 as SFM; p < 0.05: *; ns: non-significant; n = 2). | ||
Additionally, we also tested the influence of different serum concentrations in the culture medium as another critical role for cytokine secretion. The serum is supplemented with 10% v/v in routine cell culture for the majority of mammalian cells. However, reduced serum contents have been studied as a typical starvation condition to stimulate a variety of cellular metabolic activities, including VEGF expression.39,40 Therefore, we incubated HeLa cells in serum-free medium (SFM), and in media with serum concentrations of 1%, 5%, and 10%. We compared the VEGF secretion amount by analyzing the supernatants from each group with sandwiched ELISA. As shown in Fig. 3c, serum-free medium (SFM) and 1% serum induced a decrease in VEGF secretion compared to the conventional cell medium (10%), which could be attributed to cell death during incubation. However, 5% of serum showed a comparable VEGF production without adverse effects on cell viability. Moreover, reduced serum components could ameliorate microfluidic performance by reducing bubble generation, and even improve the antigen–antibody interaction with fewer interferences. Therefore, the cell culture medium supplemented with 5% of serum and 10 μM of A23187 was employed in the following experiments to induce VEGF secretion from HeLa cells (hereinafter VEGF-stimulating medium).
Evaluation of cell viability and VEGF secretion in microfluidic culture
Once the conditions to stimulate VEGF secretion were established, we examined the influence of our microfluidic culture module on the behavior of living cells, instead of the conventional multi-well plates (Fig. 4a). First, we studied the effect of constant medium flow on cell attachment, cellular morphology, and their viability in our microfluidic module. The flow is necessary to assure immediate delivery of the secreted cytokines to the optical module. The glass surface in the microfluidic chamber was coated with 0.1% gelatin to improve cell attachment. Later, HeLa cells (cell number ≈ 105) were seeded into the channel (growth area: 1 cm2) and incubated overnight at 37 °C and 5% CO2, allowing cells to attach and spread along the substrate. As shown in Fig. 4b, the cells firmly attached and spread extensively on the glass surface. The module was then placed back in the incubator (37 °C and 5% CO2), connected to a syringe pump and a flow of VEGF-stimulating medium was introduced. Different flow rates were tested (from 5 to 50 μL min−1) in order to set a suitable flow velocity. A flow rate of 10 μL min−1 was found to preserve low shear stress by preventing cell detachment (data not shown) while enabling rapid delivery of the secreted cytokines to the detection module.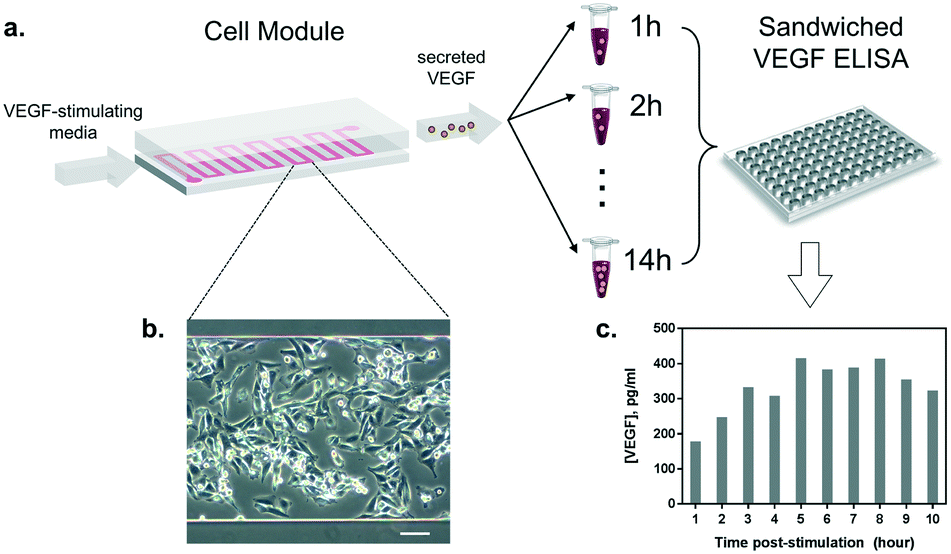 | ||
| Fig. 4 Study of the cell viability and VEGF secretion in the microfluidic culture. (a) HeLa cells were cultured in the microfluidic module with VEGF-stimulating medium and supernatants were collected every hour for 10 hours. The discrete samples were analyzed by sandwiched ELISA. (b) A magnified microscopic image of live HeLa cells cultured in the microfluidic channel. (c) Discrete time profile of VEGF secretion analyzed by ELISA, with the maximum peak reached after approximately 5 hours post-stimulation. | ||
Furthermore, we performed a preliminary study of the time profile of VEGF secretion under microfluidic flow conditions. After the injection of the stimulus at a constant flow rate through the cell module, we collected the outgoing supernatant in different vials (∼300 μL) before transferring the samples to an ELISA plate for VEGF quantification (Fig. 4a). The samples were collected every 1 hour for 10 hours. The HeLa cells constantly remained in a spindle shape, showing adequate viability throughout the test (Fig. 4b). The results in Fig. 4c show the VEGF concentrations obtained from the discrete supernatant samples. The amount of VEGF gradually increased and reached a peak level at around the 5th hour, which remained at a plateau for 4 hours. The concentration of VEGF in the plateau phase reached ∼380 pg mL−1, which is within the detection range of our label-free nanoplasmonic biosensor (Fig. 2c).
