
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimized droplet digital CFU assay (ddCFU) provides precise quantification of bacteria over a dynamic range of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) logs and beyond†
logs and beyond†
O.
Scheler
ab,
N.
Pacocha
a,
P. R.
Debski
a,
A.
Ruszczak
a,
T. S.
Kaminski
a and
P.
Garstecki
 *a
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: garst@ichf.edu.pl
bInstitute of Molecular and Cell Biology, University of Tartu, Riia 23, 51010 Tartu, Estonia
First published on 28th April 2017
Abstract
Standard digital assays need a large number of compartments for precise quantification of a sample over a broad dynamic range. We address this issue with an optimized droplet digital approach that uses a drastically reduced number of compartments for quantification. We generate serial logarithmic dilutions of an initial bacterial sample as an array of microliter-sized droplet plugs. In a subsequent step, these droplets are split into libraries of nanoliter droplets and pooled together for incubation and analysis. We show that our technology is at par with traditional dilution plate count for quantification of bacteria, but has the advantage of simplifying the experimental setup and reducing the manual workload. The method also has the potential to reduce the assay time significantly.
Introduction
Here, we demonstrate that bacteria can be quantified in droplet digital assays over a wide dynamic range of colony forming unit (CFU) densities with the use of a greatly reduced number of compartments compared to classical digital assays and in a potentially faster manner than via traditional plating methods.A precise enumeration of viable bacteria is important and often used in many different experiments in microbiology and genetics, for example, in analysis of bacterial viability following an antimicrobial treatment,1,2 evolution of mutants in bacterial population3 or analysis of bacterial physiology.4 Traditionally, microbes have been quantified in samples by plate counting, measurement of turbidity (OD) or with a cell cytometer.
Plate counting is by far the most widely used approach among these methods. In this technology, logarithmic dilutions of bacterial samples are plated on Petri dishes. The initial concentration of a sample is calculated from the number of colonies counted. While being inexpensive and widely available, the main disadvantages of this technique include: i) the requirement for piles of Petri dishes that have to be plated manually and lots of reagents and lab space for incubation, ii) long incubation time needed for colonies to grow and iii) the need for labor-intensive analysis as each Petri dish has to be analyzed separately either manually5 or by using automated readers.6
Measurement of turbidity is fast and simple. It measures the attenuation of light passing through the sample to deduct the concentration of bacteria. Its main disadvantages are: i) relative inaccuracy as the results do not show the exact amount of bacteria or their viability and ii) this technology cannot often be used for concentrations below ∼1 × 106–1 × 107 CFU mL−1 as such small densities do not scatter the light passing through the sample sufficiently for the typical sensors to measure the difference from the reference.5,7
Also, bacteria can be counted and their viability can be assessed using a flow cytometer. In this case, the results are fast and accurate, and a wide range of concentrations can be used. The main disadvantage of this technology is that it usually requires expensive, sophisticated cytometers to analyze bacterial samples. Simpler low-cost devices are often not suitable for measuring and analyzing objects in a micrometer range.
Widely popular and available quantitative PCR is a suitable tool for detection and identification of microbes. However, direct quantification of viable bacteria is complicated as the target nucleic acids can be present in the sample long after the death of bacteria.8
Droplet microfluidics has rapidly emerged as one of the key technologies opening up new experimental possibilities in microbiology.9 Examples are: i) detection and identification of pathogens,10 ii) antibiotic susceptibility testing,11–14 iii) studies of microbial physiology and interactions15,16 and iv) biotechnological selection and improvement of strains.17,18
Droplets have been shown to be an attractive technical platform for execution of digital quantification assays. In such assays, the sample is divided into a large number of small compartments (e.g. droplets) and the initial target concentration is determined by the fraction of positive droplets (that contain at least one copy of the target). The presence of the target in the compartment is usually detected by measuring a fluorescence signal.19–21
By far, the most popular field implementation of the digital assay technology has been nucleic acid quantification assays. There have been many demonstrations of droplet digital PCR19–22 and also there are several commercial platforms already available.23–25 Quantification of bacteria has also been demonstrated before with droplet technology.10,26–28 However, the technology has not gained such popularity as in the analysis of nucleic acid assays. This is plausibly because classical droplet digital assays need a very large number of compartments for operating over a wide range of target concentrations. In the classical digital assay, the number of compartments is proportional (and similar) to the dynamic range.19,29 This is especially problematic with microbiological experiments where CFU concentrations in the experiments often change from very low to very high (106–109 CFU mL−1 range).30,31 With the classical droplet digital approach, these assays would require millions of droplets to be generated and analyzed. Generation and analysis of millions of droplets in one assay has been shown in academic reports10,18,29,32,33 and in one commercial digital PCR platform,23 but it remains a time-consuming process that can be optimized further.
Here, we propose an alternative approach by implementing a rational design34 of droplet digital assays to drastically reduce the number of compartments needed for a quantitative microbial assay over a wide dynamic range. In our technology, we generate a logarithmic dilution series of samples, followed by partition of a sample and dilutions into small nL droplets. We pool these droplet libraries together for incubation and analysis.
We validate our method both theoretically and experimentally over a wide dynamic range of target bacterial concentrations. We show that such assays can be realized in a significantly shorter time than traditional plate counting. Finally, we also demonstrate ddCFU as a tool in assessing the antimicrobial efficiency of an antibiotic in a time-kill experiment.
