
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
T cell immunoengineering with advanced biomaterials
Derfogail
Delcassian
 *ab,
Susanne
Sattler
c and
Iain E.
Dunlop
*d
*ab,
Susanne
Sattler
c and
Iain E.
Dunlop
*d
aSchool of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: derfogail.delcassian@nottingham.ac.uk
bKoch Institute for Integrative Cancer Research, MIT, Massachusetts, 02139, USA
cImperial College London National Heart and Lung Institute, Du Cane Road, W12 0NN, London, UK
dDepartment of Materials, Imperial College London, SW7 2AZ, UK. E-mail: i.dunlop@imperial.ac.uk
First published on 20th February 2017
Abstract
Recent advances in biomaterials design offer the potential to actively control immune cell activation and behaviour. Many human diseases, such as infections, cancer, and autoimmune disorders, are partly mediated by inappropriate or insufficient activation of the immune system. T cells play a central role in the host immune response to these diseases, and so constitute a promising cell type for manipulation. In vivo, T cells are stimulated by antigen presenting cells (APC), therefore to design immunoengineering biomaterials that control T cell behaviour, artificial interfaces that mimic the natural APC-T cell interaction are required. This review draws together research in the design and fabrication of such biomaterial interfaces, and highlights efforts to elucidate key parameters in T cell activation, such as substrate mechanical properties and spatial organization of receptors, illustrating how they can be manipulated by bioengineering approaches to alter T cell function.
 Derfogail Delcassian | Derfogail Delcassian is an EPSRC E-TERM Landscape Fellow whose work centres on immunoengineering. Currently, she is based at both the Division of Regenerative Medicine and Cellular Therapies, University of Nottingham and the Koch Institute, MIT. Her research focuses on the interface of biomaterials and immunology, designing novel biomaterials that can interact with the immune system to re-direct cellular behaviour. Her current interests are in developing biomaterial based therapeutics for cancer, auto-immune disorders and tissue engineering applications. She completed her PhD at Imperial College London, before undertaking an EPSRC Doctoral Prize Fellowship and her current E-TERM Fellowship. |
 Susanne Sattler | Susanne Sattler's main research interest is cardiovascular immunology and immuno-modulation to improve heart regeneration. She obtained her PhD from the Department of Vascular Biology and Thrombosis Research at the Medical University Vienna, Austria in 2009 and then moved to Imperial College London for postdoctoral research on regulatory T- and B-cells in inflammatory disease. She then joined the National Heart and Lung Institute and currently leads a British Heart Foundation project investigating the adaptive immune response after cardiac tissue damage and how immune-modulatory strategies may improve the outcome of regenerative therapies in heart disease. |
 Iain E. Dunlop | Iain E. Dunlop is a Senior Lecturer in Biomaterials at the Department of Materials, Imperial College London. Previously he was an Alexander von Humboldt postdoctoral fellow at the Department of New Materials and Biosystems, Max Planck Institute for Metals Research in Stuttgart, Germany. He received his doctorate in soft matter from the Department of Chemistry, University of Oxford, UK. His current research is on bionanotechnology and cellular biophysics, with a particular focus on the immune system. |
Insight, innovation, integrationControlled immune cell activation is emerging as a powerful strategy in a range of clinical therapies. This review describes current progress in designing biomimetic structures that direct T cell behaviour. This new field integrates fundamental research in immune cell biophysics, including receptor nanopatterning and mechanotransduction, together with applied biomaterial design. We comprehensively summarize the current state of the field and advocate a future direction for innovative immunomodulatory biomaterials through the integration of spatial, temporal and mechanical cellular cues in 2- and 3-dimensional therapeutics. |
Introduction
Traditionally, biomaterials have been deliberately designed to minimize interactions with the host immune system when implanted. The desire for “immune inert” materials that avoid in vivo immune interactions, such as inflammation,1 fibrosis and encapsulation2,3 and ultimately rejection4–6 has been a major focus of tissue engineering over the last few decades. In contrast, there is a recent growth of interest in materials that achieve the opposite effect; directly influencing immune cell behaviour to control immune cell growth and differentiation, regulate inflammation or induce tolerance; effectively developing engineered materials that can direct immune cell behaviour.7–9The development of such immunoengineering biomaterials is still a nascent field; however its rise has coincided with the rapid development of biotechnology techniques that direct the immune response. Prominent examples of this technology are genetically engineered Chimeric Antigen Receptor (CAR) T cells which efficiently target leukemic cancers,10–12 and nanoparticle drug delivery strategies for targeted immune cell vaccination.13–16 These fields promise the design of advanced T cell therapeutics that can deliver appropriate stimuli to T cells to tackle a range of disease targets. Several key parameters controlling T cell activation and behaviour have been identified, and the design of biomaterials capable of manipulating these features presents a new approach to directing T cell behaviour.
In vivo, T cells are exposed to distinct stimuli that influence their behaviour, shown schematically in Fig. 1A. These stimuli, or “cellular cues,” can be loosely classified as physical (including the topographical, geometrical and mechanical nature of the cell–substrate interface) or biochemical (including engagement of cell surface receptors and response to soluble factors). To avoid aberrant immune responses under physiological conditions, in vivo T cell activation is a tightly controlled process, depending on Antigen Presenting Cells (APC) providing antigen-specific signals (signal 1) and co-stimulation (signal 2), alongside the stimulation provided by the surrounding cytokine milieu (signal 3).17
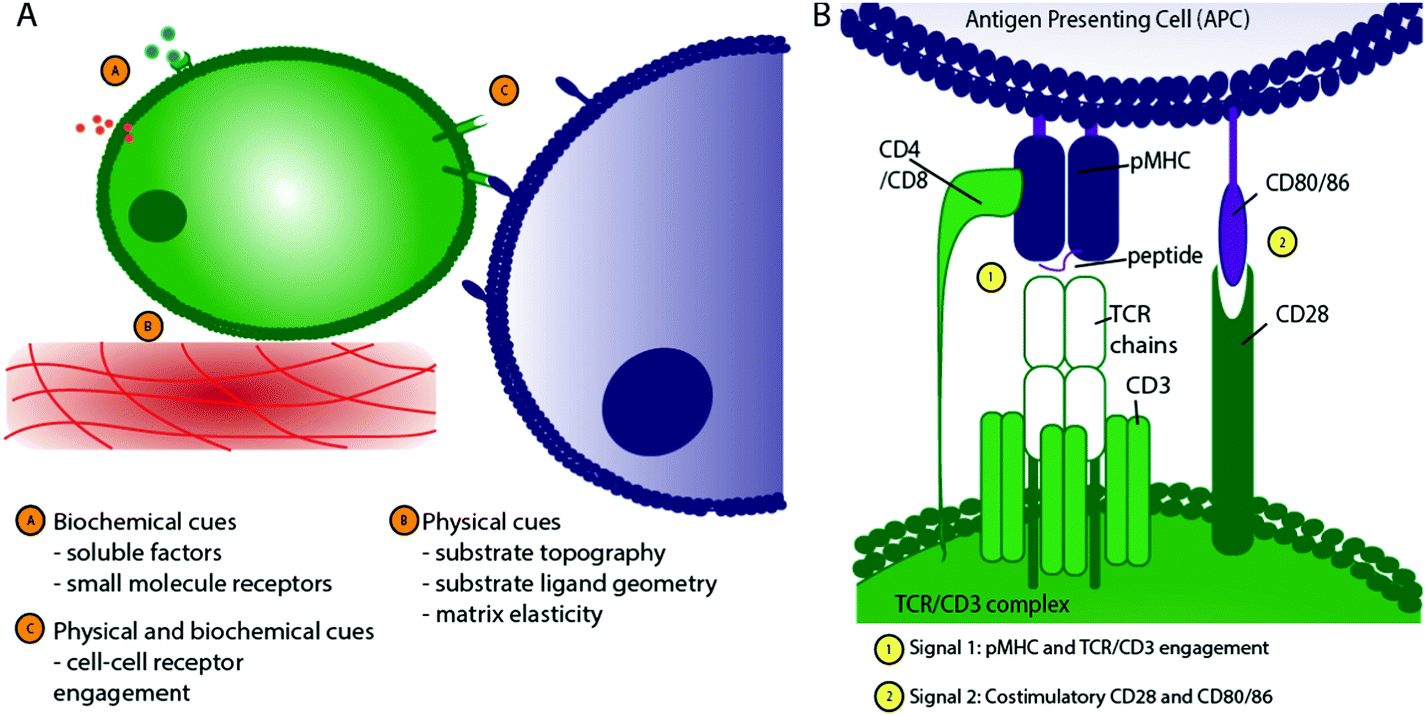 | ||
| Fig. 1 Schematic of T cell interactions. (A) T cells experience a range of stimuli in vivo that can be broadly classified into biochemical and physical cues. (B) The TCR/CD3 complex is central to T cell activation, requiring engagement of peptide-bearing MHC (pMHC) with the TCR/CD3 complex, CD4 on the T cell surface with pMHC on the APC cell surface, and stimulation through CD28 binding to CD80/CD86. | ||
Of particular importance to appropriate T cell activation is the formation of a closely-adhered immunological synapse between the T cell and APC (Fig. 2B and C) where a large number of ligand–receptor pairs combine to form a complex structure18,19 that determines the T cell's activation and downstream differentiation. Peptide-bearing Major Histocompatibility Complex (pMHC) presented by APCs engages with the T cell receptor (TCR) on T cells to create an initial activation signal. Secondly, a co-stimulatory ligand–receptor pair, such as surface receptors CD28/CD80, is engaged to support full activation, as shown in Fig. 1B.
