
Speciation of gold nanoparticles and low-molecular gold species in Wistar rat tissues by HPLC coupled to ICP-MS
Juan
Soto-Alvaredo†
a,
Carlos
López-Chaves†
b,
Cristina
Sánchez-González
b,
María
Montes-Bayón
a,
Juan
Llopis
b and
Jörg
Bettmer
*a
aDepartment of Physical and Analytical Chemistry, University of Oviedo, C/Julian Clavería 8, 33006, Oviedo, Spain. E-mail: bettmerjorg@uniovi.es
bDepartment of Physiology, University of Granada, Campus Cartuja, 18071, Granada, Spain
First published on 2nd November 2016
Abstract
Gold nanoparticles (Au NPs) are widely used today in a broad range of applications like electronics, sensors, catalysis and especially in biomedicine. However, the increase in their use has raised concerns about the possible interactions in vivo and the unexpected responses inside humans and other living organisms. Analytical tools that are able to detect their presence in biological samples and also provide information about the presence of dissolved metal ions inside biological samples are, therefore, urgently needed for a proper understanding of the behaviour of these materials. In this work, an HPLC-ICP-MS technique for the determination of Au NPs was adapted to the analysis of tissues from Wistar rats after intraperitoneal injection of 10 nm Au NPs. This technique allows the detection of Au NPs and low-molecular Au species, permitting the detection and monitoring of potential degradation processes. Alkaline and enzymatic digestions were tested to solubilize the particles and to verify their stability during the extraction process. Enzymatic digestion with proteinase K over 14 h was found to be suitable for the extraction of the Au species with recovery rates of about 91%. Finally, the method was applied to the analysis of liver and spleen samples. Significant abundances of low-molecular Au species were detected (around 30%). This observation suggested that a degradation process played a pivotal role during the transport and accumulation of Au NPs that were intraperitoneally injected into Wistar rats.
Introduction
Currently, nanoparticles are present in a broad range of scientific and industrial applications and consumer products. The special features conferred by their size and their surface to volume ratio make them very interesting for science and technology. Gold nanoparticles (Au NPs) are, among nanoparticles, widely studied and applied in electronics and sensors,1 solar cells2 or in catalysis;3 but especially in biomedical applications like radiotherapy,4 as drug carriers5 or in cancer therapies6,7 (as they are, presumably, inert and considered safe).Together with the increasing use of Au NPs in biomedical treatments and other medical applications, great concerns have arisen about the potential adverse consequences for the environment and human health of these new materials. Moreover, concern about their biological implications is also growing as there are numerous in vitro studies with Au NPs reporting mitochondrial damage, oxidative stress8 or autophagy.9 Furthermore, in vivo studies with Au NPs also report effects like bioaccumulation in important body organs,10,11 inflammation,12 apoptosis in the liver13 and genotoxic effects.14
The appearance of these, and many other, toxicological studies about the effects of metal nanoparticles in vivo has led to the foundation of a new field called “nanometallomics”. It aims for the quantification, localization and chemical speciation of the metal nanoparticles and their released ions in biological systems for the improved understanding of the biological response to metal nanoparticles.15–17 In this context, the development of suitable analytical tools for the speciation and characterization of nanoparticles is of great importance.
Herein, inductively coupled plasma-mass spectrometry (ICP-MS) plays an important role, either as a detector for separation techniques (field flow fractionation,18,19 liquid chromatography20–22 or electrophoresis23–25), as quantitative tool in total element determination10,11 or in the single-particle mode.26,27
In order to analyse biological samples, sample preparation strategies were necessary in order to maintain the species integrity. In combination with an ICP-MS based strategy, two strategies were suggested: alkaline or enzymatic digestion procedures.28,29 Alkaline digestion carried out with TMAH showed high recovery rates for Ag and Au NPs extracted from ground meat, Daphnia magna and Lumbriculus variegatus.30 High recoveries of 60 nm Au NPs were also reported for alkaline solubilization in animal tissues.31 Both approaches made use of determination by single-particle ICP-MS. Alternatively, several enzymes were suggested for the extraction of NPs from biological samples. Enzymatic digestion made use of Macerozyme R-10 enzyme for plant samples32,33 and proteinase K for samples of animal origin.31,34
In this work, a methodology based on the chromatographic separation previously developed by Helfrich et al.23,35 will be implemented to detect the potential presence of dissolved Au species and Au NPs in biological samples. For this purpose, liver and spleen tissue samples from Wistar rats will be analysed after intraperitoneal injection of solutions containing 10 nm Au NPs. Extractions procedures will be tested for the preservation of species integrity and its compatibility with the applied HPLC-ICP-MS methodology.
Experimental
Reagents and materials
The particles used in this work were 50 mg L−1 Au NPs solutions with 10 nm nominal particle diameter (according to the certificate: 8.9 ± 0.1 nm TEM), in a buffer containing sodium citrate as a stabilizer (RM 8011, NIST, Gaithersburg, USA). The Au standard for ICP (1000 mg L−1) was purchased from Sigma-Aldrich, Steinheim, Switzerland.Two Wistar rats were purchased from Charles River Laboratories (L'Arbresle, France) for the treatment with Au NPs and further tissue analysis.