Theory
The design of ddCFU assay is based on the algorithms developed for PCR-based DNA quantification assays.34,35 This approach requires preparation of a sequence of dilutions from a sample and next dividing them into libraries, i.e. sets of identical aqueous compartments. The dilution ratio between consecutive libraries is a fixed constant. Such an approach offers advantages over the classical digital assay as it enables: i) logarithmic scaling of the number of compartments (libraries) with a dynamic range of an assay, ii) constant relative precision of the assessment and iii) simple tuning of the assay's performance by changing the dilution ratio and a number of libraries. Depending on the experiment requirements, the multi-volume design allows tuning the precision of the assay and dynamic range of concentrations (or a number of targets in the sample that can be determined by the same assay) independently.The assay designed for the quantification of bacteria, which we present in this paper, allows the quantification of the concentration of the analyte over a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log range, providing precision (relative standard deviation of the estimate) better than 5%. We divided the sample into 11 libraries, each comprising approximately 3000 identical nL droplets (33000 droplets in total), with a 10-fold dilution ratio between the libraries (see Fig. 1A). The first library was not diluted. We then used the number of positive compartments counted during the end-point measurement in order to calculate the initial target concentration by means of our algorithms.
log range, providing precision (relative standard deviation of the estimate) better than 5%. We divided the sample into 11 libraries, each comprising approximately 3000 identical nL droplets (33000 droplets in total), with a 10-fold dilution ratio between the libraries (see Fig. 1A). The first library was not diluted. We then used the number of positive compartments counted during the end-point measurement in order to calculate the initial target concentration by means of our algorithms.
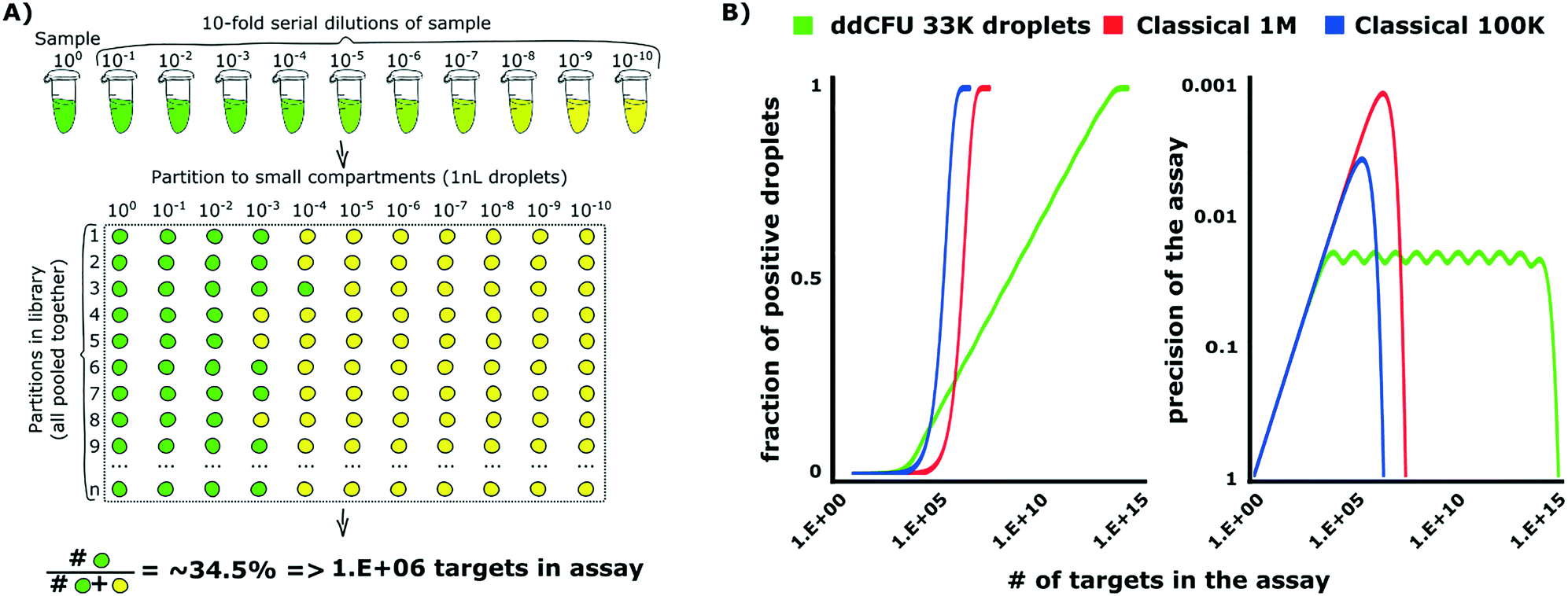 | ||
| Fig. 1 Theoretical background of optimized droplet digital counting of bacteria in ddCFU assay. A) The design of the ddCFU assay. We partition the sample into sets (libraries), each containing ∼3000 identical nanoliter partitions (compartments). The ratio of dilutions of compartments belonging to any two consecutive libraries is a fixed constant. Next, we incubate the compartments and count the positive fluorescent fraction. We use the recorded positive ratio as an input for calculating the initial number of targets in the sample via statistical algorithms based on the most probable number method and Bayes probability (see the ESI† for details). B) The comparison of the performance of the ddCFU assay with the performance of classical single-volume digital assays. The ddCFU assay responds linearly (linear increase of the fraction of positive compartments) to the logarithmically increasing number of targets in the sample, which makes it a promising analytical tool. It also offers precision of the assessment comparable to single-volume assays, even if they are composed of more compartments. Moreover, the quality of the assessment is maintained within a much wider dynamic range. The ‘waviness’ of the function describing the precision of the ddCFU assay is an artefact of the design based on the concept of libraries (please see the ESI† for details). Classical 1M and 100k stand for classical digital assays with one million and 100 thousand compartments, respectively. | ||
The algorithms are not limited to logarithmic dilutions and 11 libraries as we used in this paper. In a supplementary section, we also demonstrate an alternative assay that comprises 21 libraries, each having 1000 nanoliter droplets with a two-fold dilution sequence (21000 droplets in total). We recommend choosing the rational design that suits most the technological capabilities available in users' laboratories.34,36
We verified the performance of our assay numerically via grand canonical Monte Carlo simulations and compared it to the performance of state-of-the-art single-volume digital assays (Fig. 1B). The details of designing rational droplet digital assays and the instruction for the analysis of the outcome of such assays are given in (ESI†) section S1.