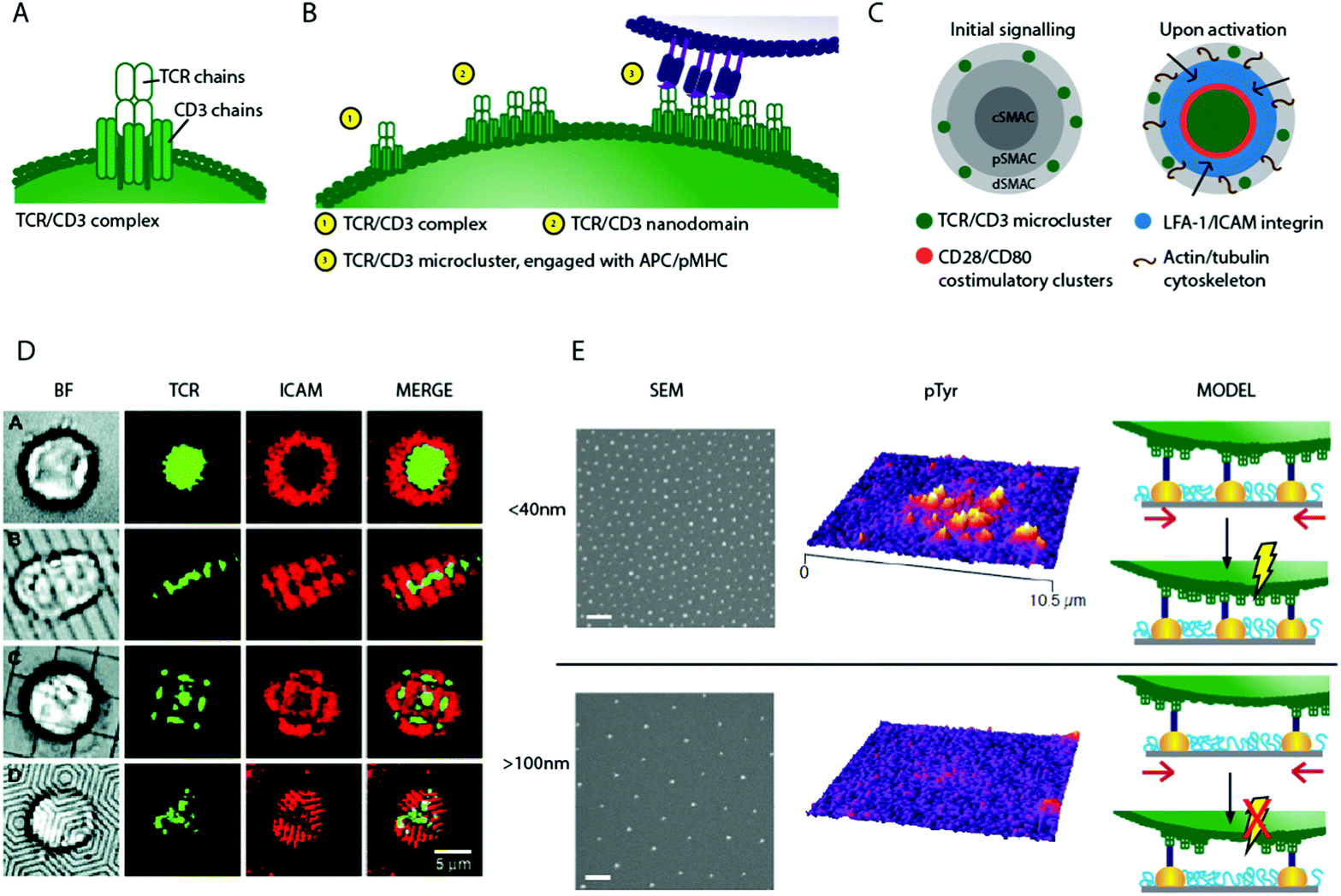 | ||
| Fig. 2 Schematic of T cell stimulation and 2D biomaterials interfaces controlling TCR movement. (A) Schematic of a single TCR/CD3 complex (B) a schematic of hypothetical micro-cluster formation; individual TCR/CD3 complexes associate to form nano-domains, which pre-exist on the T cell surface. On engagement with APC, TCR/CD3 micro-clusters form (C) a schematic of activation, where TCR/CD3 micro-clusters move from the periphery to a central Super Molecular Activation Cluster (c-SMAC), and become surrounded by rings of costimulatory and adhesion molecules. (D) Using chromium barriers on fluid lipid bilayers restricts micro-cluster formation and so synapse structure46 (E) nanopatterns fabricated using block copolymer micellar lithography present anchoring surfaces for TCR/CD3 ligands with precise inter-ligand spacing (scale bar 100 nm) such surfaces show dramatic differences in their ability to activate T cells. This may be due to bound nanoclusters being spaced too far apart on surfaces with large inter-ligand density, with nanoclusters unable to concatenate if pinned to static nanoparticles, preventing TCR/CD3 micro-cluster formation and so signalling, shown schematically (altered from ref. 39). Copyright: (D) adapted from ref. 46 Mossman et al., Science, 2005, 310(5751), 1191–1193. Reprinted with permission from AAAS; (E) adapted with permission from ref. 39 Delcassian et al. Nano Lett., 2013, 13(11), 5608–5614, Copyright 2013 American Chemical Society. | ||
Additional signals, such as those provided by the cytokine milieu, provide the third signal to allow effector T cells to expand and exhibit their full function.5,20 This tightly regulated stimulation and co-stimulation is crucial to maintaining healthy T cell function. Without efficient co-stimulation, antigen presentation can lead to T cell anergy, while inappropriate response to one or more of these signals may cause hyperinflammatory conditions, such as allergy or autoimmunity.21 Signal 1, where the T Cell Receptor (TCR)/CD3 complex recognises the pMHC on APCs, is central in initiating T cell activation. Alternatively, in the absence of pMHC, anti-CD3 can be used to engage the TCR/CD3 complex and can provide a non-antigen specific stimulus as signal 1. Most research into modulation of T cell function by biomaterials has focused on TCR- or CD3-mediated activation.22–24 The precise physical nature of pMHC presentation to the TCR is critical for T cell activation; factors including agonist valency,25–29 the mechanical properties of the activating surface30,31 and the spatial organisation of stimulatory ligands32–37 have been identified as key features modulating T cell function in response to stimulation.
Recently, a number of research groups have attempted to explore the ability of these variables to control cellular behaviour, using advanced biomaterials that replicate specific properties of the APC surface. Studies have explored a wide range of parameters, including spatial structuring by both photolithography and self-assembly methods, and the use of bio-functionalized hydrogels and rubbers to vary mechanical properties. Here, we consider materials that are designed to directly explore these parameters, and their effects on T cells. We will examine both 2D and 3D biomaterials, and consider them in order of increasing complexity. Specifically, we will focus first on materials that present a static surface to the cell, secondly on those that are responsive (interfaces the cell can manipulate or rearrange) and finally we discuss the move towards substrates that can dynamically manipulate cellular behaviour in response to external stimuli, summarized in Table 1. This focused review draws together these experiments to provide a new perspective on the design of immunomodulatory materials, and summarizes work completed so far in the move towards “immune directing” biomaterials for the control of T cell function.