Tetramethylammonium hydroxide (TMAH, Sigma-Aldrich), was used for the alkaline digestion of tissue samples of rats, while the enzymatic treatment was accomplished using proteinase K (Sigma-Aldrich, Saint Louis, MO, USA). The mobile phase consisted of 10 mmol L−1 ammonium acetate (>98%, Sigma-Aldrich) adjusted to pH 6.8 and 10 mmol L−1 sodium dodecylsulfate (SDS, 98.5%, Sigma-Aldrich). All solutions were prepared using high-purity MilliQ water (Millipore, Bedford, USA). The ICP-MS was operated with 99.999% Alphagaz Argon (Air Liquid, Valladolid, Spain).
Instruments
Separations were carried out on a Nucleosil C18 column (7 μm particle size, 1000 Å pore size, 250 × 4.6 mm, Phenomenex, Aschaffenburg, Germany) with an HPLC pump (LC-10AD, Shimadzu Corporation, Kyoto, Japan) and a 6-way injection valve (Rheodyne model 9125, Cotati, CA, USA). All connections and the injection loop (20 μL) were made of PEEK. 197Au+ detection was carried out with an inductively coupled plasma-mass spectrometer (Agilent 7700, Agilent Technologies, Waldbronn, Germany). Detailed information is summarized in Table 1.| ICP-MS | |
|---|---|
| Instrument | Agilent 7700x |
| RF power | 1500 W |
| Auxiliary gas flow | 0.87 L min−1 |
| Coolant gas flow | 15.5 L min−1 |
| Dwell time | 100 ms |
| Isotopes monitored | 197Au |
| Nebulizer | Meinhard-type |
| Spray chamber | Peltier-cooled Scott-type spray chamber |
| Nebulizer gas flow | 1.01 L min−1 |
| HPLC | |
|---|---|
| Instrument | Shimadzu LC-20AD |
| Column | Nucleosil C18, 1000 Å pore size, 250 × 4.6 mm |
| Flow rate | 0.5 mL min−1 |
| Injection volume | 20 μL |
| Mobile phase | SDS, ammonium acetate (both 10 mmol L−1) at pH 6.8 |
Methods
Animals and diets
Male Wistar rats weighing 190–220 g (Charles River Laboratories, L'Arbresle, France) were randomly divided and injected with the following solutions intraperitoneally: the first rat was injected with 1.66 mL MilliQ water per day as a control, and the second rat was injected with 1.66 mL of the 10 nm Au NPs solution per day. Both rats were fed with the semi-synthetic diet AIN93M and allowed free access to drinking water and food throughout the experimental period. The control rat consumed 22.0 ± 3.2 mL water per day and the rat with an injection of Au NPs 28.5 ± 4.4 mL water per day.On day 3, the rats were anaesthetised with a solution of ketamine (0.75 mg kg−1 body weight, Fatro Ibérica, Barcelona, Spain) and xylazine (0.10 mg kg−1 body weight, Fatro Ibérica, Barcelona, Spain), and exsanguinated by cannulating the posterior aorta. The liver and spleen were removed, weighed, placed in pre-weighed polyethylene vials, and stored at −80 °C.
From day 0 of the experiment, the animals were housed in individual metabolic cages. The cages were located in a well-ventilated, temperature-controlled room at 21 ± 2 °C with a relative humidity ranging from 40 to 60%, and a light/dark period of 12 h.
All experiments were carried out in accordance with Directional Guides Related to Animal Housing and Care36 and all procedures were approved by the Animal Experimentation Ethics Committee of the University of Granada.
TEM studies
For the studies about the fate and uptake of Au NPs into cells and tissues, a TEM LIBRA 120 PLUS microscope (Carl Zeiss SMT, Oberkochen, Germany) was used. In this sense, the tissues of the Wistar rats after injection of Au NPs solutions, as well as the control rat, were treated to allow their microscopic analysis following a modification of the method proposed by Glauert and Reid.38 Basically, liver samples were fixed with fresh primary fixative (1.5% glutaraldehyde, 1.0% formaldehyde in 0.05 mol L−1 sodium cacodylate buffer, pH 7.4) and post-fixed with a secondary fixative (1% osmium tetroxide, 1% potassium ferrocyanide in MilliQ water) followed by dehydration with an ascending series of alcohol solutions before embedding samples in epoxy resin. Ultrathin sections were cut and doubly stained with uranyl acetate and lead citrate. All samples were examined at 120 kV.Alkaline and enzymatic sample treatment for the speciation studies
Based on the work of Loeschner et al. with some modifications, two different sample treatments were tested: (i) an alkaline digestion with TMAH, and (ii) an enzymatic digestion with proteinase K.31 (i) To the freeze-dried and homogenized tissue samples (25 mg), 200 μL MilliQ water was added and the solutions were sonicated for 1 h. Then, TMAH was added to a final concentration of 5% (v/v) with a total volume of 2 mL. (ii) In the case of the enzymatic digestion, an enzyme solution containing 3 mg mL−1 of proteinase K, and 0.5% SDS in a 50 mmol L−1 ammonium bicarbonate buffer (pH 7.4) was prepared. The freeze-dried tissue (25 mg) was suspended in MilliQ water, 1.5 mL of the enzyme solution was added and filled with MilliQ water to a final volume of 2 mL. The mixture was finally agitated for 1 h and rotated mechanically at 37 °C overnight.Results and discussion
Optimization of the extraction procedure
The general objectives of an effective and efficient extraction of NPs from biological samples are the prevention of artefacts, e.g. NP degradation, and quantitative recoveries of the species in its original composition, form and size. A number of ICP-MS based approaches are described in the literature for the extraction of intact NPs from biological samples. As mentioned above, two different extractions are generally suggested: alkaline or enzymatic digestion procedures.28Our first objective was to test whether alkaline solubilization was also adaptable to the applied HPLC-ICP-MS method.23 Several experiments were carried out with rat tissues with and without addition of Au NPs. The resulting chromatograms showed an instability of the baseline, even after buffering the extract to the pH of the mobile phase. This effect impeded a correct integration of the ionic Au species peak. Additionally, during the storage of the extraction solution, a certain degradation of the particles was observed leading to a higher retention time shift (equivalent to smaller NP fractions). In conclusion, sample treatment with TMAH was found to be incompatible with the applied HPLC-ICP-MS method.
