
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Non-invasive” portable laser ablation sampling of art and archaeological materials with subsequent Sr–Nd isotope analysis by TIMS using 1013 Ω amplifiers
A. C. S.
Knaf
 *,
J. M.
Koornneef
and
G. R.
Davies
*,
J. M.
Koornneef
and
G. R.
Davies
Geology and Geochemistry Research Cluster, Vrije Universiteit Amsterdam, De Boelelaan 1085, 1081HV Amsterdam, The Netherlands. E-mail: a.c.s.knaf@vu.nl
First published on 13th September 2017
Abstract
A new integrated trace element and multi-isotope provenancing methodology is presented that uses a portable “non-invasive” pulsed laser ablation sampling technique. Samples are collected on location onto Teflon filters for return to a clean laboratory for low blank (pg) geochemical procedures. Ablation pits approximately 60 or 120 μm in width and depth remove μg amounts of material. Following dissolution, trace element ratios are determined by inductively coupled plasma mass spectrometry and combined Sr–Nd isotopes by thermal ionization mass spectrometry. Use of 1013 Ω resistors allows precise analysis of subnanogram amounts of Sr–Nd isotopes, which coupled with the trace element data, provides highly effective multi-variant discrimination for material provenance and authenticity verification. Monitoring of blank contributions is required.
Introduction
Chemical fingerprinting of art or archaeological materials can potentially determine how, when and where objects were made and hence their provenance and potentially authenticity. If public and/or private institutions allow study of art or artefacts, generally material cannot be transported for examination or analysis, nor is bulk destructive analysis permitted. Analyses need to be essentially non-invasive and preferably carried out at the institution. This reality often renders validating authenticity or linking precious artefacts to their source difficult. Consequently, portable non-destructive instrumentation and analysis is required.Commercially available non-destructive portable analytical techniques have limitations and are not capable of determining isotope compositions. Portable XRF (pXRF) and laser-induced breakdown spectroscopy (pLIBS) used for major and trace element analysis, for example, have relatively poor analytical accuracy and precision (1–10%) and require thorough calibration with matrix matched standards.1–3 These portable techniques therefore generally do not discriminate the source of most raw geological materials that comprise art and archaeological artefacts. Detection limits of conventional (non-portable) XRF or inductive coupled plasma mass spectrometry (ICPMS) are up to five orders of magnitudes lower than non-invasive techniques for trace elements resulting in higher accuracy and precisions but require sample preparation.4,5
To resolve the provenance of geological materials, ideally, a combination of elemental abundances and isotopic ratios are needed. Several recent studies have stressed that a multiple isotopic approach is most effective for the provenance of geological and biological materials.6–8 Such methods, however, along with petrological studies of the constituent mineralogy, generally require grams of material, something that is impossible to obtain from museum-grade artefacts.
This paper presents a macroscopically non-destructive sampling method for art and archaeological materials that is coupled with low blank trace element analyses by ICPMS and Sr–Nd isotope analyses by thermal ionisation mass spectrometry (TIMS) using 1013 Ω amplifiers.9,10 Precise and accurate analyses are performed on μg amounts of material ensuring that the integrity of the object is preserved.
Methodology
Analysis of μg of ablated material or ≤ng of individual elements requires rigorous control of sample contamination by minimising the input of extraneous material during sampling, sample preparation, pre-treatment and chemical separation procedures. Inter alia, Sr and Nd can potentially be introduced to a sample by airborne dust, reagents and from the surfaces of lab ware. Ideally a sample to blank ratio >100 is achieved to avoid the requirement for blank corrections. Here we developed a technique that aimed to produce accurate and precise trace element and Sr–Nd data by minimizing the blanks during all aspects of the method. Our isotope analysis techniques require a minimum of 10 pg Nd and 2 ng Sr.Portable laser ablation sampling
The essentially “non-invasive” sampling methodology incorporates a pulsed portable laser ablation sampling device (pLA)11,12 that has been further developed and optimised. The ability of the system, in combination with clean laboratory techniques, to produce reproducible coupled trace element and Sr–Nd isotope ratios is evaluated using the international glass standard BHVO-2G and jadeitite rocks that have lower trace element contents. The pLA system is transported in an airline cabin compatible suitcase (54.5 × 35.0 × 23.0 cm) weighing approximately 15 kg including a pulsed diode pumped solid state laser (Wedge HB 532, Bright Solutions SRL, Cura Carpignano, Italy), optical fibre, laser ablation module, sampling filter holder and membrane pump (Fig. 1). | ||
| Fig. 1 Assembled portable laser ablation sampling device composed of a DPSS laser (λ 532 nm), an optical fibre, a laser ablation module and a sample holder. Magnified inset image shows the sample wheel with six filters. | ||
The compact laser produces a wavelength of 532 nm with a pulse duration of <1 ns. The pulse frequency can be varied between 1–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. Sampling efficiency and elemental fractionation effects are not affected by the ablation frequency.13 The laser beam output has a diameter of 1 mm and output energy of 1.3 mJ. Before entering the fibre, the laser beam is focused by an aspheric lens mounted on a x/y translation stage. In this study two different optical fibres were used, a standard fibre (QP450-2-XSR, Ocean Optics Inc., Dunedin, FL, USA) with a constant core diameter of 450 μm, and a tapered fibre (AFT600TO200Y, Fiberguide Industries Inc., ID, USA) with a core diameter of 600 μm passing into a core diameter of 200 μm.14 Both optical fibres are 2 m in length and allow flexibility when sampling objects of different size and shape. The laser produces a homogenised energy distribution that is focused on the sample surface by two lenses in the ablation module. The standard fibre produces a beam with a 100–130 μm diameter, the tapered fibre reduces the beam diameter to 50–70 μm (Fig. 2) and maintains the maximum energy at the sample surface at >1 mJ. An open ablation cell allows aerosol extraction to a Teflon filter by suction generated by a miniature oil-free membrane pump (Fig. 1). During ablation, the sample surface is illuminated with a LED ring and observed with a monochromatic CCD camera (Chameleon, CMLN-13S2M-CS, Point Grey Research, Richmond, BC, Canada; software package Fly Capture Point Grey SDK). The eight-fold magnification allows observation of a 1 mm2 sample area, so that the exact sampling position and the focal point of the laser can be optimised. An individual ablation sample is taken in a 1 minute routine at 100 Hz frequency, i.e., 6000 pulses. To obtain enough material for combined trace element and isotopic analyses, multiple ablations of individual samples may be required, depending on the elemental concentrations of the ablated material. Samples were collected on PTFE Mitex® membrane filters (LSWP01300, Merck Millipore Corporation, MA, USA) that have a pore size of 5 μm, a porosity of 60% and are hydrophobic. Filters are 13 mm in diameter with a thickness of 170 μm. Filters were pre-cleaned by submerging them for >3 days at 120 °C in a mixture of 3 M HCl and 0.2 M HF and are transported in Milli-Q® in cleaned PTFE vials. A filter wheel contains six filter positions. The ablation chamber and tubing to the filter holder are cleaned using ethanol and compressed air before switching to a new filter position. The blank contribution during sampling is monitored on each sample wheel of six filters. After sample collection, filters are extracted from the holder on site and stored in pre-cleaned centrifuge tubes ready for processing in the low blank isotope geochemistry laboratories of the Vrije Universiteit Amsterdam.