Results and discussion
Description of ddCFU technology
We started our ddCFU assay by generating ten serial logarithmic dilutions of a bacterial sample and transferring them together with the initial sample to a 96-well plate (Fig. 2A). We used the 96-well plate to place sample dilutions onto a rotAXYS automated positioning system tray for sequential introduction into a microfluidic system. Next, we aspirated 3 μL of each of the 11 concentrations (sample and 10 dilutions) into microfluidic tubing using the positioning system and a programmable syringe pump. Between the 3 μL plugs comprising the dilutions of the sample of bacteria, we aspirated the same volume of oil to keep the plugs separated in the tubing (3 μL spacer was the smallest volume that stably guaranteed spacing during transition from tubing to the chip). In our experiments, we prepared the dilutions manually, but for further automatization, additional microfluidic modules can be used.13,37,38 | ||
| Fig. 2 Schematic of the technology for ddCFU assay. A) We prepared a logarithmic dilution series of bacterial samples consisting of the sample itself and 10 dilutions. We transferred this series to a 96-well plate where a portion of each dilution was aspirated into microfluidic tubing as 3 μL plugs separated by additional oil spacers; B) then, we split the 3 μL plugs sequentially into 1 nL daughter droplets by flow focusing and collected them all together in a 1.5 mL test tube for incubation at 37 °C; C) after incubation, we measured the droplet fluorescence one-by-one and calculated the initial sample concentration using rational algorithms. | ||
In the next step, we inserted the tubing with dilutions into a poly(dimethylsiloxane) (PDMS) microfluidic chip with a flow-focusing junction (Fig. 2B) where we compartmentalized the 3 μL plugs into ∼1 nL droplets (the exact size distribution of droplets is shown in ESI† S3). We collected all of the generated droplets together in a standard 1.5 mL test tube which was then incubated at 37 °C.
After incubation, we aspirated the nL droplets into microfluidic tubing and pushed them into the counting chip (Fig. 2C). Droplet generation and counting chips both had similar flow-focusing geometries, but with slightly different channel sizes (see ESI† S2 for a more precise layout of the chips used in this article). Then, we fixed the chip in a confocal microscope stage to measure the intensity of fluorescence emitted by each of the nL droplets. To space the droplets during the measurement, we introduced an additional stream of carrier oil into the flow-focusing junction immediately before the image acquisition area.
Comparison between ddCFU and traditional plate counting
In order to test the rational design for droplet digital quantification of bacteria, we designed an experiment, as shown in Fig. 3A. In this experiment, we used an enhanced green fluorescent protein (EGFP)-producing Escherichia coli DH5α strain. We cultured the bacteria overnight and used them the next morning to prepare six different samples. Sample concentrations ranged over six orders of magnitude – from overnight stationary culture to 6th 10-fold logarithmic dilution. | ||
| Fig. 3 A) Comparison between ddCFU assay and conventional plate counting enumeration assay. We prepared six different samples from bacteria cultured overnight ranging over concentrations of six orders of magnitude and enumerated viable bacteria in these samples with both ddCFU and conventional plate counting. B) The quantification results between ddCFU assay and traditional plate counting. Axes X and Y show the bacterial concentration in the initial sample measured by conventional plate counting and ddCFU assay, respectively, on a logarithmic scale. | ||
Then, we used our ddCFU assay to quantify the viable bacteria in these prepared samples and we incubated droplets at 37 °C for 16 hours. For comparison and reference, we ran a conventional microbiological quantification assay where samples were plated on Petri dishes and colonies were counted after overnight incubation. The same samples and their dilutions were used in both experiments.
We compared the results (i.e. assessed the number of CFUs in the sample) obtained with our ddCFU assay and conventional plate counting. As shown in Fig. 3B, the two methods yield highly correlated results. The least-squares exponential fit is almost perfectly linear (the exponent in the fitted equation is close to unity) in the entire 6log range of the measured sample concentrations. Also, the quality of the fit, measured using the Pearson coefficient (R2 = 0.9964), confirms perfect corroboration of the two methods. The ddCFU assay offers a linear response (i.e. direct proportionality between the input concentration and the calculated concentration) qualifying itself as a promising tool for the quantification of bacteria.