Monitoring the VEGF secretion in real-time
Following the characterization of our biosensor for VEGF detection and establishing the optimal secretion scheme from cells, we demonstrate its ability to perform real-time analysis of VEGF secretion from live cells. The gold NHA biosensor was functionalized as described above and integrated with the microfluidic system as the detection module. The multiplexing capability of our biosensor enables the inclusion within the same microfluidic channel of two independent NHAs for the specific detection of VEGF (sensor A and sensor B) and a reference sensor (sensor C). This ensures the reliability of the experiment by providing two assay replicates simultaneously, and guarantees the selectivity and specificity of the measurements, as the reference sensor provides an integrated negative control. To further ensure the selectivity of the biosensor to VEGF from the complex composite of cell media, the sensor surface was first blocked with the complete cell media after antibody immobilization. This blocking step prevents undesired nonspecific adsorption of other proteins present in the media, so that the sensor response can only be derived from VEGF specifically captured on the antibodies.In parallel, a microfluidic cell module was prepared by seeding the HeLa cells and incubating them overnight under the same conditions as described before. Then, the two modules were connected using microfluidic tubing and placed on the inverted microscope. A custom incubation box was put on the cell module to provide the appropriate culture conditions (37 °C and 5% CO2). The VEGF-stimulating medium was introduced at a flow rate of 10 μL min−1 throughout the measurement. The stimulus A23187 was confirmed not to interfere with the VEGF detection (Fig. S4†). The EOT spectra from the three in-line NHA sensors were simultaneously monitored for 10 hours (1 spectrum per second).
During the measurement, we observed a significant resonance shift emerging at approximately 5.5 h after the introduction of VEGF-stimulating medium (Fig. 5a), which corresponds well to the time profile obtained by ELISA analysis. The signal was obtained from the two NHA sensors functionalized with the anti-VEGF antibody (sensor A and sensor B) while the control sensor remained at a stable baseline. The absence of resonance response on the control sensor implies the lack of interference from other proteins and confirms the selectivity and specificity of our biosensor for VEGF detection. Interpolating the resonance shifts from both functionalized sensors with the VEGF calibration curve corresponded to an average VEGF concentration of 239.7 pg mL−1. Following this increase, no significant binding events were observed from the optical responses, implying that the secretion of VEGF reached a peak.
