
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-chamber microfluidic platform for high-precision skin permeation testing
M.
Alberti†
 a,
Y.
Dancik†
*bc,
G.
Sriram
b,
B.
Wu
a,
Y. L.
Teo
c,
Z.
Feng
a,
M.
Bigliardi-Qi
bc,
R. G.
Wu
a,
Z. P.
Wang‡
*a and
P. L.
Bigliardi‡
bc
a,
Y.
Dancik†
*bc,
G.
Sriram
b,
B.
Wu
a,
Y. L.
Teo
c,
Z.
Feng
a,
M.
Bigliardi-Qi
bc,
R. G.
Wu
a,
Z. P.
Wang‡
*a and
P. L.
Bigliardi‡
bc
aSingapore Institute of Manufacturing Technology, A*STAR, 2 Fusionopolis Way, Level 10, Innovis, 138634 Singapore. E-mail: yuri.dancik@imb.a-star.edu.sg; zpwang@simtech.a-star.edu.sg
bExperimental Dermatology Laboratory, Institute of Medical Biology, A*STAR, 8a Biomedical Grove, #06-06, 138648 Singapore
cClinical Research Unit for Skin, Allergy and Regeneration, Institute of Medical Biology, A*STAR, 8a Biomedical Grove, #06-06, 138648 Singapore
First published on 3rd April 2017
Abstract
The established in vitro tool used for testing the absorption and penetration of chemicals through skin in pharmacology, toxicology and cosmetic science is the static Franz diffusion cell. While widespread, Franz cells are relatively costly, low-throughput and results may suffer from poor reproducibility. Microfluidics has the potential to overcome these drawbacks. In this paper, we present a novel microfluidic skin permeation platform and validate it rigorously against the Franz cell by comparing the transport of 3 model chemicals of varying lipophilicity: caffeine, salicylic acid and testosterone. Permeation experiments through silicone membranes show that the chip yields higher sensitivity in permeant cumulative amounts and comparable or lower coefficients of variation. Using a skin organotypic culture, we show that the chip decreases the effect of unstirred water layers that can occur in static Franz cells. The validation reported herein sets the stage for efficient skin permeation and toxicity screening and further development of microfluidic skin-on-chip devices.
Introduction
High-throughput, reliable and cost-effective in vitro skin permeation assays are in high demand. In pharmaceutical science, transdermal drug delivery (TDD) is a major area of research and development due to the advantages of cutaneous over oral drug administration for certain classes of compounds. This includes drugs requiring a prolonged period of delivery, high potency drugs with a short biological half-life and those subject to a significant hepatic first-pass effect.1 The sheer number of possible drug/vehicle interactions necessitates fast and effective screening methods for drug formulation design.2 In toxicology and risk assessment, estimating toxicity following skin exposure of consumer products, pesticides or lipophilic industrial solvents is of major concern.3–5 With the European Union's 2007 Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) regulation having drastically increased the number of chemicals that need to be evaluated for toxicity6 and the European Union's 2013 ban on the use of animals in cosmetic product testing, toxicology and cosmetic science have a similar need for the development and validation of high-throughput, alternative skin permeation testing methods.The traditional systems for in vitro skin permeation testing are the static Franz and flow-through diffusion cells, in which excised skin or a skin substitute is sandwiched between a donor compartment and a receptor compartment (Fig. 1a). Sampling of the receptor solution occurs at pre-determined times following application of a donor solution. This yields profiles of the concentration or cumulative amount over time of the compound of interest, from which transport parameters can be derived. Though widely used, Franz cells' typical diffusion areas of 1 to 3 cm2, receptor volumes on the order of a few mL and time-consuming procedures render them relatively costly and low-throughput.7 In addition, an unstirred water layer (UWL) may form in the static Franz cell when highly permeable membranes and lipophilic chemicals are used. Supplementary experiments and calculation are necessary to assess the effect of the UWL on the chemical's permeability through the membrane.8,9
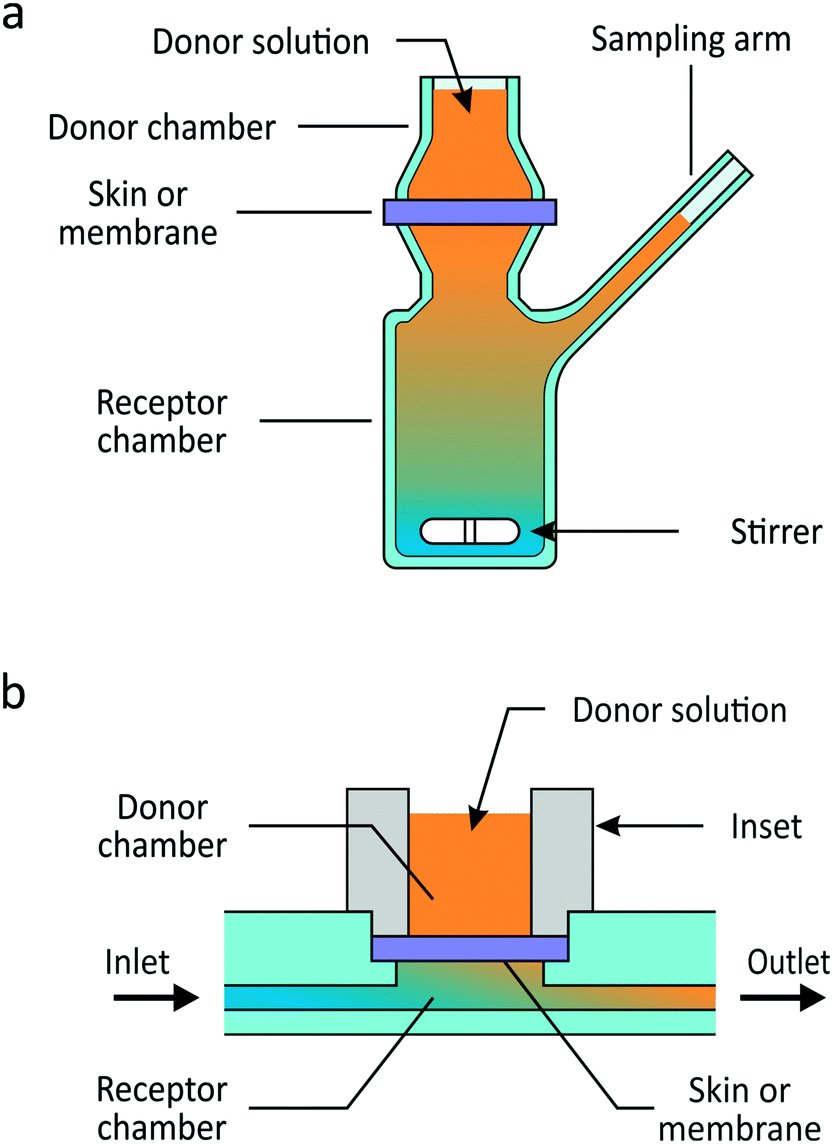 | ||
| Fig. 1 Schematic illustrations of (a) the static Franz diffusion cell and (b) a permeation unit of the microfluidic permeation array (μFPA). In the μFPA, skin or a synthetic membrane is placed in the permeation chamber, and its edges sealed by an inset defining the diffusion area. The receptor chamber is provided with inlet and outlet channels, and it is perfused with a receiver solution into which the compound of interest permeates. | ||
Microfluidics has the potential to increase throughput and improve reproducibility of experimental skin simulations by providing cost-effective platforms that mimic the cutaneous physiological environment.10 Microfluidic systems have been designed to grow a keratinocyte and dendritic cells co-culture,11 integrate in vitro skin models, cultures or biopsies and hair follicle units12 or other tissues,13,14 and to simulate and test perspiration.15–17 Systems focusing on skin permeation include Mah et al.’s flow-through cell with rat or pig skin,18 Abaci et al.’s pumpless microfluidic platform housing a human skin equivalent19 and Provin et al.’s microfluidic diffusion cell with a lipid-coated polycarbonate membrane as a skin substitute.20 Each of these permeability devices, however, was made of polydimethylsiloxane (PDMS), a material unsuitable for industrial mass production, incompatible with organic solvents and known to exhibit significant adsorption of small hydrophobic compounds.21–24 Differences in design parameters between static and flow-through cell designs can contribute to variability in the final results.8,25–28 Hence, there is a clear need for rigorous validation of novel microfluidic devices for skin permeation against the conventional systems. Given the growing number of organ-on-a-chip systems, such validation is also an important, often underestimated step towards the development of reliable skin-on-chips for in vitro permeation studies.