The first important advantage of the ddCFU assay is that it requires just a single test tube to run the quantification experiment as sample dilutions are pooled together. With traditional Petri dish-based plate counting, all sample dilutions have to be plated on separate dishes to guarantee analyzable colony density (30–300 per plate). This means that for every test tube in the ddCFU assay we need six Petri dishes in a quantification assay where possible target concentrations range over six orders of magnitude.
Secondly, the ddCFU assay reduces the volume of reagents, the use of plasticware and the requirements for lab space and equipment, contributing to an overall reduced cost and ecological footprint of the assay.
Thirdly, the ddCFU analysis is easier to automate, making it less labor-consuming than the traditional plate-counting approach. In our case, we used a custom-made microfluidic setup and a confocal microscope to determine the fluorescence signal in droplets. However, there are already relatively inexpensive commercial droplet reading platforms available that have been designed for digital analysis.39 Also, conventional cytometers can be used to measure droplet fluorescence with double-emulsions33 or gel emulsions.14
We observed that the results obtained with our ddCFU assay were often slightly lower than those for traditional plate counting (∼25% on average in our case, see the ESI† for exact details). A similar reduced count was observed when we conducted the ddCFU assay with 2-fold dilutions (see the ESI† for further information). A similar reduction of bacterial cell viability in droplet microfluidic systems has been demonstrated before with E. coli. It has been hypothesized that such cell death may be due to shear forces on the cells during encapsulation.40,41 As a matter of caution, thus, each strain should be calibrated for such effects before direct use of the ddCFU assay for bacterial cell counting.
The lowest target concentration in the sample that can be detected with ddCFU assay depends on the initial sample volume introduced into a microfluidic system. In our case, the initial volume is 3 μL. This means that the lowest bacterial concentration that we can detect is 1 target per 3 μL. In microbiological experiments, this would stand for ∼3.33 × 102 CFU mL−1. This is more than enough for an analysis of wound infections for example.42 If there is a need for precise quantification of viable bacteria below that limit, additional enrichment stages can be applied, e.g. by centrifugation. There are also several microfluidic approaches described for sample enrichment.43–46 For example, such enrichment would be needed to detect bloodstream infections where it is common to find only a few pathogens per mL.47
Assay time in our droplet microfluidic system
Next, we investigated how fast we can detect viable bacteria with our droplet microfluidic approach. We tested two different bacteria and three different fluorescent labelling systems. In this experiment, we measured the minimum time of detection of fluorescence signals in bacteria-containing droplets with: a) intrinsic plasmid-encoded EGFP or b) metabolic marker dyes resazurin and C12-resazurin. These metabolic dyes show the presence of viable aerobic microorganisms in a sample and are therefore popular marker molecules that have been used in microbiological experiments with a droplet platform before. Aerobic organisms metabolically convert (reduce) resazurin to fluorescent resorufin (C12-resazurin to C12-resorufin, respectively).11,48–50 Their working principle is similar, but C12-resorufin has been shown before to be retained in droplets for an extended period of time.49We encapsulated bacteria in 1 nL droplets and monitored the emergence of a fluorescence signal over time. We adjusted the initial bacterial concentration so that most of the bacteria-containing droplets contained just a single CFU in the beginning (please see Scheler et al.49 for a detailed description). First, we tested EGFP-producing E. coli that was described and used in the ddCFU assay in the previous part of this report (Fig. 4). With EGFP, we could distinguish positive bacteria-containing droplets from the empty ones in six hours. Metabolic activity signals for resorufin and C12-resorufin were detected in four and five hours, respectively. Secondly, we tested Enterobacter aerogenes, the bacteria which has over the last few decades been increasingly associated with nosocomial infections.51 In our case, the fluorescence signals of E. aerogenes with resorufin and C12-resorufin were detected in three and four hours, respectively. Such detection time is a norm for resazurin assays in nL droplets as a similar detection time has been shown before for Staphylococcus aureus.11 Although the detection was faster with resazurin, with C12-resazurin we had higher sensitivity, specificity, and signal-to-noise ratio over time.49
 | ||
| Fig. 4 Graph presenting the incubation time needed for different bacteria and labelling methods to distinguish between positive (bacteria-containing) and negative (empty) droplets. | ||
These results demonstrate another advantage that our ddCFU assay presents over the traditional plate counting protocol. With the traditional approach, the incubation on the Petri dishes always includes a lengthy incubation period of at least ∼16–20 hours. In droplet assays, the small volume enables target molecules, cells and signals to accumulate faster to detectable concentrations.
In our experiments here, we tested only fluorescently labelled bacteria or fluorescent metabolic marker dyes. This does not mean that our ddCFU assay or droplet microfluidic technology in general is limited to fluorescence technologies. While the field is certainly dominated by fluorescence technologies,9–11,14,26,41,48,49 mostly because of their speed, there have also recently been rapid developments in label-free detection methods that include measuring bacteria-induced scattering or transmission changes.52–54 Further advances in label-free detection and monitoring of bacteria in droplets would make our ddCFU assay usable with greater selection of microbes in the future.