Biomaterials for T cell activation
2-Dimensional interfaces
The region of contact between an immune cell and an antigen presenting cell (APC) is termed the immunological synapse. Usually this interface includes known activatory, inhibitory or adhesion molecules arranged in an organised manner.19,53–55In vitro, this interaction can be replicated by creating advanced biomaterials that replicate specific properties of the APC surface, functionalized with ligands that bind specifically to known receptors on the immune cell surface. These materials can be used to form an artificial model of the immune synapse that can engage with cells. Here, we review artificial interfaces that explore the spatial and physical presentation of T cell ligands in 2D.The movement of protein and lipid molecules across the cell surface into central c-, peripheral p- and distal d-rings surrounding a central feature point is a key step in T cell activation mediated by the intracellular cytoskeleton.59–65 Early artificial immune synapses used fluid lipid bilayers to explore the formation and movement of these dynamic protein domains.33,66 Fluid lipid bilayer substrates are coated with phospholipid molecules which mimic the natural bilayer found in the cell membrane, and can be loaded with proteins that can engage with cell surface receptors. On activation, the cell may rearrange proteins expressed on the cell surface into activatory clusters, causing the proteins embedded in the fluid bilayer, and those engaged with receptors on the cell surface, to be redistributed. This allows the cell to re-arrange the proteins embedded in the fluid bilayer interface. To explore activation through the TCR, pMHC molecules specific to the cognate TCR can be introduced into the lipid bilayer. Alternatively, anti-CD3, which does not require a specific match to bind to the TCR/CD3 complex, can be used. By using these TCR agonists in conjunction with advanced microscopy techniques, the formation and movement of individual TCRs and TCR protein micro-clusters during cellular activation have been explored.
These studies showed that during T cell activation, TCR/CD3 micro-clusters form in the p-SMAC and are transported to the c-SMAC,60,67 as illustrated in Fig. 2C. The movement of TCR clusters across the cell surface to the c-SMAC on activation is now known to propagate and aid sustained signalling in T cells, and is thought to be mediated by F-actin61,68 and myosin II.69 By adding nanoscale chromium barriers to the fluid bilayer, more advanced substrates prevented the movement of TCRs in certain directions (Fig. 2D). Through directing and constraining the TCR micro-cluster mobility, signalling in the cell can be controlled; signalling is maximised if TCR micro-clusters are constrained to the p-SMAC using these devices.46,70 In an alternative approach, synthetic polymer chains which present anti-CD3 and anti-CD28 at specific sites on a semi-flexible poly(isocyano peptide) polymer chain have been used to activate T cells. The polymers present multiple copies of the antibodies on a single chain, and so enable multivalent receptor interactions. These are thought to lead to surface receptor rearrangements and activation of the T cell.71
Intriguingly, T cells display distinct patterns of micro-cluster proteins both before and during activation.72,73 These patterns typically demonstrate a TCR/CD3 enriched protein domain in the c-SMAC surrounded by a ring dominated by adhesion receptor LFA-1. The TCR/CD3 domains may display a bulls-eye or multi-focular pattern within the LFA-1 domain, depending on cell phenotype, maturity and activation status.32,36,74–76
To explore the importance of these micro-features, and the ways in which changes in protein distribution can control cellular activation, planar substrates with a range of protein micropatterns have been fabricated by microcontact printing. In one example, anti-CD3 was micropatterned in a focal or annular patch, and the activation response of T cells on these substrates measured by cytokine secretion. Much higher secretion of the cytokine IFN-γ occurred on patterns with focal anti-CD3 protein organisation compared to annular rings, even when the amount of antigen presented was the same. The inclusion of co-stimulatory agents such as anti-CD28 augmented activation, however positioning anti-CD3 and anti-CD28 in separate distinct focal points (rather than a singular focal point patterned with both stimuli) resulted in much higher IL-2 production.47 Separately, recent work has shown that regulatory T cells (Treg) display a reduced sensitivity to micropatterns in comparison to effector T cells,76 whilst functionally distinct subsets of CD4+ effector T cells (Th1 and Th2) form distinct patterns in their immunological synapse with APCs.74,75
Clearly, T cell activation is partly driven by spatial TCR micro-cluster distribution and movement; thus a logical next step explored whether controlling the dynamics of formation of these TCR micro-clusters could control T cell behaviour. Many studies indicate that smaller TCR nano-domains exist prior to ligand engagement, and that these nano-domains may play a significant role in the process of micro-cluster formation. Super-resolution microscopy has been used to explore the size of these pre-clustered TCR nano-domains on the surface of T cells; individual TCR complexes (10–20 nm in size) have been recorded24 and TCR nano-domains of 35–70 nm in size have been reported on non-activated T cells.56,60,77–80 On activation, these nano-domains are hypothesized to associate, transitioning from individual nanoscale units to larger structures, as illustrated in Fig. 2B.81 Indeed, such pre-clustering of TCR complexes may facilitate T cell activation by easing micro-cluster formation, and similar pre-clustering is also seen in other immune cell types.79,80,82–86
To explore the relationship between individual TCR receptors, potential pre-clustered nano-domains and micro-clusters, several research groups developed nano-interfaces fabricated using the self-assembly-based block copolymer micellar lithography technique. This allowed the precise positioning of gold nanoparticles (Fig. 2E) with controllable inter-particle distance that were subsequently functionalised with TCR agonists such as anti-CD3 or pMHC. Using this fabrication technique, we and others showed that early stage activation in human and mouse CD4+ T cells was a function of inter-particle spacing.39–42 As inter-particle spacing was increased from 25 to 100 nm, activation decreased, and above an inter-ligand threshold of approximately 100–150 nm, T cells were unable to activate despite the presence of stimulatory receptors. These observations are in line with the biological observation that triggering full T cell activation requires activation signals to exceed a certain threshold, in which TCR ligand density plays a key role.25,26,87,88 There are several potential explanations for this observation; primarily we consider a minimum density of receptor engagement, receptor cluster concatenation and/or minimum force requirements to be likely candidate models.
Fig. 2E illustrates one hypothesis; that pre-clustered TCR nano-domains are required to concatenate before micro-cluster formation, transport and sustained signalling can occur. If receptor nano-domains are bound too far apart, they are unable to concatenate and induce signalling. This is supported by the observed spacing activation threshold of ∼70 nm, corresponding well to the nano-domain length scale of 35–70 nm as measured by Lillemeier et al.81 Further supporting the role of clustering in cellular activation, interfaces fabricated using a single walled carbon nanotube composite have been used to activate T cells. Nanotubes coated with anti-CD3 protein and embedded in a polymer matrix enhanced T cell activation compared to cells stimulated using soluble agonist at the same concentration.89–91 The induction of efficient T cell proliferation for immunological assays has long relied on coating anti-CD3 and anti-CD28 onto standard tissue culture plates or onto beads to induce TCR crosslinking, as soluble antibodies do not induce efficient activation, suggesting that pre-clustering antibodies enhances stimulation.
A separate hypothesis considers engagement of a minimum number of receptors within a particular sub-cellular location, which may be required to allow signalling and stimulation to overcome the intrinsic activation threshold in immune cells necessary to protect from aberrant activation.92,93 Although receptor–receptor proximity, and even the proximity of several different receptor types to one another, clearly plays an important role in controlling cellular activation, such spatial effects are intrinsically coupled to receptor and cytoskeletal mechanics. In positioning receptor ligands precisely, nano- and micro-patterned interfaces also alter the position and number of traction anchoring points for the cell. This means that the observed effects of nanoscale ligand patterning could arise directly from the mechanical activation of the TCR, hinting that T cell stimulation requires a certain force to be applied through each TCR to allow conformational changes, and activation, downstream of TCR engagement.20,31,37,63,94,95 Interestingly, an intact actin cytoskeleton appears to be crucial for T cell–APC interactions; without actin, several receptors are unable to form the described micro-clusters, and protein rearrangement crucial for signalling cannot occur.59,62 Both F-actin and myosin IIA are thought to play a key role in the movement of cell surface proteins during TCR activation, and the dynamic polarisation/depolarisation of intracellular actin is key to the directional formation of the SMAC.59,61,68,69 Despite these observations, the nature of the dynamic relationship between applied mechanical force, cytoskeletal rearrangement, TCR stimulation and the level of downstream cell activation is not yet fully elucidated.96,97 In order to fully understand such effects, more complex interfaces that orthogonally vary ligand patterning and substrate mechanical properties are needed. Typically, silicon or glass materials have been used as underlying substrates for the fabrication of advanced micro- and nano-patterned interfaces, however neither material facilitates modulation of substrate mechanical properties in a biologically relevant range. The next section discusses biomechanical aspects of T cell biomaterial interface design, including the mechanical properties of biomaterial substrates.