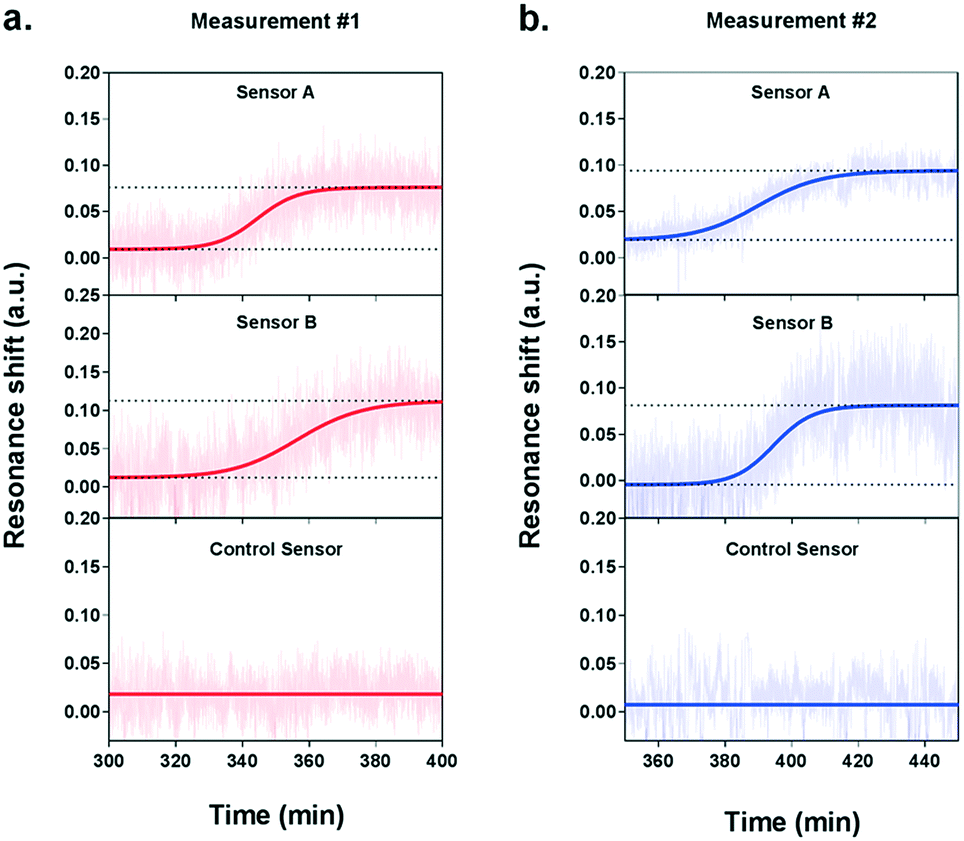 | ||
| Fig. 5 Real-time detection of VEGF secretion from live HeLa cells with the plasmonic nanohole array biosensor. The real-time measurements obtained from two independent experiments are showed in (a) and (b). Each graph shows the three sensorgrams acquired from the three individual sensor arrays in the detection module: sensor A and sensor B were functionalized with anti-VEGF antibodies, while sensor C without antibodies serves as a negative control. In both experiments, sensorgrams obtained from sensors A and B showed a clear increase of the signal while the control sensor remains at a low flat baseline. | ||
In order to ensure the reliability of our results, we performed a second independent measurement with new cells and a different plasmonic microarray but employed the same conditions as described before. A similar shift emerged at approximately 6 hours of monitoring, which corresponded to an average VEGF concentration of 235.8 pg mL−1 (Fig. 5b). Again, this time, the signal was clearly seen for the two specific anti-VEGF sensors while the control reference showed a flat baseline. The two concentrations for VEGF secretion were equivalent, which demonstrates the good reproducibility of our biosensor. Besides, the values obtained with our plasmonic microarray were in the same concentration range as those measured by ELISA with samples at discrete time points, therefore proving that our integrated biosensor can be used for continuous real-time monitoring and label-free quantification of cell secretion with good accuracy. It is worth mentioning that in this case, we employed ELISA to primarily confirm the time profile of VEGF secretion, rather than a validation method in terms of quantification due to the different working principle and sample collection manner. For ELISA, each sample was harvested after one hour, thus the VEGF is accumulated resulting in a relatively high concentration. In our plasmonic biosensing, the supernatant is not accumulated but it is immediately and continuously flowed over the sensor surface. Thereby, the VEGF does not accumulate and its concentration may be lower. Furthermore, we checked the viability and number of cells after both experiments, which were well maintained after the long-term measurements, confirming the reliability of the obtained signals.
Our experiments demonstrated the significant advantages that a nanoplasmonic label-free biosensor offers versus traditional enzymatic immunoassay techniques (e.g. ELISA), regarding simplicity, reagent consumption, and especially the real-time monitoring capability. Poor time resolution is inevitable with conventional analysis as samples are harvested at discreet time points and each sample has to be accumulated for a certain duration, causing laborious experimental operation for long-term analysis. However, label-free nanoplasmonic biosensors do not require pre-harvested samples and are capable of operating continuously for the analysis, which greatly enhance the temporal resolution and enable cost-effective and effortless biodetection. This label-free biosensing platform paves a new avenue for quick evaluation of the secretion ability among different cells (e.g. VEGF-secreting vs. non-secreting cells) or potent chemical agents for drug development.