Antimicrobial time-kill analysis with ddCFU assays
As a demonstration of our ddCFU technology, we conducted an experiment where we measured the fraction of viable bacteria over time in a culture that was treated with an antibiotic. Time-kill tests are often conducted in studies to evaluate the efficiency and speed of antimicrobials to kill bacteria.30,31In our experiment, we used ∼30 mL of E. coli DH5α (EGFP) with ∼2 × 106 CFU mL−1 concentration as a test culture (Fig. 5). We started the experiment by adding one minimum inhibitory final concentration (1 MIC) of antibiotic norfloxacin to the bacterial solution. Then, we collected the samples after designated time intervals ranging from 0 to 25 hours, transferred the bacteria to fresh media without antibiotic and used the ddCFU assay to quantify viable bacteria.
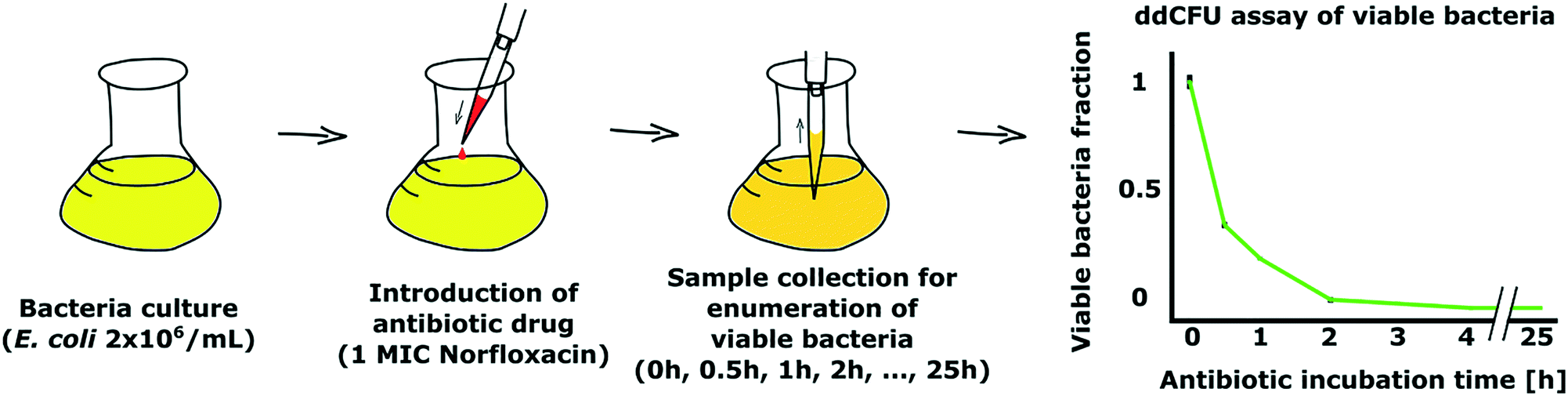 | ||
| Fig. 5 Demonstration of our ddCFU assay as a suitable tool to assess the bactericidal effect of antibiotics. We added an antibiotic, norfloxacin, to bacterial culture and collected samples after designated time intervals to measure the viability of bacteria (left). Graph showing the rapid decline of viability of bacteria treated with antibiotic over time (right). Viable fraction value 1 on the Y-axis stands for the concentration of bacteria at the beginning of the treatment, which was ∼2 × 106 CFU mL−1. | ||
Norfloxacin belongs to the antibiotic class of quinolones that target DNA gyrase in bacteria. DNA gyrase is an enzyme catalyzing structural changes in DNA topology. Norfloxacin disrupts the action of DNA gyrase that in turn leads to death of the bacterial cells over time.55
We observed that norfloxacin treatment rapidly started reducing the fraction of viable bacteria in our test solution (Fig. 5). In 30 minutes, more than half of the bacteria were killed by the antibiotic. The decrease in viability continued rapidly and we could not detect any viable bacteria in the sample after 4 h of incubation with the drug.
In a similar time-kill experiment, we tested the ddCFU assay in 2-fold dilution format that was described in the theory section. In this experiment, we investigated the effect of antimicrobial peptide p4 from chemerin protein. Chemerin is a multifunctional protein that has also been shown to have antimicrobial properties.56–59 In our experiment, we observed a rapid decrease of bacterial viability with addition of p4 at a final concentration of 100 μm. More detailed explanation of the experiment can be found in ESI† S6.
Conclusions
The traditional bacterial quantification approach by plating dilutions of a sample on Petri dishes is time- and labor-consuming. Classical droplet digital quantification provides a convenient alternative that offers great potential to save incubation time and design highly automated assays. The drawback of the classical design is that it needs a vast number of compartments (up to millions) for samples with high target concentrations and requires testing of multiple samples with presumed concentrations ranging over many orders of magnitude. Albeit technically possible, it greatly increases the time needed for droplet generation and analysis.Here, we both theoretically and experimentally demonstrated a rational approach to design a droplet digital quantification assay for viable bacteria. To our knowledge, this is the first demonstration of how to reduce drastically the number of compartments in a digital assay for quantification of target bacteria over a wide dynamic range of concentrations. Our approach enables designing (droplet) digital assays with a relatively small number of compartments. This simplifies the technology and reduces the time that is used to generate and analyze compartments.