000 Hz. Sampling efficiency and elemental fractionation effects are not affected by the ablation frequency.13 The laser beam output has a diameter of 1 mm and output energy of 1.3 mJ. Before entering the fibre, the laser beam is focused by an aspheric lens mounted on a x/y translation stage. In this study two different optical fibres were used, a standard fibre (QP450-2-XSR, Ocean Optics Inc., Dunedin, FL, USA) with a constant core diameter of 450 μm, and a tapered fibre (AFT600TO200Y, Fiberguide Industries Inc., ID, USA) with a core diameter of 600 μm passing into a core diameter of 200 μm.14 Both optical fibres are 2 m in length and allow flexibility when sampling objects of different size and shape. The laser produces a homogenised energy distribution that is focused on the sample surface by two lenses in the ablation module. The standard fibre produces a beam with a 100–130 μm diameter, the tapered fibre reduces the beam diameter to 50–70 μm (Fig. 2) and maintains the maximum energy at the sample surface at >1 mJ. An open ablation cell allows aerosol extraction to a Teflon filter by suction generated by a miniature oil-free membrane pump (Fig. 1). During ablation, the sample surface is illuminated with a LED ring and observed with a monochromatic CCD camera (Chameleon, CMLN-13S2M-CS, Point Grey Research, Richmond, BC, Canada; software package Fly Capture Point Grey SDK). The eight-fold magnification allows observation of a 1 mm2 sample area, so that the exact sampling position and the focal point of the laser can be optimised. An individual ablation sample is taken in a 1 minute routine at 100 Hz frequency, i.e., 6000 pulses. To obtain enough material for combined trace element and isotopic analyses, multiple ablations of individual samples may be required, depending on the elemental concentrations of the ablated material. Samples were collected on PTFE Mitex® membrane filters (LSWP01300, Merck Millipore Corporation, MA, USA) that have a pore size of 5 μm, a porosity of 60% and are hydrophobic. Filters are 13 mm in diameter with a thickness of 170 μm. Filters were pre-cleaned by submerging them for >3 days at 120 °C in a mixture of 3 M HCl and 0.2 M HF and are transported in Milli-Q® in cleaned PTFE vials. A filter wheel contains six filter positions. The ablation chamber and tubing to the filter holder are cleaned using ethanol and compressed air before switching to a new filter position. The blank contribution during sampling is monitored on each sample wheel of six filters. After sample collection, filters are extracted from the holder on site and stored in pre-cleaned centrifuge tubes ready for processing in the low blank isotope geochemistry laboratories of the Vrije Universiteit Amsterdam.
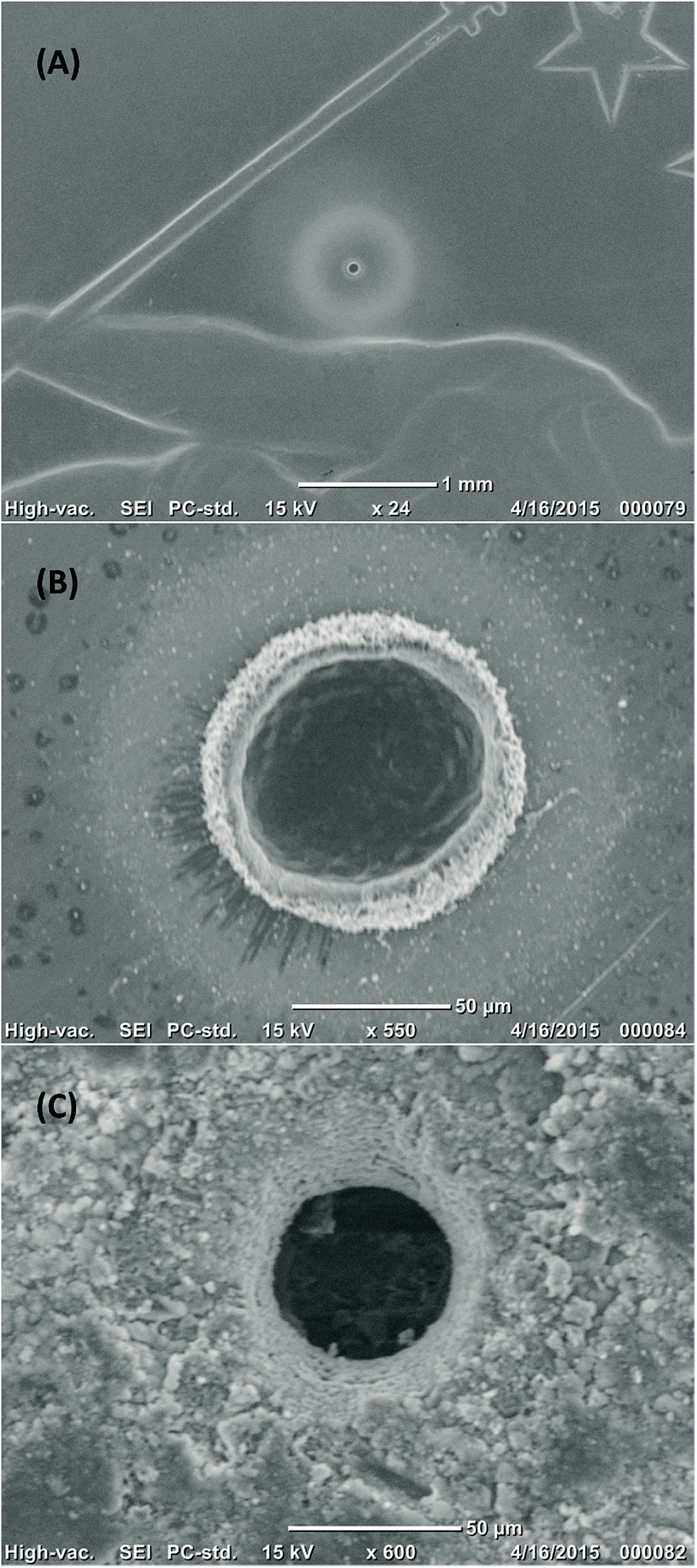 | ||
| Fig. 2 SEM images of ablation pits generated with a tapered fibre; crater dimensions are ∼60 μm in width and depth. (A) and (B) craters in half a Swiss franc; note that the pale halo around the pit is debris which can be easily removed by cleaning with water. (C) Crater in a natural jadeite bearing rock from the Rio San Juan Complex in the Dominican Republic.15 | ||
Sample dissolution
In this study trace element and isotopic composition analyses were sampled separately onto different sample filters. Filters with sampled material were placed in 7 ml ultra-clean PTFE beaker and dissolved by adding ∼1.0 ml of concentrated HF and ∼0.5 ml of concentrated HNO3. Closed beaker were placed on a hotplate at 120 °C for 3 days and placed in an ultra-sonic bath twice a day in order to disaggregate the material and promote acid attack. Following filter removal, solutions were evaporated to dryness. For trace element analyses by ICPMS samples were taken up in 1 ml of 5% HNO3. To prepare for column chemistry and subsequent isotope composition analyses by TIMS, dried down samples were nitrated with two drops of concentrated HNO3.Filter blanks (see results section) were determined on unclean and cleaned filters by isotope dilution (ID) following the procedure used for sample dissolution. After filter removal, the solution was centrifuged (4 min/12000 U) and ∼0.1 ml of spike (84Sr spike = 10.443 ppb, 150Nd spike = 35.52 ppb) was added for Sr and Nd. The solution was evaporated to dryness and nitrated with two drops of concentrated HNO3.
