
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the oxidative transformation of roxarsone by electrochemistry coupled to hydrophilic interaction liquid chromatography/mass spectrometry†
Lisa M.
Frensemeier
,
Lars
Büter
,
Martin
Vogel
and
Uwe
Karst
 *
*
University of Münster, Institute of Inorganic and Analytical Chemistry, Corrensstraße 30, 48149 Münster, Germany. E-mail: uk@uni-muenster.de; Fax: +49-251-8336013; Tel: +49-251-8333141
First published on 5th December 2016
Abstract
The oxidative transformation of the animal feed additive roxarsone (3-nitro-4-hydroxyphenylarsonic acid) was investigated using electrochemistry (EC) coupled to hydrophilic interaction liquid chromatography (HILIC) and mass spectrometry. Although roxarsone is claimed to be relatively stable in the organism, only little is known about its metabolism. By means of high-resolution electrospray (ESI)-MS analysis of electrochemically oxidized roxarsone, potential transformation products were identified including the toxic metabolite arsenate (As(V)). Thus, an oxidative carbon–arsenic bond cleavage was observed. For the first time, not only arsenic containing products were considered, but also non-arsenic species which were formed during As(V) generation and which could also be toxic metabolites of roxarsone. A hydrophilic interaction liquid chromatographic (HILIC) separation coupled to electrospray (ESI)-MS enabled polarity estimation and the identification of isomeric products. Additionally, hyphenation of HILIC to inductively coupled plasma (ICP)-MS allowed speciation analysis and quantification of arsenic containing products, revealing As(V) to be the main species formed under the applied oxidative conditions. The reactivity of the TPs towards biomolecules was investigated by means of adduct formation experiments. It has been found that two electrochemically generated species, one quinone and one quinone imine, showed reactivity towards free thiol groups of the tripeptide glutathione and the proteins β-lactoglobulin A and human serum albumin, which were used as model compounds for adduct formation.
Introduction
Phenylarsonic acids such as the animal feed additive roxarsone (3-nitro-4-hydroxyphenylarsonic acid, ROX) have been used in agriculture for decades.1,2 Roxarsone is administered to poultry and swine in order to prevent parasitic diseases such as coccidiosis, to promote animal growth and to improve tissue pigmentation.1,3 Despite to the veterinarian advantages of ROX application, there are concerns about its health risks. For this reason, roxarsone was banned in the European Union already in 1999.4 In the U.S., it has not been officially banned by the Food and Drug Administration (FDA), but the ROX manufacturer Pfizer stopped the production in 2011.2,5 However, the use of roxarsone and other phenylarsonic acid derivates still continues in many parts of Asia, in Canada and in South American countries.5 Generally, toxicity of arsenic is highly species dependent. Organoarsenicals as arsenobetain or arsenocholin are considered to be least toxic, whereas the inorganic species As(V) and As(III) have been classified as carcinogens by the International Agency for Research on Cancer (IARC) already in 1980.6 Roxarsone is considered to be relatively inert towards degradation and is non-toxic. It is presumed to be excreted mainly unmetabolized from the animal body. First investigations on ROX metabolism found only one major metabolite, 3-amino-4-hydroxyphenylarsonic acid (3-AHPAA), in fresh urine and feces.7,8 More recently, Conklin et al. analyzed liver tissue extracts of roxarsone treated chicken and of non-treated control chicken by means of ion chromatography coupled to inductively coupled plasma mass spectrometry (IC-ICP-MS).9 They found a variety of different arsenic species which were detected only in the extract of ROX-fed chicken. Only a few of the species were identified by analytical standards and up to 80% of the compounds remained unidentified. In addition, the simultaneous combination of IC with element specific ICP-MS and molecular ESI-MS/MS enabled Peng et al. to identify six different potential metabolites in chicken liver extracts of ROX-treated animals, namely As(III), dimethylarsinic acid (DMA), monomethylarsonic acid (MMA), As(V), 3-AHPAA, and N-acetyl-4-hydroxyphenylarsonic acid (N-AHPAA).10 Nachman et al. performed the first investigations on arsenic accumulation in chicken breast meat sold in U.S. supermarkets.2 By means of HPLC-ICP-MS, they determined the amounts of inorganic arsenic (iAs), DMA, roxarsone and unknown arsenic species in meat extracts. They suggested that at least iAs or the unknown species could be metabolites of roxarsone. However, not only direct metabolic reactions in animals and the accumulation in meat should be critically investigated, but also the biotransformation of roxarsone after excretion. Since the majority of roxarsone is excreted in its native form, it is widely present in animal based fertilizers or litter, which are spread on fields. This route of exposure is crucial for arsenic entering the environment since more than 90% of produced poultry litter are applied on land.11 It has been shown that composting, photodegradation as well as microbial transformation promote the degradation of roxarsone into iAs.12–15 The main transformation products (TPs) of roxarsone in animal manure are As(III), As(V), DMA, MMA, 3-AHPAA, 4-hydroxyphenylarsonic acid (4-HPAA) and further unknown species.4,5,15–17 Most TPs (up to 75%) are more water soluble than roxarsone itself, thus being more susceptible to leaching processes, which can cause adsorption to soil, runoff to groundwater or uptake by plants.4,18,19Since the metabolism and biotransformation of roxarsone has not been fully elucidated yet, electrochemistry (EC) is presented as a simple, purely instrumental approach to study the oxidative transformation in this work. In the last years, EC has turned out to be a powerful tool especially for the simulation of metabolic reactions.20–24 By the hyphenation of EC with MS, TPs can directly be detected after their generation and even short-lived, reactive intermediates can be identified.25 In conventional in vitro or in vivo assays based on liver cell incubations or the analysis of urine and feces, the detection of reactive species is difficult due to matrix influence and rapid binding to cell constituents such as proteins or DNA. Therefore, conventional analysis of roxarsone metabolites in tissue or urine extracts may not reveal all arsenic containing species because of matrix trapping. Furthermore, no investigations on the identity and potential toxicity of non-arsenic containing TPs resulting from the cleavage of As(V) from phenylarsonic acids have been carried out so far. Only Peng et al. detected one non-arsenic compound in the extract of chicken liver by speciation analysis using IC-ESI-MS/MS, although they did not elucidate its structure.10 Usually, separation of the highly polar arsenicals is performed using anion exchange columns and non-volatile solvents such as phosphate buffers as the mobile phase.4,13,26 Thus, compatibility with ESI-MS is limited. Therefore, a method for the separation of the TPs using hydrophilic interaction liquid chromatography (HILIC) was developed. Thereby, compatibility of the applied solvents with both ICP-MS and ESI-MS for speciation analysis was achieved. In addition, quantification of arsenic containing species was performed by means of EC-HILIC-ICP-MS in order to specify the amount of the generated products and elucidate the major products formed.