To explore the role of rigidity on cellular activation, biomaterial surfaces with varied mechanical properties have been fabricated. Using planar poly(dimethyl siloxane) (PDMS) substrates functionalised with stimulatory anti-CD3 antibodies, maximal proliferation in CD4+ and CD8+ T cells has been observed in cells cultured on surfaces with a rigidity of <100 kPa compared to 2 MPa.30 However, other studies on poly(acrylamide) (PA) surfaces describe maximal T cell responses on substrates with rigidity of 200 kPa31 compared to softer substrates at 10 kPa. Separately, B cells showed preferential activation on PA gels at around 20 kPa (compared to softer 2 kPa substrates)98 and hematopoietic stem cells demonstrated increased cell spreading on much stiffer PA substrates100 of around 200 kPa compared to less rigid matrices. The differences in these results pose interesting questions on how cells sense rigidity, and whether various material chemistries may play a role. It has been suggested that cells may “feel” the rigidity of the chemical bond connecting the ligand of interest to the substrate109,110 rather than the rigidity of the substrate itself.
Force transduction by the TCR/CD3 complex has been investigated using flexible micropillars coated with anti-CD3 to measure forces applied by the cell to the substrate.111,112 These experiments showed that T cells apply forces via the TCR/CD3 complex to such bio-functionalized substrates, and implicitly therefore to APC. In contrast, CD28 was shown to affect force transduction only indirectly by influencing intracellular signalling, with no evidence of significant traction forces being applied through the CD28 receptor itself. TCR-mediated traction forces were of the order of 100 pN per 1 μm-diameter pillar, much smaller than the forces observed in integrin-mediated cell adhesion on similar substrates.
A plausible hypothesis to explain the mechanosensitivity of the T cell is that mechanically-induced conformational changes in TCR/CD3 are needed for activation. Such changes could be induced by cytoskeletal pulling forces.84 It is well known that cytoskeletal function is important to T cell function; surface bound triggering requires an intact cytoskeleton,28 and changes in the actin cytoskeleton compromise signalling in T cells as discussed earlier, supporting this hypothesis.37,59,62–64,113,114
In several receptor types, such as selectins, there is a transition between catch and slip bond systems at specific force values, implying that the cell can sense force over a dynamic range and that force can regulate conformational changes in these systems.105,106In vivo, these mechanisms are crucial for immune cell extravasation from blood vessels to allow migration into inflamed tissues. Immune cell extravasation is largely dependent on L- and E/P-selectin adhesion receptors, which allow leukocytes to first tether and roll along the activated endothelium and then firmly adhere before squeezing into the tissue through gaps between endothelial cells. Stabilisation of leukocyte adhesion to the blood vessel wall requires a minimum level of fluid shear stress in order to avoid inappropriate aggregation or adhesion outside the vasculature. This has been called catch–slip transition and explains how increasing levels of shear stress and thus force applied to a specific receptor–ligand pair, can stabilize leukocyte adhesion under flow.115–117
A similar mechanism could occur within the TCR, for example exposing cryptic binding sites downstream of the TCR/CD3 machinery that are force sensitive, using a similar mechanism to cellular force-sensing in the formation of focal adhesions.118,119 Additionally, the mechanical force applied through the TCR could correlate with viral or infectious load, so that the number of anchoring points and/or force loading provides the T cell with additional information about the status of an APC. Intriguingly, studies have demonstrated a link between the spatial distribution of proteins on T cell surfaces, the intrinsic mechanical properties of T cell, and the activation level of the cells.120
Importantly, a ‘receptor deformation model’ has been proposed, which provides an explanation for TCR triggering under physiological conditions, where the amount of one specific peptide available is likely too small to allow activation based on TCR crosslinking.94,95 The model suggests that the pulling force associated with T cell motility induces a TCR conformation that favours induction of downstream signalling. To fully explore these mechanisms, advanced materials that vary rigidity simultaneously with other variables (such as ligand chemical linkage and spatial nanopatterning) are required.
3-Dimensional interfaces
The natural T cell–APC interaction occurs in three dimensions (3D) between two cells which are themselves 3D objects. In addition to their overall shape, natural APCs and immune cell surface membranes exhibit a range of micro topographical features including invaginations, protrusions, and extending nanotubes.121,122 Given the inherently 3D nature of immune cells, and the wealth of 3D features present on their surfaces, more complex biomaterial interfaces are starting to be developed to explore the cellular response to altering these features. Here, we describe interfaces that replicate the 3D aspects of cell surface features, and move towards more complex systems.Using advanced microfabrication techniques, micro-topographical features have been patterned on planar substrates to explore their effects on T cell stimulation. Large micro-pits have been used to trap individual T cells122 and investigate the morphology of the APC interface, however studies into the effect of topography itself are extremely limited. Although the effect of surface topography on T cell behaviour has not been well studied, evidence from other immune cells suggests this may be an important parameter to consider in stimulatory biomaterial interface design. Neutrophil activation is enhanced on budding, as opposed to smooth, microparticles.123 Similarly, surface topography has been shown to induce phenotypic shifts towards distinct subtypes in macrophages; titanium surfaces with smooth or micro pitted features were able to induce macrophage differentiation towards an “inflammatory” or “regenerative” phenotype, respectively.124
A range of materials have been used to fabricate fully synthetic spherical constructs including polystyrene,127 PLGA,43 polymer emulsions,128 and synthetic liposomes.48,49 These materials are usually functionalized with proteins, either chemically linked or adsorbed onto the material surface. Importantly, both the size and shape of the material play an important role in cellular activation.13,126,129 Non-spherical, micro-scale ellipsoid shapes have been shown to enhance T cell activation when compared to their spherical129 counterparts, possibly due to changes in presented surface area or T cell membrane disruption. A range of studies have explored the role of particle size in both vaccine delivery strategies and, independently, phagocytosis of nanoparticles in several immune cell types.13,130 For T cells, it has previously been shown that nanoscale particles are less effective than their microscale counterparts in stimulating T cells towards activation.131,132 Particles of at least 4–5 μm diameter are thought to be required to provide the minimum contact area133 needed for full activation. The reasons for this are unclear, but are likely to include the need for microscale contact areas to enable receptor segregation (SMAC formation), or the inability of smaller interfaces to establish long-range mechanical coupling across distinct parts of the T cell as discussed in other sections of this review.
Spherical interfaces with diameters above this 4–5 μm threshold are therefore strong potential candidates for in vivo therapeutic applications, and indeed polystyrene beads bio-functionalized with anti-CD3 and anti-CD28 have been used to stimulate T cells in clinical trials.12,134 Moving to more flexible self-assembled materials, PEGylated liposomes have recently proven successful in targeting up to 95% of circulating adoptively transferred T cells48,49 offering great promise for future in vivo therapies using multicomponent systems that are able to engage selectively with target T cell sub-populations.
Interestingly, recent work in this area has outlined more advanced materials for stimulation in 3D; Janus particles135 presenting focular CD3 protein micropatterns have been used to stimulate T cells representing a move towards 3D materials with controlled surface patterning. The design of 3D interfaces that move from simple, single parameter interfaces towards more complex architectures that consider several parameters at a time will be the next step in T cell biomaterial design.