We present a novel microfluidic platform enabling high-throughput skin permeation testing in a flow-through design at a fraction of the material cost of Franz diffusion cells (Fig. 1b). This microfluidic permeation array (μFPA) is made of thermoplastic material, suitable for mass production. Synthetic membranes, excised animal or human skin, and skin organotypic cultures (skin-OTCs) can be easily integrated. The device's current design allows for six permeation experiments to be conducted simultaneously, important for reproducibility. The flow rate can be tuned to guarantee minimal permeant concentration in the receptor compartment in order to mimic the in vivo sink condition due to cutaneous blood flow.29 We validated the μFPA against static Franz cells by comparing the steady-state permeation of caffeine, salicylic acid and testosterone. These chemicals cover a range of lipophilicities as recommended by the OECD guideline 428.30,31 In a first instance, silicone membranes were used to avoid variability associated with skin. These experiments show that the μFPA yields lower limits of detection as well as lower coefficients of variation than the Franz cells. The μFPA was further tested for permeation through full-thickness skin-OTCs developed in-house. This latter experiment shows that the μFPA lessens the effect of unstirred water layers, a potentially significant pitfall of static Franz cells.8
Materials and methods
μFPA design and fabrication
The μFPA is composed of a multi-chamber microfluidic chip and a set of self-locking hollow cylindrical insets that define six independent permeation units symmetrically arranged in a 50 mm × 75 mm format (Fig. 2a). Six synthetic membrane disks or circular skin punches (7 mm in diameter) can be accommodated in the open chambers of the microfluidic chip. The edges of the membranes or skin pieces are sealed by the self-locking insets, if necessary equipped with elastomeric annuli. Each inset defines the donor chamber and the diffusion area of the permeation unit. The assembled device is a 6-chamber μFPA with diffusion areas of 0.2 cm2, donor capacities of up to 300 μL and receptor volumes of 28 μL.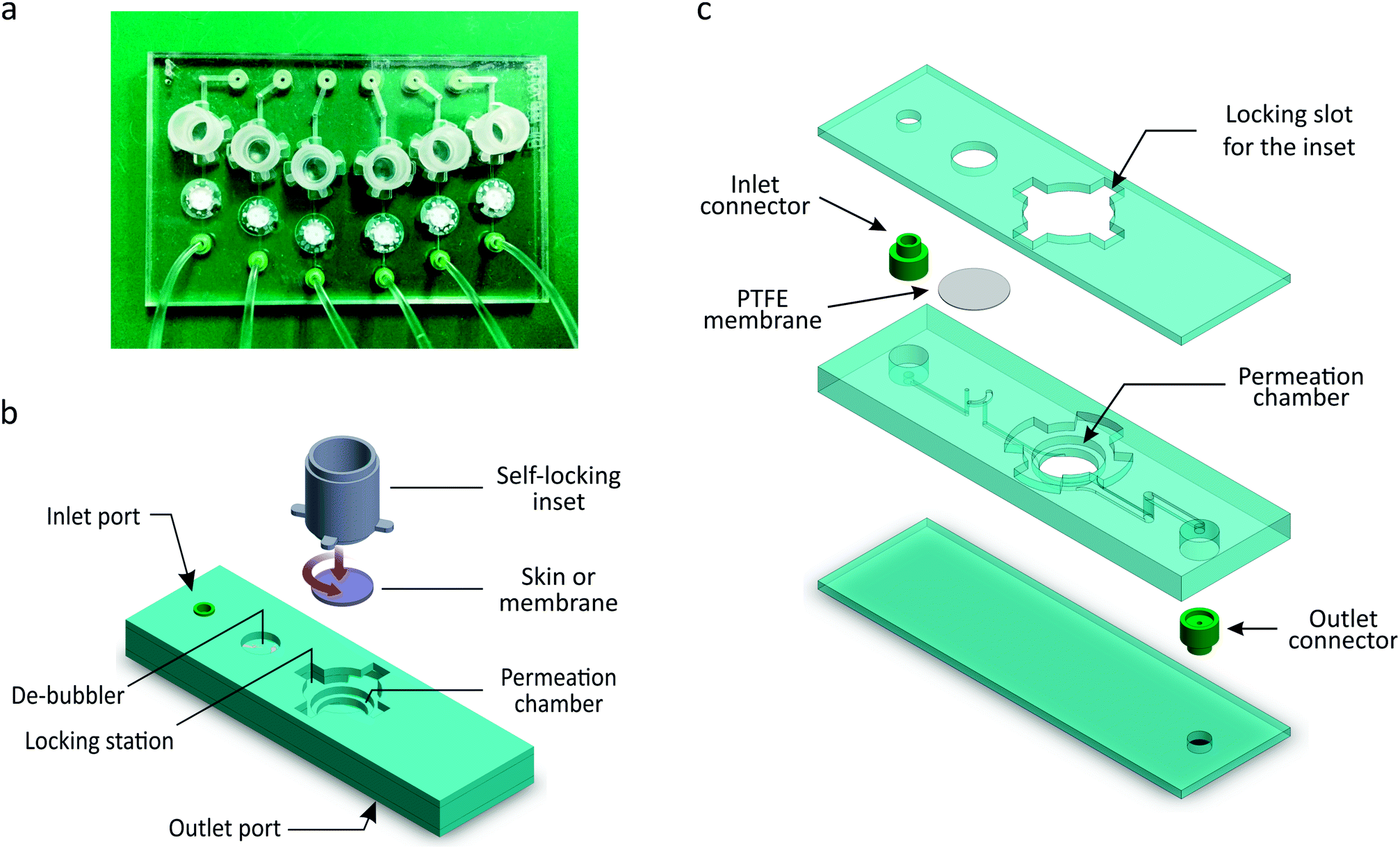 | ||
| Fig. 2 (a) Top view image of the 6-chamber μFPA. (b) 3D illustration of a single permeation unit composing the μFPA and of the self-locking inset mechanism. The inset enables the sealing of the skin tissue or membrane into the permeation chamber. (c) Exploded view of the permeation microfluidic unit composing the chip. | ||
As shown in Fig. 2b, each microfluidic permeation unit is composed of a cylindrical permeation chamber; a locking station for the inset; a set of inlet and outlet channels; a de-bubbling unit integrated along the inlet channel; and embedded connectors. The receptor's inlet and outlet channels are 0.5 mm and 1 mm wide, respectively; all channels are 0.3 mm high. The inlet port is located in the upper surface of the chip in order to connect the inlet channel to the inlet tubing. The outlet port is located in the lower surface of the chip so that the rim of the embedded connector protrude out from the chip surface: this feature allows the outflow to drip directly into the collecting well without touching the chip surface, thus avoiding the spreading of the drops on the chip lower surface and compromising the collection volumes. The six outlet ports are regularly spaced as the wells of a 96-well plate.