Conclusions
We introduced a novel nanoplasmonic biosensor platform for the monitoring of live cell secretion activity in a real-time and label-free configuration. By incorporating a microfluidic cell culture module and an optofluidic biosensor equipped with robust and specific surface chemistry, we performed the first reported label-free and long-term analysis of cytokine secretion events by using plasmonic NHA-based biosensors. The biosensor functions based on the plasmonic EOT principle, enabling effortless device operation on a basic inverted microscopy set-up and circumventing the needs of complex prism-coupled configuration in SPR. We achieved an outstanding sensitivity of 145 pg mL−1 for VEGF detection in complex media. Additionally, the NHA biosensor showed excellent agreement with measurements performed using the current gold-standard, the ELISA technique, validating our results and demonstrating the reliability of our biosensor. The presented nanoplasmonic biosensor also represents a promising multiplexed sensing platform due to the high-throughput format of nanohole microarrays and the versatile surface chemistry on individual sensors. With these capabilities, the proposed label-free plasmonic NHA biosensor system provides a powerful analytical tool to monitor live cellular activities in a label-free and real-time manner for various fundamental biological studies, such as the analysis of tumor microenvironments or the evaluation of new therapies.Acknowledgements
We acknowledge the École Polytechnique Fédérale de Lausanne, the Switzerland Commission for Technology and Innovation, the NATO Public Diplomacy Division in the framework of “Science for Peace”, and the European Union Horizon 2020 research and innovation program under grant agreement no. 644956 for financial support. We acknowledge the Laboratory of Nanoscale Biology in EPFL for kindly providing the HeLa cell line. We also thank the Center of MicroNano Technology at EPFL for their contribution in nanofabrication.References
-
Z. Dembic, in The Cytokines of the Immune System, Academic Press, Amsterdam, 2015, pp. 263–281 Search PubMed
.
- G. Dranoff, Nat. Rev. Cancer, 2004, 4, 11–22 CrossRef CAS PubMed
- J. A. Ludwig and J. N. Weinstein, Nat. Rev. Cancer, 2005, 5, 845–856 CrossRef CAS PubMed
- S. Sethi, S. Ali, P. A. Philip and F. H. Sarkar, Int. J. Mol. Sci., 2013, 14, 14771–14784 CrossRef PubMed
- C. Sima, J. Hua, M. Cypert, T. Miller, H. M. Wilson-Robles, J. M. Trent, E. R. Dougherty and M. L. Bittner, Cancer Inf., 2016, 14, 33–43 CrossRef PubMed
- M. S. Wang, Z. Luo and N. Nitin, Anal. Bioanal. Chem., 2014, 406, 4195–4206 CrossRef CAS PubMed
- S. Sang, Y. Wang, Q. Feng, Y. Wei, J. Ji and W. Zhang, Crit. Rev. Biotechnol., 2016, 36, 465–481 CAS
- G. Acharya, C.-L. Chang and C. Savran, J. Am. Chem. Soc., 2006, 128, 3862–3863 CrossRef CAS PubMed
- J. M. Pérez, M. Jofre, P. Martínez, M. A. Yáñez, V. Catalan and V. Pruneri, Analyst, 2015, 140, 7734–7741 RSC
- A. B. González-Guerrero, S. Dante, D. Duval, J. Osmond and L. M. Lechuga, Procedia Eng., 2011, 25, 71–75 CrossRef
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed
- L. Xie, X. Yan and Y. Du, Biosens. Bioelectron., 2014, 53, 58–64 CrossRef CAS PubMed