Dynamic interfaces
Natural T cell–APC interactions are dynamic in nature, with distinct events occurring over a timescale that starts with cellular stimulation in the first few seconds, followed by a complex cascade of activation events occurring over several hours.136–138 Fully interactive innate and adaptive immune responses often occur over several months, and in some cases, protective immune response can last a lifetime. The sequence, duration and nature of each step in the stimulation pathway can drastically affect cellular outcome. In moving to more advanced biomaterial interfaces, it will be important to develop materials that can dynamically control cellular behaviour. These materials should controllably alter their properties in response to externally applied stimuli, and therefore provide a range of cues to the cell that are conditioned to specific environments, effectively modulating T cell behaviour.An example includes the temporal delivery of stimulatory signals to T cells, explored using biodegradable polymers. Fig. 3A shows a schematic where polymer particles have been functionalized with anti-CD3 and anti-CD28, and modified to contain the cytokine IL2 that is either chemically linked to the particle surface or encapsulated within the polymer matrix and released as the polymer degrades.50
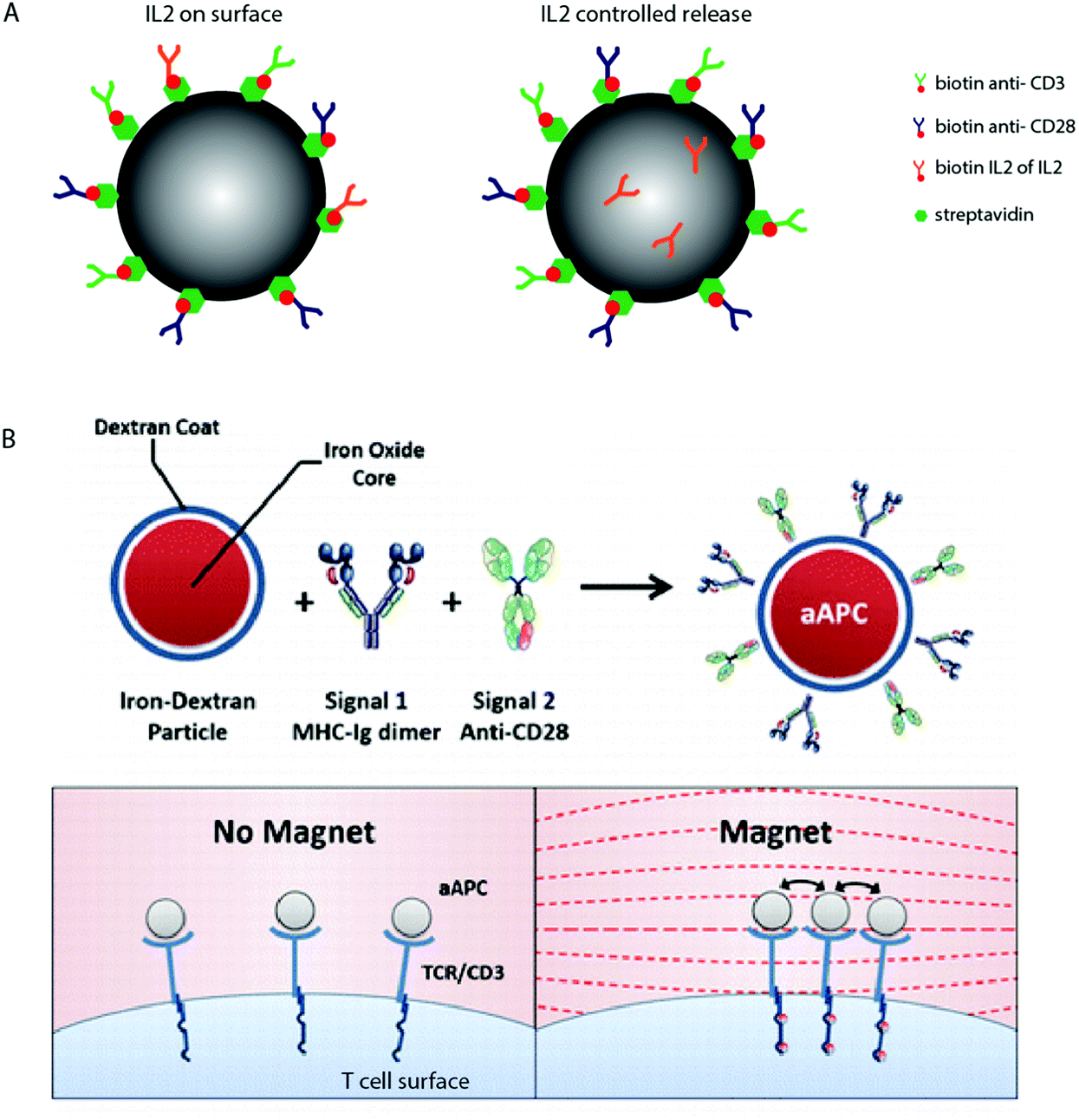 | ||
| Fig. 3 Dynamic T cell interfaces with APC-mimicking biomaterials. (A) Biodegradable PLGA particles have been used to deliver surface bound and soluble T cell signals, paracrine IL2 delivery seems to result in increased activation.50 (B) A schematic of paramagnetic nanoparticles functionalized with anti-CD3 and anti-CD28, which represent the move towards dynamic biomaterials that are able to induce T cell activation in response to specific stimuli. Copyright: (B) adapted with permission from ref. 51 Perica et al. ACS Nano, 2014, 8(3), 2252–2260, Copyright 2014 American Chemical Society. | ||
These materials have shown that paracrine release of IL2 results in enhanced T cell activation compared to materials that present IL2 directly on the surface. In silico experiments which model the release of protein and cytokines from biodegradable materials50,132 suggest that the kinetics of release may drastically affect cellular behaviour.
In other fields, the manipulation of material physical properties using external stimuli, such as temperature, pH, or UV light, has been widely explored as a tool to regulate kinetic drug dosing, ligand presentation and subsequent cell behaviour139–141 however these studies are limited in respect to T cell stimulatory biomaterial design. One notable example involves the manipulation of TCR nanoclusters using functionalized para-magnetic particles.51 These particles bind to TCR receptors on the cell surface, and naturally aggregate in an applied magnetic field. The aggregation of these particles induces TCR clustering, which leads to magnetically-induced cell activation, shown schematically in Fig. 3B. We anticipate that using a similar principle, TCR aggregation (and so activation) could be induced using a range of stimuli response biomaterials in future dynamic artificial immune synapse design.
Conclusions
The biomaterials described here exemplify a paradigm shift in biomaterial design away from traditional “immune inert” materials towards materials that are specifically designed to control, and perhaps elicit, specific immune cell behaviour. A range of factors controlling T cell behaviour, such as spatial patterning, substrate geometry and substrate mechanical properties have been identified and individually explored using increasingly complex static, responsive and dynamic biomaterial substrates. Table 1 highlights the breadth of materials that have already been fabricated to explore these parameters, but also indicates that there is huge scope for development of more complex materials capable of supporting artificial immune synapses, with many possible parameter combinations as yet unaddressed.The high level of interest in T cell biomaterial design arises not only from the fundamental importance of modulating T cell activation in human biology but also from the potential of these materials to contribute to current therapeutic aims. Determining the mechanisms that underpin phenotype specific induced activation pathways will have important implications in future immunotherapeutic design. The long-term aim is for advanced biomaterial interfaces to facilitate the generation or delivery of precisely-controlled T cell populations of desired phenotypes for each specific therapeutic application.
Static biomaterial interfaces, including nanopatterned and rigidity-controlled substrates, present precisely-controllable stimuli designed to induce a specific response in T cells, and have delivered fundamental insights into the thresholds for different parameters to stimulate T cells. Observations from these works highlight that the nano- and micro-patterned distribution of TCR/CD3 ligands on stimulatory surfaces play a key role in T cell activation, with signalling thresholds directly related to the precise positioning of TCR/CD3 complexes and co-stimulatory receptors.
Other studies have focused on the role of substrate mechanical properties in controlling T cell activation. These studies are limited in number, but indicate that force appears to affect activation through the TCR/CD3 complex. It is important to note that many of the variables investigated, including ligand–receptor spacing and protein patterning, may be intrinsically linked to adhesion force. To fully understand the importance of these parameters, more complex biomaterials will be needed to isolate and re-combine them, allowing an exploration of the relative contributions of each of these factors.
Responsive materials present stimuli to cells, but can be remodelled by the responding cells to allow modulation of behaviour driven by the cell. An important example is the fluid lipid bilayer system, which has provided a wealth of information about the rearrangement of ligands following activation. A variety of ligand protein distribution sizes and patterns have been observed on the cell both before and during activation. Moving forward in the design of immune cell–biomaterial interfaces, it will be important to understand the nature of these differences, and whether they may be due to the behaviour of distinct T cell phenotypes.
Finally, dynamic materials are those that respond to external stimuli to induce a specific state in the cell only under certain conditions. Research on dynamic biomaterials for the manipulation of immune cell behaviour is still in its infancy, but offers great promise, using varying kinetic and stimuli responsive biomaterials to fine tune immune responses and potentially enable localized, rather than systemic, control of immune behaviour. Coupled with cell engineering strategies, these materials could enable a more complex temporal and spatial regulation of immune cell behaviour to deliver individually engineered immune therapies.
Acknowledgements
DD acknowledges an EPRSC E-TERM Landscape Fellowship scheme (EP/I017801/1). SS was supported by the BHF Cardiovascular Regenerative Medicine Centre (RM/13/1/30157 to Prof. Sian E. Harding).References
- Z. Sheikh, et al., Macrophages, foreign body giant cells and their response to implantable biomaterials, Materials, 2015, 8(9), 5671–5701 CrossRef.
- S. Jhunjhunwala, et al., Neutrophil responses to sterile implant materials, PLoS One, 2015, 10(9), e0137550 Search PubMed.
- A. J. Vegas, et al., Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates, Nat. Biotechnol., 2016, 34(3), 345 CrossRef CAS PubMed.
- T. Bergler, et al., Infiltration of Macrophages Correlates with Severity of Allograft Rejection and Outcome in Human Kidney Transplantation, PLoS One, 2016, 11, 6 Search PubMed.
- S. Goral, The three-signal hypothesis of lymphocyte activation/targets for immunosuppression, Dial. Transplant., 2011, 40(1), 14–16 CrossRef.
- F. C. Edozie, et al., Regulatory T-Cell therapy in the induction of transplant tolerance: The issue of subpopulations, Transplantation, 2014, 98(4), 370–379 CrossRef CAS PubMed.