The ddCFU technology is useful for microbiologists as we demonstrate a novel bacterial quantification technology that is easier to handle and potentially faster than conventional plate counting. From a more general droplet microfluidic engineering perspective, we demonstrate further the capabilities of rational design to simplify the needs for digital assay developments. Compared to traditional plate counting, our technology is less labor-consuming and potentially faster. Its advantage over classical droplet digital assays is the radically reduced number of compartments needed for the assay.
At the current stage, the demonstrated technology is not optimized for very low microbiological sample concentrations and additional calibration is needed to determine if test samples have reduced viability in the system. Also, non-fluorescent bacterial detection technologies in droplets have to be adapted to widen the scale of possible target bacteria. We foresee all of these technological hurdles being solved in the near future.
Experimental
Microfluidics
In our experiments, we used two different microfluidic chips, one for the generation of droplets and the second one for droplet analysis. Both chips were made of a poly(dimethylsiloxane) (PDMS) fluidic layer that was plasma-bonded to a 1 mm-thick glass slide (see ESI† S2 for details). The fabrication of the fluidic layer was as follows: in the first step, we fabricated polycarbonate (PC) molds in 5 mm-thick plates of PC (Macroclear, Bayer, Germany) using a CNC milling machine (MSG4025, Ergwind, Poland). Next, we poured PDMS (Sylgard 184, Dow Corning, USA) onto the PC chip and subsequently polymerized the mold at 95 °C for 45 min. In the following step, we silanized the PDMS mold for 3 h under 10 mbar pressure with vapors of tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane (United Chemical Technologies, USA). Cleaned PDMS negative masters were employed for molding of the positive PDMS replicas, which we subsequently bonded to a glass slide by exposing both parts for 30 s to oxygen plasma and placing them together. We modified the microfluidic channels hydrophobically by filling the chip with Novec 1720 (3M, United States) for 10 min. After fluid evaporation at room temperature, the chips were baked at 135 °C for 1 h to enhance bonding and preserve the modification. We controlled the flow of the oil and reagents in the microfluidic experiments with a rotAXYS positioning system and neMESYS syringe pumps (both from Cetoni, Germany). Droplets were generated with ∼600 Hz frequency and collected for off-chip incubation in 15 mL test tubes.Bacteria
We used two different bacteria in our experiments: Escherichia coli DH5α placEGFP (a kind gift from Prof. Douglas Weibel, Department of Biochemistry, University of Wisconsin-Madison, USA) and Enterobacter aerogenes PCM183 (Polish Collection of Microorganisms). For culturing bacteria in bulk and droplets, we used LB-Lennox media (Roth, Germany). With E. coli, the media contained 100 μg mL−1 ampicillin and 1 mM isopropyl β-D-1-thiogalactopyranoside IPTG (both from Thermo Fischer Scientific, USA) for EGFP plasmid retention and expression, respectively.Reagents
We used Novec HFE-7500 fluorocarbon oil (3M, USA) with 2% PFPE–PEG–PFPE surfactant (synthesized according to the protocol published by Holtze et al.60). The dye substrates resazurin sodium salt (Sigma-Aldrich, Germany) and C12-resazurin (part of Vybrant Cell Metabolic Assay Kit, Life Technologies, USA) were used as a 50 μM final solution. In the antibiotic kill test, we used 0.2 μg mL−1 (1 MIC) of norfloxacin (Sigma-Aldrich, Germany).Fluorescence measurement and data analysis
We measured the fluorescence of droplets using a droplet reading chip, which was mounted on the stage of an A1R confocal microscope (Nikon, Japan). We acquired the fluorescence of EGFP at excitation/detection wavelengths of 488 nm/500–550 nm and, in the case of resorufin/C12R, at 561 nm/570–620 nm, respectively. We analyzed the raw fluorescence data using MS Office Excel (Microsoft, USA) with Real Statistics Resource Pack (http://www.real-statistics.com/). Droplet signals stand for the peak relative fluorescence intensities allocated to each droplet. The numerical simulations testing the performance of ddCFU assays were executed by means of the ROOT Environment (https://root.cern.ch/).Acknowledgements
The project was co-financed by the European Research Council Starting Grant 279647 and National Science Centre funding based on decision number DEC-2014/12/W/NZ6/00454 (Symfonia). This work was also supported by the Estonian Research Council grant PUTJD589 (to O. S.). T. S. K. was supported by the Ministry of Science and Higher Education through the scholarship for outstanding young researchers (agreement 0722/E 64/STYP/10/295). This project was partially performed in the laboratories funded by NanoFun POIG.02.02.00-00-025/09. P. G. acknowledges support from the Foundation for Polish Science via the Idee dla Polski program.References
- G. L. Drusano, M. Neely, M. Van Guilder, A. Schumitzky, D. Brown, S. Fikes, C. Peloquin and A. Louie, PLoS One, 2014, 9, e101311 Search PubMed.
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190ra81 CrossRef PubMed.
- G. G. Zhanel, M. Mayer, N. Laing and H. J. Adam, Antimicrob. Agents Chemother., 2006, 50, 2228–2230 CrossRef CAS PubMed.
- K. E. Kram and S. E. Finkel, Appl. Environ. Microbiol., 2014, 80, 1732–1738 CrossRef PubMed.
- A. Brown, in Benson's Microbiological applications, 12th edition, 2012, pp. 143–154 Search PubMed.