The microfluidic chips were fabricated by thermally bonding together three microstructured thermoplastics layers, six circular polytetrafluoroethylene (PTFE) filter membranes (Fluoropore™, Merck KGaA, Germany), and twelve silicone rubber connectors (Fig. 2c).
All the chips used for permeation tests on silicone membranes were made of polycarbonate (PC). One chip made of PC and one made of poly(methyl methacrylate) (PMMA) were used for the permeation tests on skin-OTCs to check for variability due to the material. The microfluidic features were microstructured in the polymer layers by micromilling. The PTFE filter membranes (8 mm in diameter) were placed between the second and the third layers, aligned with the microstructured features of the de-bubbling unit. The de-bubblers prevent bubbles from reaching the receptor chambers and affecting permeation. The tubing connectors were cast in silicone rubber (XIAMETER® RTV-4130-J, Dow Corning, USA) from a micromilled PMMA mould. The de-bubblers and connectors were fabricated according to designs and processes developed by SIMTech Microfluidic Foundry.32
The self-locking insets were fabricated in PC by micromilling. The inset has a hollow cylindrical body and four thin teeth (0.6 mm thick, 1.6 mm wide) protruding 1.8 mm from its lateral surface, perpendicularly to the cylinder axis (Fig. 2a). The locking station on the chip is composed of four docking slots arranged in a geometry corresponding to the inset's teeth. Each slot is composed of a vertical entrance for the tooth and a lateral (1 mm high) locking chamber.
The base of the inset is meant to clamp and seal the edges of the membrane or the skin tissue to avoid undesired flow of the donor solution through eventual gaps between the membrane and the walls of the diffusion chamber. When using skin-OTCs, a gasket annulus made of silicone (HT-6240 BISCO® transparent, Rogers Corporation, USA) is placed between the inset and the skin tissue in order to guarantee an adequate sealing. The inset may also allow placing a protective material on the membrane or skin and studying its influence on the permeation results.
Working principle
Once the membrane (or the skin tissue and a gasket annulus) is placed in the permeation chamber, the inset is mounted in the locking structure of the chip by aligning its teeth with the corresponding vertical entrances in the locking station, pressing it down and turning it counterclockwise to position and maintain the teeth inside the lateral slots (Fig. 2b). Once in the locking position, the inset compresses the edges of the membrane. This compression seals the system and at the same time generates a counterforce from the inset's teeth against the upper surface of the locking chambers that keeps the inset in place.The inlets are connected to six 2-stop SC0002 Tygon® ST R-3607 tubings (Cole-Parmer GmbH, Germany) of an Ismatec® IPCN-12 peristaltic pump (Cole-Parmer GmbH, Germany) to supply the receptor buffer to each diffusion unit. The μFPA is positioned above a UV transparent 96-well plate so that each outlet port aligns with one row of microplate wells. The outflow is collected directly in the wells and, at the end of each time interval, the μFPA is slid above the plate until the outlet ports align to the wells of the next row (Fig. 3).
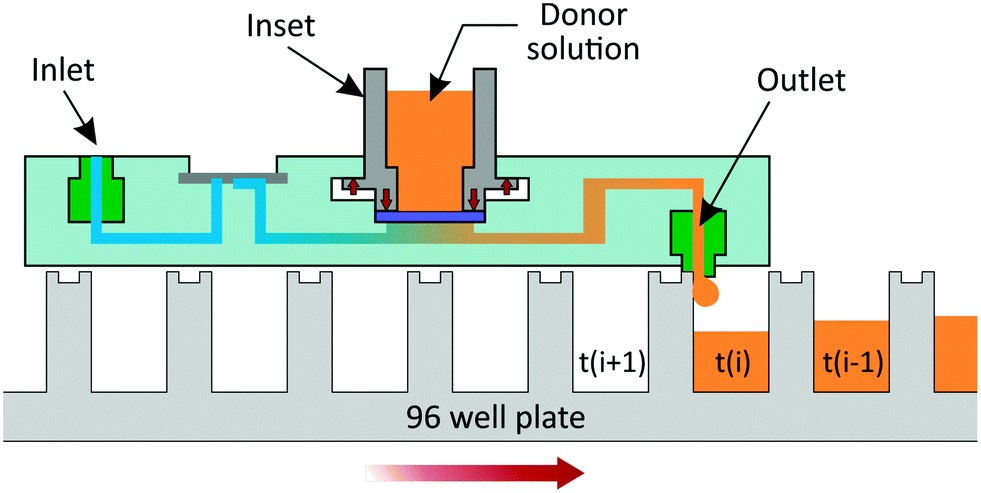 | ||
| Fig. 3 Schematic illustration of the μFPA's working principle. The μFPA is placed on a 96-well plate such that the 6 outlet connectors are aligned with 6 wells of a given row. At the end of each time interval, the plate is moved until the outlet ports align with the next row of wells. The receptor solution is collected in each well from time t(i − 1) to t(i). As the outlet connector protrudes slightly from the chip's bottom surface, contact between the connector and the edge of the well at time t(i), when the plates is moved, prevents undesired spillage of the perfusate outside of the well. | ||
The pump flow rate and collection time intervals are tuned so that, unlike conventional flow-through systems, the entire perfusate is fractioned and collected directly in 96-well plates and easily assayed for concentrations.