- A. F. Chrimes, K. Khoshmanesh, S.-Y. Tang, B. R. Wood, P. R. Stoddart, S. S. E. Collins, A. Mitchell and K. Kalantar-zadeh, Biosens. Bioelectron., 2013, 49, 536–541 CrossRef CAS PubMed
- A. G. Brolo, Nat. Photonics, 2012, 6, 709–713 CrossRef CAS
- D. R. Shankaran, K. V. Gobi and N. Miura, Sens. Actuators, B, 2007, 121, 158–177 CrossRef CAS
- J. Schmidt, P. Guillaume, M. Irving, P. Baumgaertner, D. Speiser and I. F. Luescher, J. Biol. Chem., 2011, 286, 41723–41735 CrossRef CAS PubMed
- M.-C. Estevez, M. A. Otte, B. Sepulveda and L. M. Lechuga, Anal. Chim. Acta, 2014, 806, 55–73 CrossRef CAS PubMed
- C. Escobedo, Lab Chip, 2013, 13, 2445 RSC
- A. E. Cetin, A. F. Coskun, B. C. Galarreta, M. Huang, D. Herman, A. Ozcan and H. Altug, Light: Sci. Appl., 2014, 3, e122 CrossRef CAS
- T. W. Ebbesen, H. J. Lezec, H. F. Ghaemi, T. Thio and P. A. Wolff, Nature, 1998, 391, 667–669 CrossRef CAS
- A. A. Yanik, M. Huang, O. Kamohara, A. Artar, T. W. Geisbert, J. H. Connor and H. Altug, Nano Lett., 2010, 10, 4962–4969 CrossRef CAS PubMed
- S. H. Lee, N. C. Lindquist, N. J. Wittenberg, L. R. Jordan and S.-H. Oh, Lab Chip, 2012, 12, 3882 RSC
- A. Fanget, F. Traversi, S. Khlybov, P. Granjon, A. Magrez, L. Forró and A. Radenovic, Nano Lett., 2014, 14, 244–249 CrossRef CAS PubMed
- H. Im, H. Shao, Y. I. Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed
- T.-Y. Chang, M. Huang, A. A. Yanik, H.-Y. Tsai, P. Shi, S. Aksu, M. F. Yanik and H. Altug, Lab Chip, 2011, 11, 3596 RSC
- H. H. Nguyen, J. Park, S. Kang and M. Kim, Sensors, 2015, 15, 10481–10510 CrossRef CAS PubMed
- A. F. Coskun, A. E. Cetin, B. C. Galarreta, D. A. Alvarez, H. Altug and A. Ozcan, Sci. Rep., 2014, 4, 6789 CrossRef CAS PubMed
- B.-R. Oh, N.-T. Huang, W. Chen, J. H. Seo, P. Chen, T. T. Cornell, T. P. Shanley, J. Fu and K. Kurabayashi, ACS Nano, 2014, 8, 2667–2676 CrossRef CAS PubMed
- U. Y. Lau, S. S. Saxer, J. Lee, E. Bat and H. D. Maynard, ACS Nano, 2016, 10, 723–729 CrossRef CAS PubMed
- M. P. Raphael, J. A. Christodoulides, J. B. Delehanty, J. P. Long and J. M. Byers, Biophys. J., 2013, 105, 602–608 CrossRef CAS PubMed
- S. S. Aćimović, M. A. Ortega, V. Sanz, J. Berthelot, J. L. Garcia-Cordero, J. Renger, S. J. Maerkl, M. P. Kreuzer and R. Quidant, Nano Lett., 2014, 14, 2636–2641 CrossRef PubMed
- H. L. Goel and A. M. Mercurio, Nat. Rev. Cancer, 2013, 13, 871–882 CrossRef CAS PubMed
- S. Zhan, X. Wang and Y. Liu, Meas. Sci. Technol., 2011, 22, 25201 CrossRef
- T. McPherson, A. Kidane, I. Szleifer and K. Park, Langmuir, 1998, 14, 176–186 CrossRef CAS
- D. MacDougall and W. B. Crummett, Anal. Chem., 1980, 52, 2242–2249 CrossRef CAS
- G. Delfino, L. Di Costanzo, A. Paulis, G. Fabbrocini and G. Monfrecola, Photodermatol., Photoimmunol. Photomed., 2012, 28, 165–168 CrossRef CAS PubMed
- S. F. Abcouwer, P. L. Marjon, R. K. Loper and D. L. Vander Jagt, Invest. Ophthalmol. Visual Sci., 2002, 43, 2791–2798 Search PubMed
- C. Liu, T. Lei, K. Ino, T. Matsue, N. Tao and C.-Z. Li, Chem. Commun., 2012, 48, 10389 RSC
- P. Page, J. DeJong, A. Bandstra and R. A. Boomsma, Int. J. Cell Biol., 2014, 2014, e601063 Search PubMed
- S. Dai, A. Gocher, L. Euscher and A. Edelman, FASEB J., 2016, 30, 714.9 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc00277g |
| This journal is © The Royal Society of Chemistry 2017 |