As an alternative, proteinase K was tested for the extraction of differently sized Au NPs from rat tissues followed by HPLC-ICP-MS analysis. Initial experiments were directed to observe the impact of the enzyme on the stability of Au NPs in an aqueous solution. Fig. 1a shows the obtained chromatograms for 10 nm Au NPs as function of the incubation time with proteinase K. In all cases, the NPs eluted as a symmetric peak at 4.7 min with repeatable peak areas (RSD < 6%). Apparently, the chosen conditions maintained intact NPs as ascertained by the fact that no low-molecular Au species were present (absence of peaks at a retention time of 5.7 min). As shown in Fig. 1b, the incubation of the same particles with proteinase K in the presence of a control liver tissue did not give any evidence of a significant decomposition and formation of ionic species during the first hours of treatment. After 5 h and 14 h incubation, minor contributions of ionic species were monitored. This observation might be the result of degradation processes occurring in the sample independent of the presence of proteinase K. Additionally, the presence of the complex matrix did have impact on the peak width and symmetry of the NP fraction. The peak areas for the incubation under these conditions remained relatively constant (RSD ∼ 4.5%) between 0.5 and 14 h. From these experiments, we concluded that the use of proteinase K overnight (14 h) was feasible for the extraction of the applied Au NPs without any remarkable artefact formation and degradation to the ionic Au species.
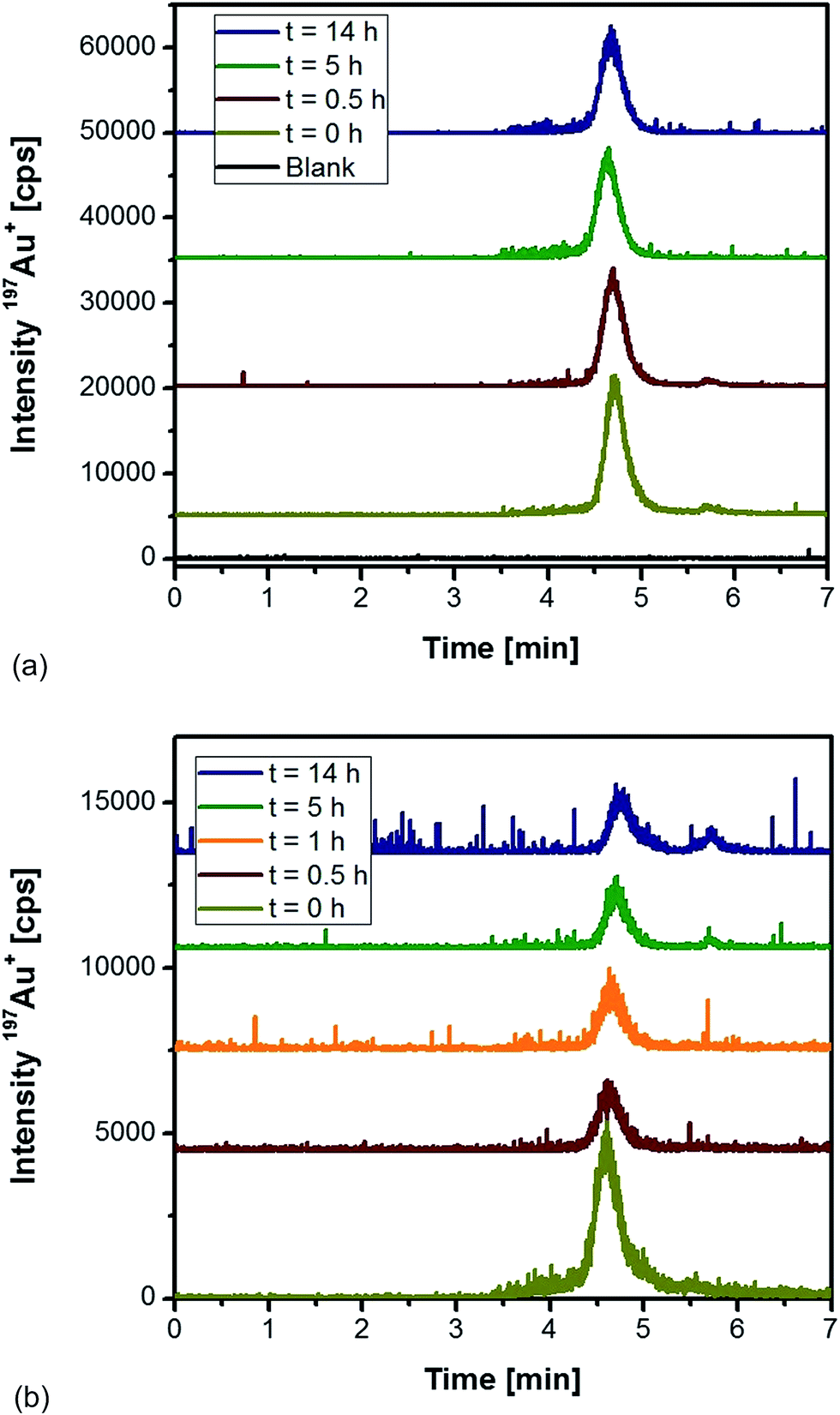 | ||
| Fig. 1 HPLC-ICP-MS chromatograms of 10 nm Au NPs as a function of the incubation time (a) with proteinase K, and (b) with proteinase K in the presence of a control rat liver tissue. | ||
Based on these initial observations, liver tissue from a Wistar rat treated intraperitoneally with 10 nm Au NPs was homogenized, and the extract was analysed under the same conditions. Fig. 2 represents the chromatograms of the extracted Au species as function of time. Within the first hour of extraction, a relatively broad and noisy signal for the nanoparticle fraction was observed. The peak width might originate from the formation of the “protein corona”38 that can significantly contribute to the hydrodynamic diameter of the NPs. Especially in the case of smaller Au NPs, the formation of the protein corona can significantly change the hydrodynamic diameter as observed in previous studies on Au NPs in human serum.39 Although denaturing conditions and a protease were applied during the extraction, the resulting peak broadening seemed to reveal the presence of remaining proteins on the surface of the NPs.21 Moreover, the observed signal noise during the elution of the NP fraction might indicate the presence of agglomerated particles. In any case, with increasing incubation time the nanoparticle fraction grew after 14 h by roughly a factor of three (peak areas) indicating a more efficient extraction. After 5 h incubation, the extraction conditions also permitted the observation of low-molecular, non-particulate bound Au species. These species had a maximum peak intensity after 14 h incubation. Taking into account that protease K did not degrade the Au NPs during the previous experiments, this observation indicated the release of these Au species from the NP intraperitoneally administered to the rats. Longer incubation times did not increase the extraction yield of the species, so that further experiments made use of a 14 h extraction period.
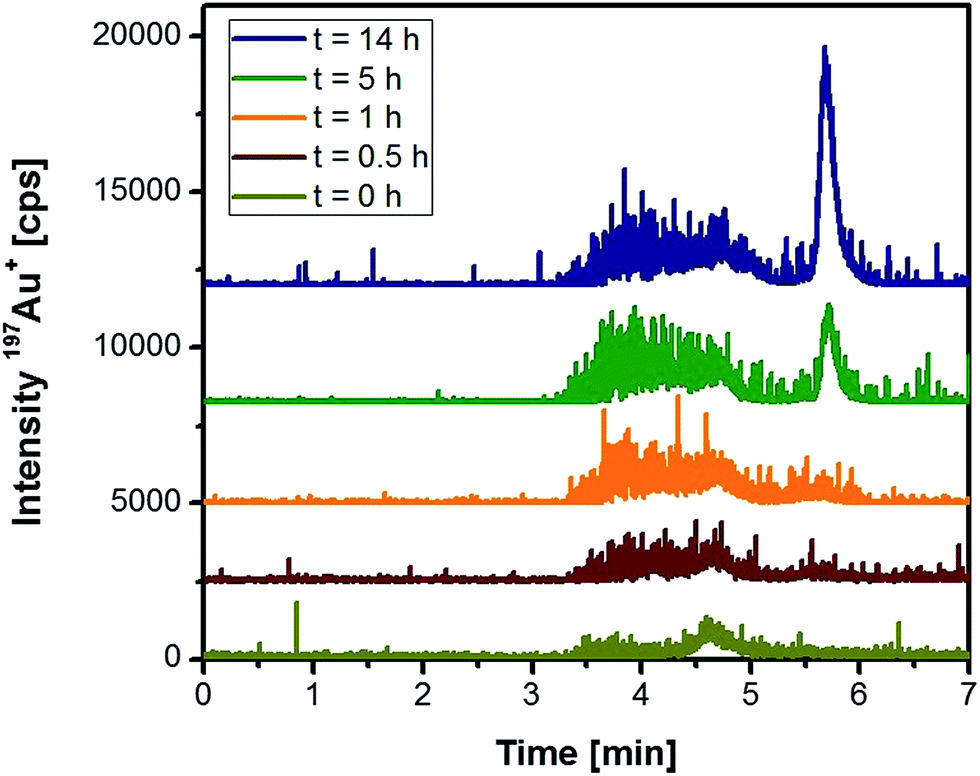 | ||
| Fig. 2 HPLC-ICP-MS chromatograms of rat liver tissue as a function of the incubation time with proteinase K (rats were injected 10 nm Au NPs intraperitoneally). | ||
In order to determine the recovery for 10 nm Au NPs and for Au3+, different concentrations (0, 0.1, 0.5 and 1 μg L−1 as Au) of these two species were added independently to a freeze-dried control rat liver tissue. These tissues were homogenised and extracted as described above. The obtained peak areas were compared with those observed for the standard solutions without any incubation in order to construct a recovery curve for Au concentrations between 0 and 1 μg L−1. The recovery rates were 91% ± 6% (N = 3) for both analytes (10 nm Au NPs and for Au3+). However, the artificial addition of the species could only mimic the actual circumstances in the tissue. The recoveries obtained in this work were quite higher than those reported by Loeschner et al. for 60 nm Au NPs (∼33%).31 According to the authors, inferior transport efficiency of the Au NPs was responsible for these low values in their single-particle ICP-MS experiments. In this work, however, the NPs were chromatographically separated under denaturing conditions and the mobile phase was basically the medium for introducing them into the plasma source. Potential transport or nebulization interferences induced by proteinase K might, therefore, be reduced.
Localization of Au NPs and speciation of Au in rat tissues
For the evaluation of the fate and the uptake of the nanoparticles into the rat liver tissue, a transmission electron microscope (TEM) at 120 kV was used. The image in Fig. 3a shows a hepatocyte sample of a control sample, and Fig. 3b illustrates a hepatocyte sample after intraperitoneal injection of 10 nm Au NPs. As reported also by other groups,40–42 the nanoparticles were present and isolated in vesicles, probably after entering the cells by endocytosis.43 The observed diameters were around 8 ± 1 nm, which were slightly smaller, but very close to the diameter of the NPs injected (8.9 ± 0.1 nm). In order to prove whether the particles underwent a partial degradation during the transport and/or cell uptake, the extracts of the liver samples were subject to HPLC-ICP-MS analysis.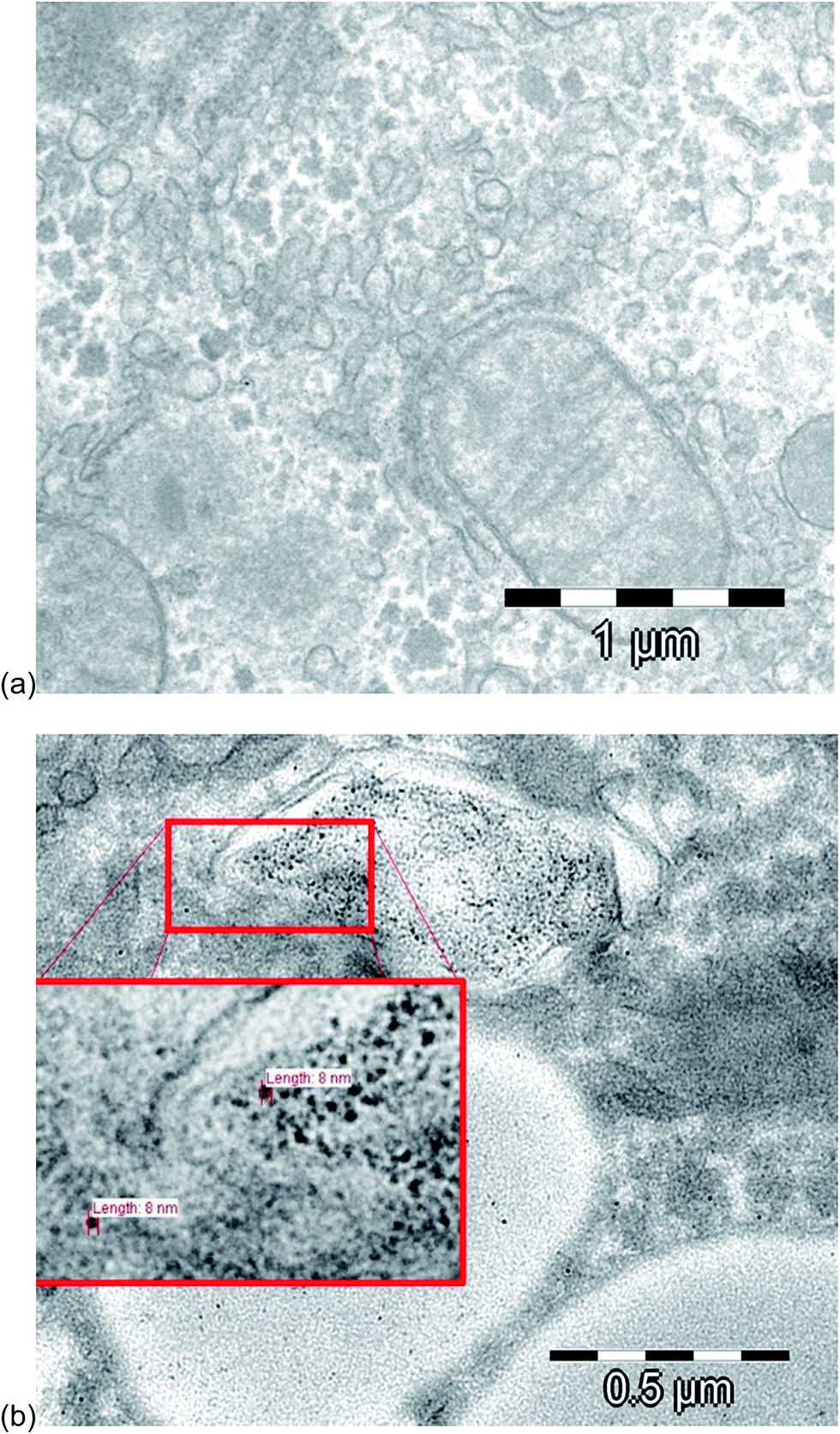 | ||
| Fig. 3 TEM images of (a) a control hepatocyte sample and of (b) a hepatocyte sample after intraperitoneal injection of 10 nm Au NPs. | ||
Fig. 4a represents the chromatogram obtained from the analysis of liver sample. The distribution of different Au species was very similar to the ones observed during the optimization of the extraction procedure. A broad signal between 3.5 and 5 min corresponded to the nanoparticle fraction as discussed above, but an additional signal appeared at 5.7 min related to the occurrence of low-molecular Au species. This observation proved that the Au NPs suffered a kind of degradation, possibly as a consequence of a digestion process in vivo. Using TEM as a unique analytical technique, such a process of deliberating metal ions from the NPs – in general – is not detectable. Here, a significant advantage can be demonstrated for using the proposed HPLC-ICP-MS approach as a complementary tool as it allowed the detection of released non-particulate bound Au species.