- S. D. Brugger, C. Baumberger, M. Jost, W. Jenni, U. Brugger and K. Mühlemann, PLoS One, 2012, 7, e33695 CAS.
- J. Campbell, Curr. Protoc. Chem. Biol., 2010, 2, 195–208 Search PubMed.
- S. Aellen, Y. A. Que, B. Guignard, M. Haenni and P. Moreillon, Antimicrob. Agents Chemother., 2006, 50, 1913–1920 CrossRef CAS PubMed.
- T. S. Kaminski, O. Scheler and P. Garstecki, Lab Chip, 2016, 16, 2168–2187 RSC.
- D.-K. Kang, M. M. Ali, K. Zhang, S. S. Huang, E. Peterson, M. A. Digman, E. Gratton and W. Zhao, Nat. Commun., 2014, 5, 5427 CrossRef CAS PubMed.
- J. Q. Boedicker, L. Li, T. R. Kline and R. F. Ismagilov, Lab Chip, 2008, 8, 1265–1272 RSC.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- L. Derzsi, T. S. Kaminski and P. Garstecki, Lab Chip, 2016, 16, 893–901 RSC.
- Y. Eun, A. S. Utada, M. F. Copeland, S. Takeuchi and D. B. Weibel, ACS Chem. Biol., 2011, 6, 260–266 CrossRef CAS PubMed.
- S. M. Bjork, S. L. Sjostrom, H. Andersson Svahn and H. N. Joensson, Biomicrofluidics, 2015, 9, 44128 CrossRef PubMed.
- S. Huang, J. K. Srimani, A. J. Lee, Y. Zhang, A. J. Lopatkin, K. W. Leong and L. You, Biomaterials, 2015, 61, 239–245 CrossRef CAS PubMed.
- S. L. Sjostrom, Y. Bai, M. Huang, Z. Liu, J. Nielsen, H. N. Joensson and H. Andersson Svahn, Lab Chip, 2014, 14, 806–813 RSC.
- J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J. C. Baret, M. Marquez, A. M. Klibanov, A. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6550–6550 CrossRef CAS PubMed.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- M. M. Kiss, L. Ortoleva-donnelly, N. R. Beer, J. Warner, C. G. Bailey, B. W. Colston, J. M. Rothberg, D. R. Link, H. Leamon, R. Technologies and H. Ave, Anal. Chem., 2008, 80, 8975–8981 CrossRef CAS PubMed.
- J. Madic, A. Zocevic, V. Senlis, E. Fradet, B. Andre, S. Muller, R. Dangla and M. E. Droniou, Biomol. Detect. Quantif., 2016, 10, 34–46 CrossRef CAS PubMed.
- R. T. Hayden, Z. Gu, S. S. Sam, Y. Sun, L. Tang, S. Pounds and A. M. Caliendo, J. Clin. Microbiol., 2016, 54, 2602–2608 CrossRef CAS PubMed.
- http://raindancetech.com/digital-pcr-tech/ .
- http://www.stilla.fr/ .
- http://www.bio-rad.com/en-us/category/digital-pcr .
- F. Lyu, M. Xu, Y. Cheng, J. Xie, J. Rao and S. K. Y. Tang, Biomicrofluidics, 2015, 9, 44120 CrossRef PubMed.
- P. R. Marcoux, M. Dupoy, R. Mathey, A. Novelli-Rousseau, V. Heran, S. Morales, F. Rivera, P. L. Joly, J. P. Moy and F. Mallard, Colloids Surf., A, 2011, 377, 54–62 CrossRef CAS.
- M. Najah, A. D. Griffiths and M. Ryckelynck, Anal. Chem., 2012, 84, 1202–1209 CrossRef CAS PubMed.
- A. C. Hatch, J. S. Fisher, A. R. Tovar, A. T. Hsieh, R. Lin, S. L. Pentoney, D. L. Yang and A. P. Lee, Lab Chip, 2011, 11, 3838 RSC.
- M. M. Sopirala, J. E. Mangino, W. A. Gebreyes, B. Biller, T. Bannerman, J. M. Balada-Llasat and P. Pancholi, Antimicrob. Agents Chemother., 2010, 54, 4678–4683 CrossRef CAS PubMed.
- M. Albur, A. Noel, K. Bowker and A. MacGowan, Antimicrob. Agents Chemother., 2012, 56, 3441–3443 CrossRef CAS PubMed.
- M. Kim, M. Pan, Y. Gai, S. Pang, C. Han, C. Yang and S. K. Y. Tang, Lab Chip, 2015, 15, 1417–1423 RSC.
- S. W. Lim and A. Abate, Lab Chip, 2013, 13, 4563–4572 RSC.
- P. R. Debski, K. Gewartowski, M. Sulima, T. S. Kaminski and P. Garstecki, Anal. Chem., 2015, 87, 8203–8209 CrossRef CAS PubMed.
- P. R. Debski and P. Garstecki, Biomol. Detect. Quantif., 2016, 10, 24–30 CrossRef PubMed.
- J. E. Kreutz, T. Munson, T. Huynh, F. Shen, W. Du and R. Ismagilov, Anal. Chem., 2011, 83, 8158–8168 CrossRef CAS PubMed.
- M. Ruan, C.-G. Niu, P.-Z. Qin, G.-M. Zeng, Z.-H. Yang, H. He and J. Huang, Anal. Chim. Acta, 2010, 664, 95–99 CrossRef CAS PubMed.
- S. Jakiela, T. S. Kaminski, O. Cybulski, D. B. Weibel and P. Garstecki, Angew. Chem., Int. Ed., 2013, 52, 8908–8911 CrossRef CAS PubMed.
- http://www.elveflow.com/microfluidic-flow-control-products/microfluidic-flow-control-module/optical-reader-for-microfluidics/ .
- S. Cho, D. Kang, S. Sim, F. Geier, J. Kim, X. Niu, J. B. Edel, S. Chang, R. C. R. Wootton, K. S. Elvira and J. Andrew, Anal. Chem., 2013, 85, 8866–8872 CrossRef CAS PubMed.
- D.-K. Kang, X. Gong, S. Cho, J.-Y. Kim, J. B. Edel, S.-I. Chang, J. Choo and A. J. DeMello, Anal. Chem., 2015, 87, 10770–10778 CrossRef CAS PubMed.
- P. G. Bowler, B. I. Duerden and D. G. Armstrong, Clin. Microbiol. Rev., 2001, 14, 244–269 CrossRef CAS PubMed.
- J. Y. Zhang, J. Do, W. R. Premasiri, L. D. Ziegler and C. M. Klapperich, Lab Chip, 2010, 10, 3265–3270 RSC.
- D. Puchberger-Enengl, S. Podszun, H. Heinz, C. Hermann, P. Vulto and G. A. Urban, Biomicrofluidics, 2011, 5, 1–10 Search PubMed.
- H. W. Hou, H. Y. Gan, A. A. S. Bhagat, L. D. Li, C. T. Lim and J. Han, Biomicrofluidics, 2012, 6, 024115 Search PubMed.
- P. Ohlsson, M. Evander, K. Petersson, L. Mellhammar, A. Lehmusvuori, U. Karhunen, M. Soikkeli, T. Seppä, E. Tuunainen, A. Spangar, P. Von Lode, K. Rantakokko-Jalava, G. Otto, S. Scheding, T. Soukka, S. Wittfooth and T. Laurell, Anal. Chem., 2016, 88, 9403–9411 CrossRef CAS PubMed.
- M. S. Moore, M. G. McCarroll, C. D. McCann, L. May, N. Younes and J. A. Jordan, J. Clin. Microbiol., 2016, 54, 99–105 CrossRef CAS PubMed.
- M. Pan, L. Rosenfeld, M. Kim, M. Xu, E. Lin, R. Derda and S. K. Y. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 21446–21453 CAS.
- O. Scheler, T. S. Kaminski, A. Ruszczak and P. Garstecki, ACS Appl. Mater. Interfaces, 2016, 8, 11318–11325 CAS.
- Y. Chen, W. A. Gani and S. K. Y. Tang, Lab Chip, 2012, 12, 5093–5103 RSC.
- A. Davin-Regli and J.-M. Pagès, Front. Microbiol., 2015, 6, 392 Search PubMed.
- X. Liu, R. E. Painter, K. Enesa, D. Holmes, G. Whyte, C. G. Garlisi, F. J. Monsma, M. Rehak, F. F. Craig and C. A. Smith, Lab Chip, 2016, 16, 1632–1643 Search PubMed.
- E. Zang, S. Brandes, M. Tovar, K. Martin, F. Mech, P. Horbert, T. Henkel, M. T. Figge and M. Roth, Lab Chip, 2013, 13, 3707–3713 RSC.
- J. Q. Yu, W. Huang, L. K. Chin, L. Lei, Z. P. Lin, W. Ser, H. Chen, T. C. Ayi, P. H. Yap, C. H. Chen and A. Q. Liu, Lab Chip, 2014, 14, 3519 RSC.
- F. Collin, S. Karkare and A. Maxwell, Appl. Microbiol. Biotechnol., 2011, 92, 479–497 CrossRef CAS PubMed.
- M. Banas, K. Zabieglo, G. Kasetty, M. Kapinska-Mrowiecka, J. Borowczyk, J. Drukala, K. Murzyn, B. A. Zabel, E. C. Butcher, J. M. Schroeder, A. Schmidtchen and J. Cichy, PLoS One, 2013, 8, e58709 CAS.
- M. Banas, A. Zegar, M. Kwitniewski, K. Zabieglo, J. Marczynska, M. Kapinska-Mrowiecka, M. LaJevic, B. A. Zabel and J. Cichy, PLoS One, 2015, 10, e0117830 Search PubMed.
- P. Kulig, T. Kantyka, B. A. Zabel, M. Banas, A. Chyra, A. Stefanska, H. Tu, S. J. Allen, T. M. Handel, A. Kozik, J. Potempa, E. C. Butcher and J. Cichy, J. Immunol., 2011, 187, 1403–1410 CrossRef CAS PubMed.
- B. A. Zabel, M. Kwitniewski, M. Banas, K. Zabieglo, K. Murzyn and J. Cichy, Am. J. Clin. Exp. Immunol., 2014, 3, 1–19 Search PubMed.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angilè, C. H. J. Schmitz, S. Köster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc00206h |
| This journal is © The Royal Society of Chemistry 2017 |