ICPMS analyses
Trace element analyses were performed on a Thermo Fisher X-series-II ICP-MS following a modified analytical protocol from Eggins et al.16 The limit of detection (LOD) and limit of quantification (LOQ) of the ICPMS were determined by analysing an acid blank (5% HNO3) 10 times. A three point calibration curve was applied containing 0.1 ppb, 0.2 ppb and 0.5 ppb solutions of provenance relevant elements like REE, HFSE, LILE and additional elements (Th, U, Sr, Pb, Ba, Rb, Sc). For most of the elements the LOQ is below 2 ppt, exceptions are Ba (4 ppt), Zr (5 ppt) and Sc (45 ppt). When elemental blanks were below LOQ, data were not blank corrected. Calibration and instrumental drift were corrected using a BHVO-2 solution. Repeat analysis of a USGS reference material BCR-2 yield better than 10% (2 RSD) for all reported trace elements.Determination of mass removal
The mass removal of four BHVO-2 glass samples was determined based on ICPMS analysis from known element concentrations in the reference glass (USGS values). Samples of BHVO-2 glass were obtained by combining 10 individual ablations at 100 Hz using a normal optical fibre.Miniaturized low blank chemical separation for Sr and Nd
Chemical separation of the element of interest is required prior to TIMS analysis to avoid isobaric interferences and optimise ionisation efficiency. Here we utilise a combined miniaturised low blank chemical separation methodology for sub-nanogram amounts of strontium and neodymium.9,10,17 Sample preparation was conducted in a class 1000 clean laboratory equipped with class 100 laminar flow hoods. Low blanks were achieved by purification of reagents, thorough cleaning of lab ware and by minimizing the volume of chromatographic columns and reagents. Analytical reagent grade acids (HCl, HNO3, HF) were purified by double sub-boiling distillation in silica glass or PTFE stills. Water was purified using a Milli-Q® integral water purification system, Merck Millipore Corporation (resistivity 18.2 MΩ cm at 25 °C).Thermal ionization mass spectrometry
Sr and Nd isotope ratios were determined on a Thermo Fisher Scientific TRITON Plus TIMS in static mode on outgassed single and double Re filaments, respectively. The instrument is equipped with a multi-collector assembly featuring one fixed central and eight movable Faraday cups. All the presented Sr isotope data were collected using default 1011 Ω amplifiers (>ng), whilst Nd isotope ratios were analysed employing 1013 Ω amplifiers (<ng).9,10 Cup configurations for isotope ratio analyses of Sr and Nd using a relay matrix (“virtual amplifier” system) are shown in Table 1. The 143Nd/144Nd and 87Sr/86Sr ratios were corrected for instrumentation mass fractionation using an exponential law by normalization to 146Nd/144Nd = 0.721903 and 86Sr/88Sr = 0.1194. Over the period of this study, the long term values for 200 ng loads of the in-house CIGO Nd standard yielded 0.511328 ± 0.000010 (2SD, n = 29) and for 200 pg loads 0.511371 ± 0.000065 (2SD, n = 4); the JNdI measured on 100 ng gave 0.512096 ± 0.000013 (2SD, n = 4); and the NBS987 Sr standard resulted in 0.710242 ± 0.000025 (200 ng, 2SD, n = 45).| Cup | L4 | L3 | L2 | L1 | C | H1 | H2 | H4 |
|---|---|---|---|---|---|---|---|---|
| Sr-routine mass amplifier | 84Sr | 85Rb | 86Sr | 87Sr | 88Sr | |||
| 1011 | 1011 | 1011 | 1011 | 1011 | ||||
| Nd-routine mass amplifier | 142Ce, Nd | 143Nd | 144Nd | 145Nd | 146Nd | 147Sm | 148Nd | 150Nd |
| 1011 | 1013 | 1013 | 1013 | 1013 | 1011 | 1011 | 1011 |
Results
Mass removal
The sample volume taken depends on the fibre used, the stability of the laser beam and the material itself. Crater geometries determined by a VK-9710K Series 3D Laser Scanning Confocal Microscope (Keyence, Osaka, Japan) at the Leibniz University in Hannover, Germany, range from cylinder to cone, including transitional shapes.The four BHVO-2 glass analyses (10 ablations each) yielded on average 14.6 ng Sr (11.7–15.7 ng) and 0.85 ng Nd (0.63–0.95 ng). Thus, the amount of material sampled varied between 25.5 to 40.0 μg (36.3%) for the 10 ablations based on Sr and Nd abundances in BHVO-2 glass of 393 and 24.9 ppm respectively (USGS values).
Trace element data
Due to the uncertainty in sampling volumes, precise trace element abundances in the sampled material cannot be determined. Importantly, however, trace element ratios will be unaffected and hence can be used for provenance and authenticity studies.To validate the reproducibility of trace element ratios when sampling with the portable laser, four BHVO-2G analyses were conducted. The resulting trace element ratios are generally in agreement (for most of the ratios 2RSD of <10%) with trace element ratios of recommended USGS and GeoReM18 BHVO-2G values (Table 2; Fig. 3) even when considering elements with differing volatility.
| BHVO-2G | I | II | III | IV | Average | 2SD | % RSD | USGS | Georem |
|---|---|---|---|---|---|---|---|---|---|
| Ba/Nb | 5.39 | 5.58 | 6.17 | — | 5.71 | 0.82 | 14.4 | 6.25 | 7.22 |
| Rb/Sr | 0.0227 | 0.0222 | 0.0227 | 0.0224 | 0.0225 | 0.0005 | 2.4 | 0.0226 | 0.0239 |
| Lu/Hf | 0.0628 | 0.0667 | 0.0654 | 0.0674 | 0.0656 | 0.0040 | 6.1 | 0.0683 | 0.0650 |
| Sm/Nd | 0.239 | 0.233 | 0.238 | 0.243 | 0.238 | 0.008 | 3.5 | 0.249 | 0.249 |
| Cr/Ti | 0.0195 | 0.0191 | 0.0193 | 0.0197 | 0.0194 | 0.0005 | 2.6 | 0.0176 | 0.0182 |
| V/Ti | 0.0196 | 0.0197 | 0.0195 | 0.0197 | 0.0196 | 0.0002 | 1.0 | 0.0192 | 0.0194 |
| Co/Ti | 0.0028 | 0.00281 | 0.00287 | 0.00283 | 0.00283 | 0.00005 | 1.7 | 0.00273 | 0.00281 |
| Hf/Zr | 0.0246 | 0.0245 | 0.0264 | 0.0255 | 0.0253 | 0.0017 | 6.9 | 0.0252 | 0.0261 |
| Nb/Zr | 0.111 | 0.107 | 0.112 | 0.112 | 0.111 | 0.004 | 3.9 | 0.123 | 0.110 |
| Ta/Zr | 0.0077 | 0.0057 | 0.0069 | 0.0069 | 0.0068 | 0.0017 | 24.2 | 0.0086 | 0.0068 |
| Dy/Yb | 2.664 | 2.660 | 2.747 | 2.752 | 2.706 | 0.101 | 3.7 | 2.498 | 2.594 |
| Ta/Th | 1.069 | 0.776 | 0.947 | 0.924 | 0.929 | 0.240 | 25.8 | 1.176 | 0.923 |
| Nb/U | 35.78 | 38.15 | 39.52 | 38.27 | 37.93 | 3.13 | 8.2 | 47.62 | 41.65 |
| Nb/Ta | 14.33 | 18.79 | 16.10 | 16.21 | 16.36 | 3.67 | 22.4 | 14.29 | 16.07 |
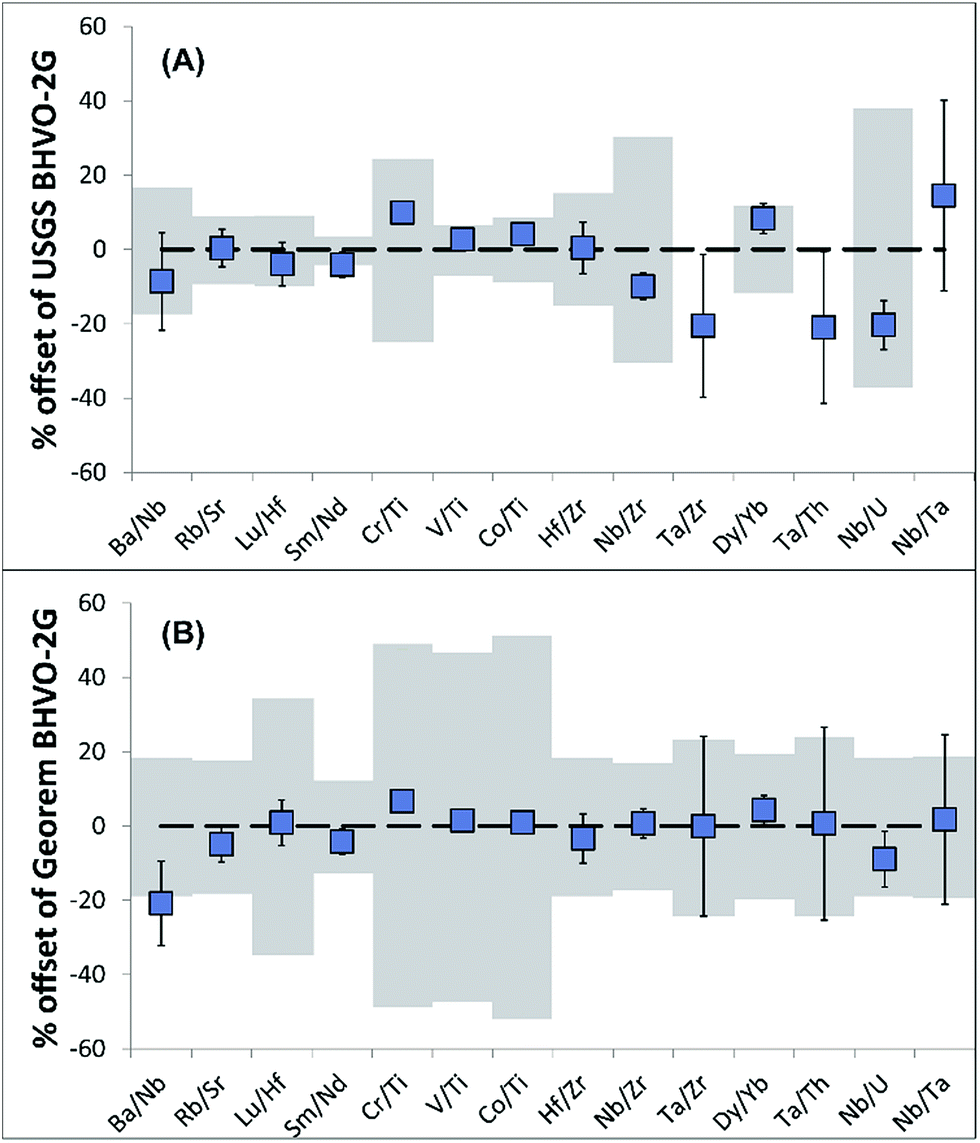 | ||
| Fig. 3 Trace element ratio reproducibility and 2SD (which may be smaller than symbols) of four BHVO-2G samples (excluding outliers) compared to ratios calculated from (A) recommended BHVO-2G USGS values and (B) GeoReM BHVO-2G data. Note grey shaded area displays propagated error of the USGS and GeoReM ratios. Note no error is available for USGS Ta data. | ||
Filter blanks
Analysis of filters before cleaning yielded average blanks of 90.3 pg for Sr (41.2–145.6 pg, n = 2) and 1.5 pg for Nd (1.4–1.6 pg, n = 2). In contrast, cleaned filters yielded average blanks of 28.9 pg of Sr (22.4–37.3 pg; n = 8) and <0.5 pg of Nd (n = 5). Thus, Nd filter blanks are negligible, whereas Sr may contribute significantly to the total procedural blank (Fig. 4 and 5).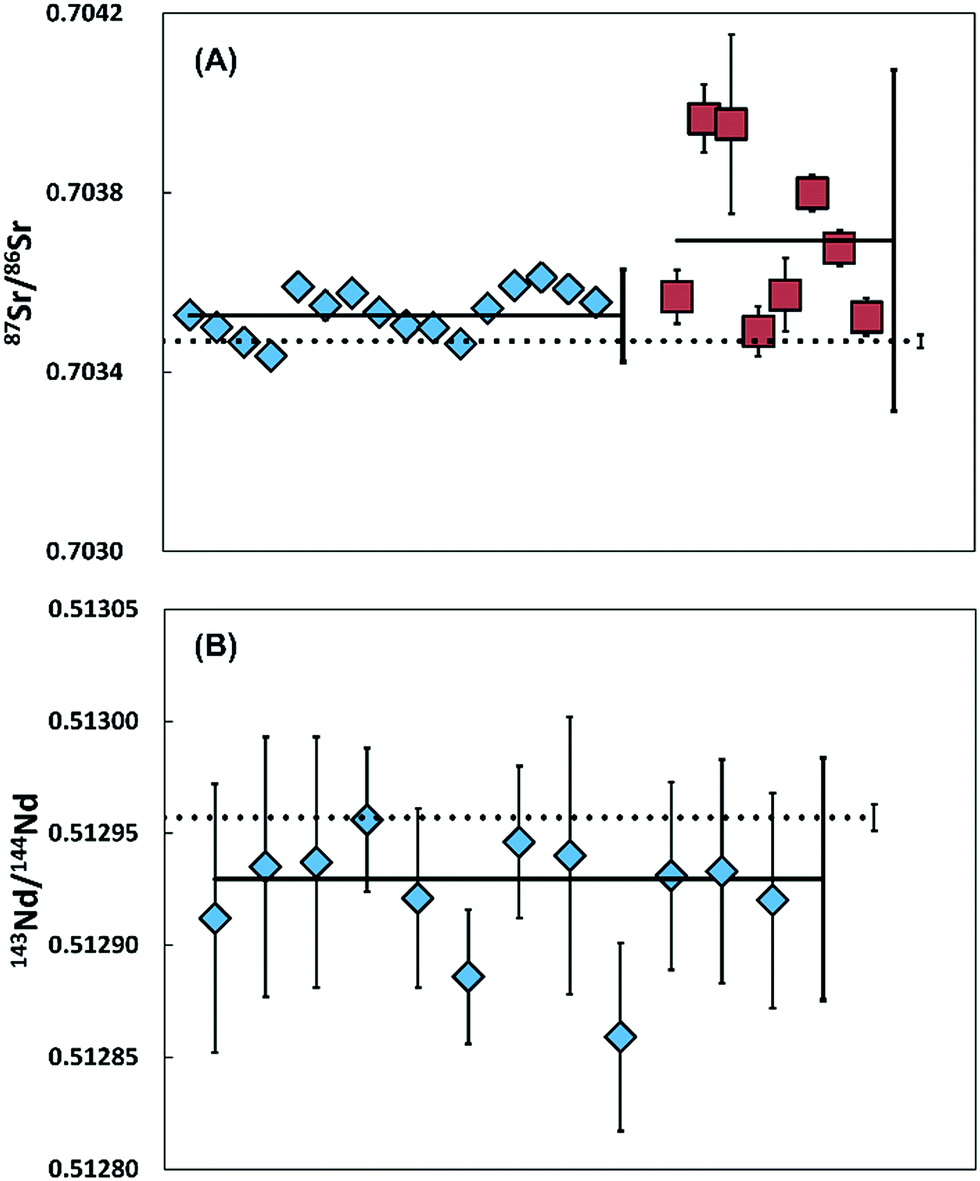 | ||
| Fig. 4 Sr and Nd isotope data on multiple ablated samples of BHVO-2 glass (A) 87Sr/86Sr ratios with 2SD error after 10 (diamonds, 2SE smaller than symbols) and 5 (squares) ablations, measured using 1011 Ω amplifiers. The dashed line represents the average (87Sr/86Sr = 0.703469 ± 0.000014, 2SD) measured on large aliquots of BHVO-2G.20 (B) 143Nd/144Nd ratios of a BHVO-2 glass with 2SD error after 10 ablations, measured using 1013 Ω amplifiers. The dashed line represents the average (143Nd/144Nd = 0.512957 ± 0.000006, 2SD) measured on large aliquots of BHVO-2G.21 | ||
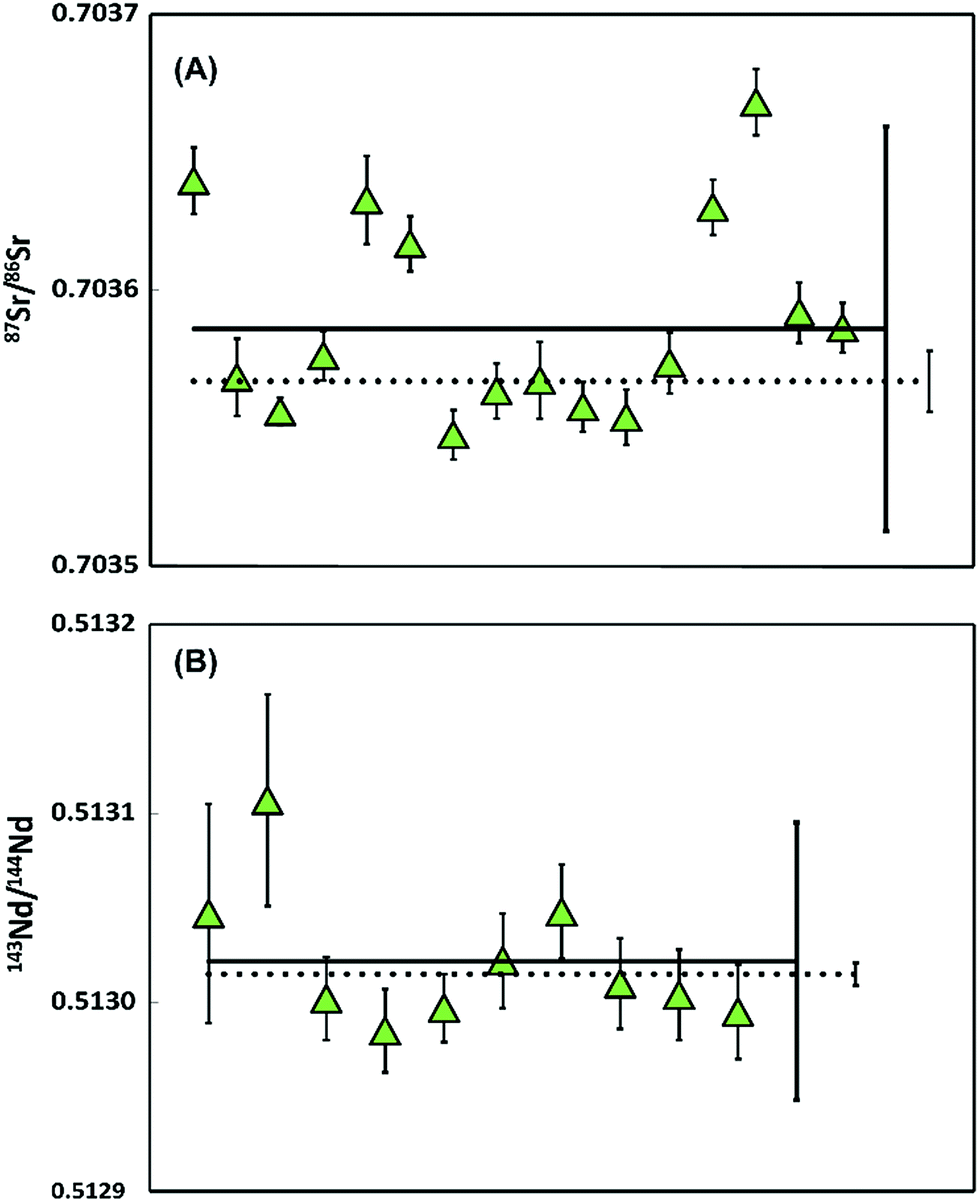 | ||
| Fig. 5 Sr and Nd isotope data on multiple ablated samples of jadeitite after 20 ablations (A) 87Sr/86Sr ratios with 2SD error, measured using 1011 Ω amplifiers. The dashed line represents the 87Sr/86Sr and 2SE (87Sr/86Sr = 0.703517 ± 0.000011) measured on a large aliquot (376 ng). (B) 143Nd/144Nd ratios with 2SD error, measured using 1013 Ω amplifiers. The dashed line represents the 143Nd/144Nd and 2SE (143Nd/144Nd = 0.512965 ± 0.000006) measured on a large aliquot of the sample (262 ng). | ||
Clean laboratory blanks
Nd and Sr clean laboratory blanks, including sample digestion, chemical separation and TIMS loading blank, are 2.1 pg for Nd (n = 3, ±1.3 pg 2SD) and 50.7 pg for Sr (n = 5, ±36.1 pg 2SD). These blanks exclude the sampling process and the contribution of the filter. To allow precise clean-lab induced blank corrections to be applied if required, the isotopic compositions (Sr–Nd) of the lab blanks were determined by Koornneef et al.10 by upscaling the separation procedure by a factor of 30. The isotopic composition of the in-house total procedural chemical blanks, were 87Sr/86Sr = 0.711120 ± 0.000050 (2SD, n = 3) and 143Nd/144Nd = 0.511856 ± 0.000090 (2SD, n = 3) (Fig. 5).Total procedural blanks
Blanks determined on Teflon filters that were exposed to 10 minutes of air sampling with subsequent column chemistry yielded 78.4 pg Sr (n = 11, ± 50.6 pg 2SD) and 4.6 pg Nd (n = 11, ± 5.2 pg 2SD). These total procedural blanks include contamination introduced by sampling, the filters, the sample dissolution, chemical separation and loading. Based on the highest measured blanks we conclude that for Nd the sampling process approximately contributes 67.3%, filter 4.3% and the chemical procedure 28.4%. For Sr the pLA sampling adds 13.8%, filter 29.4% and the chemical procedure 56.8%.87Sr/86Sr and 143Nd/144Nd reproducibility: BHVO-2G and jadeitite
Isotopic composition reproducibility of the entire methodology was evaluated using a polished sample of BHVO-2 glass and two pure jadeitite samples from the Sierra del Convento Complex in Cuba.19When using 1013 Ω amplifiers on a TIMS, accurate and precise Sr–Nd isotope ratios can be determined on as little as 10 pg but potential blank contributions suggest >100 pg of an element is required.9 To provide enough material and a representative sample, the BHVO-2 glass was ablated 10 times using a normal optical fibre. To validate the minimum amount of material needed for Sr isotope analyses when sampling with the pLA device, a further 8 samples were collected with only 5 ablations. The jadeitites were each ablated 20 times.
The reproducibility and internal precision of Sr and Nd isotope ratios produced from BHVO-2 glass and two pure jadeitite samples are shown in Fig. 4 and 5.
Repeated analyses of aliquots of Sr (∼10–15 ng), when ablating the BHVO-2 glass 10 times, yielded an external reproducibility of 87Sr/86Sr of 0.703533 ± 0.000103 (2SD, n = 16) with internal precisions of <±0.000033 (2SE). The average is in agreement with the 87Sr/86Sr obtained by Elburg et al.20 on large sample aliquots (87Sr/86Sr = 0.703469 ± 0.000014, 2SD). Although within error, the highest absolute 87Sr/86Sr may imply a blank contribution of 1.9%. For samples obtained using 5 ablations (Sr < 5–7.5 ng), 87Sr/86Sr ratios vary significantly and all except two samples are outside the external reproducibility reported by Elburg et al.20 We conclude that for the 5 ablation approach, insufficient material was collected to overcome the influence of the variable total procedure blank and that currently a minimum of 10 ablations is required for a accurate analysis.
Multiple aliquots of Nd (<1 ng, n = 12) from BHVO-2G were analysed using 1013 ohm resistors. The analyses yield an average internal precision of ±0.000046 (2SE) and an external reproducibility of 143Nd/144Nd 0.512923 ± 0.000054 (2SD). The data is within error of 143Nd/144Nd = 0.512957 ± 0.000006 (2SD) measured on large aliquots of BHVO-2G.21
Multiple 87Sr/86Sr analyses of jadeitite sample containing 13.7–21. 9 ng gave 87Sr/86Sr = 0.703536 ± 0.000073 (2SD, n = 16), with an average internal precision of ±0.000011 (2SE). Eleven of the 16 samples are within error of the ratio determined on a 376 ng aliquot processed through conventional wet chemistry techniques on the powdered sample (0.703517 ± 0.000011, 2SE). Five samples have elevated values that could be explained by a maximum blank contribution of 1.3%. Alternatively, the Sr isotope variability could reflect sample heterogeneity within this natural jadeitite rock.
The repeated 143Nd/144Nd analyses of jadeitite sample, determined on 169–270 pg, gave 0.512972 ± 0.000073 (2SD, n = 10) with an average internal precision of ±0.000030 (2SE), are within error of the 262 ng aliquot of the sample (0.512965 ± 0.000006, 2SE).
Discussion
To optimise the efficiency of sample collection, focussing is necessary during ablation. As a consequence the laser beam may move, resulting in cylindrical instead of conical shaped pits. This effect produces a larger crater (up to 37.5%) than observed when sampling without focussing. Differences in mass removal are also a consequence of laser beam stability and variation in the absorption coefficient of the ablated mineral(s). Therefore, estimates of the amount of material sampled come with large (<36.3%) uncertainties and we are unable to determine trace element concentrations accurately. Note, however, that elemental ratios will be essentially unaffected by uncertainties in sample volume (see Table 2). Trace element ratios potentially used for provenancing show good reproducibility (<10% for most ratios, Fig. 3). Ideally, if samples have high concentrations of the elements of interest, samples should be aliquoted for trace element and isotopic analysis to minimise the ablated surface and volume. If, however, the available sample surface is limited and concentrations low, trace element fractions can be collected from the whole rock matrix during column chemistry. In such an approach element yields will be variable and need quantification. Previous studies have shown that such an approach can successfully discriminate different sample populations.22,23The total procedural blanks were 78.4 pg for Sr and 4.6 pg for Nd and represent the accumulated effect of contamination during sampling, filter dissolution, geochemical procedures and loading. The relatively high and variable Sr blanks represent the limiting factor in the successful application of the pLA sampling technique to small Sr samples. The tests with 5 ablation craters clearly showed a variable off-set of the Sr data towards the higher ratios recorded in the blank. Due to the potential influence of the blank on the isotopic analyses, it is imperative to determine the total amount of Sr and Nd in an ablated sample. Isotope dilution is routinely conducted yielding Sr and Nd concentrations to better than 0.1%, allowing blank corrections to be performed. A blank correction is possible provided the isotope composition of the total procedural blank is known. Consequently, we recommend obtaining dust samples from sampling locations for Sr- and Nd-isotope composition determination to establish if the sampling blank differs markedly from the laboratory blank. In order to minimise blanks during future sampling, a portable clean unit will be used.
The accuracy of 87Sr/86Sr and 143Nd/144Nd of the BHVO-2 glass and the two jadeitite samples validates the techniques and confirms that there is no detectable laser-induced isotopic fractionation associated with ablation. In general, external reproducibility is better for Nd than for Sr due to blank influences. The external precisions obtained (given as 2RSD) for both materials were <0.17‰ for 87Sr/86Sr and <0.14‰ for 143Nd/144Nd. This reproducibility is sufficient to detect Sr and Nd isotope variation in natural samples to within the fourth decimal place and can thus be used for provenancing or potentially validating the authenticity of materials.
Conclusion
A modified portable laser ablation sampling device allows in situ sampling of art and archaeological objects and artefacts independent of size and shape that cannot be transported to the laboratory. Laser ablation sampling removes a volume equivalent to ∼2.5–4 μg of sample material generating a well-defined cylindrical to conical crater, invisible to the naked eye. The presented technique thus leaves minimal damage and preserves the integrity of the objects on a macroscopic scale. Depending on the elemental concentration within a sample, the methodology currently requires multiple ablations to collect sufficient material for the analyses (10 for BHVO-2G, 20 for jadeite bearing rocks). Low blank analyses of the sampled materials using TIMS and ICPMS allow the determination of precise and reproducible Sr–Nd isotope compositions and trace element ratios. The technique can resolve isotopic variability for Sr isotope and Nd within the fourth decimal place. Compared to other portable techniques, such as pLIBS and pXRF, two to three orders of magnitude lower limits of detection were reached allowing the determination of trace element ratios for elements present in the sub-μg/g range. Currently the overall precision of the technique is not limited by instrumental measurements but controlled by sampling blanks. Determination of elemental amounts by ID and knowledge of the isotopic composition of the total procedural blank can be adopted to correct for the effect of blank contributions, however, the preferred strategy will be to reduce blanks by undertaking sampling using a portable clean unit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 319209 and the European Union's Horizon 2020 research and innovation programme under grant agreement no. 654208 (Europlanet 2020 RI). Thanks to Richard Smeets, Bas van der Wagt and Kirsten van Zuilen who provided analytical assistance. Bram Mooij and Judith van Santen are acknowledged for advice on ablation pit volume determination. Detlef Günther, Bodo Hattendorf, Joachim Koch, Debora Käser and Marcel Burger provided invaluable input on the laser modification and usage. We are grateful to Martin Oeser-Rabe, Mona Weyrauch and Stefan Dultz for conducting the 3D laser scanning microscopy.References
- A. Heginbotham, A. Bezur and 18 others, in METAL 2010 (CD), ed. P. Mardikian, C. Chemello, C. Watters and P. Hull, 2010, pp. 178–188 Search PubMed.
- A. Heginbotham, J. Bassett, D. Bourgarit, C. Eveleigh, T. Frantz, L. Glinsman, D. Hook, D. Smith, R. J. Speakman, R. Sugar and R. Van Langh, Archaeometry, 2015, 57(5), 856–868 CrossRef CAS.
- R. S. Harmon, R. E. Russo and R. R. Hark, Spectrochim. Acta, Part B, 2013, 87, 11–26 CrossRef CAS.
- X. Hou, Y. He and B. T. Jones, Appl. Spectrosc. Rev., 2004, 39(1), 1–25 CrossRef CAS.
- P. J. Potts, A Handbook of Silicate Rock Analyses, Springer Science & Business Media, 2012 Search PubMed.
- P. Degryse and J. Schneider, J. Archaeol. Sci., 2008, 35, 1993–2000 CrossRef.
- A. Blomme, P. Degryse, E. Dotsika, D. Ignatiadou, A. Longinelli and A. Silvestri, J. Archaeol. Sci., 2017, 78, 134–146 CrossRef CAS.
- L. Font, G. Van der Peijl, C. Van Leuwen, I. van Wetten and G. R. Davies, Sci. Justice, 2015, 55, 34–42 CrossRef PubMed.
- J. M. Koornneef, C. Bouman, J. B. Schwieters and G. R. Davies, Anal. Chim. Acta, 2014, 819, 49–55 CrossRef CAS PubMed.
- J. M. Koornneef, I. Nikogosian, M. J. van Bergen, R. Smeets, C. Bouman and G. R. Davies, Chem. Geol., 2015, 397, 14–23 CrossRef CAS.
- R. Glaus, J. Koch and D. Günther, Anal. Chem., 2012, 84, 5358–5364 CrossRef CAS PubMed.
- R. Glaus, L. Dorta, Z. Zhang, Q. Ma, H. Berke and D. Günther, J. Anal. At. Spectrom., 2013, 28, 801–809 RSC.
- R. Glaus, Doctoral Dissertation No. 21161, ETH Zurich, 2013.
- D. Käser, Redesign of a portable laser ablation setup to allow sampling of ancient Chinese Jade and Porcelain, Master thesis, ETH Zurich, 2014.
- H.-P. Schertl, W. V. Maresch, K. P. Stanek, A. Hertwig, M. Krebs, R. Baese and S. S. Sergeev, Eur. J. Mineral., 2012, 24, 199–216 CrossRef CAS.
- S. M. Eggins, J. D. Woodhead, L. P. J. Kinsley, G. E. Mortimer, P. Sylvester, M. T. McCulloch, J. M. Hergt and M. R. Handler, Chem. Geol., 1997, 134, 311–326 CrossRef CAS.
- J. M. Koornneef, C. Bouman, J. B. Schwieters and G. R. Davies, J. Anal. At. Spectrom., 2013, 28, 749–754 RSC.
- http://georem.mpch-mainz.gwdg.de/sample_query_pref.asp .
- A. García-Casco, A. Rodriguez Vega, J. Cardenas Parraga, M. A. Iturralde-Vinent, C. Lazaro, I. Blanco Quintero, Y. Rojas Agramonte, A. Kroener, K. Nunez Cambra, G. Millan, R. L. Torres-Roldan and S. Carrasquilla, Contrib. Mineral. Petrol., 2009, 158, 1–16 CrossRef.
- M. Elburg, P. Vroon, B. van der Wagt and A. Tchalikian, Chem. Geol., 2005, 223(4), 196–207 CrossRef CAS.
- I. Raczek, K. P. Jochum and A. W. Hofmann, Geostand. Newsl., 2003, 27(2), 173–179 CrossRef CAS.
- S. Timmerman, J. M. Koornneef, I. L. Chinn and G. R. Davies, Earth Planet. Sci. Lett., 2017, 463, 178–188 CrossRef CAS.
- J. M. Koornneef, M. U. Gress, I. L. Chinn, H. A. Jelsma, J. W. Harris and G. R. Davies, Nat. Commun., 2017 Search PubMed , in press.
| This journal is © The Royal Society of Chemistry 2017 |