- J. I. Andorko, K. L. Hess and C. M. Jewell, Harnessing biomaterials to engineer the lymph node microenvironment for immunity or tolerance, AAPS J., 2015, 17(2), 323–338 CrossRef CAS PubMed.
- N. A. Hotaling, et al., Biomaterial strategies for immunomodulation, in Annu. Rev. Biomed. Eng., ed. M. L. Yarmush, 2015, Annual Reviews, Palo Alto, pp. 317–349 Search PubMed.
- P. S. Hume, et al., Strategies to reduce dendritic cell activation through functional biomaterial design, Biomaterials, 2012, 33(14), 3615–3625 CrossRef CAS PubMed.
- C. H. June, B. R. Blazar and J. L. Riley, Engineering lymphocyte subsets: tools, trials and tribulations, Nat. Rev. Immunol., 2009, 9, 704–716 CrossRef CAS PubMed.
- M. C. Milone and C. H. June, Adoptive immunotherapy: New ways to skin the cat?, Clin. Immunol., 2005, 117, 101–103 CrossRef CAS PubMed.
- D. L. Porter, et al., Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia, Sci. Transl. Med., 2015, 7(303), 12 Search PubMed.
- D. J. Irvine, et al., Synthetic Nanoparticles for Vaccines and Immunotherapy, Chem. Rev., 2015, 115(19), 11109–11146 CrossRef CAS PubMed.
- J. Lee, et al., T Cell-Specific siRNA Delivery Using Antibody-Conjugated Chitosan Nanoparticles, Bioconjugate Chem., 2012, 23(6), 1174–1180 CrossRef CAS PubMed.
- M. T. Stephan, et al., Therapeutic cell engineering with surface-conjugated synthetic nanoparticles, Nat. Med., 2010, 16(9), 1035–U135 CrossRef CAS PubMed.
- M. T. Stephan, et al., Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles, Biomaterials, 2012, 33(23), 5776–5787 CrossRef CAS PubMed.
- C. R. F. Monks, et al., Three-dimensional segregation of supramolecular activation clusters in T cells, Nature, 1998, 395(6697), 82–86 CrossRef CAS PubMed.
- A. Grakoui, et al., The immunological synapse: a molecular machine controlling T cell activation, Science, 1999, 285(5425), 221–227 CrossRef CAS PubMed.
- D. M. Davis and M. L. Dustin, What is the importance of the immunological synapse?, Trends Immunol., 2004, 25(6), 323–327 CrossRef CAS PubMed.
- K. Perica, A. K. Kosmides and J. P. Schneck, Linking form to function: Biophysical aspects of artificial antigen presenting cell design, Biochim. Biophys. Acta, 2015, 1853(4), 781–790 CrossRef CAS PubMed.
- C. G. Fathman and N. B. Lineberry, Molecular mechanisms of CD4(+) T-cell anergy, Nat. Rev. Immunol., 2007, 7, 599–609 CrossRef CAS PubMed.
- G. P. Morris and P. M. Allen, How the TCR balances sensitivity and specificity for the recognition of self and pathogens, Nat. Immunol., 2012, 13(2), 121–128 CrossRef CAS PubMed.
- L. P. Kane, J. Lin and A. Weiss, Signal transduction by the TCR for antigen, Curr. Opin. Immunol., 2000, 12(3), 242–249 CrossRef CAS PubMed.
- K. C. Garcia, et al., An alpha beta T cell receptor structure at 2.5 angstrom and its orientation in the TCR-MHC complex, Science, 1996, 274(5285), 209–219 CrossRef CAS PubMed.
- Y. Sykulev, N. Anikeeva and D. Gakamsky, The role of peptide-MHC ligand density in stimulating T-cell receptor signaling, J. Immunol., 2012, 188(Suppl. 1), 58.21 Search PubMed.
- N. Anikeeva, et al., Evidence that the Density of Self Peptide-MHC Ligands Regulates T-Cell Receptor Signaling, PLoS One, 2012, 7(8), e41466 CAS.
- J. D. Stone and L. J. Stern, CD8 T cells, like CD4 T cells, are triggered by multivalent engagement of TCRs by MHC-Peptide ligands but not by monovalent engagement, J. Immunol., 2006, 176, 1498–1505 CrossRef CAS.
- Z. Ma, K. A. Sharp, P. A. Janmey and T. H. Finkel, Surface- anchored monomeric agonist pMHC's alone trigger TCR with high sensitivity, PLoS Biol., 2008, 6(2), 328–342 CAS.
- G. P. O'Donoghue, et al., Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells, eLife, 2013, 2, e00778 Search PubMed.
- R. S. O'Connor, et al., Substrate rigidity regulates human T cell activation and proliferation, J. Immunol., 2012, 189(3), 1330–1339 CrossRef PubMed.
- E. Judokusumo, et al., Mechanosensing in T lymphocyte activation, Biophys. J., 2012, 102(2), L5–L7 CrossRef CAS PubMed.
- J. Tsai, et al., Microscale colocalization of CD3 and CD28 is required for activation of human CD4 + T Cells, Biophys. J., 2010, 98(3), 406A CrossRef.
- K. Shen, et al., Self-Aligned Supported Lipid Bilayers for Patterning the Cell-Substrate Interface, J. Am. Chem. Soc., 2009, 131(37), 13204–13205 CrossRef CAS PubMed.
- K. Shen, et al., Micropatterning of costimulatory ligands enhances CD4+ T cell function, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(22), 7791–7796 CrossRef CAS PubMed.
- J.-H. Lee, M. Dustin and L. Kam, Micron-scale exclusion of CD45 enhances activation of TCR zeta associated protein of 70 kDa tyrosine kinase, J. Immunol., 2013, 190, P1192 CrossRef PubMed.
- D. Dutta and L. C. Kam, Micropatterned, Multicomponent Supported Lipid Bilayers for Cellular Systems, in Micropatterning in Cell Biology, Pt B., ed. M. Piel and M. Thery, 2014, pp. 53–67 Search PubMed.
- W. Chen and C. Zhu, Mechanical regulation of T-cell functions, Immunol. Rev., 2013, 256(1), 160–176 CrossRef CAS PubMed.
- K. T. Bashour, et al., Cross talk between CD3 and CD28 is spatially modulated by protein lateral mobility, Mol. Cell. Biol., 2014, 34(6), 955–964 CrossRef PubMed.
- D. Delcassian, et al., Nanoscale ligand spacing influences receptor triggering in T cells and NK cells, Nano Lett., 2013, 13(11), 5608–5614 CrossRef CAS PubMed.
- J. Matic, et al., Fine tuning and efficient T cell activation with stimulatory aCD3 nanoarrays, Nano Lett., 2013, 13(11), 5090–5097 CrossRef CAS PubMed.
- J. Deeg, et al., T cell activation is determined by the number of presented antigens, Nano Lett., 2013, 13(11), 5619–5626 CrossRef CAS PubMed.
- H. Cai, et al., Probing the minimum geometric requirements for T-cell stimulation, Biophys. J., 2015, 108(2), 631A CrossRef.
- S. L. Demento, et al., Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype, Biomaterials, 2012, 33(19), 4957–4964 CrossRef CAS PubMed.
- N. Anikeeva, et al., Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(45), 16846–16851 CrossRef CAS PubMed.
- R. S. O'Connor, et al., Substrate Rigidity Regulates Human T Cell Activation and Proliferation, J. Immunol., 2012, 189(3), 1330–1339 CrossRef PubMed.
- K. D. Mossman, et al., Altered TCR signaling from geometrically repatterned immunological synapses, Science, 2005, 310(5751), 1191–1193 CrossRef CAS PubMed.
- J. Doh and D. J. Irvine, Immunological synapse arrays: Patterned protein surfaces that modulate immunological synapse structure formation in T cells, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(15), 5700–5705 CrossRef CAS PubMed.
- Z. Liu, et al., Ganoderma lucidum polysaccharides encapsulated in liposome as an adjuvant to promote Th1-bias immune response, Carbohydr. Polym., 2016, 142, 141–148 CrossRef CAS PubMed.
- Y. Zheng, et al., In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes, J. Controlled Release, 2013, 172(2), 426–435 CrossRef CAS PubMed.
- E. R. Steenblock, et al., An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response, J. Biol. Chem., 2011, 286(40), 34883–34892 CrossRef CAS PubMed.
- K. Perica, et al., Magnetic field-induced T cell receptor clustering by nanoparticles enhances T cell activation and stimulates antitumor activity, ACS Nano, 2014, 8(3), 2252–2260 CrossRef CAS PubMed.
- K. Perica, et al., Enrichment and expansion with nanoscale artificial antigen presenting cells for adoptive immunotherapy, ACS Nano, 2015, 9(7), 6861–6871 CrossRef CAS PubMed.