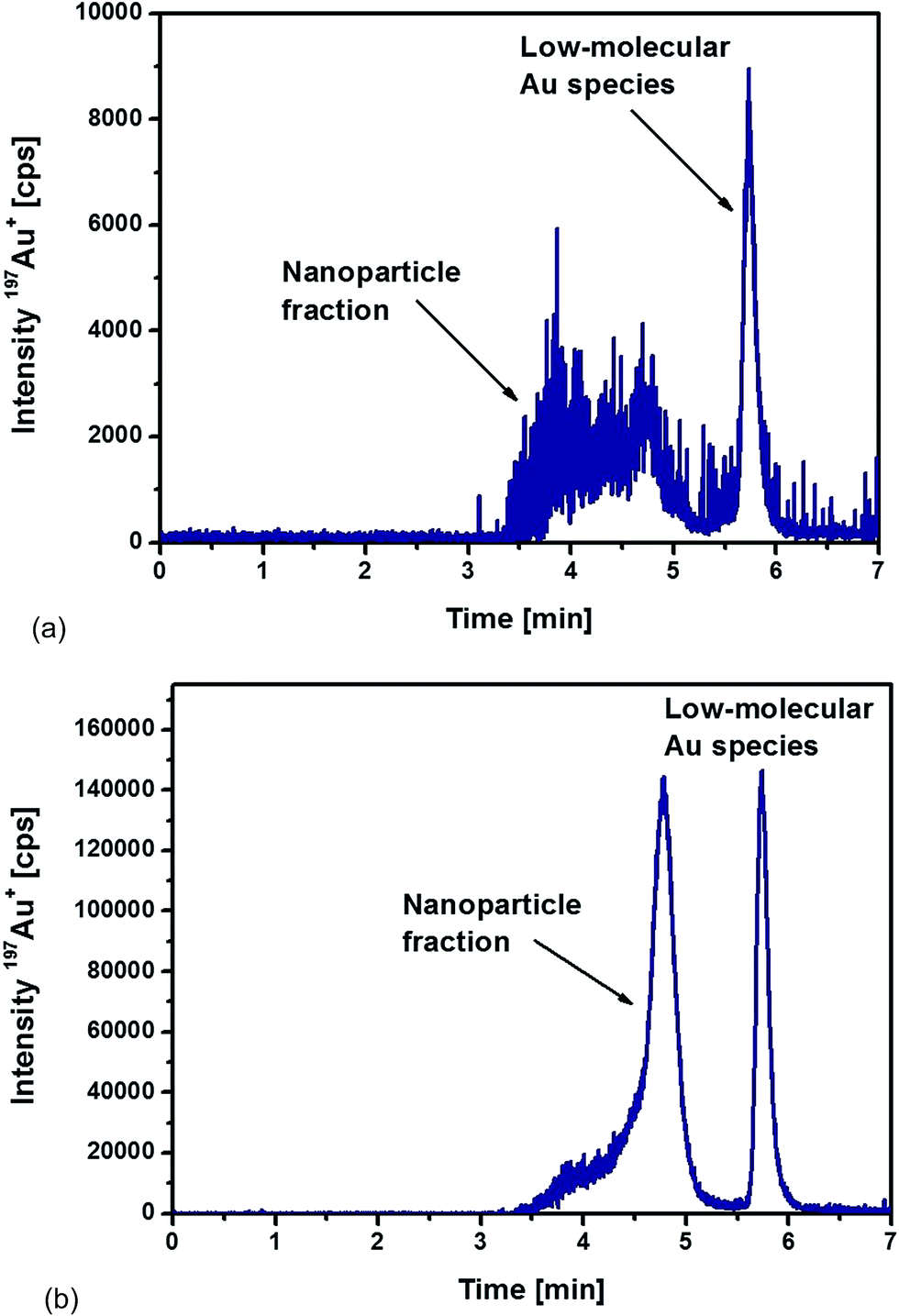 | ||
| Fig. 4 HPLC-ICPMS chromatograms of (a) liver and (b) spleen samples digested with proteinase K overnight from a rat injected intraperitoneally with 10 nm Au NPs. | ||
In the case of the analysed spleen samples, a representative chromatogram is shown in Fig. 4b. Again, a significant contribution of low-molecular Au species appeared. However, the release mechanism of these Au species from NPs remains unknown and will require further investigation. In comparison to the results obtained from the analysis of the liver samples, two significant differences could be observed for the spleen samples. Whereas the detected nanoparticle fraction in the liver samples was relatively broad, the spleen samples revealed a more homogeneous size distribution of the Au NPs. Fig. 4b demonstrates that the most abundant fraction, eluted at a retention time of 4.8 min, corresponded to a particle size of about 10 nm. The peak fronting indicated, again, that part of the particles remained with associated compounds, probably proteins that were not completely degraded during the proteolysis.
The second observation was the significantly higher signal intensities for the spleen sample. Total Au concentrations were determined after acidic digestion of the samples followed by ICP-MS analysis with internal standardisation. The determined Au concentrations were 2.6 ± 0.1 μg g−1 dry weight in the liver sample and 46.3 ± 2.0 μg g−1 dry weight in the spleen sample (total injected amount of Au as 10 nm NPs: 250 μg). The significant difference between the liver and spleen sample was confirmed by the chromatograms recorded of the corresponding extracts. Relative concentrations of each fraction were determined by the use of external calibrations (standard solutions of ionic Au and 10 nm Au NPs). Total Au was distributed as follows among the NP fraction and the low-molecular size (LM) fraction (% Au): liver (64% NP fraction, 36% LM fraction) and spleen (72% NP fraction, 28% LM fraction). In both tissues, roughly 30% was present as low-molecular Au species. This observation revealed that the partial dissolution and degradation of the Au NPs played a significant role during their transport and accumulation. The degradation mechanism and the significance of this effect remains unclear during these experiments and will require further investigations in the future.
Conclusions
The differentiation between Au NPs and low-molecular size Au species and their detection in biological samples was aimed at by a suitable extraction procedure followed by an HPLC-ICP-MS method. Proteinase K, for the deliberation of the sought species from Wistar rat tissues, proved to be a suitable candidate with good recoveries of about 91% after 14 h incubation. The procedure was applied to the analysis of liver (total Au: 2.6 μg g−1 dry weight) and spleen tissues (total Au: 46.3 μg g−1 dry weight) of Wistar rats after previous intraperitoneal injection of 10 nm Au NPs (total Au injected: 250 μg). The resulting chromatograms showed about 30% of the total Au as low-molecular sized Au species in both tissues. This indicated that the degradation of the Au NPs was clearly identified as an important process in the transport and/or accumulation of the used NPs. The diminished NP diameter observed during the TEM experiments confirmed this suggestion. In conclusion, the developed method can be generally considered as a useful tool for monitoring the degradation of Au NPs in biological samples, especially for smaller NPs (e.g. 10 nm Au NPs).Future work will have to be directed at exploring the mechanisms leading to the degradation of Au NPs in biological systems in detail. Furthermore, adaptation of the developed methodology to other nanoparticle systems, like silver-containing nanomaterials, is planned.
Acknowledgements
The authors gratefully acknowledge the financial support from the Spanish MICINN (Spanish ministry for science and innovation, Grant Number CTQ2011-23038) and MECD (Spanish ministry for education culture and sports, Grant Number FPU13/00062). Some of the presented results are included in the PhD thesis of Carlos López Chaves from the University of Granada, Nutrition and Food Sciences doctoral program.References
- Y. D. Han, Y. M. Park, H. J. Chun and H. C. Yoon, Sens. Actuators, B, 2015, 220, 233–242 CrossRef CAS.
- L. Chen, S. Wang, C. Han, Y. Cheng and L. Qian, Synth. Met., 2015, 209, 544–548 CrossRef CAS.
- R. Nita, S. A. Trammell, G. A. Ellis, M. H. Moore, C. M. Soto, D. H. Leary, J. Fontana, S. F. Talebzadeh and D. A. Knight, Chemosphere, 2016, 144, 1916–1919 CrossRef CAS PubMed.
- W. Ngwa, R. Kumar, S. Sridhar, H. Korideck, P. Zygmanski, R. A. Cormack, R. Berbeco and G. M. Makrigiorgos, Nanomedicine, 2014, 9, 1063–1082 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. Kim and V. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- M. Yamada, M. Foote and T. W. Prow, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 428–445 CrossRef CAS PubMed.
- O. S. Muddineti, B. Ghosh and S. Biswas, Int. J. Pharm., 2015, 484, 252–267 CrossRef CAS PubMed.
- Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann, G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076 CrossRef CAS PubMed.
- J. J. Li, D. Hartono, C.-N. Ong, B.-H. Bay and L.-Y. L. Yung, Biomaterials, 2010, 31, 5996–6003 CrossRef CAS PubMed.
- W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. A. M. Sips and R. E. Geertsma, Biomaterials, 2008, 29, 1912–1919 CrossRef CAS PubMed.
- C. Lasagna-Reeves, D. Gonzalez-Romero, M. A. Barria, I. Olmedo, A. Clos, V. M. Sadagopa Ramanujam, A. Urayama, L. Vergara, M. J. Kogan and C. Soto, Biochem. Biophys. Res. Commun., 2010, 393, 649–655 CrossRef CAS PubMed.
- M. A. K. Abdelhalim and B. M. Jarrar, Lipids Health Dis., 2011, 10, 133 CrossRef CAS PubMed.
