
Disposable cartridge platform for rapid detection of viral hemorrhagic fever viruses†

Abstract
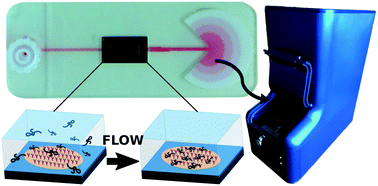
Light microscopy is a straightforward and highly portable imaging approach that is used for the detection of parasites, fungi, and bacteria. The detection of individual virus particles has historically not been possible through this approach. Thus, characterization of virus particles is typically performed using high-energy approaches such as electron microscopy. These approaches require purification of virions away from its normal milieu, significant levels of expertise, and only count a small number of particles at a time. To correct these deficiencies we created a platform that allows label-free, point-of-need virus imaging and counting. We adapted a multiplex-capable, interferometric imaging technique to a closed-system that allows real-time particle detection in complex mixtures. To maximize virus particle binding we constructed a disposable device with a constant flow rate of ∼3 μl min−1. Biosafety was achieved by having a sealable sample addition port. Using this platform we were able to readily identify virus binding in a 20 minute experiment. Sensitivity was comparable to laboratory-based assays such as ELISA and plaque assay, and showed equal or better sensitivity against paper-based assays designed for point-of-need use. Our results demonstrate a platform that can be used for rapid multiplexed detection and visualization of whole virus particles. We envision this technology as a sample-to-answer platform for detection and visualization of viruses without the need for prior labeling. This would enable both research investigation of virus particle behavior and morphology and have the potential to be used in a diagnostic context, where direct imaging from samples such as blood and urine would be valuable.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