- J. Xie, C. M. Tato and M. M. Davis, How the immune system talks to itself: the varied role of synapses, Immunol. Rev., 2013, 251(1), 65–79 CrossRef PubMed.
- S. Valitutti, D. Coombs and L. Dupre, The space and time frames of T cell activation at the immunological synapse, FEBS Lett., 2010, 584(24), 4851–4857 CrossRef CAS PubMed.
- M. L. Dustin and D. Depoil, New insights into the T cell synapse from single molecule techniques, Nat. Rev. Immunol., 2011, 11(10), 672–684 CrossRef CAS PubMed.
- T. J. Crites, L. Chen and R. Varma, A TIRF microscopy technique for real-time, simultaneous imaging of the TCR and its associated signaling proteins, J. Visualized Exp., 2012,(61), e3892 Search PubMed.
- B. Alarcon, et al., Initiation of TCR signaling: regulation within CD3 dimers, Immunol. Rev., 2003, 191(1), 38–46 CrossRef CAS PubMed.
- T. Yokosuka, et al., Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76, Nat. Immunol., 2005, 6(12), 1253–1262 CrossRef CAS PubMed.
- Y. Yu, A. A. Smoligovets and J. T. Groves, Modulation of T cell signaling by the actin cytoskeleton, J. Cell Sci., 2013, 126(5), 1049–1058 CrossRef CAS PubMed.
- R. Varma, et al., T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster, Immunity, 2006, 25(1), 117–127 CrossRef CAS PubMed.
- S. Valensin, et al., F-actin dynamics control segregation of the TCR signaling cascade to clustered lipid rafts, Eur J. Immunol., 2002, 32(2), 435–446 CrossRef CAS PubMed.
- A. S. Sechi and J. Wehland, Interplay between TCR signalling and actin cytoskeleton dynamics, Trends Immunol., 2004, 25(5), 257–265 CrossRef CAS PubMed.
- E. Klotzsch, et al., Do mechanical forces contribute to nanoscale membrane organisation in T cells?, Biochim. Biophys. Acta, 2015, 1853(4), 822–829 CrossRef CAS PubMed.
- J. Dinic, P. Ashrafzadeh and I. Parmryd, Actin filaments attachment at the plasma membrane in live cells cause the formation of ordered lipid domains, Biochim. Biophys. Acta, 2013, 1828(3), 1102–1111 CrossRef CAS PubMed.
- G. Campi, R. Varma and M. L. Dustin, Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling, J. Exp. Med., 2005, 202(8), 1031–1036 CrossRef CAS PubMed.
- J. C. Sunshine and J. J. Green, Nanoengineering approaches to the design of artificial antigen-presenting cells, Nanomedicine, 2013, 8(7), 1173–1189 CrossRef CAS PubMed.
- A. M. Beal, et al., Kinetics of Early T Cell Receptor Signaling Regulate the Pathway of Lytic Granule Delivery to the Secretory Domain, Immunity, 2009, 31(4), 632–642 CrossRef CAS PubMed.
- W. Lin, et al., Morphological change of CD4(+) T cell during contact with DC modulates T-cell activation by accumulation of F-actin in the immunology synapse, BMC Immunol., 2015, 16, 15 CrossRef PubMed.
- Y. Yu, et al., Myosin IIA Modulates T Cell Receptor Transport and CasL Phosphorylation during Early Immunological Synapse Formation, PLoS One, 2012, 7(2), e30704 CAS.
- T. Lohmuller, et al., Supported membranes embedded with fixed arrays of gold nanoparticles, Nano Lett., 2011, 11(11), 4912–4918 CrossRef CAS PubMed.
- S. Mandal, et al., Polymer-based synthetic dendritic cells for tailoring robust and multifunctional T cell responses, ACS Chem. Biol., 2015, 10(2), 485–492 CrossRef CAS PubMed.
- D. J. Irvine, J. Doh and B. Huang, Patterned surfaces as tools to study ligand recognition and synapse formation by T cells, Curr. Opin. Immunol., 2007, 19(4), 463–469 CrossRef CAS PubMed.
- D. J. Irvine and J. Doh, Synthetic surfaces as artificial antigen presenting cells in the study of T cell receptor triggering and immunological synapse formation, Semin. Immunol., 2007, 19(4), 245–254 CrossRef CAS PubMed.
- T. J. Thauland and D. C. Parker, Diversity in immunological synapse structure, Immunology, 2010, 131(4), 466–472 CrossRef CAS PubMed.
- T. J. Thauland, et al., Th1 and Th2 cells form morphologically distinct immunological synapses, J. Immunol., 2008, 181(1), 393–399 CrossRef CAS.
- J.-H. Lee, M. L. Dustin and L. C. Kam, A microfluidic platform reveals differential response of regulatory T cells to micropatterned costimulation arrays, Integr. Biol., 2015, 7(11), 1442–1453 RSC.
- J. L. Zhang, et al., Novel method to load nanoparticles into mesoporous materials: Impregnation of MCM-41 with ZnS by compressed CO(2), Langmuir, 2003, 19(18), 7616–7620 CrossRef CAS.
- P. Sharma, et al., Nanoscale organization of multiple GPI-anchored proteins in living cell membranes, Cell, 2004, 116(4), 577–589 CrossRef CAS PubMed.
- J. Rossy, et al., Conformational states of the kinase Lck regulate clustering in early T cell signaling, Nat. Immunol., 2013, 14(1), 82–89 CrossRef CAS PubMed.
- A. Hashimoto-Tane and T. Saito, Dynamic Regulation of TCR-Microclusters and the Microsynapse for T Cell Activation, Front. Immunol., 2016, 7, 8 Search PubMed.
- B. F. Lillemeier, et al., TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation, Nat. Immunol., 2010, 11(1), 90–96 CrossRef CAS PubMed.
- D. J. Williamson, et al., Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events, Nat. Immunol., 2011, 12(7), 655–662 CrossRef CAS PubMed.
- B. Treanor, et al., Microclusters of inhibitory killer immunoglobulin like receptor signaling at natural killer cell immunological synapses, J. Cell Biol., 2006, 174(1), 153–161 CrossRef CAS PubMed.
- S. Minguet, et al., Full activation of the T cell receptor requires both clustering and conformational changes at CD3, Immunity, 2007, 26(1), 43–54 CrossRef CAS PubMed.
- M. S. Fassett, et al., Signaling at the inhibitory natural killer cell immune synapse regulates lipid raft polarization but not class I MHC clustering, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(25), 14547–14552 CrossRef CAS PubMed.
- D. Depoil, et al., CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand, Nat. Immunol., 2008, 9(1), 63–72 CrossRef CAS PubMed.
- H. A. Anderson, E. M. Hiltbold and P. A. Roche, Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation, Nat. Immunol., 2000, 1(2), 156–162 CrossRef CAS PubMed.
- H. A. Anderson and P. A. Roche, A role for membrane rafts in antigen presentation by MHC class II molecules, FASEB J., 2000, 14(6), A1195 Search PubMed.
- T. R. Fadel, et al., A carbon nanotube-polymer composite for T-cell therapy, Nat. Nanotechnol., 2014, 9(8), 639–647 CrossRef CAS PubMed.
- T. R. Fadel, et al., Clustering of Stimuli on Single-Walled Carbon Nanotube Bundles Enhances Cellular Activation, Langmuir, 2010, 26(8), 5645–5654 CrossRef CAS PubMed.
- T. R. Fadel, et al., Adsorption of multimeric T cell antigens on carbon nanotubes: Effect on protein structure and antigen-specific T cell stimulation, Small, 2013, 9(5), 666–672 CrossRef CAS PubMed.
- H. A. van den Berg and D. A. Rand, Dynamics of T cell activation threshold tuning, J. Theor. Biol., 2004, 228(3), 397–416 CrossRef CAS PubMed.
- A. Kaplan, et al., Simulations of the NK cell immune synapse reveal that activation thresholds can be established by inhibitory receptors acting locally, J. Immunol., 2011, 187(2), 760–773 CrossRef CAS PubMed.
- Z. Y. Ma, P. A. Janmey and T. H. Finkel, The receptor deformation model of TCR triggering, FASEB J., 2008, 22(4), 1002–1008 CrossRef CAS PubMed.
- Z. Y. Ma and T. H. Finkel, T cell receptor triggering by force, Trends Immunol., 2010, 31(1), 1–6 CrossRef CAS PubMed.
- P. Beemiller and M. F. Krummel, Regulation of T-cell receptor signaling by the actin cytoskeleton and poroelastic cytoplasm, Immunol. Rev., 2013, 256(1), 148–159 CrossRef CAS PubMed.