Skin organotypic cultures
Immortalized human N/TERT-1 keratinocytes33 were maintained in keratinocyte serum free medium (K-SFM, Gibco, ThermoFisher Scientific) supplemented with 0.09 mM calcium, 0.2 ng mL−1 epidermal growth factor (EGF, Gibco, ThermoFisher Scientific), 25 μg mL−1 bovine pituitary extract (Gibco, ThermoFisher Scientific) and 1% penicillin–streptomycin (PAN Biotech GmbH). Human primary foreskin-derived dermal fibroblasts were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin.Full-thickness skin-OTCs were fabricated using a fibrin-based dermal matrix as previously described by Toh et al.34 Briefly, dermal equivalents were generated by encapsulating human dermal fibroblasts (2.5 × 105 cells per mL of matrix) within a fibrin-based matrix in a 6-well Falcon® cell culture insert (polyethylene terephthalate (PET) membrane, pore size 1 μm, Corning Life Sciences, MA, USA). The dermal equivalents were cultured in serum-free OTC medium-A comprising of Opti-MEM (Gibco, ThermoFisher Scientific), supplemented with 0.1% bovine serum albumin (BSA), hydrocortisone (50 μg mL−1), ascorbic acid (10 mg mL−1), 1% SITE supplement containing selenium (5 ng mL−1), insulin (10 μg mL−1), transferrin (5.5 μg mL−1), and ethanolamine (2 μg mL−1), 200 KIU mL−1 aprotinin (MP Biomedicals, CA, USA) and 1% penicillin/streptomycin. After 4 days, N/TERT-1 keratinocytes (4 × 105 cm−2) were seeded on top of fibroblast-populated dermal equivalents and cultured under submerged conditions using serum-free OTC medium-B comprising of K-SFM supplemented with 0.1% BSA, hydrocortisone (50 μg mL−1), ascorbic acid (10 mg mL−1), 1% SITE supplement, 0.2 ng mL−1 EGF, 200 KIU mL−1 aprotinin, 1.2 mM calcium chloride and 1% penicillin/streptomycin. After 2 days, the organotypic cultures were moved to deep-well plates (Corning Life Sciences, MA, USA) and cultured at air–liquid interface for 2 weeks to allow differentiation, stratification, and cornification. During the air–liquid interface culture, serum-free OTC media-C comprised of K-SFM supplemented with 0.1% BSA, hydrocortisone (50 μg mL−1), ascorbic acid (10 mg mL−1), 1% SITE+3 supplement containing selenium (5 ng mL−1), insulin (10 μg mL−1), transferrin (5.5 μg mL−1), ethanolamine (2 μg mL−1), linoleic acid (4.7 μg mL−1) and oleic acid (4.7 μg mL−1), 200 KIU mL−1 aprotinin, 1.2 mM calcium chloride (MP Biomedicals, CA, USA) and 1% penicillin/streptomycin. All the media supplements were obtained from Sigma-Aldrich, unless otherwise specified.
Chemicals
Caffeine, salicylic acid, testosterone, isopropyl myristate (IPM), propylene glycol (PG) were obtained from Sigma-Aldrich, Singapore; ethanol (EtOH) from Merck KGaA (Germany), Dulbecco's phosphate buffered saline (PBS) from GE healthcare Life Sciences (USA). Solvents and concentrations (Table 1) were selected to obtain solutions corresponding to ∼90% saturation, ensuring infinite applied doses and the onset of steady-state transport while allowing for mass balances to be measured.35| Permeant | Donor concentration 103 [μg mL−1] | Vehicle | Membrane treatment | Receptor solution |
|---|---|---|---|---|
| Caffeine | 13.5 | PBS | IPM | PBS |
| Salicylic acid | 2.92 | PG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBS (20:80) :PBS (20:80) |
PBS | PBS |
| Testosterone | 7.07 | EtOH:PBS (50:50) |
IPM | EtOH:PBS (50:50) |
Silicone membrane and organotypic skin preparation
Prior to the permeation experiments, 2 × 2 cm2 pieces of 0.05 cm-thick silicone membrane (HT-6240 BISCO® transparent, Rogers Corporation, USA) were equilibrated overnight in IPM for the caffeine and testosterone experiments, and in PBS for the salicylic acid experiments. The skin-OTCs attached to their support membranes were carefully cut out from the cell culture inserts using a scalpel on the day of the permeation experiment. Thicknesses of the silicone membrane and skin-OTCs pieces were measured using a digital caliper. Penetration experiments were also run on the PET membranes of the cell culture inserts to account for any diffusive resistance of the support membrane. For each compound, permeation experiments in the μFPA and in the Franz cells were conducted on the same day under the same environmental conditions.Permeation experiments
The skin-OTCs experiment was performed with two different chips, one made of PC, the other of PMMA. In this way, the effect of material on inter-chip variability was assessed. Six circular pieces of skin-OTCs (7 mm in diameter) attached to support membranes were punched out of two cell culture inserts and integrated into the μFPAs such that each chip contained skin-OTCs from each insert. Six PC support membrane disks alone were inserted into the remaining permeation chambers. In each experiment, 275 μL of donor solution was applied to the silicone membrane or skin. To minimize evaporation during the experiments, the top openings of the self-locking insets (Fig. 3) were occluded with sealing tape (Petri-Seal™, Diversified Biotech, USA). Immediately after each experiment, the solutions remaining in the donor and in the receptor were collected for the mass balance. The receptor compartment was emptied by occluding the de-bubbler with sealing tape and infusing air into it. The sealing tape, the silicone membranes or skin pieces, the silicone annulus used in the skin experiments as well as the PBS used to wash the μFPA components were assayed.
Analytical method
Concentrations were determined by UV spectroscopy using a microplate reader (Synergy™ H1, BioTek Instruments Inc., VT, USA). Caffeine, salicylic acid and testosterone peak absorbance readings were at 272 nm, 296 nm and 245 nm, respectively.Data analysis
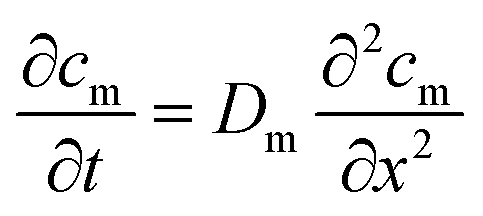 | (1) |
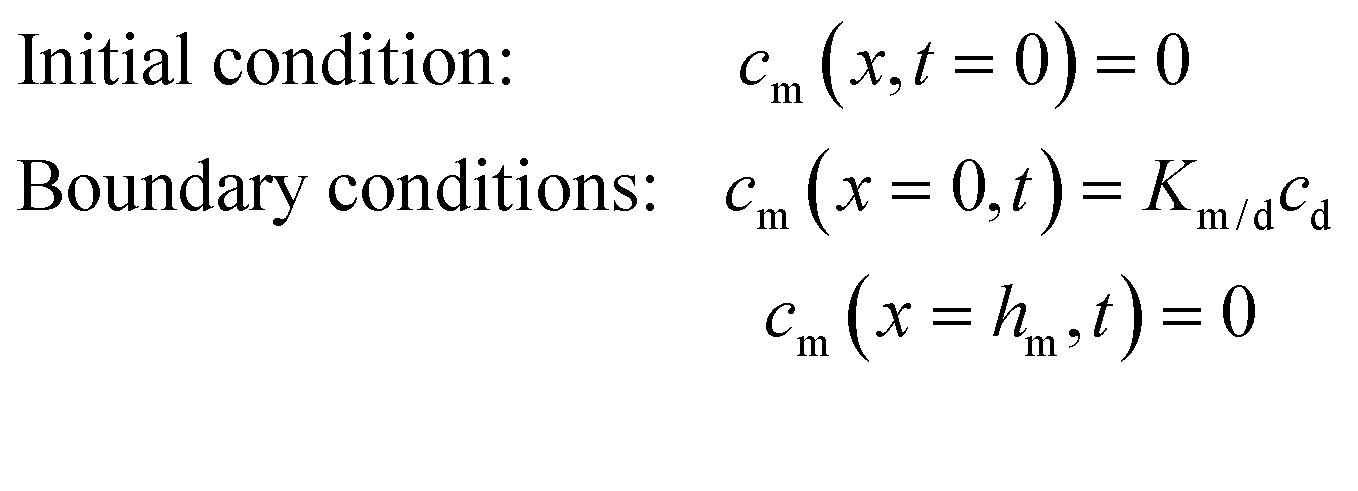 | (2) |
The solution to eqn (1) and (2) is often expressed as the cumulative amount of permeant in the receptor, Q(t):
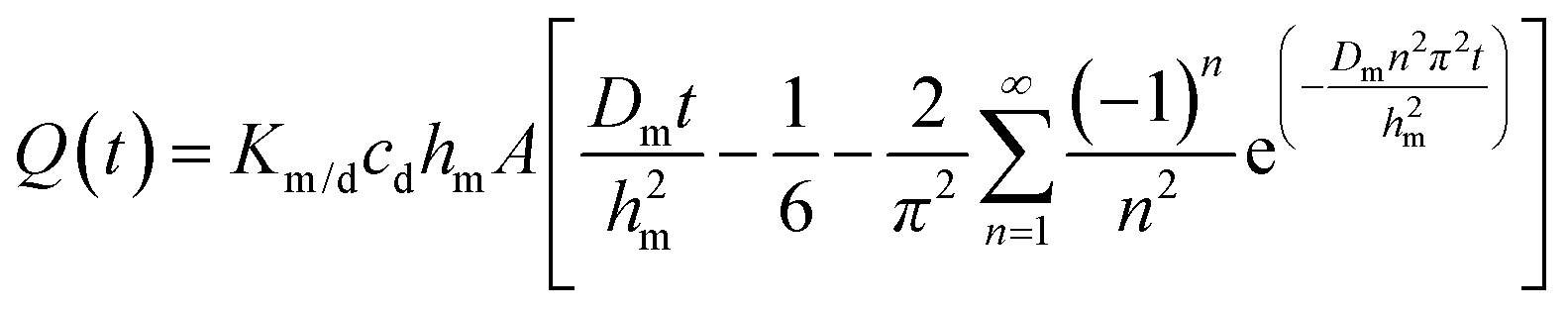 | (3) |
In eqn (3), hm designates the membrane thickness, Km/d the membrane/donor equilibrium partition coefficient and A the diffusion area. At steady state, the cumulative amount is given by
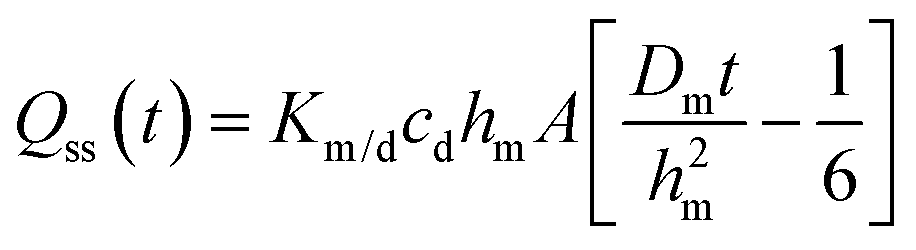 | (4) |
| Qss(t) = kpcdA(t − tlag) = JssA(t − tlag) | (5) |
The receptor solution concentrations obtained from the Franz cell and μFPA permeation experiments were converted to cumulative amounts. In the case of the μFPA results, this conversion accounts for the receptor flow rate. Regression of eqn (5) against the linear part of the experimental Qss(t) profiles yields kp, tlag and Jss.
The steady state 1D model can be extended to incorporate unstirred water layers (UWLs) above and below the membrane by considering the membrane and UWL steady state permeability coefficients as inverses of serial diffusive resistances. The total diffusive resistance is thus the sum of the resistances due to the membrane and the UWLs:
| Rtotal = Rm + Ruwl | (6) |
In the UWL experiments, linear regression of Rtotal against resistances from i dialysis membranes is conducted to obtain Ruwl according to eqn (7):8
| Rtotal = iRdialysis m + Ruwl | (7) |
The total UWL thickness huwl is estimated from Ruwl and the diffusivity of the permeant of interest in dilute aqueous solution, D∞w:
| huwl = D∞wRuwl | (8) |
To estimate D∞w, the Wilke–Chang correlation36 is used,
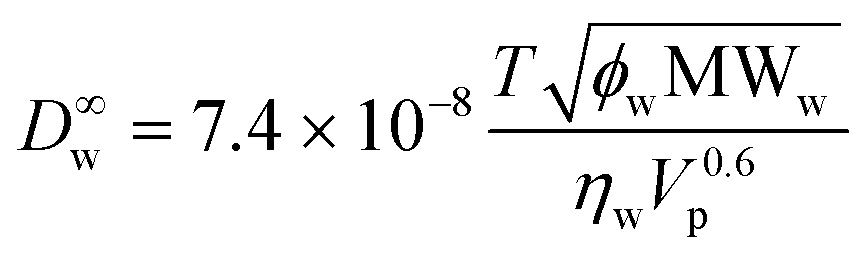 | (9) |
Numerical simulation
In the μFPA, permeant accumulation in the receptor chamber may be expected. It is described by the left-hand term of the general mass balance equation for flow-through diffusion cells:26,28,38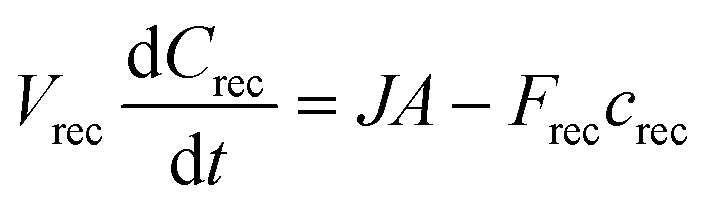 | (10) |
To evaluate the relevance of the accumulation for the three compounds, a finite element analysis of the 3D transport in the μFPA was implemented in COMSOL Multiphysics® (COMSOL Inc., USA). The membrane diffusion and partition coefficients (Dm, Km/d) were estimated from the Franz cells experiments.
Caffeine and salicylic acid diffusivities in the receptor solution (PBS) were estimated using the Wilke–Chang correlation36 (eqn (9)). Testosterone diffusivity in the 1:1 v/v ethanol:PBS (water) mixture (D∞m) was estimated from the Leffler–Cullinan correlation:39
 | (11) |
Results and discussion
Flow rate
Flow-through system parameters such as receptor cell volume, flow rate, sampling frequency and collector tube volume have been reported to modify the apparent flux41 and the onset of steady state.26 The flow rates in the μFPA were set at 4 μL min−1 in the silicone membrane experiments and 8 μL min−1 in those with skin-OTCs, where higher fluxes were expected. These flow rates maintained receptor sink conditions over the course of the experiment, avoided excessive permeant dilution in the outflow volumes, and allowed for collection volumes smaller than the maximum 96-well plate capacity (<300 μL) and minimal lower limits of detection (LOD). In order to collect the outflow during each time interval in a single well, maximum time intervals of 1 h and 30 min were allowed with receptor flow rates of 4 and 8 μL min−1, respectively. These flow rates determine an outflow volume of 240 μL at these maximum time intervals, which enables LODs of 0.1 μg mL−1 for caffeine and salicylic acid, and 0.4 μg mL−1 for testosterone. The limits of quantitation (LOQ) are 0.4 μg mL−1 and 1.2 μg mL−1, respectively.Permeation in μFPA vs. Franz cell
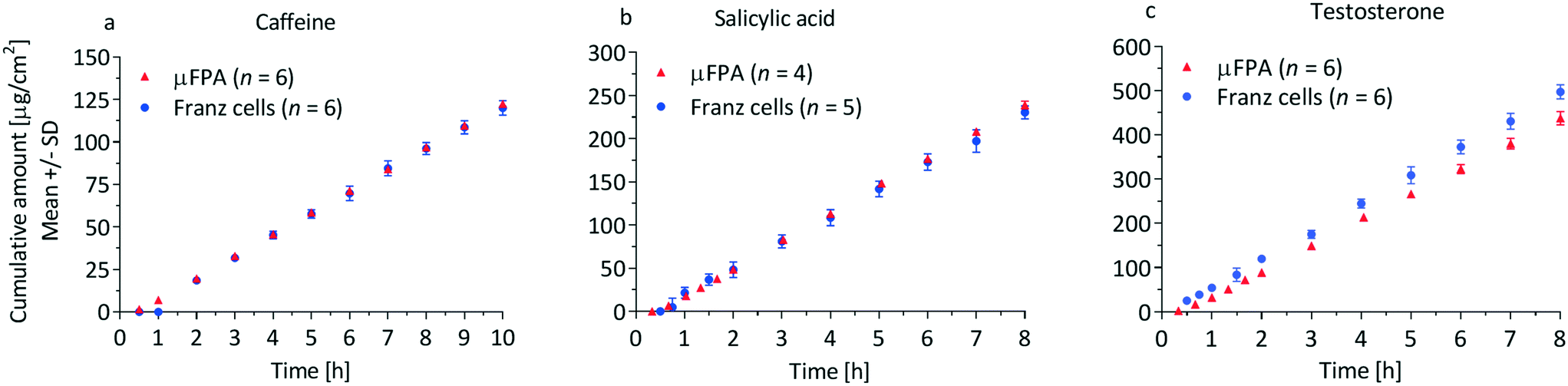 | ||
| Fig. 4 Cumulative amount profiles of (a) caffeine, (b) salicylic acid and (c) testosterone through silicone membranes in the μFPA and the Franz cells. | ||
| μFPA | Franz cells | |
|---|---|---|
| Caffeine | n = 6 | n = 6 |
| J ss [μg cm−2] | 13 ± 0.13 | 13 ± 0.36 |
| t lag [h] | 0.43 ± 0.022 | 0.45 ± 0.072 |
| k p [10−3 cm h−1] | 0.93 ± 0.03 | 0.94 ± 0.01 |
| Permeant recovered [%] | 97.1 ± 1.1 | 100.8 ± 1.3 |
| Salicylic acid | n = 4 | n = 5 |
| J ss [μg cm−2] | 32 ± 0.60 | 30 ± 0.34 |
| t lag [h] | 0.43 ± 0.03 | 0.32 ± 0.11 |
| k p [10−3 cm h−1] | 11 ± 0.21 | 10 ± 0.12 |
| Permeant recovered [%] | 99.8 ± 0.8 | 103.2 ± 2.2 |
| Testosterone | n = 6 | n = 6 |
| J ss [μg cm−2] | 57 (62) ± 4.9 | 64 ± 2.0 |
| t lag [h] | 0.36 ± 0.11 | 0.22 ± 0.16 |
| k p [10−3 cm h−1] | 8.2 (8.9) ± 0.70 | 9.2 ± 0.29 |
| Permeant recovered [%] | 98.5 ± 4.5 | 103.4 ± 1.7 |
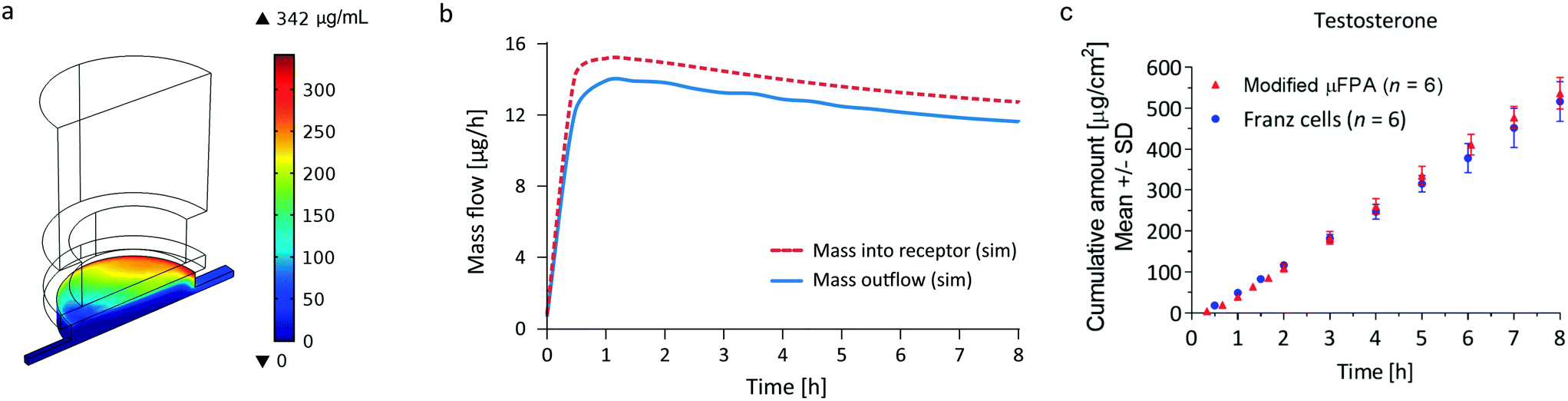 | ||
| Fig. 5 Simulation results for the determination of testosterone accumulation in the μFPA's receptor chamber. (a) Simulated testosterone concentration in the μFPA: the mass accumulates in the receptor towards the outlet channel. (b) Comparison of the simulated testosterone mass flowing from the membrane into the receptor (dashed line) with the mass flowing out of the outlet channel (continuous line). (c) Cumulative amount profiles of testosterone through silicone membranes in the μFPA modified to eliminate permeant accumulation in the receptor compartment and Franz cells. | ||
| Modified μFPA (n = 6) | Franz cells (n = 6) | |
|---|---|---|
| J ss [μg cm−2] | 70.4 ± 2.8 | 68 ± 7.2 |
| t lag [h] | 0.28 ± 0.19 | 0.33 ± 0.16 |
| k p [10−3 cm h−1] | 10.2 ± 0.40 | 9.8 ± 0.10 |
| Permeant recovered [%] | 91.4 ± 5.2 | 101.7 ± 2.1 |
The precision of the two systems was assessed from the coefficients of variation (CV) of the steady state cumulative amount values. Sources of variations may be related to the manufacturing tolerances for the Franz cell orifice diameter and to small deformations of the clamped membranes. For caffeine and salicylic acid, the steady state cumulative amount CVs range from 1 to 2% in the μFPA, and 3 to 8% in the Franz cells, indicating higher measurement precision in the former (Fig. 4a and b). For testosterone, CVs are 6 to 8% in the unmodified μFPA vs. 3 to 6% in the Franz cells (Fig. 4c). The somewhat lower precision in the μFPA's testosterone test is due to cumulative amounts obtained from one of the six permeation chambers. In the comparison of the modified μFPA to Franz cells (Fig. 5c), CVs are 5 to 8% in the μFPA, comparable to the 5 to 10% obtained in the Franz cells.
Khan et al. reported high CVs for caffeine (64.9%) and for testosterone (32.3%) permeation through PDMS membranes in Franz cells.42 Provin et al. tested their microfluidic diffusion cell with caffeine through a lipid coated membrane, reporting an average CV of 52.3%.20 Ng et al. achieved a CV of 6% for ibuprofen permeation through PDMS membranes on Franz cells.43 Bosman et al. obtained CVs around 10% in a flow-through cell for transport of [3H]dexetimide through Silastic® membranes.44 None of these studies reached CVs as low as the ones achieved by our μFPA. Overall, the μFPA enables higher precision when compared directly to the Franz cell or to other systems for permeation testing.
A possible explanation for the lower variability observed in the μFPA is the fact that the whole perfusate is assayed, while in the static Franz cell only samples of the receptor solution are used for quantification of the permeate. Moreover, the higher reproducibility of the geometrical features in the μFPA due to the computer numerical control machining aids minimizing the variability. It is expected that fabrication by injection molding may enhance this effect.
The cumulative amount profiles in Fig. 4 also show that, with the analytical method used in this study, concentrations of the initial permeation time points can be measured only in the μFPA experiments. This higher sensitivity is due to the smaller volumes in which the permeated compounds are diluted. Since the initial permeation typically follows an exponential profile, the μFPA may allow a more accurate estimate of the lag time if the entire experimental cumulative amount curve is fit directly to eqn (3). Consequently the use of the μFPA can also be advantageous for finite dose experiments, for which sharper changes in the flux need to be detected and smaller permeation rates need to be quantified.
 | ||
| Fig. 6 (a) Representative hematoxylin and eosin-stained section of the full-thickness skin organotypic culture (skin-OTC) used in the caffeine permeation experiment. (b) Cumulative amount profiles of caffeine through the skin-OTC in the μFPA vs. Franz cells. (c) Total resistances to caffeine diffusion in the unstirred water layer (UWL) experiments. | ||
| μFPA (n = 6) | Franz cells (n = 3) | |
|---|---|---|
| J ss [μg cm−2] | 670 (788) ± 23 | 600 (836) ± 26 |
| t lag [h] | ∼0 | ∼0 |
| k p [10−3 cm h−1] | 51 (59) ± 1.7 | 45 (62) ± 1.9 |
| Permeant recovered [%] | 95.6 ± 1.8 | 90.9 ± 2.1 |
The CV of the cumulative amounts in the steady state regime stabilizes around 3% in the μFPA experiments, and around 5% in the Franz cells, indicating higher measurement precision in the former. These CV values are calculated from the combined results of both μFPAs and of both skin tissues used in this test. The μFPA's inter-chip and inter-OTC variability for caffeine permeating through the skin-OTC is very low. Similarly, the bulk materials of the chip (PC and PMMA) did not affect the reproducibility or the precision, with steady state cumulative amounts in the PC and PMMA chips differing by less than 4% and the derived flux and permeability coefficients by less than 2%.
Conclusions and outlook
In this study, we presented and validated a microfluidic platform for improved, cost-effective in vitro skin permeation testing. This μFPA yields comparable or lower variability in the results, allows quantitation of lower permeation rates and reduces the problem of UWLs. In contrast to other miniaturization attempts,9–11 the μFPA was developed following the principles of design for manufacturing, with a standard format size and a body made of a thermoplastic material; thus it is suitable for mass production techniques. The possibility of using PC, PMMA or cyclic olefin copolymer for the bulk of the μFPA overcomes issues related to adsorption of lipophilic compounds usually associated with PDMS. The six-chamber version of the μFPA described here allows for an adequate number of replicates as recommended by OECD guidelines for skin absorption30 and by the FDA's Guidance for Industry for in vitro release testing of semisolid dosage form.47 The throughput can be easily increased by using more μFPAs in parallel and is only limited by the number of available perfusion channels in the pumps. The design of the μFPA can also be modified to integrate more permeation chambers, further reducing the cost of a replicate. In addition, maintenance of the skin surface temperature at 32 °C can be achieved by either placing the μFPA on a hotplate or pre-heating the receptor solution.The cost of the experiment is significantly reduced as the μFPA requires ten-fold smaller skin tissues and lower amounts of tested drugs compared to Franz diffusion cells. Six glass-made Franz diffusion cells with large fabrication tolerances are replaced by an equivalent multi-chamber disposable plastic chip. As the proposed analytical method only requires a microplate reader, the μFPA is a convenient option for those laboratories where expensive and sophisticated instrumentation such as high-performance liquid chromatography or mass spectroscopy is not available.
Because of the simplicity of the μFPA, the fraction collection process and the procedural steps required for a valid mass balance can be automated by means of a downstream collecting system and an upstream fluidic control system, respectively. The downstream collecting system will automatically slide the 96-well plates placed under the chip at predetermined time intervals, enabling long-term experiments with minimal manual operations. The upstream fluidic control system should be programmable and its operation coordinated with the downstream collecting system. For finite dose protocols48 involving liquid or gaseous substances, exposure to the skin could be automated in the μFPA by integrating microfluidics on the donor side.
The validation of this μFPA is a critical step towards the development of a skin-on-chip device for reliable, standardized and high-throughput skin permeation and toxicity assays.
Acknowledgements
This research was financially supported by Singapore A*STAR's Joint Council Office, grant no. 1334 K00081. We thank Dr. Bhimsen Rout (Institute of Medical Biology, A*STAR) and Matthew A. Miller (James L. Winkle College of Pharmacy, University of Cincinnati) for helpful discussions.Notes and references
- S. Wiedersberg and R. H. Guy, J. Controlled Release, 2014, 190, 150–156 CrossRef CAS PubMed.
- D. Karadzovska and J. E. Riviere, Eur. J. Pharm. Sci., 2013, 50, 569–576 CrossRef CAS PubMed.
- H. E. Buist, G. Schaafsma and J. J. M. van de Sandt, Regul. Toxicol. Pharmacol., 2009, 54, 221–228 CrossRef CAS PubMed.
- M. A. Ngo, M. O'Malley and H. I. Maibach, J. Appl. Toxicol., 2010, 30, 91–114 CAS.
- C. J. Weschler and W. W. Nazaroff, Indoor Air, 2012, 22, 356–377 CrossRef CAS PubMed.
- S. Schreiber, A. Mahmoud, A. Vuia, M. Rübbelke, E. Schmidt, M. Schaller, H. Kandarova, A. Haberland, U. Schäfer and U. Bock, Toxicol. In Vitro, 2005, 19, 813–822 CrossRef CAS PubMed.
- M. R. Prausnitz, S. Mitragotri and R. Langer, Nat. Rev. Drug Discovery, 2004, 3, 115–124 CrossRef CAS PubMed.
- M. A. Miller and G. Kasting, Pharm. Dev. Technol., 2012, 17, 705–711 CrossRef CAS PubMed.
- P. E. Nielsen and A. Avdeef, Eur. J. Pharm. Sci., 2004, 22, 33–41 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nature, 2014, 201, 4 Search PubMed.
- Q. Ramadan and F. C. W. Ting, Lab Chip, 2016, 16, 1899–1908 RSC.
- B. Ataç, I. Wagner, R. Horland, R. Lauster, U. Marx, A. G. Tonevitsky, R. P. Azar and G. Lindner, Lab Chip, 2013, 13, 3555–3561 RSC.
- I. Maschmeyer, A. K. Lorenz, K. Schimek, T. Hasenberg, A. P. Ramme, J. Hübner, M. Lindner, C. Drewell, S. Bauer and A. Thomas, Lab Chip, 2015, 15, 2688–2699 RSC.
- I. Wagner, E.-M. Materne, S. Brincker, U. Süßbier, C. Frädrich, M. Busek, F. Sonntag, D. A. Sakharov, E. V. Trushkin and A. G. Tonevitsky, Lab Chip, 2013, 13, 3538–3547 RSC.
- L. Hou, J. Hagen, X. Wang, I. Papautsky, R. Naik, N. Kelley-Loughnane and J. Heikenfeld, Lab Chip, 2013, 13, 1868–1875 RSC.
- R. Peng, Z. Sonner, A. Hauke, E. Wilder, J. Kasting, T. Gaillard, D. Swaille, F. Sherman, X. Mao and J. Hagen, Lab Chip, 2016, 16, 4415–4423 RSC.
- Z. Sonner, E. Wilder, J. Heikenfeld, G. Kasting, F. Beyette, D. Swaile, F. Sherman, J. Joyce, J. Hagen and N. Kelley-Loughnane, Biomicrofluidics, 2015, 9, 031301 CrossRef CAS PubMed.
- C. S. Mah, J. Singh Kochhar, P. S. Ong and L. Kang, Int. J. Pharm., 2013, 441, 433–440 CrossRef CAS PubMed.
- H. E. Abaci, K. Gledhill, Z. Guo, A. M. Christiano and M. L. Shuler, Lab Chip, 2015, 15, 882–888 RSC.
- C. Provin, A. Nicolas, S. Grégoire and T. Fujii, Pharm. Res., 2015, 32, 2704–2712 CAS.
- N. Li, M. Schwartz and C. Ionescu-Zanetti, J. Biomol. Screening, 2009, 14, 194–202 CrossRef CAS PubMed.
- K. Ren, J. Zhou and H. Wu, Acc. Chem. Res., 2013, 46, 2396–2406 CrossRef CAS PubMed.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- P. M. van Midwoud, A. Janse, M. T. Merema, G. M. M. Groothuis and E. Verpoorte, Anal. Chem., 2012, 84, 3938–3944 CrossRef CAS PubMed.
- R. Chilcott, N. Barai, A. Beezer, S. Brain, M. Brown, A. Bunge, S. Burgess, S. Cross, C. Dalton and M. Dias, J. Pharm. Sci., 2005, 94, 632–638 CrossRef CAS PubMed.
- M. Córdoba-Díaz, M. Nova, B. Elorza, D. Córdoba-Díaz, J. Chantres and M. Córdoba-Borrego, J. Controlled Release, 2000, 69, 357–367 CrossRef.
- S.-F. Ng, J. J. Rouse, F. D. Sanderson, V. Meidan and G. M. Eccleston, AAPS PharmSciTech, 2010, 11, 1432–1441 CrossRef PubMed.
- W. G. Reifenrath, B. Lee, D. R. Wilson and T. S. Spencer, J. Pharm. Sci., 1994, 83, 1229–1233 CrossRef CAS PubMed.
- L. Bartosova and J. Bajgar, Curr. Med. Chem., 2012, 19, 4671–4677 CrossRef CAS PubMed.
- OECD, Test No. 428. OECD Guideline for Testing of Chemicals: Skin Absorption: In Vitro Method, OECD Publishing, 2004 Search PubMed.
- J. J. M. van de Sandt, J. A. van Burgsteden, S. Cage, P. L. Carmichael, I. Dick, S. Kenyon, G. Korinth, F. Larese, J. C. Limasset, W. J. M. Maas, L. Montomoli, J. B. Nielsen, J. P. Payan, E. Robinson, P. Sartorelli, K. H. Schaller, S. C. Wilkinson and F. M. Williams, Regul. Toxicol. Pharmacol., 2004, 39, 271–281 CrossRef CAS PubMed.
- R. G. Wu, Z. P. Wang, H. Xia, W. Fan, W. C. Wang, M. K. Teo, J. Salmon and J. O'Halloran, Lab on a chip Asia, Singapore, November, 2014 Search PubMed.
- M. A. Dickson, W. C. Hahn, Y. Ino, V. Ronfard, J. Y. Wu, R. A. Weinberg, D. N. Louis, F. P. Li and J. G. Rheinwald, Mol. Cell. Biol., 2000, 20, 1436–1447 CrossRef CAS PubMed.
- P. P. C. Toh, M. Bigliardi-Qi, A. M. Y. Yap, G. Sriram and P. Bigliardi, Exp. Dermatol., 2016, 25, 1002–1005 CrossRef CAS PubMed.
- J. Krüse, D. Golden, S. Wilkinson, F. Williams, S. Kezic and J. Corish, J. Pharm. Sci., 2007, 96, 682–703 CrossRef PubMed.
- C. Wilke and P. Chang, AIChE J., 1955, 1, 264–270 CrossRef CAS.
- E. Baum, Chemical Property Estimation: Theory and Application, Lewis Publishers, 1997 Search PubMed.
- D. J. Harrison and K. Knutson, Pharm. Res., 1995, 12, 2003–2011 CrossRef CAS.
- A. Safi, C. Nicolas, E. Neau and J. Escandell, J. Chem. Eng. Data, 2010, 55, 5449–5452 CrossRef CAS.
- I. S. Khattab, F. Bandarkar, M. A. A. Fakhree and A. Jouyban, Korean J. Chem. Eng., 2012, 29, 812–817 CrossRef CAS.
- J. Sclafani, J. Nightingale, P. Liu and T. Kurihara-Bergstrom, Pharm. Res., 1993, 10, 1521–1526 CrossRef CAS.
- G. M. Khan, Y. Frum, O. Sarheed, G. M. Eccleston and V. M. Meidan, Int. J. Pharm., 2005, 303, 81–87 CrossRef CAS PubMed.
- S.-F. Ng, J. Rouse, D. Sanderson and G. Eccleston, Pharmaceutics, 2010, 2, 209–223 CrossRef CAS PubMed.
- I. J. Bosman, S. R. Avegaart, A. L. Lawant, K. Ensing and R. A. de Zeeuw, J. Pharm. Biomed. Anal., 1998, 17, 493–499 CrossRef CAS PubMed.
- W. J. Addicks, G. L. Flynn and N. Weiner, Pharm. Res., 1987, 4, 337–341 CrossRef CAS.
- T. Korjamo, A. T. Heikkinen and J. Mönkkönen, J. Pharm. Sci., 2009, 98, 4469–4479 CrossRef CAS PubMed.
- SUPAC-SS, Guidance for industry: nonsterile semisolid dosage forms, scale-up and post approval changes: chemistry, manufacturing and controls; in vitro release testing and in vivo bioequivalence documentation, 1997 Search PubMed.
- Y. Dancik, P. L. Bigliardi and M. Bigliardi-Qi, Reprod. Toxicol., 2015, 58, 252–281 CrossRef CAS PubMed.
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Equal contribution as supervising authors. |
| This journal is © The Royal Society of Chemistry 2017 |