Furthermore, the reactivity of the electrogenerated TPs towards biomolecules has been investigated. The phase II metabolism of roxarsone was simulated and toxicity estimation was performed. As this was carried out in the absence of complex matrix, the identification of reactive species by means of EC/(LC/)MS could be achieved as well.27
Experimental section
Chemicals
Roxarsone (ROX, 98.1%) and formic acid (99–100%) were purchased from Fluka (Buchs, Switzerland). Ammonium formate (≥99.995%, trace metal basis), glutathione (99%), guanidine hydrochloride (≥99%), bovine β-lactoglobulin A (≥90%) and human serum albumin (>99%) were obtained from Sigma Aldrich (Steinheim, Germany) and acetonitrile (≥99.9%, hypergrade) from Merck (Darmstadt, Germany). Water for sample preparation and HPLC was purified using an Aquatron A4000D system (Barloworld Scientific, Nemour, France). All solutions were degassed prior to application using ultrasonication for 15 minutes (Sonorex, Bandelin, Berlin, Germany).Electrochemical oxidation
Electrochemical oxidation of roxarsone was carried out by means of an electrochemical thin-layer cell (μPrepCell™, Antec Leyden, Zoeterwoude, The Netherlands). The cell consisted of a boron-doped diamond (BDD) working electrode, a titanium auxiliary electrode and a Pd/H2 reference electrode. A potential ramp from 0 to 2500 mV (10 mV s−1) vs. Pd/H2 was applied for oxidation. ROX solution (100 μM) was prepared in 10 mM ammonium formate (NH4FA) adjusted to pH 7.4 with ammonia and acetonitrile (50/50, v/v). The solution was pumped through the electrochemical cell under continuous flow (20 μL min−1) delivered by a syringe pump (Model 74900, Cole Parmer, Vernon Hills, IL, USA). The effluent of the cell was introduced into an Exactive™ mass spectrometer (Thermo Fisher Scientific, Bremen, Germany), where the negative ionization mode (ESI(−)) was applied. The mass spectrometric parameters are listed in Table S-1 (ESI†). Post-processing of the data was carried out with the software XCalibur 2.1 (Thermo Fisher Scientific). For visualization, the data were depicted in form of a three-dimensional mass voltammogram by plotting the mass spectra in dependency of the applied potential with the software Origin 9.1 (OriginLab, Northhampton, MA, USA).Separation of the transformation products
In order to separate the electrochemically generated TPs, an HPLC system (Shimadzu, Kyoto, Japan) was integrated between EC and MS. The system was equipped with two LC-10ADVP pumps, a SIL-10A autosampler, a SCL-10AVP system controller, a DGC-14A degasser and a CTO-10ASVP column oven. LCSolution software version 1.2.2 (Shimadzu) was used for control. Roxarsone was oxidized at the potential showing the highest conversion rate (2200 mV vs. Pd/H2) and analyzed by means of HILIC-ESI-MS (Exactive™) and complementarily by means of HILIC-ICP-MS (iCAP Qc, Thermo Fisher Scientific). ICP-MS measurements were performed in kinetic energy discrimination (KED) mode in order to minimize isobaric interferences of 75As. All other parameters applied for both ESI-MS and ICP-MS detection are listed in the ESI† (Tables S-1 and S-2). The column used for the separation was a Trinity P2 (Thermo Fisher Scientific, Bremen, Germany) with 100 mm length and 2.1 mm inner diameter, particle size of 3 μm and pore size of 120 Å. The mobile phase consisted of 50 mM NH4FA adjusted to pH 3.0 with formic acid and AcN. The gradient program is listed in Table S-3a (ESI†). The injection volume was 8 μL for HILIC-ESI-MS and 3 μL for HILIC-ICP-MS analysis.Quantification of arsenic species
Quantification of arsenic species was performed by external calibration after recording a response function of the ICP-MS in dependency of the changing solvent compositions during LC gradient separation. Therefore, a solution of ROX (5 μM in NH4FA (50 mM, pH 3.0)/AcN, 40/60, v/v) was spiked in a post-column approach every minute into the mobile phase during continuous ICP-MS detection. Spiking was also performed in the last three minutes of the equilibration step during HILIC-ICP-MS analysis of roxarsone and its TPs in order to ensure plasma stability. External six-point calibration was carried out by flow-injection analysis of roxarsone in different concentrations between 0.5 μmol L−1 and 60 μmol L−1 representing 75As. Column recovery was found to be 99% with a relative standard deviation (RSD) of 2.5% after threefold analysis of roxarsone in different concentrations (1 μM, 5 μM, 10 μM) by means of HPLC-ICP-MS with and without column.GSH adduct formation
The phase II metabolism of roxarsone was simulated with the tripeptide glutathione (GSH). Therefore, a solution of 100 μM GSH in 10 mM NH4FA (pH 7.4) was added to the effluent of the electrochemical cell via a T-piece using a second syringe pump (Model 74900, Cole Parmer, flow: 20 μL min−1). Roxarsone (100 μM) in NH4FA (10 mM, pH 7.4)/AcN (50/50, v/v) was oxidized by applying a potential ramp from 0 to 2500 mV vs. Pd/H2. After a reaction time of 5 minutes, detection was performed by means of ESI-MS (Exactive™, for mass spectrometric parameters see Table S-1, ESI†).Protein adduct formation
Adduct formation with proteins was investigated with β-lactoglobulin A (β-LGA) and human serum albumin (HSA). A 20 μM solution of the respective protein dissolved in guanidine hydrochloride (6 M in water) was added to the effluent of the cell. A 1 mM roxarsone solution in NH4FA (10 mM, pH 7.4)/AcN (50/50, v/v) was oxidized by applying a constant potential of 2200 mV vs. Pd/H2. After a reaction time of 10 minutes, the reaction mixture was collected in a 5 μL injection loop of a ten-port valve (Antec Leyden, Zoeterwoude, The Netherlands). By switching the valve, the reaction mixture was transferred onto a reversed-phase column (Discovery BIO WidePore C5, 150 mm × 2.1 mm, 5 μm, 300 Å, Supelco, Bellefonte, PA, USA). Separation was carried out using an HPLC system from Antec Leyden (Zoeterwoude, The Netherlands) and 0.1% formic acid in water and acetonitrile as mobile phase. The system comprised two LC 100 pumps, an OR 110 degasser, an AC 100 controller, an AS 100 autosampler and a ROXY™ column oven. The software Alexys was used for system control. Gradient elution was performed by applying the parameters listed in Table S-3b (ESI†). For detection, a time-of-flight ESI-MS (micrOToF, Bruker Daltonics, Bremen, Germany) in positive ion mode was used (mass spectrometric parameters are listed in Table S-4, ESI†). In order to prevent salts from entering the MS, the elution volume of the first 2.5 minutes of the LC separation was discarded and not transferred into the micrOToF.Results and discussion
Electrochemical oxidation
In order to obtain a general overview on the oxidation behavior and the oxidation products of roxarsone, a potential ramp from 0 to 2500 mV vs. Pd/H2 was applied to the electrochemical cell. The effluent was directly transferred into an Orbitrap-based ESI-MS in order to detect accurate masses of roxarsone and the obtained products. Exact mass measurements and/or fragmentation experiments are obligatory for the identification of potential products in order to enable reliable sum formula and structure prediction. The obtained mass spectra were plotted against the applied potential resulting in a three-dimensional mass voltammogram (Fig. 1). Roxarsone itself was detected at m/z 262 as [M − H]−. The oxidation of roxarsone can be easily identified by the decreasing signal intensity at potentials higher than approximately 1500 mV. Simultaneously, transformation products are generated, which show increasing signal intensities at increasing potential. The determined mass deviations of roxarsone and its transformation products to the calculated values were in the range of 0.0–5.7 ppm, allowing reliable sum formula proposals. Products formed by electrochemical oxidation of roxarsone can generally be divided into different groups: the first group includes those products, which were either hydroxylated, aminated and/or dehydrogenated at the phenyl ring (M1). The second group comprises all TPs, where the arsonic acid functionality has been cleaved off (M2) under formation of arsenate (As(V)). The products of the third group are the result of reduction reactions (Mred), which occur due to the use of an undivided cell, which allows a simultaneous reduction at the counter electrode. Moreover, dimers of roxarsone were detected. An overview of all products is depicted in Fig. 2. Enlarged sections of the mass voltammogram for all species as well as exact masses, sum formulae and structure proposals are shown in the ESI† (Fig. S-1–S-5 and Table S-5). In the following, the m/z of the oxidation products are presented as integers, for reasons of simplicity, without decimals even if accurate mass spectra were recorded. The first constituent of M1 products is the monohydroxylated species of roxarsone (M1.1) with m/z 278. Originating from M1.1, a second hydroxylation yielding M1.2 (m/z 294) or an amination yielding M1.5 (m/z 293) takes place. Amination was obtained due to the use of ammonium formate as electrolyte in the solvent. The formation of aminated products during electrochemical treatment should be considered as a side reaction, which is not relevant since it does not occur in vivo. In a follow-up reaction, both, M1.2 and M1.5, are dehydrogenated leading to electrophilic quinoid structures with m/z 292 (M1.3) and m/z 291 (M1.6). M1.2 undergoes reactions yielding the trihydroxylated species of roxarsone (M1.4, m/z 310) and the dehydrogenated form of dihydroxylated, mono-aminated roxarsone (M1.7, m/z 307).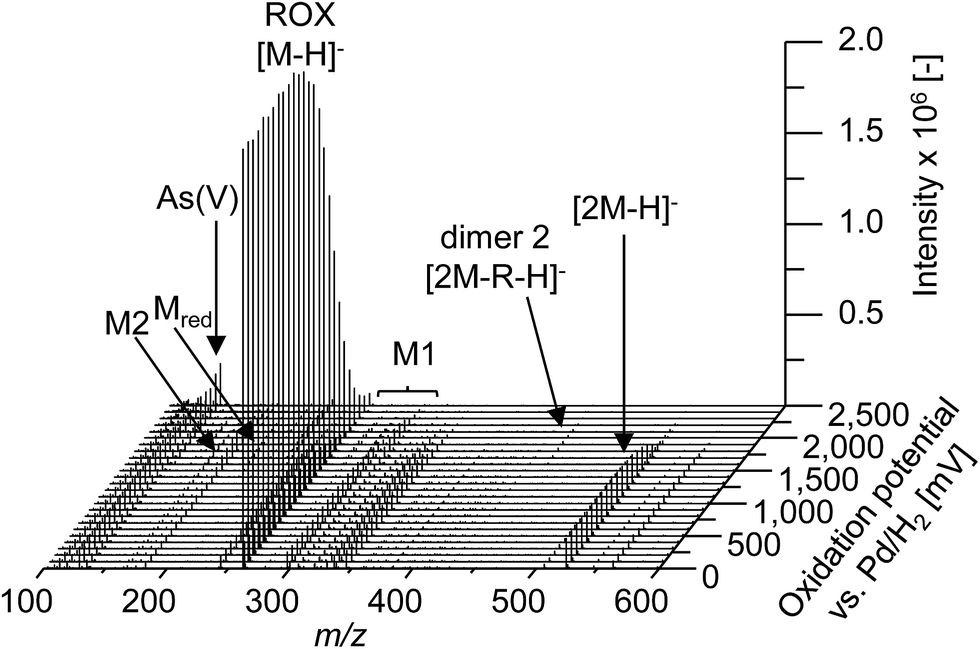 | ||
| Fig. 1 Mass voltammogram (m/z 100–600) of roxarsone obtained with ESI(−)-MS in the potential range 0–2500 mV vs. Pd/H2. R = +2O + NH − 4H − HAsO3. | ||
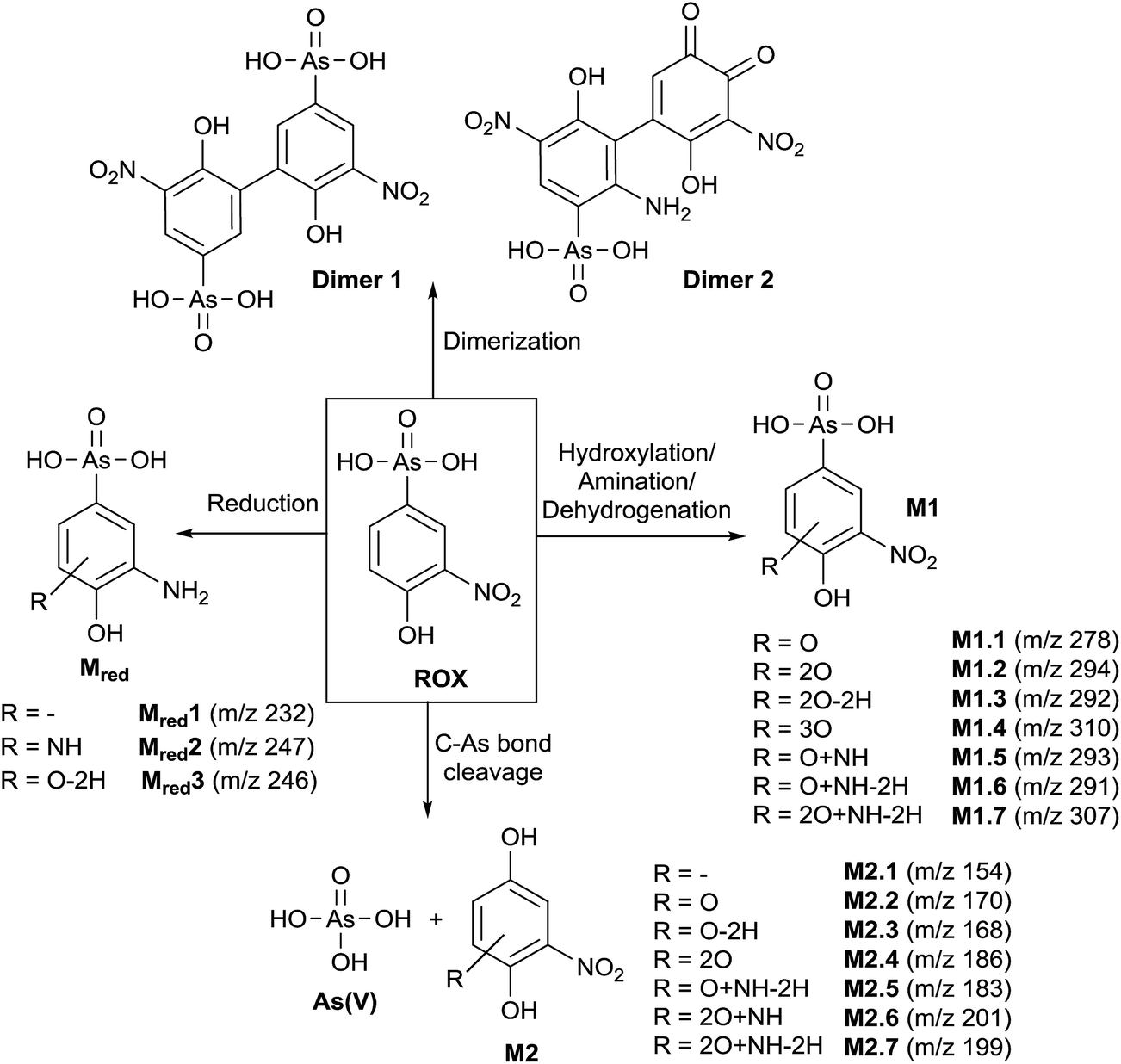 | ||
| Fig. 2 Reaction pathway of the TPs obtained from electrochemical oxidation of roxarsone. | ||
Starting from roxarsone and products found for M1, the arsonic acid functionality can be cleaved off under formation of a substituted phenyl compound (M2) and As(V). The mechanism of the oxidative carbon–arsenic bond cleavage of phenylarsonic acid was described by Xu et al.28 in 2007. They postulated an ipso addition of a hydroxyl radical to C1, which is also the binding site of the arsonic acid group, by both experimental data and computational calculations. Fig. 2 shows the structure proposals of the oxidative C–As bond cleavage of roxarsone under formation of products found for M2 and As(V) after having transferred the mechanism described by Xu et al. to substituted phenylarsonic acids.
Additionally, dimer formation of roxarsone was observed during electrochemical oxidation. Fig. 2 presents only one possibility of a roxarsone dimer, where the monomeric units are bound via C5–C5 (dimer 1). Starting from this, or by combining one intact molecule of ROX and one oxidation product or two oxidation products, respectively, a modified dimer (dimer 2) is formed, where several isomeric forms are possible. Both dimeric forms can be seen as by-products of the electrochemical oxidation since dimer formation in vivo is unusual.
The last class of transformation products of roxarsone generated and detected by means of EC/ESI-MS comprises the reduced form of roxarsone or its oxidation products (Mred). Since working and counter electrode are not spatially separated in the used electrochemical cell, the analytes can diffuse to the cathode, where reduction takes place.29 For this reason, the reduction of the nitro functionality to an amino group could be observed. In a first step, roxarsone directly is reduced yielding Mred1. Starting from this, either amination to Mred2 or hydroxylation (not determined, n.d.) followed by dehydrogenation (Mred3), takes place (illustrated in Fig. S-5, ESI†). Thus, the undivided cell not only induces reduction of roxarsone itself but also leads to the formation of products, which have undergone both, oxidation and reduction.
Next to the identified transformation products M1, M2, dimers and Mred, some unknown species (U) were detected, where no indication of the structure and/or sum formula could be found based on their exact masses. However, because of the high number of transformation products of roxarsone being electrochemically formed, the lack of information for these species does not significantly affect the study performed in this work. The exact masses of the unidentified products are listed in the ESI† (Table S-5).
To conclude, investigations of the oxidative metabolism of roxarsone by means of EC/MS identified 25 different transformation products. Those species include arsenic containing oxidation products, products where the arsonic acid functionality has been cleaved off under simultaneous formation of As(V), dimers as well as reduction products. For the first time, not only potential arsenic TPs of roxarsone were detected, but also non-arsenic containing compounds. The knowledge about TPs observed by means of electrochemical oxidation provides helpful information about the class of compounds which could be also found in real samples. All species are likely to be also formed in vivo since electrochemistry provides a broad range of reactions taking place also in biological systems. In order to determine the presence of such species in real samples, such as poultry urine or litter extracts, the subsequently developed HILIC-ESI-MS and HILIC-ICP-MS analysis should be transferred to those.
Separation and quantification of transformation products
In order to characterize the TPs of roxarsone in more detail regarding their polarity and possible isomer formation, a separation method based on HILIC has been developed. By means of HILIC, separation of the highly polar phenylarsonic acid compounds was enabled and compatibility with both ESI-MS and ICP-MS could be achieved. In order to obtain molecular information about the TPs, the separation system was directly coupled to ESI-MS. For elemental analysis of arsenic (75As), ICP-MS was used as detection method. Different HILIC columns including zwitterionic, polyethylenimine-, diol- and amino-based stationary phases were screened for their separation efficiency. A trimodal mixed-mode column (Trinity P2) turned out to yield best separation results. Next to its hydrophilic interaction properties, the column has additional anion-exchange as well as cation-exchange functionalities in order to provide retention for highly polar and ionic species. Fig. 3 shows the extracted ion chromatograms of M1, M2, Mred and unknown species (U) detected by means of ESI-MS. Not all compounds, which had been previously found in the mass voltammogram, were detected after HILIC separation, likely due to the slightly inferior limit of detection and/or limited chemical stability. Roxarsone elutes at tR = 14.7 min, while the more polar As(V) was detected at 20.6 min. For M1 products, only M1.7 was detected at tR = 27.5 min. Moreover, Mred1 was detected after 10.8 min. No further arsenic-containing products were detected thus indicating that most of these TPs described before are present in fairly low concentrations. All non-arsonic acid and therefore much less polar products (M2) elute close to the void volume of the column between 0.9 and 3.4 min. M2.1 and M2.2 were not detected. M2.3 shows two isomeric forms, which elute at tR1 = 1.6 min and tR2 = 2.2 min. For M2.4 and M2.5, only one respective isomer was found. Both are coeluting at tR = 1.7 min. M2.6 shows five different isomers (tR1 = 0.9 min, tR2 = 1.6 min, tR3 = 1.8 min, tR4 = 2.5 min, tR5 = 3.4 min), whereas for M2.7, six different isomeric forms were detected (tR1 = 1.6 min, tR2 = 1.9 min, tR3 = 2.1 min, tR4 = 2.3 min, tR5 = 2.6 min, tR6 = 2.9 min). Furthermore, a signal for the unknown species U4 was detected at tR = 1.6 min. The early elution of U4 indicates that this species does not contain an arsonic acid functionality and thus supports the proposed sum formula of C6H3O7N3 (Table S-5, ESI†).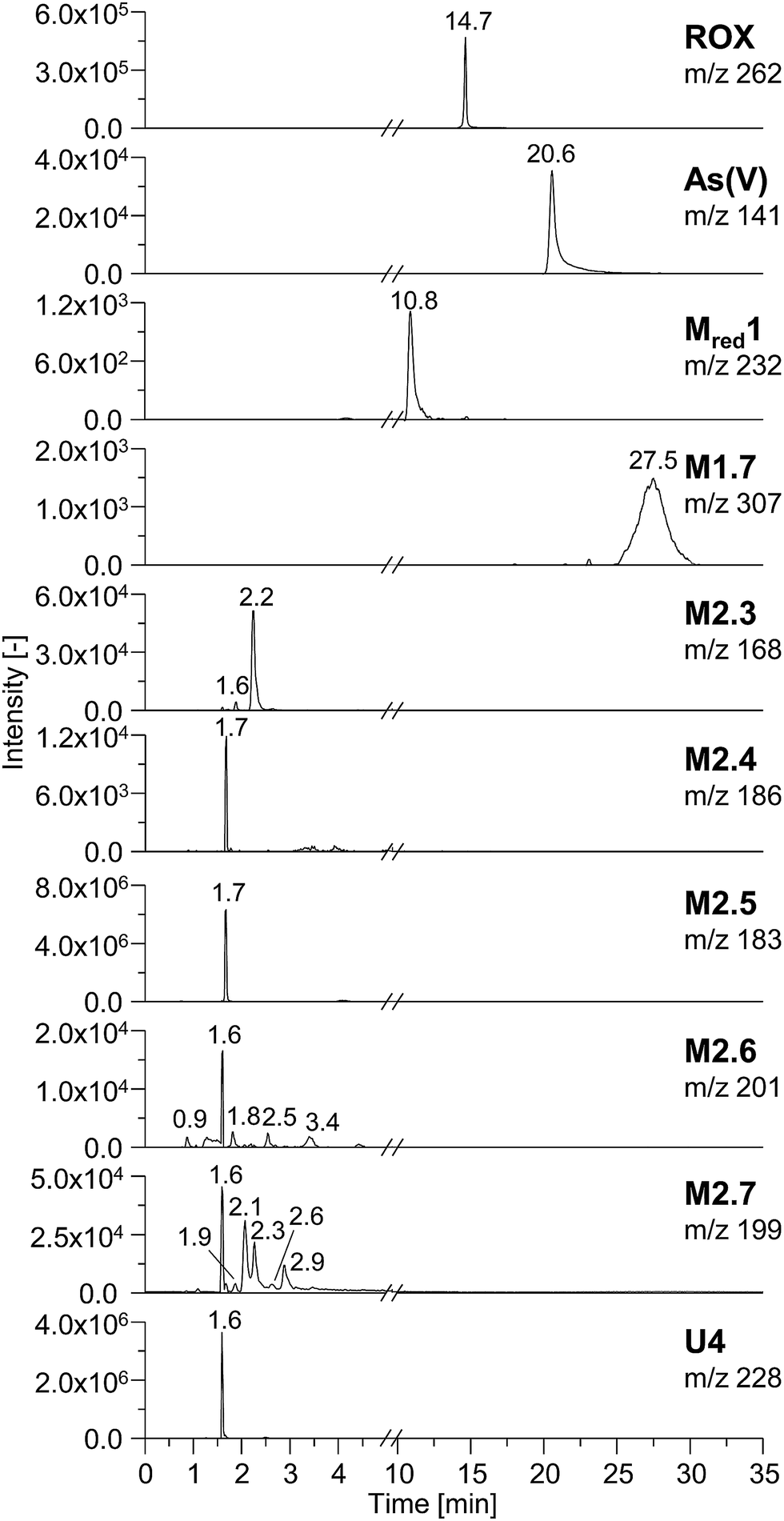 | ||
| Fig. 3 HILIC separation of electrochemically generated TPs of roxarsone (2200 mV vs. Pd/H2). Depicted are the extracted ion chromatograms of all detectable products as well as their retention times (tR). | ||
Comparing the chromatographic information obtained by means of HILIC-ESI-MS with information obtained by means of HILIC-ICP-MS for 75As, speciation analysis was carried out. The molecular and elemental chromatograms are compared in Fig. 4. In the ESI-MS chromatogram (Fig. 4a), only the arsenic containing species are illustrated in order to simplify matters. The comparison with the ICP-MS chromatogram (Fig. 4b) shows that Mred1, roxarsone as well as As(V) were detected by both methods. At the retention time of M1.7, no peak was observed in the HILIC-ICP-MS chromatogram, most likely due to the limit of detection (LOD), which for arsenic was determined in triplicate and found to be 197.8 nmol L−1 (RSD 6%) for the used system. The LOD was determined based upon the 3σ criterion. The ICP-MS chromatogram shows additional peaks, which were not detected by means of HILIC-ESI-MS. The retention times for those species are listed in Table 1. All species are numbered in order of elution.
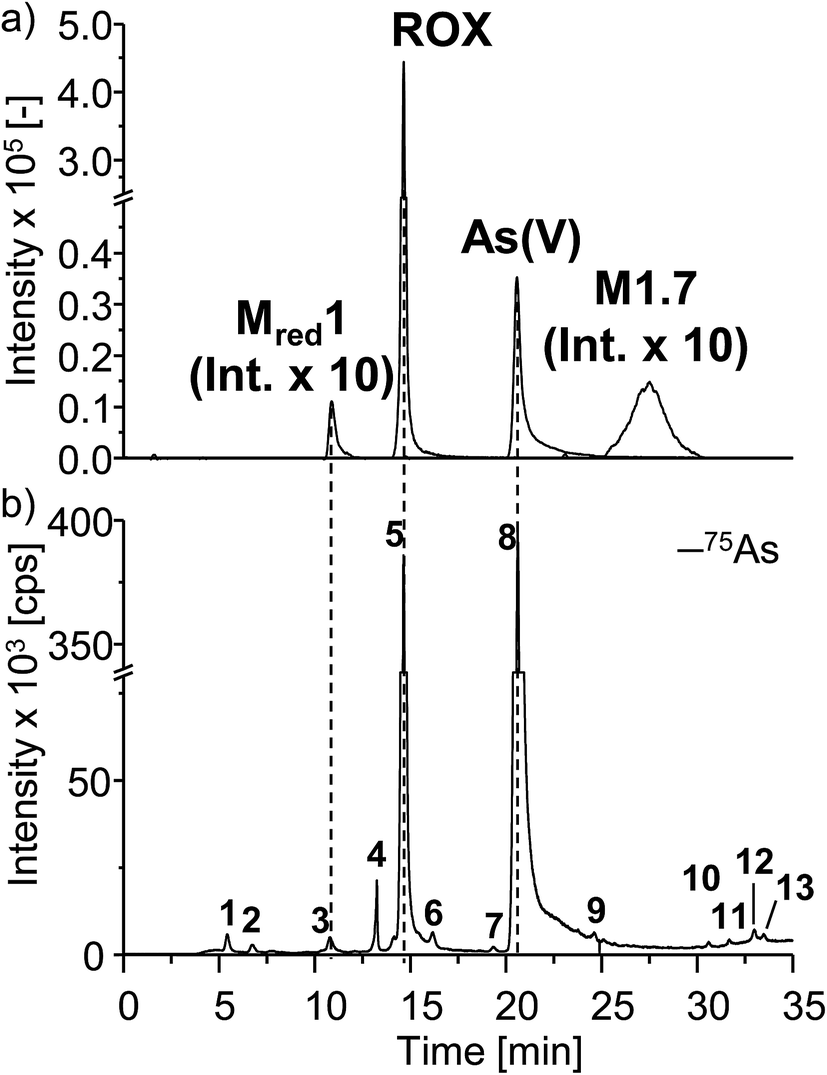 | ||
| Fig. 4 HILIC separation of electrochemically generated arsenic containing TPs of roxarsone. For speciation analysis, (a) the ESI-MS chromatogram including the extracted ion chromatograms of the respective TPs as well as (b) the complementary ICP-MS chromatogram for 75As are directly compared to each other. | ||
| Retention time tR [min] | Species | c(75As) [μmol L−1] | RSD [%] | |
|---|---|---|---|---|
| 1 | 5.6 | Unknown | 0.94 | 1.4 |
| 2 | 6.9 | Unknown | 0.54 | 1.1 |
| 3 | 10.8 | Mred1 | 1.15 | 1.2 |
| 4 | 13.5 | Unknown | 1.54 | 0.6 |
| 5 | 14.7 | ROX | 51.5 | 2.3 |
| 6 | 16.2 | Unknown | 1.07 | 0.8 |
| 7 | 19.4 | Unknown | <LOQ | — |
| 8 | 20.6 | As(V) | 96.67 | 2.0 |
| 9 | 24.6 | Unknown | <LOQ | — |
| 10 | 30.6 | Unknown | <LOQ | — |
| 11 | 31.7 | Unknown | <LOQ | — |
| 12 | 33.0 | Unknown | <LOQ | — |
| 13 | 33.5 | Unknown | <LOQ | — |
Quantification of arsenic containing transformation products of roxarsone was carried out after HILIC separation using ICP-MS detection. In order to correct for plasma changes originating from gradient elution, a response function was recorded (see ESI Fig. S-6†) by a post-column addition of roxarsone solution as 75As standard. After an external six-point calibration, the concentrations of the three identified species roxarsone, As(V) and reduced roxarsone (Mred1) were determined to be 51.5 μmol L−1, 96.6 μmol L−1 and 1.15 μmol L−1 in a threefold analysis. Due to the lack of complementary molecular information for all other compounds, no species concentration could be determined. Nevertheless, the concentration of 75As for all of them was found to be below 1.5 μmol L−1 and is listed in Table 1, where also the relative standard deviations are presented. Thus, As(V) is the main oxidation product being electrochemically formed under the applied conditions. The LOQ was determined according to the 10σ criterion and found to be 440.8 nmol L−1 (RSD = 6%) for 75As.
Summing up all concentrations found for arsenic by means of HILIC-ICP-MS and comparing them with the initial concentration, an 75As recovery of 77% was obtained. This value represents a reasonable amount considering the number of peaks detected. Furthermore, it needs to be considered that 15% of the analytes may remain inside the electrochemical cell due to adsorption effects, e.g., on the electrode surface.
To conclude, the performed separation enabled the identification of three arsenic containing species by both elemental and molecular MS. Furthermore, five of seven non-arsenicals were determined by means of HILIC-ESI-MS. Some of them showed several isomers. HILIC-ICP-MS analysis revealed some additional arsenic containing species for which no complementary molecular information was observed. HILIC separation with volatile buffer systems is fully compatible with both ICP- and ESI-MS. Quantification of the arsenic products identified As(V) to be the main oxidation product of roxarsone under the applied electrochemical conditions. Since As(V) was also found to be the main TP of roxarsone, e.g., in soil, further investigations of ROX TPs should not only consider arsenic containing species but also non-arsenic ones resulting from C–As cleavage. The developed method can be transferred to real sample analysis of, e.g., soil or liver extracts in order to detect and identify TPs of roxarsone in biological matrices.
Adduct formation with biomolecules
Adduct formation with biomolecules was carried out in order to investigate the reaction behavior of roxarsone and its transformation products towards endogenous nucleophiles such as cysteine residues in peptides or proteins. By means of this, the phase II metabolism can be simulated and toxicity estimation can be performed.27Adduct formation with glutathione
Glutathione (GSH) is an important trapping agent for reactive intermediates formed in vivo during the phase II metabolism. Therefore, the reactivity of electrochemically generated ROX TPs towards GSH was investigated. GSH was added to the oxidized solution of roxarsone (0–2500 mV vs. Pd/H2) via a T-connection immediately after the electrochemical cell. After a reaction time of 5 minutes, accurate mass spectra were recorded using ESI-MS on an Orbitrap-based instrument. By exact mass analysis, two different adducts with GSH were identified. The extracted ion traces of roxarsone and GSH show decreasing signal intensities whereas increasing intensities for the found adducts were observed (see ESI†, Fig. S-7). Adduct formation of reactive species corresponds to a Michael-type addition of GSH to, e.g., quinones (M2.3, Δm = 168 Da, Fig. 5a) or quinone imines (Mred1-2H, Δm = 230 Da, Fig. 5b). Autoxidation takes place after rearomatization leading again to a quinoid structure with m/z 473 (Δm = 166 Da, Fig. 5a) and m/z 535 (Δm = 228 Da, Fig. 5b). However, no second addition of GSH to the quinoids was found. The detected exact masses of the adducts as well as the relative deviations to the calculated exact masses are listed in Table S-6 (ESI†). Autoxidation of the two adducts formed with glutathione can likely be attributed to oxidation inside the ESI interface taking place due to a lowered oxidation potential by electron withdrawal from GSH.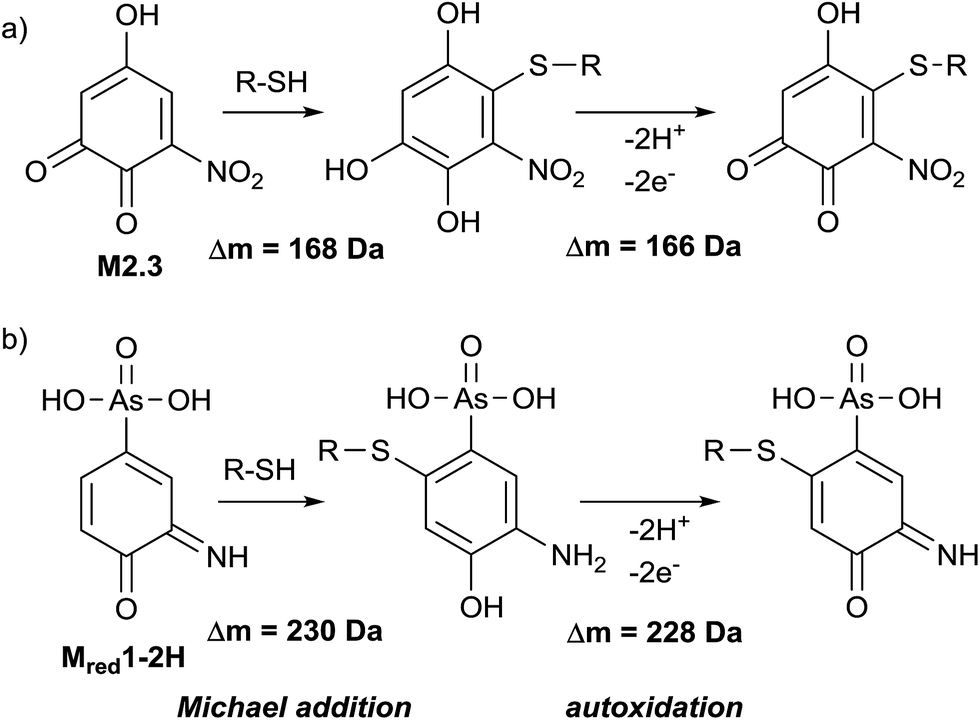 | ||
| Fig. 5 Adduct formation of TPs with cysteine containing biomolecules. | ||
Adduct formation with β-lactoglobulin A
First investigations of the reactivity of electrochemically generated ROX transformation products towards proteins were performed using β-lactoglobulin A (β-LGA) as a model protein. β-LGA is a relatively small protein of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 363 Da and contains 162 amino acids, including five cysteine residues. Four of those are disulfide bond and thus, one free cysteine is available for nucleophilic reactions. The free cysteine moiety is located inside β-LGA; therefore the protein was denatured prior to application using guanidine hydrochloride. A solution of denatured β-LGA was added to the effluent of the electrochemical cell. Roxarsone was continuously pumped through the cell and in a first experiment, no potential (0 mV) was applied in order to identify the signals resulting from the native protein. Then, 2200 mV vs. Pd/H2 was applied to the cell. The reaction mixtures were collected in an injection loop of a ten-port valve after 10 minutes and were injected on a Discovery BIO WidePore C5 column by switching the valve. By discarding the first 2.5 minutes of the separation, the protein fraction was separated from salts and excessive TPs to avoid ion suppression and contamination of the ESI-ToF-MS.
363 Da and contains 162 amino acids, including five cysteine residues. Four of those are disulfide bond and thus, one free cysteine is available for nucleophilic reactions. The free cysteine moiety is located inside β-LGA; therefore the protein was denatured prior to application using guanidine hydrochloride. A solution of denatured β-LGA was added to the effluent of the electrochemical cell. Roxarsone was continuously pumped through the cell and in a first experiment, no potential (0 mV) was applied in order to identify the signals resulting from the native protein. Then, 2200 mV vs. Pd/H2 was applied to the cell. The reaction mixtures were collected in an injection loop of a ten-port valve after 10 minutes and were injected on a Discovery BIO WidePore C5 column by switching the valve. By discarding the first 2.5 minutes of the separation, the protein fraction was separated from salts and excessive TPs to avoid ion suppression and contamination of the ESI-ToF-MS.
The neutral mass spectrum for the native protein (Fig. 6a) was extracted by deconvolution of the charge distribution protein spectrum. The nominal mass of β-LGA is found at 18363 Da. Furthermore, an isoform of β-LGA was detected at 18687 Da. The mass spectra obtained with oxidized roxarsone (Fig. 6b) show that two adducts were formed. The first mass difference of 167 Da indicates an adduct formation with M2.3 under consideration of a mass variation of 1–2 Da for protein spectra. This difference was also found for the isomeric form of the protein, which confirms the result. The other mass difference of 228 Da corresponds to an adduct formation with the dehydrogenated species of reduced roxarsone (Mred1-2H). Thus, it could be shown that β-LGA shows reactivity towards the same transformation products as GSH. Structure proposals for both are illustrated in Fig. 5.
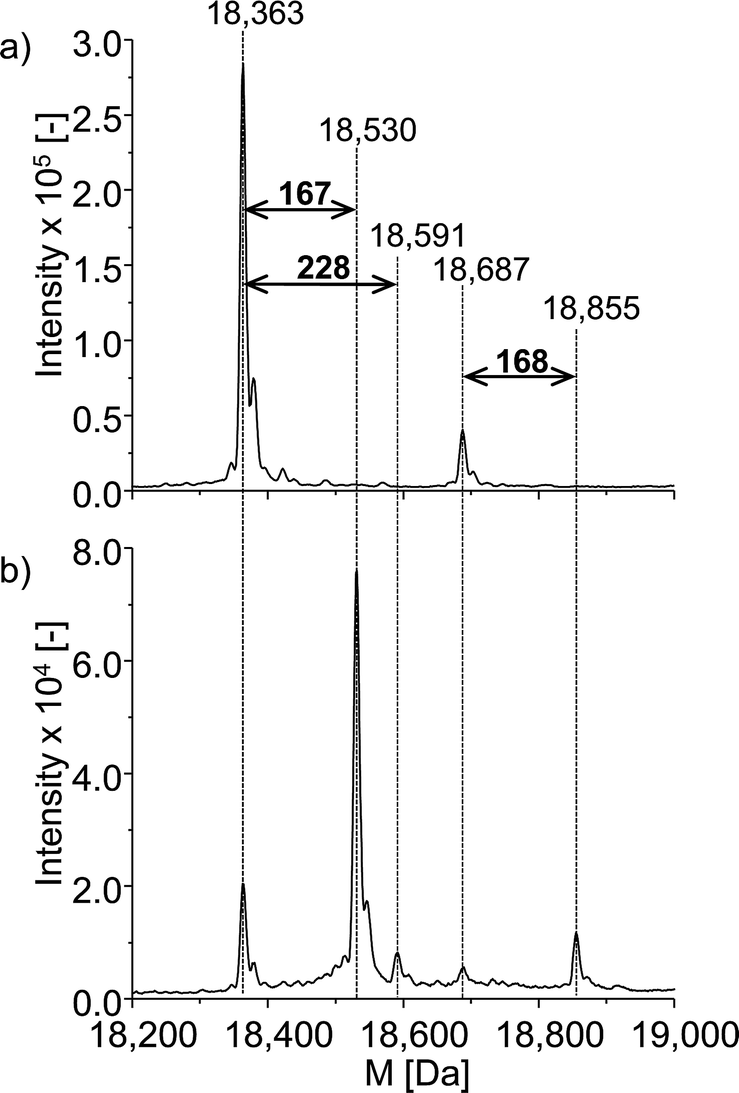 | ||
| Fig. 6 ESI-ToF-MS investigation of the adduct formation of ROX with β-LGA. (a) Deconvoluted spectrum of native β-LGA, (b) deconvoluted spectrum of the adduct formation with oxidized ROX. | ||
Adduct formation with human serum albumin
Next to the investigations of GSH and β-LGA, the reactivity towards the more relevant protein human serum albumin (HSA) was studied. HSA is highly abundant in human blood plasma and is a complex protein containing 585 amino acids, including 35 cysteine residues, 34 of which are bound in disulfide bridges and one is free. It has a nominal mass of 66555 Da. The experimental procedure was analogous to that used for β-LGA adduct formation. The denatured protein was added to the effluent of the electrochemical cell. Afterwards, LC-ESI-ToF-MS analysis was performed with and without applying an oxidative potential of 2200 mV vs. Pd/H2. Fig. 7a shows the deconvoluted spectrum of native HSA. Two major isoforms of the protein can be identified, namely human mercaptalbumin (HMA) with 66440 Da, and human non-mercaptalbumin (HNA) with 66555 Da. HNA is the oxidized form where the previously free cysteine residue is blocked by posttranslational modification and is thus not accessible for adduct formations. In HMA, this cysteine moiety is not conjugated and therefore, this isoform can undergo nucleophilic reactions with reactive intermediates. The adduct formation between HSA and oxidized roxarsone is shown in Fig. 7b. Again, a mass shift of 167 Da and 229 Da was detected, which, can be traced back to an adduct formation with M2.3 and Mred1-2H in (see Fig. 5).
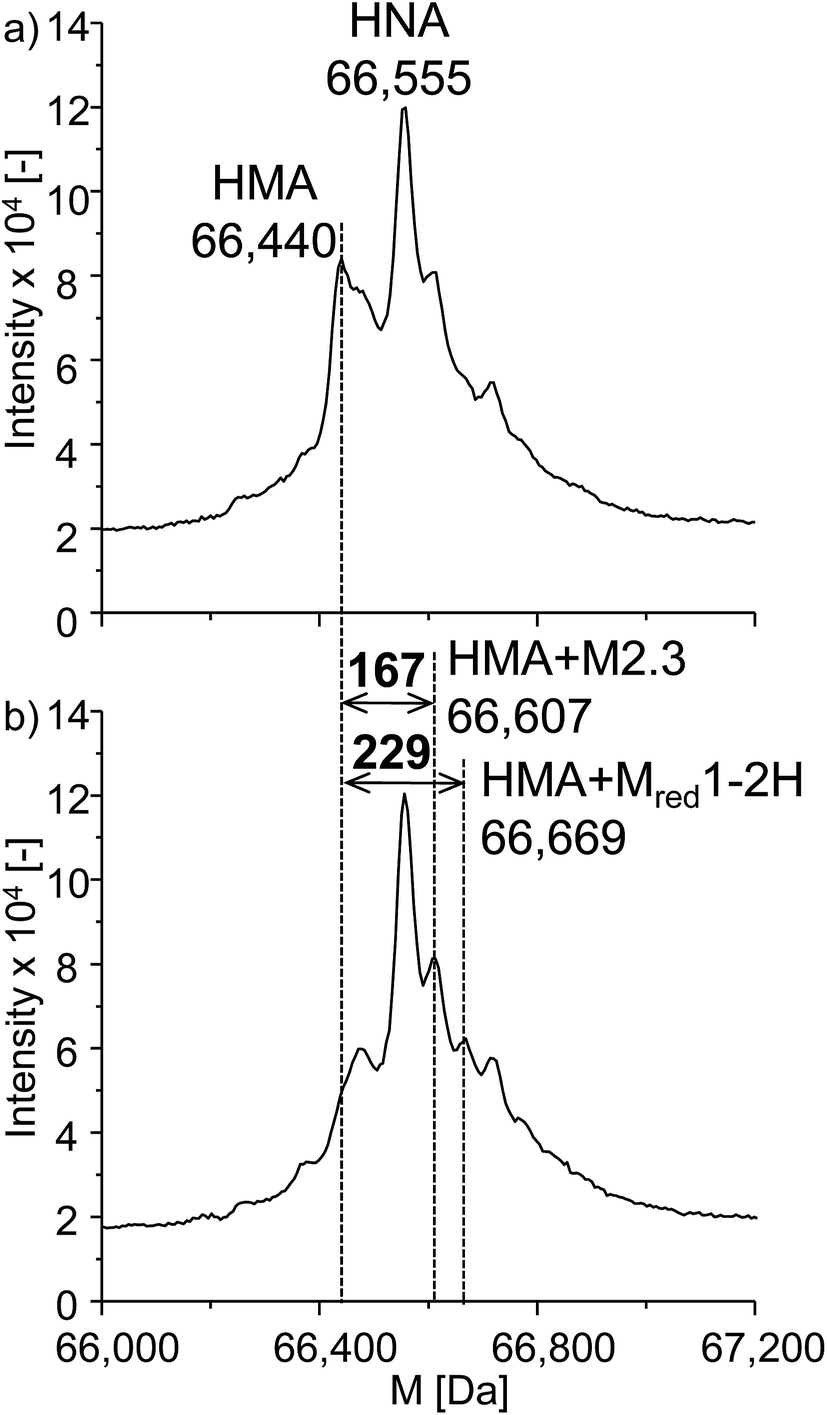 | ||
| Fig. 7 Adduct formation of ROX with HSA. Illustrated are the deconvoluted spectra of (a) the native protein and (b) the adduct formation between HSA and oxidized ROX (2.2 V vs. Pd/H2). | ||
To conclude, two electrochemically generated oxidation products of roxarsone formed adducts with the tripeptide glutathione and the proteins β-LGA and HSA. Thereby, the reactivity is related to the formation of electrophilic quinoid structures, which can undergo reactions towards nucleophilic groups in biomolecules such as cysteines. One of the reactive species contains arsenic, the other one is a non-arsenical. Consequently, arsenic, e.g. As(V), originating from roxarsone is not solely a potential health risk of the drug but also the formation of reactive, (non-)arsenic containing intermediates.
Conclusions
The application of EC/MS as an alternative approach for investigations concerning oxidative transformations of roxarsone has proven to be a powerful tool for an easy generation and identification of potential metabolites. Using EC/ESI-MS, numerous products were detected and separation by means of HILIC provided information about polarity and isomer formation. Furthermore, hyphenation of HILIC to ICP-MS and ESI-MS enabled species analysis and the quantification of arsenic-containing products revealing toxic As(V) as main metabolite. In addition, EC/MS as a tool for reactivity estimation towards free cysteines in biomolecules by means of adduct formation revealed two potential reactive intermediates of roxarsone. Thus, discussions about toxic effects resulting from transformation products of roxarsone and the continuing application of the drug in animal industries are justified.References
- E. K. Silbergeld and K. Nachman, Ann. N. Y. Acad. Sci., 2008, 1140, 346–357 CrossRef CAS PubMed.
- K. E. Nachman, P. A. Baron, G. Raber, K. A. Francesconi, A. Navas-Acien and D. C. Love, Environ. Health Perspect., 2013, 121, 818–824 CrossRef PubMed.
- H. D. Chapman and Z. B. Johnson, Poultry Sci., 2002, 81, 356–364 CrossRef CAS PubMed.
- L. X. Huang, L. X. Yao, Z. H. He, C. M. Zhou, G. L. Li, B. M. Yang and X. C. Deng, Chemosphere, 2014, 100, 57–62 CrossRef CAS PubMed.
- E. D'Angelo, G. Zeigler, E. G. Beck, J. Grove and F. Sikora, Sci. Total Environ., 2012, 438, 286–292 CrossRef PubMed.
- M. F. Hughes, Toxicol. Lett., 2002, 133, 1–16 CrossRef CAS PubMed.
- J. P. Moody and R. T. Williams, Food Cosmet. Toxicol., 1964, 2, 707–710 CrossRef CAS PubMed.
- J. L. Morrison, J. Agric. Food Chem., 1969, 17, 1288–1290 CrossRef CAS.
- S. D. Conklin, N. Shockey, K. Kubachka, K. D. Howard and M. C. Carson, J. Agric. Food Chem., 2012, 60, 9394–9404 CrossRef CAS PubMed.
- H. Y. Peng, B. Hu, Q. Q. Liu, Z. L. Yang, X. F. Lu, R. F. Huang, X. F. Li, M. J. Zuidhof and X. C. Le, J. Chromatogr. A, 2014, 1370, 40–49 CrossRef CAS PubMed.
- P. A. Moore, T. C. Daniel, A. N. Sharpley and C. W. Wood, J. Soil Water Conserv., 1995, 50, 321–327 Search PubMed.
- J. R. Garbarino, A. J. Bednar, D. W. Rutherford, R. S. Beyer and R. L. Wershaw, Environ. Sci. Technol., 2003, 37, 1509–1514 CrossRef CAS PubMed.
- W. Zhang, F. Xu, J. Han, Q. Sun and K. Yang, J. Hazard. Mater., 2015, 286, 171–178 CrossRef CAS PubMed.
- A. J. Bednar, J. R. Garbarino, I. Ferrer, D. W. Rutherford, R. L. Wershaw, J. F. Ranville and T. R. Wildeman, Sci. Total Environ., 2003, 302, 237–245 CrossRef CAS PubMed.
- C. G. Rosal, G. M. Momplaisir and E. M. Heithmar, Electrophoresis, 2005, 26, 1606–1614 CrossRef CAS PubMed.
- B. P. Jackson and P. M. Bertsch, Environ. Sci. Technol., 2001, 35, 4868–4873 CrossRef CAS PubMed.
- D. W. Rutherford, A. J. Bednar, J. R. Garbarino, R. Needham, K. W. Staver and R. L. Wershaw, Environ. Sci. Technol., 2003, 37, 1515–1520 CrossRef CAS PubMed.
- F. X. Han, W. L. Kingery, H. M. Selim, P. D. Gerard, M. S. Cox and J. L. Oldham, Sci. Total Environ., 2004, 320, 51–61 CrossRef CAS PubMed.
- A. J. Bednar, J. R. Garbarino, J. F. Ranville and T. R. Wildeman, J. Agric. Food Chem., 2002, 50, 7340–7344 CrossRef CAS PubMed.
- S. Jahn, A. Baumann, J. Roscher, K. Hense, R. Zazzeroni and U. Karst, J. Chromatogr. A, 2011, 1218, 9210–9220 CrossRef CAS PubMed.
- A. Baumann, A. Faust, M. P. Law, M. T. Kuhlmann, K. Kopka, M. Schäfers and U. Karst, Anal. Chem., 2011, 83, 5415–5421 CrossRef CAS PubMed.
- W. Lohmann, B. Meermann, I. Möller, A. Scheffer and U. Karst, Anal. Chem., 2008, 80, 9769–9775 CrossRef CAS PubMed.
- H. Faber, D. Melles, C. Brauckmann, C. A. Wehe, K. Wentker and U. Karst, Anal. Bioanal. Chem., 2012, 403, 345–354 CrossRef CAS PubMed.
- U. Bussy, M. Delaforge, C. El-Bekkali, V. Ferchaud-Roucher, M. Krempf, I. Tea, N. Galland, D. Jacquemin and M. Boujtita, Anal. Bioanal. Chem., 2013, 405, 6077–6085 CrossRef CAS PubMed.
- W. Lohmann and U. Karst, Anal. Chem., 2007, 79, 6831–6839 CrossRef CAS PubMed.
- T. Liang, Z. Ke, Q. Chen, L. Liu and G. Chen, Chemosphere, 2014, 112, 128–133 CrossRef CAS PubMed.
- W. Lohmann, H. Hayen and U. Karst, Anal. Chem., 2008, 80, 9714–9719 CrossRef CAS PubMed.
- T. Xu, P. V. Kamat, S. Joshi, A. M. Mebel, Y. Cai and K. E. O'Shea, J. Phys. Chem. A, 2007, 111, 7819–7824 CrossRef CAS PubMed.
- C. Zettersten, R. Lomoth, L. Hammarstrom, P. J. R. Sjoberg and L. Nyholm, J. Electroanal. Chem., 2006, 590, 90–99 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ja00277c |
| This journal is © The Royal Society of Chemistry 2017 |