- J. H. Hwang, S. J. Kim, Y.-H. Kim, J.-R. Noh, G.-T. Gang, B. H. Chung, N. W. Song and C.-H. Lee, Toxicology, 2012, 294, 27–35 CrossRef CAS PubMed.
- E. Cardoso, E. Londero, G. K. Ferreira, G. T. Rezin, E. T. Zanoni, F. de Souza Notoya, D. D. Leffa, A. P. Damiani, F. Daumann, P. Rohr, L. da Silva, V. M. Andrade and M. M. da Silva Paula, J. Nanopart. Res., 2014, 16, 2727 CrossRef.
- Y. Li, Y. Gao and C. Chen, Metallomics, 2014, 6, 220–232 RSC.
- F. Benetti, L. Bregoli, I. Olivato and E. Sabbioni, Metallomics, 2014, 6, 729–747 RSC.
- S. Sharifi, S. Behzadi, S. Laurent, M. Laird Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC.
- B. Schmidt, K. Loeschner, N. Hadrup, A. Mortensen, J. J. Sloth, C. Bender Koch and E. H. Larsen, Anal. Chem., 2011, 83, 2461–2468 CrossRef CAS PubMed.
- A. R. Poda, A. J. Bednar, A. J. Kennedy, A. Harmon, M. Hull, D. M. Mitrano, J. F. Ranville and J. Steevens, J. Chromatogr. A, 2011, 1218, 4219–4225 CrossRef CAS PubMed.
- K. Tiede, A. B. A. Boxall, D. Tiede, S. P. Tear, H. David and J. Lewis, J. Anal. At. Spectrom., 2009, 24, 964–972 RSC.
- J. Soto-Alvaredo, M. Montes-Bayón and J. Bettmer, Anal. Chem., 2013, 85, 1316–1321 CrossRef CAS PubMed.
- X.-X. Zhou, R. Liu and J. F. Liu, Environ. Sci. Technol., 2014, 48, 14516–14524 CrossRef CAS PubMed.
- A. Helfrich, W. Brüchert and J. Bettmer, J. Anal. At. Spectrom., 2006, 21, 431–434 RSC.
- B. Franze and C. Engelhard, Anal. Chem., 2014, 86, 5713–5720 CrossRef CAS PubMed.
- H. Qu, T. K. Mudalige and S. W. Linder, Anal. Chem., 2014, 86, 11620–11627 CrossRef CAS PubMed.
- C. Degueldre and P.-Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- C. Degueldre, P.-Y. Favarger and S. Wold, Anal. Chim. Acta, 2006, 555, 263–268 CrossRef CAS.
- C.-K. Su and Y.-C. Sun, J. Anal. At. Spectrom., 2015, 30, 1689–1705 RSC.
- F. Laborda, E. Bolea, G. Cepriá, M. T. Gòmez, M. S. Jiménez, J. Pérez-Arantegui and J. R. Castillo, Anal. Chim. Acta, 2016, 904, 10–32 CrossRef CAS PubMed.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Environ. Sci. Technol., 2013, 47, 14315–14323 CrossRef CAS PubMed.
- K. Loeschner, M. S. J. Brabrand, J. J. Sloth and E. H. Larsen, Anal. Bioanal. Chem., 2014, 406, 3845–3851 CrossRef CAS PubMed.
- Y. Dan, W. Zhang, R. Xue, X. Ma, C. Stephan and H. Shi, Environ. Sci. Technol., 2015, 49, 3007–3014 CrossRef CAS PubMed.
- D. Bao, Z. G. Oh and Z. Chen, Front. Plant Sci., 2016, 7, 32 Search PubMed.
- K. Loeschner, J. Navratilova, C. Købler, K. Mølhave, S. Wagner, F. Von der Kammer and E. H. Larsen, Anal. Bioanal. Chem., 2013, 405, 8185–8195 CrossRef CAS PubMed.
- A. Helfrich and J. Bettmer, Int. J. Mass Spectrom., 2011, 307, 92–98 CrossRef CAS.
- European Union, Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals used for Scientific Purposes, Official Journal of the European Union, 2010, pp. 33–79 Search PubMed.
- A. M. Glauert and N. Reid, Fixation, Dehydration and Embedding of Biological Specimens, Elsevier, New York, 1975 Search PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- N. Fernández-Iglesias and J. Bettmer, Nanoscale, 2015, 7, 14324–14331 RSC.
- J. J. Li, L. Zou, D. Hartono, C. N. Ong, B. H. Bay and L. Y. Lanry Yung, Adv. Mater., 2008, 20, 138–142 CrossRef CAS.
- B. C. Nelson, E. J. Petersen, B. J. Marquis, D. H. Atha, J. T. Elliott, D. Cleveland, S. S. Watson, I.-H. Tseng, A. Dillon, M. Theodore and J. Jackman, Nanotoxicology, 2011, 7, 21–29 CrossRef PubMed.
- J. Soto-Alvaredo, E. Blanco, J. Bettmer, D. Hevia, R. M. Sainz, C. López Cháves, C. Sánchez, J. Llopis, A. Sanz-Medel and M. Montes-Bayón, Metallomics, 2014, 6, 1702–1708 RSC.
- N. Oh and J. H. Park, Int. J. Nanomed., 2014, 9(S1), 51–63 Search PubMed.
Footnote |
| † These authors contributed equally in this work. |
| This journal is © The Royal Society of Chemistry 2017 |