- W. A. Comrie and J. K. Burkhardt, Action and Traction: Cytoskeletal Control of Receptor Triggering at the Immunological Synapse, Front. Immunol., 2016, 7, 1–25 Search PubMed.
- Z. Wan, et al., B cell activation is regulated by the stiffness properties of the substrate presenting the antigens, J. Immunol., 2013, 190(9), 4661–4675 CrossRef CAS PubMed.
- C. A. Muth, et al., Regulation of Hematopoietic Stem Cell Behavior by the Nanostructured Presentation of Extracellular Matrix Components, PLoS One, 2013, 8(2), e54778 CAS.
- J. S. Choi and B. A. C. Harley, The combined influence of substrate elasticity and ligand density on the viability and biophysical properties of hematopoietic stem and progenitor cells, Biomaterials, 2012, 33(18), 4460–4468 CrossRef CAS PubMed.
- A. J. Engler, H. L. Sweeney and D. E. Discher, Substrate elasticity alters human mesenchymal stem cell differentiation, Biophys. J., 2005, 88(1), 500A Search PubMed.
- A. J. Engler, et al., Matrix elasticity directs stem cell lineage specification, Cell, 2006, 126(4), 677–689 CrossRef CAS PubMed.
- K. Konstantopoulos, W. D. Hanley and D. Wirtz, Receptor-ligand binding: 'Catch' bonds finally caught., Curr. Biol., 2003, 13(15), R611–R613 CrossRef CAS PubMed.
- B. T. Marshall, et al., Direct observation of catch bonds involving cell-adhesion molecules, Nature, 2003, 423(6936), 190–193 CrossRef CAS PubMed.
- T. Yago, et al., Catch bonds govern adhesion through L-selectin at threshold shear, J. Cell Biol., 2004, 166(6), 913–923 CrossRef CAS PubMed.
- V. A. Barsegov, Two conformational state model for P-selectin catch-slip bond character, Biophys. J., 2004, 86(1), 152A CrossRef.
- G. Plesa, et al., TCR affinity and specificity requirements for human regulatory T-cell function, Blood, 2012, 119(15), 3420–3430 CrossRef CAS PubMed.
- A. Varela-Rohena, et al., Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor, Nat. Med., 2008, 14(12), 1390–1395 CrossRef CAS PubMed.
- B. Trappmann, et al., Extracellular-matrix tethering regulates stem-cell fate, Nat. Mater., 2012, 11(7), 642–649 CrossRef CAS PubMed.
- V. Vogel and M. P. Sheetz, Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways, Curr. Opin. Immunol., 2009, 21(1), 38–46 CrossRef CAS PubMed.
- S. Ghassemi, et al., Gold-tipped elastomeric pillars for cellular mechanotransduction, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2009, 27(6), 3088–3091 CrossRef CAS PubMed.
- S. Ghassemi, et al., Fabrication of elastomer pillar arrays with modulated stiffness for cellular force measurements, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2008, 26(6), 2549–2553 CAS.
- L. J. Standeven, et al., The actin cytoskeleton controls the efficiency of killer Ig-like receptor accumulation at inhibitory NK cell immune synapses, J. Immunol., 2004, 173(9), 5617–5625 CrossRef CAS.
- L. C. Santos, et al., Rigidity sensing in T cells by actin-dependent phosphorylation of Src family kinase substrate Cas-L, Mol. Biol. Cell, 2012, 23 Search PubMed , abstract no. 2031.
- M. B. Lawrence, et al., Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E), J. Cell Biol., 1997, 136(3), 717–727 CrossRef CAS PubMed.
- E. B. Finger, et al., Adhesion through L-selectin requires a threshold hydrodynamic shear, Nature, 1996, 379(6562), 266–269 CrossRef CAS PubMed.
- R. Alon, et al., Interactions through L-selectin between leukocytes and adherent leukocytes nucleate rolling adhesions on selectins and VCAM-1 in shear flow, J. Cell Biol., 1996, 135(3), 849–865 CrossRef CAS PubMed.
- B. Geiger, A role for p130Cas in mechanotransduction, Cell, 2006, 127(5), 879–881 CrossRef CAS PubMed.
- Y. Sawada, et al., Force sensing by mechanical extension of the Src family kinase substrate p130Cas, Cell, 2006, 127(5), 1015–1026 CrossRef CAS PubMed.
- Y. Sun, et al., Phosphatidylinositol (4,5) Bisphosphate Controls T Cell Activation by Regulating T Cell Rigidity and Organization, PLoS One, 2011, 6(11), e27227 CAS.
- A. Chauveau, et al., Membrane nanotubes facilitate long-distance interactions between natural killer cells and target cells, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(12), 5545–5550 CrossRef CAS PubMed.
- M. J. P. Biggs, et al., High-resolution imaging of the immunological synapse and T-cell receptor microclustering through microfabricated substrates, J. R. Soc., Interface, 2011, 8(63), 1462–1471 CrossRef CAS PubMed.
- C. A. Vaine, et al., Tuning innate immune activation by surface texturing of polymer microparticles: The role of shape in inflammasome activation, J. Immunol., 2013, 190(7), 3525–3532 CrossRef CAS PubMed.
- K. M. Hotchkiss, et al., Titanium surface characteristics, including topography and wettability, alter macrophage activation, Acta Biomater., 2016, 31, 425–434 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll and L. Zhang, Nano-immunoengineering: Nanoparticle-based manipulation of antigen-presenting cells for cancer immunotherapy, Small, 2015, 11(41), 5466 CrossRef.
- R. A. Meyer, et al., Immunoengineering: biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation, Small, 2015, 11(13), 1612 CrossRef.
- J. Curtsinger, et al., Artificial cell surface constructs for studying receptor-ligand contributions to lymphocyte activation, J. Immunol. Methods, 1997, 209(1), 47–57 CrossRef CAS PubMed.
- I. Platzman, J.-W. Janiesch and J. P. Spatz, Synthesis of Nanostructured and Biofunctionalized Water-in-Oil Droplets as Tools for Homing T Cells, J. Am. Chem. Soc., 2013, 135(9), 3339–3342 CrossRef CAS PubMed.
- J. C. Sunshine, et al., Particle shape dependence of CD8 + T cell activation by artificial antigen presenting cells, Biomaterials, 2014, 35(1), 269–277 CrossRef CAS PubMed.
- M. O. Oyewumi, A. Kumar and Z. Cui, Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses, Expert Rev. Vaccines, 2010, 9(9), 1095–1107 CrossRef CAS PubMed.
- E. R. Steenblock, et al., Antigen presentation on artificial acellular substrates: modular systems for flexible, adaptable immunotherapy, Expert Opin. Biol. Ther., 2009, 9(8), 1115 CrossRef.
- M. Labowsky, J. Lowenthal and T. M. Fahmy, An in silicoanalysis of nanoparticle/cell diffusive transfer: Application to nano-artificial antigen-presenting cell:T-cell interaction, Nanomedicine, 2015, 11(4), 1019–1028 CAS.
- M. F. Mescher, Surface-contact requirements for activation of cytotoxic lymphocytes-T, J. Immunol., 1992, 149(7), 2402–2405 CAS.
- S. A. Grupp, et al., Chimeric antigen receptor-modified T cells for acute lymphoid leukemia, N. Engl. J. Med., 2013, 368(16), 1509–1518 CrossRef CAS PubMed.
- B. Chen, et al., Janus particles as artificial antigen-presenting cells for T cell activation, ACS Appl. Mater. Interfaces, 2014, 6(21), 18435–18439 CAS.
- G. Iezzi, D. Scheidegger and A. Lanzavecchia, Migration and function of antigen-primed nonpolarized T lymphocytes in vivo, J. Exp. Med., 2001, 193(8), 987–993 CrossRef CAS PubMed.
- P. A. Gonzalez, et al., T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(13), 4824–4829 CrossRef CAS PubMed.
- R. V. Blanden, T-cell response to viral and bacterial infection, Transplant. Rev., 1974, 19, 56–88 CAS.
- J.-W. Yoo, N. Doshi and S. Mitragotri, Adaptive micro and nanoparticles: Temporal control over carrier properties to facilitate drug delivery, Adv. Drug Delivery Rev., 2011, 63(14-15), 1247–1256 CrossRef CAS PubMed.
- M. Wirkner, et al., Photoactivatable Caged Cyclic RGD Peptide for Triggering Integrin Binding and Cell Adhesion to Surfaces, ChemBioChem, 2011, 12(17), 2623–2629 CrossRef CAS PubMed.
- C. A. Custodio, et al., Photopatterned Antibodies for Selective Cell Attachment, Langmuir, 2014, 30(33), 10066–10071 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |